
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dually sensitive dextran-based micelles for methotrexate delivery†
B. Blanco-Fernandez‡
 *ab,
A. Concheiroa,
H. Makwanab,
F. Fernandez-Trillo§
b,
C. Alexanderb and
C. Alvarez-Lorenzo*a
*ab,
A. Concheiroa,
H. Makwanab,
F. Fernandez-Trillo§
b,
C. Alexanderb and
C. Alvarez-Lorenzo*a
aDepartamento de Farmacia y Tecnología Farmacéutica, R+DPharma Group (GI-1645), Facultad de Farmacia, Universidade de Santiago de Compostela, 15782 Santiago de Compostela, Spain. E-mail: carmen.alvarez.lorenzo@usc.es; Web: barbara.blanco.fernandez@gmail.com
bSchool of Pharmacy, University of Nottingham, University Park, Boots Science Building, Nottingham NG7 2RD, UK
First published on 6th March 2017
Abstract
Temperature-sensitive polymeric micelles were prepared from dextran grafted with poly(N-isopropylacrylamide) (PNIPAAm) or polyethylene glycol methyl ether (PEGMA) via controlled radical polymerization and evaluated as delivery systems of the anticancer drug methotrexate (MTX). Polymer-grafting was carried out after introduction of initiating groups onto the polysaccharide backbone, without the need for protection of hydroxyl groups and avoiding the use of toxic solvents. Temperature-responsive dextran-based copolymers were designed to exhibit self-aggregation behaviour, affinity for MTX and high cellular internalization. In addition, some grafted polymers incorporated 2-aminoethyl methacrylate to reinforce MTX encapsulation in the micelles by means of ionic interactions. Dextran-based micelles were cytocompatible and had an appropriate size to be used as drug carriers. MTX release was dependent on the pH and temperature. The combination of poly(2-aminoethylmethacrylate) and PNIPAAm with the dextran backbone permitted the complete release of MTX at normal physiological temperature. Co-polymer micelles were highly internalized by tumour cells (CHO-K1) and, when loaded with MTX, led to enhanced cytotoxicity compared to the free drug.
Introduction
Methotrexate (MTX) is one of the most effective drugs in the treatment of a variety of solid tumours, hematologic malignancies, psoriasis and rheumatoid arthritis.1 This folic acid analogue inhibits dihydrofolate reductase and other folate-dependent enzymes, resulting in the interruption of DNA and RNA synthesis and adenosine overproduction. Importantly, MTX exploits folate-receptors to allow absorption across the gut after oral administration and for intracellular accumulation in tumour tissue. However, folate-receptors can become saturated before the uptake of a required therapeutic dose.2 This problem, together with certain side effects, such as bone marrow suppression, hepatotoxicity, leukopenia and nephrotoxicity, have prompted the search for nanocarriers that use other pathways for cell internalization and which selectively deliver MTX to target cells. Nanoemulsions,3 nanoparticles,4 liposomes,5 and polymeric micelles6,7 have been tested as suitable MTX delivery systems. If adequately designed, polymeric micelles can encapsulate large amounts of poorly soluble drugs,8–11 prevent premature leakage in the blood stream,12–15 and facilitate accumulation in tumour and inflamed tissues as a consequence of the enhanced permeation and retention (EPR) effect.16–18 Furthermore, amphiphilic copolymers can be adapted with stimuli-responsiveness to afford better control of drug loading and release processes.15,19,20 Established tumour tissues are in general characterized by a decrease in pH, an increase in glutathione levels, and a slightly higher temperature than healthy tissues, which can be exploited for enhanced delivery of anticancer drugs.21,22Hybrid materials that combine natural and synthetic polymers are receiving increasing attention as components of drug and gene delivery systems. Polysaccharides are good candidates for biomaterials due to their biocompatibility, solubility in water, degradability in the environment, low cost and availability from renewable sources.23 However, not all polysaccharides are readily amenable for systemic drug delivery owing to a lack of affinity for drug molecules or functionality for amphiphile formation. Growing a synthetic polymer which is reversibly amphiphilic from a polysaccharide backbone potentially enables formation of much more versatile drug carriers. Controlled radical chemistries, such as atom transfer radical polymerization (ATRP), reversible addition–fragmentation chain transfer (RAFT), nitroxide-mediated polymerization (NMP) or cyanoxyl-mediated free radical polymerization, allows tuneable and repeatable preparation of a variety of amphiphilic derivatives of polysaccharides.24 As a polysaccharide core dextran is a highly suitable candidate for chemical derivatization, as it is biocompatible and is already used as a plasma volume expander and antithrombotic agent. Dextran consists of glucose units linked through α-1,6 bonds with some α-1,2, α-1,3 and α-1,4 modifications, providing numerous hydroxyl groups for derivatization. Hydrophobic chains,25–27 stimuli-sensitive chains/groups28–31 and a number of drugs32 have been grafted to dextran to obtain self-assembling derivatives. So far, attempts to apply controlled radical polymerisation for the preparation of amphiphilic copolymers of dextran have required the use of toxic solvents or the protection of specific hydroxyl groups to introduce initiating groups onto the polysaccharide backbone.33–35 For example, synthesis of poly(N-isopropylacrylamide) (PNIPAAm)-graft dextran involved dimethylformamide and LiCl.36 RAFT-grafting of poly(ethylene glycol) methyl ether methacrylate (PEGMA) onto dextran has been explored to obtain micelles able to encapsulate doxorubicin for treatment of neuroblastoma cancer cells.29 Copolymers of dextran grafted with poly(lactobionamidoethyl methacrylate) and di(ethylene glycol) methyl ether methacrylate were able to form micelles which recognized a lectin for targeted delivery.37
In the present work, PNIPAAm-grafted dextran and PEGMA-grafted dextran copolymers were synthesised using controlled radical polymerisation following the introduction of initiating groups into the polysaccharide backbone without the protection of hydroxyl groups and under mild conditions. The final aim of the work was to obtain temperature-responsive copolymers that exhibit self-aggregation features, affinity for MTX and high cellular internalization. PNIPAAm was chosen due to its widely-used lower critical solution temperature (LCST) properties in aqueous medium (close to 32 °C).38 For comparison, PEGMA macromonomers were selected to enable the preparation of more cytocompatible copolymers with tuneable LCST by combining macromonomers of different molar masses.39,40 2-Aminoethyl methacrylate (AEM) was also copolymerized as a way to endow the copolymers with enhanced affinity for MTX in order to overcome the low loading content (LC 2–5%) that is commonly observed in polymeric micelles for this drug.15,41,42 Moreover, polyAEM components were also expected to enhance cellular uptake due to the presence of positive charges in the polymer chains. The obtained polymeric micelles were characterized regarding their suitability as MTX delivery systems, critical micellar concentration, micelle size, surface charge and cytocompatibility. The effects of temperature and pH on drug release were evaluated, and the effectiveness of the MTX-loaded micelles and their cellular internalization in cancer cell lines were investigated.
Materials and methods
Materials
4-(Dimethylamino) pyridine (DMAP) was obtained from Fluka (China). L-Ascorbic acid sodium salt (99%) and methotrexate (MTX) were from Acros Organics (Belgium) and AK Scientific (USA) respectively. Dextran from Leuconostoc spp. (40 kDa), dimethyl sulfoxide anhydrous (DMSO), phosphate buffer saline pH 7.4 (PBS), α-bromoisobutyryl bromide (BiBB), copper(II) bromide (CuBr2, 99%), copper(I) bromide (CuBr, 99%), pyrene, Dulbecco's modified Eagle medium F-12 Ham (DMEM-F12 Ham), fetal bovine serum (FBS), 2,2′-dipyridyl (Bpy, 99%), 2-aminoethyl methacrylate (AEM, 99%), N-isopropylacrylamide (NIPAAm), di(ethylene glycol) methyl ether methacrylate (PEGMA 188 monomer, Mn 188.22), poly(ethylene glycol) methyl ether methacrylate (PEGMA 475 monomer, Mn 475) and fluorescein isothiocyanate (FITC) were from Sigma Aldrich (St. Louis, MO, USA). NIPAAm was recrystallized with hexane prior use. PEGMA 188 and 475 monomers were purified before use by passing through a column of neutral alumina. Tris(2-pyridyl) methylamine (TPMA) was prepared as described previously.43 Dialysis membranes (MWCO 50 kDa, 25 kDa, 15 kDa and 3.5 kDa) and Float-A-Lyzer G2 (8–10 kDa, 1 mL, cellulose ester) were from Spectra/Por (Fisher Scientific, USA). RPMI 1640 without folic acid and penicillin (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 units per mL)/streptomycin (10000 μg mL−1) were from Invitrogen (USA). ProLong® Gold Antifade Reagent with DAPI and Phalloidine (Biodipy® 650/665) were from Life Technologies (USA). Phosphate buffer pH 5.5 was prepared as described in the European Pharmacopeia. Purified water (Milli-Q system, Millipore; resistivity > 18.2 MΩ cm) was used in all the experiments. Other solvents were analytical grade and purchased from Sigma Aldrich (St. Louis. MO, USA).
000 units per mL)/streptomycin (10000 μg mL−1) were from Invitrogen (USA). ProLong® Gold Antifade Reagent with DAPI and Phalloidine (Biodipy® 650/665) were from Life Technologies (USA). Phosphate buffer pH 5.5 was prepared as described in the European Pharmacopeia. Purified water (Milli-Q system, Millipore; resistivity > 18.2 MΩ cm) was used in all the experiments. Other solvents were analytical grade and purchased from Sigma Aldrich (St. Louis. MO, USA).
Synthesis and characterization of dextran copolymers
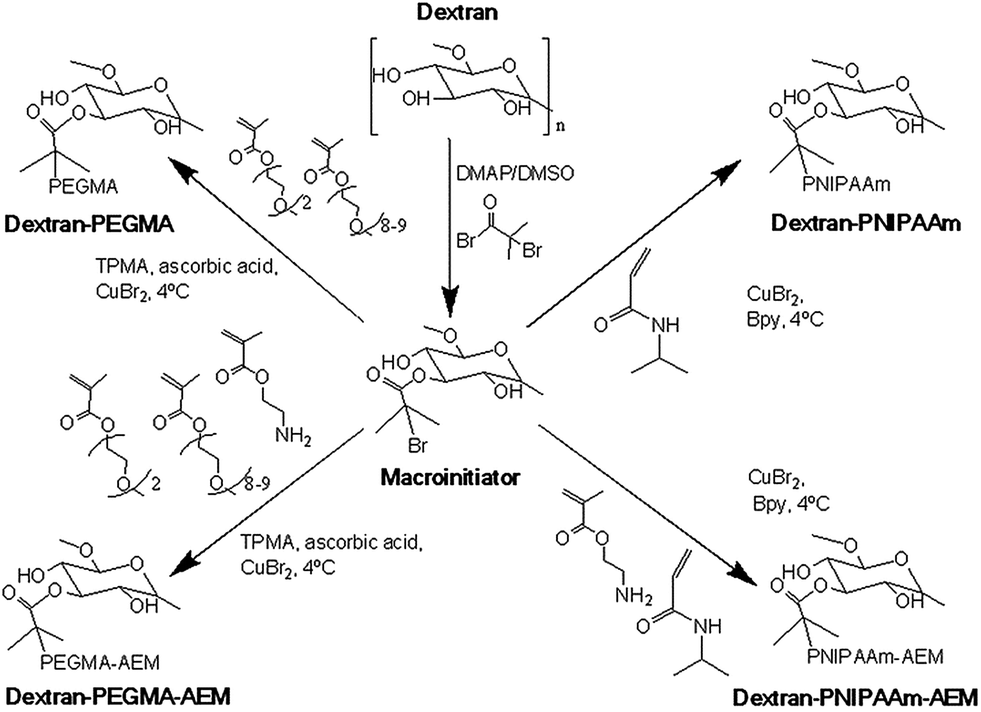 | ||
| Fig. 1 Schematic of the synthesis of dextran grafted with thermoresponsive polymers (PEGMA, PEGMA–AEM, PNIPAAm and PNIPAAm–AEM) by means of controlled radical polymerization starting from a dextran macroinitiator. | ||
 | ||
| Fig. 2 1H-NMR spectra of dextran and dextran macroinitiators (dextBr0.2 eq., dextBr0.4 eq. and dextBr0.8 eq.) in DMSO-d6. | ||
Cloud points of polymers in solution were used as approximations of the lower critical solution temperatures (LCST) of the polymers. Absorbances of aqueous solutions of the macroinitiator and copolymers in PBS pH 7.4 (1 mg mL−1) were recorded at 550 nm (Beckman DU 640 UV, USA) in the 20–50 °C range. The temperature was controlled using a Peltier plate heating system and it was increased at a rate of 1 °C min−1. LCST was estimated as the temperature at which the transmittance was 50% of the initial one.
Micelle characterization
The zeta-potentials of the copolymers (1 mg mL−1) in water were measured using a Zetasizer (Nano series, Malvern, UK). Critical micellar concentrations (CMC) of dextBr0.4 eq. and copolymers was determined using pyrene as a fluorescent probe. Aliquots of pyrene solution in acetone (50 μL, 50 mg mL−1) were added to Eppendorf vials and acetone was allowed to evaporate. Then, solutions (1 mL) of each polymer prepared in PBS pH 7.4 at various concentrations (1, 5, 10, 25, 50, 75, 100, 250, 500, 750 and 1000 μg mL−1) were added. The solutions were kept at 37 °C overnight and the fluorescence was measured using an emission wavelength of 390 nm and recording the excitation in the 250–360 nm range (Cary Eclipse, Varian, USA, equipped with a thermostated cell compartment). The same solutions were kept for a further 12 h at room temperature and the fluorescence was measured again.The hydrodynamic radii of the polymeric micelles (0.5 mg mL−1) were measured using dynamic light scattering (DLS, Viscotek DLS, Malvern Omnisize 3.0, UK) with an incident laser (50 mW, 830 nm) at 90° angle at 20, 37 and 50 °C. The radius was calculated from the measured diffusions coefficients (D) applying the Stokes–Einstein equation (eqn (1)) assuming spherical shape and non-interaction between the micelles:
 | (1) |
In this equation, RH represents the hydrodynamic radius, k the Boltzmann constant, T absolute temperature, and η solvent viscosity. Measurements quoted are the averages of triplicate measurements of 3 different samples with at least 10 readings of particle size of copolymer solutions in PBS pH 7.4. The radii of the self-assembled structures correspond to those scattering more than 75% of the light.
Atomic force microscopy (AFM) images of dextran–PNIPAAm–AEM micellar solution were recorded using liquid imaging on a MultiMode 8 Scanning Probe Station with nanoScopeIIIa controller (Bruker, USA) at room temperature and 37 °C. The surface was coated with 10 mM magnesium chloride prior to sample deposition. Copolymer solutions (10 μg mL−1) were made up in filtered (0.45 μm) PBS at pH 7.4. Images were analysed using particle size analysis by NanoScope Analysis software (version 1.20, Bruker, USA).
In vitro cell viability studies were performed using BALB/3T3 (clone A31, ATCC® CCL-163™) cells. Aliquots (100 μL) of a cell suspension (200000 cell per mL) in DMEM-F12 Ham, supplemented with 10% FBS and 1% solution of penicillin (10000 units per mL)/streptomycin (10000 μg mL−1), were seeded into 96-well plates and incubated over 24 h at 37 °C, 5% CO2 and 90% RH. Solutions of dextran, dextran–PNIPAAm, dextran–PNIPAAm–AEM, dextran–PEGMA and dextran–PEGMA–AEM in cell medium were prepared, and 100 μL of each one was added to the cells to have final concentrations of 0.001, 0.01, 1, 2.5, 10, 25, 50, 100, 200, 300, 400, 500, 750 and 1000 μg mL−1 in the wells. Plates were incubated at 37 °C for 24 and 48 h in 5% CO2 and 90% RH. Cell viability was analyzed following the instructions of a MTT kit supplier (Roche, Switzerland) using an ELISA plate reader (BIORAD Model 680 Microplate Reader, USA) with a 550 nm filter (Abssample). Negative controls were also tested by adding fresh medium to the wells and treating them in the same way as the samples (Abscontrol). The tests were carried out in triplicate. The metabolic activity as a proxy for cell viability (%) was quantified as follows (eqn (2)):
 | (2) |
Preparation of methotrexate-loaded micelles and “in vitro” drug release
MTX-loaded micelles were prepared applying two different approaches: dialysis or solvent evaporation. All tests were performed in triplicate. The dialysis method consisted in incubating each copolymer solution (0.5 mL, 30 mg mL−1) with MTX (0.5 mL, 6 mg mL−1) in PBS pH 7.4 or phosphate buffer pH 5.5 for 2.5 h at 37 °C. Afterwards, solutions were poured in Float-A-Lyzer dialysis devices (MWCO 8–10 kDa) and dialyzed against water at 37 °C during 24 h, replacing the water four times (after 2, 4, 6 and 10 h). Then, samples of 125 μL were removed from the content of the dialysis tubes, diluted with 375 μL of PBS pH 7.4, and 0.5 mL of DMSO was added to disrupt the micelles. Drug loading content (L.C.) and encapsulation efficiency (E.E.) were determined as follows:
 | (3) |
 | (4) |
For the solvent evaporation approach, copolymer solutions in THF (1 mL, 60 mg mL−1) were stirred over 2 h with 12 mg of MTX at room temperature. Then, THF was allowed to evaporate overnight, and the formed film was incubated in 1 mL of PBS pH 7.4 at 37 °C for 2.5 h under magnetic stirring. The systems were centrifuged (8500 rpm, 20 min) to remove non-dissolved drug, and then the supernatant was freeze-dried. Freeze-dried samples (1 mg) were used to estimate the encapsulation efficiency (eqn (4)) and the loading content (L.C.; eqn (5)).
 | (5) |
MTX release experiments were carried out at various pH and temperature conditions. In order to check the effect of the pH, dispersions (2 mL) of MTX-loaded micelles (prepared applying the dialysis method) were added into Float-A-Lyzer dialysis devices (8–10 kDa), which were then placed in 80 mL of PBS pH 7.4 or phosphate buffer pH 5.5 at 37 °C. Regarding the effect of the temperature, freeze-dried MTX-loaded micelles (5 mg; solvent evaporation method) were reconstituted in 2 mL of PBS pH 7.4 and added into Float-A-Lyzer dialysis devices (8–10 kDa). The devices were immersed in 30 mL of PBS pH 7.4 at 37 or 40 °C, under magnetic stirring. At preestablished times, samples (1 mL) of the release medium were taken to determine MTX concentration and replaced with the same volume of fresh medium. Amounts of MTX loaded and released were determined using a HPLC equipment fitted with a Controller 600 pump, an autosampler 717, photodiode array detector (996 PDA detector) and Empower 2000 software (Waters, USA). An Atlantis® dC18 (5 μm, 4.6 × 250 mm, Waters, Ireland) column maintained at 40 °C was used. The mobile phase was water:acetonitrile mixture (90:10 v/v) at 1.2 mL min−1. MTX was quantified at 302 nm.
Cytotoxicity of methotrexate-loaded and non-loaded micelles
HeLa (ATCC® CCL-2™) and CHO-K1 (ATCC® CCL-61™) cells suspensions (200000 cells per mL) in RPMI 1640 without folic acid, supplemented with 10% FBS and 1% solution of penicillin (10000 units per mL)/streptomycin (10000 μg mL−1) were seeded in 96-well plates (100 μL) and incubated over 24 h at 37 °C, 5% CO2 and 90% RH. Micellar solutions were prepared with and without MTX using copolymer concentration 2-fold above CMC (CMCx2). Pristine dextran was tested at 400 μg mL−1. MTX-loaded micelles were prepared following the two methods described above, with an additional step of freeze-drying in all cases, and reconstitution in the culture medium. The cell medium of the wells was replaced by the micellar solutions (200 μL). Plates were incubated at 37 °C for 24 and 48 h in 5% CO2 and 90% RH. Cell viability was analyzed following the instructions of a MTT kit supplier (Roche, Switzerland) using an ELISA plate reader (BIORAD Model 680 Microplate Reader, USA) with a 550 nm filter. Cytocompatibility (%) was quantified using eqn (2). All tests were carried out in triplicate. Negative controls were also performed by adding fresh medium alone to the wells and treating them in the same way as the samples.
Cellular uptake
FITC-labeled polymers were prepared as described before for dextran micelles.45 Briefly, FITC (2 mg mL−1 in ethanol, 0.5 mL) was added to copolymer (dextran–PNIPAAm, dextran–PNIPAAm–AEM, dextran–PEGMA or dextran–PEGMA–AEM) solutions in water (10 mg in 1 mL). The reaction mixture was stirred at room temperature over 24 h, and dialyzed against water (MWCO 3.5 kDa) for 48 h to remove ethanol and unreacted FITC. Labelled copolymers were freeze-dried. The whole experiment was done protecting samples from the light.CHO-K1 cells (500000 cell per mL, 100 μL) in RPMI without folic acid supplemented with 10% FBS and 1% solution of penicillin (10000 units per mL)/streptomycin (10000 μg mL−1) were seeded in 8-well glass plates (0.7 cm2, Millicell® EZ slide, Millipore, USA) and 338 μL of fresh medium was added. After 24 h of incubation (37 °C, 5% CO2 and 90% RH), the cells medium was removed and replaced by 438 μL of the FITC-labelled copolymer solutions in culture medium (500 μg mL−1 or 2-fold CMC) or fresh medium. The cells were incubated with the micelles for 2 h and 24 h at 37 °C. Subsequently, the micellar solutions were removed, and the cells were washed three times with PBS pH 7.4. Then, cells were fixed with paraformaldehyde (100 μL, 4% w/v in PBS pH 7.4) over 20 min at room temperature and washed again with PBS three times. Cells were incubated with Triton X-100 (50 μL, 0.2% w/v in PBS) for 5 min at room temperature, and washed again with PBS pH 7.4 three times. Phalloidin solution (30 μL, 75 nM in PBS pH 7.4) was added and cells were incubated for 20 minutes at room temperature. Afterwards, cells were washed again with PBS pH 7.4 three times and the upper part of the well glass was removed ready for being visualized under confocal microscopy. A drop of ProLong® Gold Antifade Reagent with DAPI was added to each well, and the glass slide was frozen at −20 °C over night to enhance the fluorescent dye penetration inside cells. Confocal images of samples incubated for 2 h and for 24 h were recorded using a 63× objective in a TCS SP5 AOBS and in a TCS SP2 spectral confocal system (Leica Microsystems, Germany), respectively. Fluorescent signals were collected using a sequential acquisition mode. Green channel was used for FITC-labeled micelles (λexc = 488 nm, λem = 511–563 nm), red channel for phalloidin (λexc = 561 nm, λem = 577–700 nm) and blue channel for DAPI (λexc = 405 nm, λem = 425–475 nm).
Results and discussion
Synthesis of dextran copolymers
Comb-shaped temperature-sensitive dextran copolymers were synthesized according to a “grafting from” technique. First, bromoisobutyrate initiating groups were introduced onto the dextran backbone to obtain a dextran macroinitiator. Unlike previous attempts to prepare dextran macroinitiators that required protection of hydroxyl groups,33,46 the polysaccharide macroinitiator was prepared by direct esterification using BiBB in the presence of DMAP in DMSO (Fig. 1). This solvent was previously used to introduce ATRP initiating groups in dextran particles using BiBB and triethylamine as a base35 or in soluble dextran polymer using 2-bromo-2-methylpropionic acid and 1,1′ carbonyldiimidazole/DMAP.37 Triethylamine was tested initially as the base in this study, but in our case greater substitution was achieved with DMAP. Different BiBB ratios (0.2, 0.4 or 0.8 eq. per hydroxyl group) were tested in order to have different proportions of alkyl halide initiators on the backbone. Grafting was expected to occur at the hydroxyl groups in positions 2 and 3 due to the higher basicity; however, steric repulsion may have facilitated the substitution in position 3. The degree of molar substitution (the number of BiBB groups per 100 glucose units) of each macroinitiator was calculated from 1H-NMR spectra in DMSO-d6, by integrating the signal at 1.9 ppm (A1.9) corresponding to the six protons of the methyl groups of BiBB and the signal at 4.4–5.7 (A4.4–5.7) which corresponds to the anomeric proton and the three hydroxyl protons of the dextran (Fig. 2). However, due to the disappearance of the signal of hydroxyl protons after reaction with BiBB, this value had to be corrected with the signal from the BiBB. The degree of substitution was calculated as indicated in eqn (6).
 | (6) |
Substitution values were 9% for dextBr0.2 eq., 21% for dextBr0.4 eq. and 31% for dextBr0.8 eq. DextBr0.2 eq. and dextBr0.8 eq. were discarded for the synthesis of the temperature-sensitive polymers due to the small proportion of initiating groups grafted and the poor aqueous solubility of the macroinitiator, respectively.
The molecular weights of the polysaccharide and dextBr0.4 eq. were determined by means of GPC using DPBS and DMF as solvents, respectively. Dextran molecular weight estimated by GPC was around 23 kDa with moderate polydispersity (Mw 23.0 kDa, Mn 13.0 kDa; Ð = 1.8). The GPC-derived molecular weight of the macroinitiator in DMF was slightly lower than that of the pristine polysaccharide (Mw = 17.0 kDa; Mn = 9.9 kDa; Ð = 1.7), which may have been a consequence either of a different chain conformation in solution compared to the unsubstituted dextran or some partial chain cleavage due to acid generation during substitution.
Synthesis of temperature-sensitive dextran-based copolymers
Temperature-sensitive copolymers were prepared as summarized in Fig. 1. For ATRP, DextrB0.4 eq. was used as macroinitiator. For preparing dextran–PNIPAAm, CuBr was used as a catalyst, Bpy as ligand and water as solvent. The reaction was monitored by 1H-NMR in D2O until 70% conversion was achieved (ca. 5 h), aimed to provide PNIPAAm graft chains with a theoretical molecular weight of 4.7 kDa. The weight average molecular weight of the resultant dextran–PNIPAAm conjugate determined by GPC in DMF was 110 kDa (Mw = 110.3 kDa; Mn = 36.8 kDa; Ð = 3.0). The high polydispersity was likely a consequence of inherent molecular weight range of the native polysaccharide, as well as limited control of the graft polymerization. It should also be noted that the molecular weight of comb- and brush-type polymers is often underestimated by GPC because linear standards are generally used for calibration,47 thus, the true molecular weight of the copolymer was likely be higher than that estimated by GPC. However, as we were interested in relative rather than absolute molecular weights, further characterisation was not considered necessary.The synthesis of poly(ethylene glycol) polymers by ATRP using poly(ethylene glycol) alkyl ether methacrylates as starting materials has been previously reported.41 We adapted this methodology, but using dextBr0.4 eq. as initiator. TPMA, CuBr, ascorbic acid and PBS pH 7.4 were used as ligand, catalyst, reducing agent and solvent, respectively. The reaction was also monitored by 1H-NMR in chloroform-d, until 70% conversion was achieved (approximately 3.5 h) to obtain theoretical PEGMA grafts of 14 kDa. The molecular weight could not be determined by GPC due to the poor solubility in chloroform and DMF, and the high affinity of the polymers for the stationary phase of the aqueous GPC. Attempts to determine molecular weight by MALDI-TOF were also not successful (data not shown).
Temperature-responsiveness of the grafted-copolymers was demonstrated by monitoring the transmittance of the solutions with increasing temperature (Fig. 3). Both pristine dextran and dextran macroinitiator did not show temperature-responsiveness, while dextran–PNIPAAm showed a cloud point of 35 °C, which is slightly higher than the reported LCST of PNIPAAm. The difference in phase transition temperature of the conjugate compared to that expected for pure PNIPAAm can be attributed partially to the hydrophilic environment provided by dextran.47 This was likely to have altered the extent to which the PNIPAAm chains were able to associate and thus self-aggregate, and it is important to note that cloud point is a concentration-dependent phenomenon, whereas the true LCST of a polymer chain is concentration-independent. Therefore, even if the PNIPAAm grafted chains were exhibiting an LCST at 32 °C, it may only have been apparent at several degrees above this temperature that the conjugate chains could associate sufficiently to reach a cloud point. The measured cloud point of the analogous dextran–PEGMA conjugate was 34 °C, again indicative of temperature-induced self-association of hydrophobic chains, and again within a temperature region appropriate for biomedical purposes. Dextran–PEGMA–AEM and dextran–PNIPAAm–AEM were prepared by controlled free radical polymerisation following the same protocol used for the synthesis of dextran–PEGMA and dextran–PNIPAAm respectively, but incorporating AEM in the reaction mixture (Fig. 2). ATRP of AEM to produce homopolymers has been previously reported;48 thus, AEM monomers were expected to be incorporated into the polymeric chains randomly and as a function of its reactivity ratios in the same way as PEGMA and NIPAAm. Different proportions of AEM vs. PEGMA or PNIPAAm were tested. Using a 3.1% AEM feed ratio with respect to PEGMA, the obtained dextran–PEGMA–AEM showed a cloud point of 38 °C (Fig. 3). Although various ratios of AEM were tested to synthesize dextran–PNIPAAm–AEM, none of the obtained PNIPAAm copolymers showed a demonstrable cloud-point when heated (Fig. 3), although some temperature-responsiveness was observed using DLS and AFM (see below). A proportion of 7.1% AEM feed ratio with respect to PNIPAAm was chosen to evaluate the effect of this monomer on the polymer uptake by cells. The molecular weight of dextran–PNIPAAm–AEM as determined from GPC was apparently lower than that of dextran–PNIPAAm, being around 72 kDa (Mw; Mn = 20.8 kDa; Ð = 3.5), suggesting a change in solution conformation in DMF/LiBr compared to that for the dextran–PNIPAAm precursor.47
 | ||
| Fig. 3 Temperature-transmittance curves of dextran macroinitiator (dextBr0.4 eq.), dextran–PNIPAAm, dextran–PNIPAAm–AEM, dextran–PEGMA and dextran–PEGMA–AEM (1000 μg mL−1) in PBS pH 7.4. The cloud points were defined as the onset of a non-linear change in turbidity. | ||
Micelles characterization
The charges of the copolymers were measured below (25 °C) and above (50 °C) the LCST in water. Dextran macroinitiator and copolymers without AEM were negatively charged at both temperatures. As expected, AEM copolymers presented positive charges at 25 °C, which were more abundant in dextran–PNIPAAm–AEM due to its higher content in AEM (Table 1). The experiment was also done at 50 °C to ensure micelle formation; in every case the negative or the positive charge was retained at the higher temperatures.| Copolymer | LCST (°C) | Zeta-potential (mV) | CMC (μg mL−1) | Hydrodynamic radius (nm) | ||||
|---|---|---|---|---|---|---|---|---|
| 25 °C | 50 °C | 20 °C | 37 °C | 20 °C | 37 °C | 55 °C | ||
| DextBr0.4 eq. | — | −5.7 (4.4) | −5.9 (4.3) | — | — | 0.99 (0.05) | 1.48 (0.06) | 2.62 (0.19) |
| Dextran–PNIPAm | 35 | −3.9 (4.5) | −16.6 (4.7) | — | 80 | 8.7 (0.8)–93.9% | 88 (21) | 80 (10) |
| 52 (8)–6.1% | ||||||||
| Dextran–PNIPAAm–AEM | >60 | 14.8 (3.6) | 12.6 (7.1) | 380 | 200 | 4.7 (0.4) | 3.8 (0.4)–76%% | 4.5 (0.2)–44% |
| 12.3 (2.1)–12% | 82 (17)–56% | |||||||
| Dextran–PEGMA | 34 | −22.4 (5.8) | −25.7 (7.8) | 16 | 16 | 8.2 (2.7) | 14.1 (0.9)–76% | 24.1 (1.5)–87% |
| 190 (42)–24% | 306 (48)–13% | |||||||
| Dextran–PEGMA–AEM | 38 | 6.5 (3.4) | 9.3 (5.7) | 16 | 13 | 6.5 (1.3) | 10.2 (0.5)–91% | 367 (48) |
| 321 (68)–9% | ||||||||
The self-assembly properties of each copolymer in aqueous media were evaluated using pyrene as a fluorescent probe. Pyrene solutions in PBS pH 7.4 showed a maximum in fluorescence excitation spectra at 338 nm. In the presence of micelles, pyrene moved from the aqueous environment to the micelle core and a shift of the mean excitation peak to 334 nm was observed. To calculate the CMC, the ratio between these two intensities (I338/I334) was plotted against the logarithm of the copolymer concentration. The CMC was estimated as the intersection point between the line that joins the points of flat region and the line that joins the points at the increasing region49 (Table 1, Fig. 4). The CMC of dextran–PNIPAAm was estimated to be 80 μg mL−1 at 37 °C, and no micelle formation was observed at 20 °C in agreement with the hydrophilic character of both polymers at low temperature.34 This CMC value was lower than those previously reported for other dextran–PNIPAm copolymer28 probably because in our case dextran was of lower molecular weight. The presence of AEM in the copolymer constrained micelle formation, increasing the CMC up to 200 μg mL−1 at 37 °C, although allowing self-assembly at room temperature at higher concentrations (CMC = 380 μg mL−1).
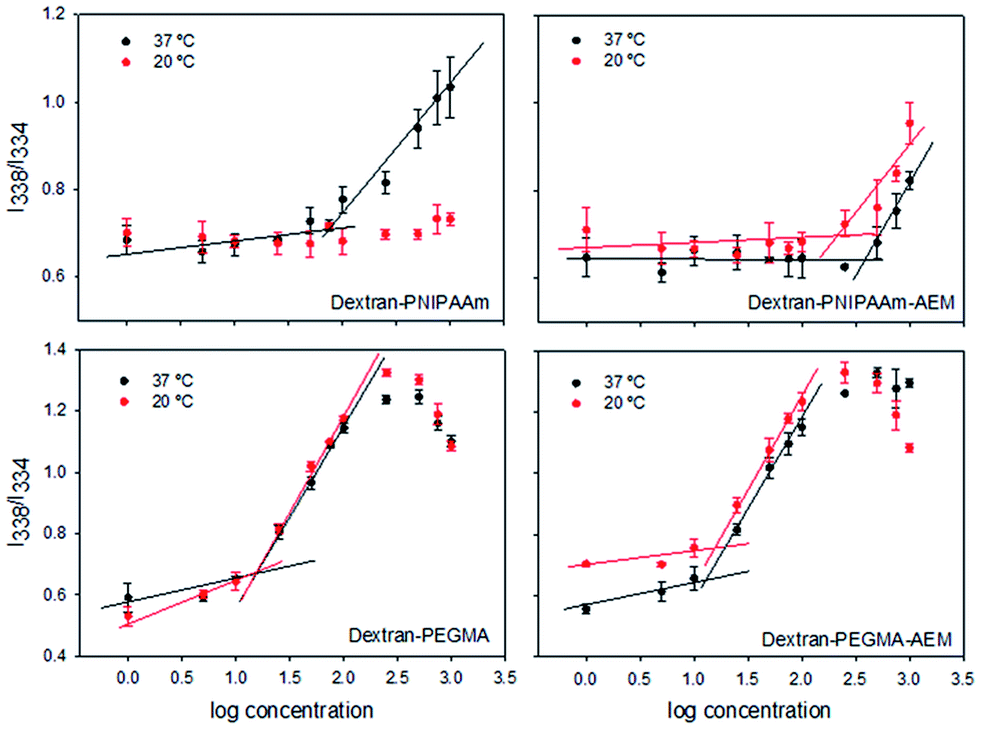 | ||
| Fig. 4 Critical micellar concentrations of the different dextran copolymers. I338/I334 ratio of pyrene emission spectra as a function of the logarithm of the concentration (μg mL−1) of dextran–PNIPAAm, dextran–PNIPAAm–AEM, dextran–PEGMA and dextran–PEGMA–AEM obtained at 37 °C and room temperature (20 °C). | ||
PEGMA-based copolymers had lower CMCs: 16 μg mL−1 for dextran–PEGMA at both temperatures, and 13 or 16 μg mL−1 for dextran–PEGMA–AEM at 37 and 20 °C, respectively. The differences in CMC between copolymers bearing PNIPAAm and PEGMA can be attributed to the longer chains of the grafted PEGMA.50 Moreover, the lower CMC values of polymers bearing PEGMA suggest that micelles may have a better physical stability against dilution owing to a more hydrophobic core.51 A previous work reported on a cellulose derivative bearing PEGMA chains with even lower CMC (0.12–0.65 μg mL−1),47 probably because of the less hydrophilic character of cellulose compared to dextran. In the case of oral administration, it has been pointed out that a CMC lower than 135 μg mL−1 is adequate to prevent rapid dissociation in the gastrointestinal tract and may provide sustained release/absorption of chemotherapeutics.52
The hydrodynamic diameters were measured using DLS (Table 2, Fig. S1 in ESI†). The macroinitiator dextBr0.4 eq. showed a quite small size (∼2 nm), which slightly increased with the increase in temperature probably due to minor aggregation. The diameter of dextran–PNIPAAm micelles was around 190 nm at 37 °C, and slightly decreased at 55 °C. However, at room temperature aggregates smaller than 20 nm were observed. The size of these micelles was greater than those previously reported for other dextran-based copolymer prepared by ATRP (80–100 nm),36 probably due to differences in dextran molecular weight and MS.53 The incorporation of AEM in the copolymer led to smaller micelles (around 10 nm) at room temperature. Upon increasing the temperature, a population of larger size appeared. Micelles of PEGMA-grafted dextrans were around 20–30 nm at 37 °C, in agreement with the data of previous micelles of PEGMA-grafted cellulose.54 The size of the micelles increased with the temperature as the copolymer became more hydrophobic and the self-aggregation was facilitated.55 It has been previously reported that a suddenly increase of the temperature may cause a reduction in the size of PEGMA copolymer micelles.56 Two populations were observed for some copolymers, probably due to the molecular weight dispersity of the polysaccharide and the copolymer and differences in self-assembly. In any case, micelles were close to or lower than 200 nm at 37 °C, which was a key design feature to make them suitable for in vivo use where they would need to avoid the recognition by the reticuloendothelial system and to be directed to tumour tissues by passive targeting.57
| Copolymer | Loading content (%) | Encapsulation efficiency (%) | ||||
|---|---|---|---|---|---|---|
| Dialysis method | Evaporation method (reconstitution at pH 7.4) | Dialysis method | Evaporation method (reconstitution at pH 7.4) | |||
| pH 7.4 | pH 5.5 | pH 7.4 | pH 5.5 | |||
| Dextran | 7.4 (1.5) | 3.8 (1.5) | 0.93 (0.29) | 32.4 (1.0) | 19.2 (7.5) | 4.6 (1.5) |
| Dextran–PNIPAAm | 9.7 (1.1) | 5.6 (1.4) | 1.9 (0.3) | 48.7 (5.6) | 27.9 (7.0) | 9.3 (1.7) |
| Dextran–PNIPAAm–AEM | 13.5 (0.3) | 8.7 (0.2) | 2.9 (0.1) | 67.5 (1.5) | 43.6 (0.9) | 14.3 (0.8) |
| Dextran–PEGMA | 6.0 (0.7) | 4.6 (0.3) | 4.3 (0.8) | 30.0 (3.6) | 23.3 (1.7) | 21.3 (2.4) |
| Dextran–PEGMA–AEM | 15.6 (1.3) | 7.5 (2.5) | 4.6 (0.2) | 77.8 (6.7) | 37.7 (1.2) | 22.9 (1.2) |
AFM images (Fig. S2 in ESI†) corroborated the temperature-sensitiveness of dextran–PNIPAAm–AEM; particles of 20–25 nm in diameter at room temperature and of 40–47 nm at 37 °C were observed. The differences in size when comparing to the DLS data could be associated to the lower copolymer concentrations tested in AFM and thus micelles may be formed with less unimers.
Cytocompatibility of the copolymers was tested against a murine fibroblast cell line (BALB/3T3) (Fig. 5), a non-tumour cell line typically used due to its sensitivity to toxic agents. As expected, pristine dextran was highly cytocompatible in the range of concentrations assayed. PEGMA-bearing copolymers also showed good cytocompatibility (>60%) at 1 mg mL−1 after 48 h of incubation. Cell viability was lowest in the presence of PNIPAAm-bearing copolymers, although dextran–PNIPAAm showed an IC50 above 1 mg mL−1 after 24 h. Dextran–PNIPAAm-AEM was the most cytotoxic copolymer, with an IC50 around 25 μg mL−1 (below the CMC). This toxicity can be associated to its higher cationic content, in agreement with previous reports that indicated that AEM polymers have some cytotoxicity.48
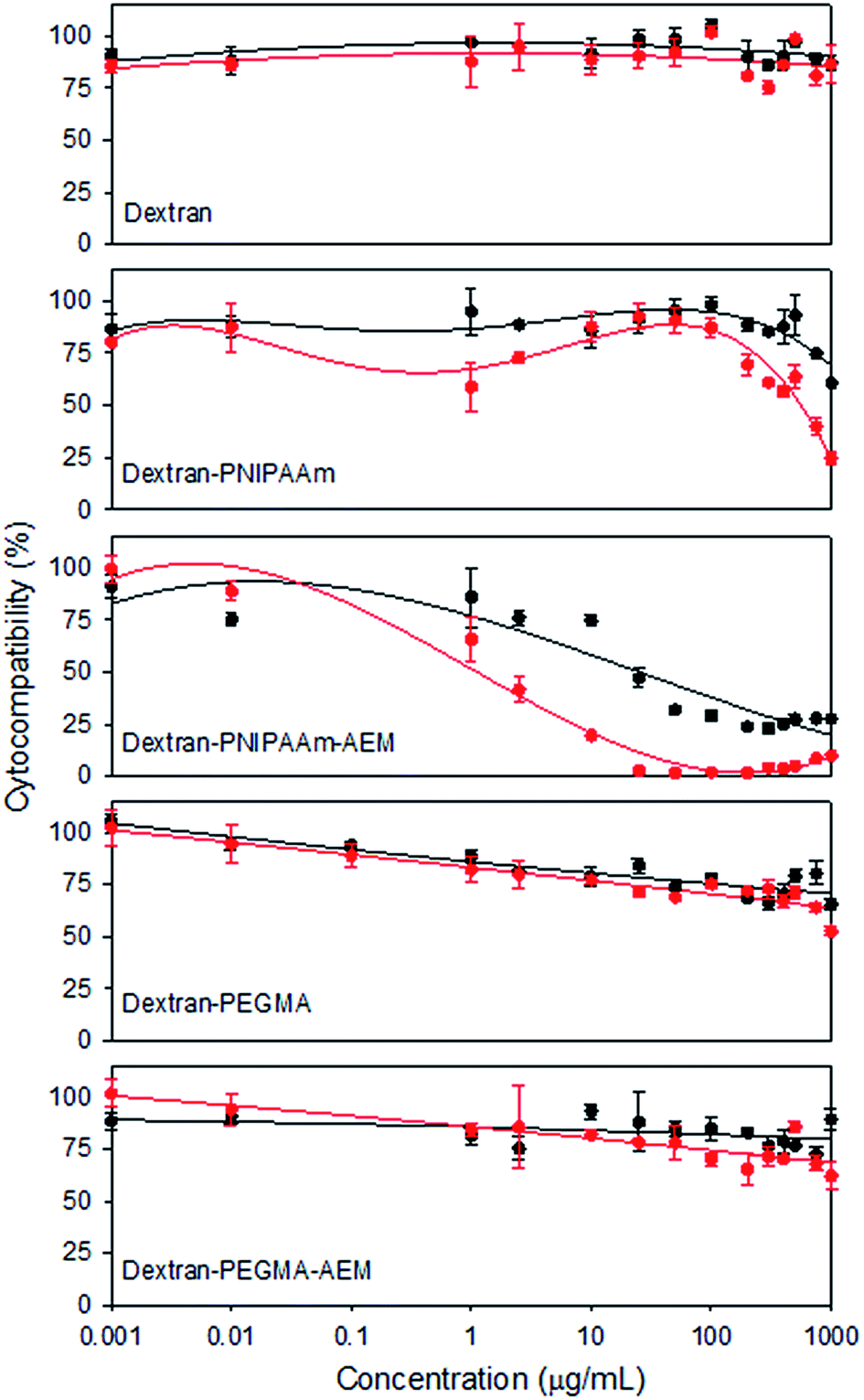 | ||
| Fig. 5 Cell compatibility of the polymers as indicated by metabolic activity (MTT assay) in BALB/3T3 cells after incubation for 24 h (black line) and 48 h (red line) with dextran, dextran–PNIPAAm, dextran–PNIPAAm–AEM, dextran–PEGMA and dextran–PEGMA–AEM at different concentrations. | ||
Overall, the cytocompatibility results pointed out that dextran–PEGMA, dextran–PEGMA–AEM and dextran–PNIPAAm might be safely used as MTX nanocarriers. Although some cytotoxicity was observed for dextran–PNIPAAm–AEM, this copolymer was also included in subsequent experiments in order to compare its performance as an MTX carrier with the other copolymers.
Methotrexate-loaded micelles and in vitro drug release
The preparation of micelles loaded with cytostatic drugs generally involves the dissolution of the polymer and the drug in an organic solvent, and then, an aqueous buffer or water is added to allow the micelle formation. Afterwards, solvent and free drug are removed by dialysis. Dextran micelles grafted with different hydrophobic and stimuli-sensitive chains, like poly(lactide-co-glycolide), polylactide, polyhistidine or PNIPAAm, have been shown able to encapsulate different amounts of drugs depending on the nature of the hydrophobic drug and the polymer chain; the L.C. being between 6 and 13%.26–28,58 In the case of MTX, L.C. is commonly in the 2–5% range.15,41,42 We tested two approaches to prepare MTX-loaded micelles. The dialysis method consisted in adding an excess of MTX to the copolymer solution followed by incubation at 37 °C to promote the micellisation and subsequent dialysis to remove the non-encapsulated drug. Two different pH values were tested to check the effect of the AEM on the copolymers. MTX loading was higher when the pH of the copolymer solutions was set at 7.4, compared to 5.5 (Table 2). At pH 7.4 the carboxylic and the amine groups of the MTX are deprotonated, presenting a negative charge, favouring interactions with the AEM co-monomer. As expected, the L.C. values for the non-ionic copolymers were similar to those recorded for other drugs in dextran-based micelles26,58,59 but even using an AEM feed ratio as low as 3.1%, the dextran–PEGMA–AEM copolymers showed much higher MTX loadings than those previously reported.7,9,10 Controls carried out with dextran revealed that MTX is also loaded by the pristine polysaccharide, probably through non-specific hydrophobic interactions as observed for loading of hydrophobic drugs by other polysaccharides.A solvent evaporation method was also tested for MTX encapsulation, using the same copolymer:drug feed ratio as in the dialysis method. Each copolymer and MTX were dissolved in THF and, then, the solvent was allowed to evaporate. The film formed was dissolved in PBS pH 7.4 at 37 °C and the non-encapsulated MTX was removed by centrifugation. Lower encapsulation efficiencies and loading contents were recorded using this approach, although the copolymers ranked in a similar order to that recorded in the case of the dialysis method (Table 2). The presence of AEM on the grafts slightly increased the loading by the polymeric micelles, especially in the case of dextran–PNIPAAm–AEM.
Release studies were carried out in phosphate buffer pH 5.5, which simulates the endosomal pH, and PBS pH 7.4, which mimics extracellular conditions (Fig. 6).
 | ||
| Fig. 6 MTX release profiles from dextran dispersions and polymeric micelles (7.5 mg mL−1) at 37 °C in PBS pH 7.4 and phosphate buffer pH 5.5. Release profiles at 40 °C are indicated by triangle symbols and dash lines. | ||
Pristine dextran dispersions released 60–70% MTX in the first 24 h, which indicates that the interaction with the drug was relatively weak. In contrast, polymeric micelles showed sustained release profiles. The highest release rate was recorded for dextran–PNIPAAm at pH 5.5. Also dextran–PEGMA copolymers released MTX faster than dextran–PEGMA–AEM counterparts. In general, the slowest release was observed for copolymers bearing AEM at pH 5.5, which can be explained by the decrease in solubility of MTX when partly protonated and the retention of some ionic interactions with AEM.60 Sustained release from polymeric micelles was prolonged for more than four days, in agreement with previous studies carried out with other dextran-based systems.45
The effect of the temperature on the drug release was also investigated (Fig. 6). Established solid tumour tissues can present an increase in temperature of 1–3 °C, so the experiment was carried out at 37 °C and also 40 °C (as a mimic of solid tumour temperature). These temperatures were above the predicted LCST for the polymer side-chains, and above the cloud-points of all the copolymers, except for dextran–PEGMA–AEM. In our case, both copolymers bearing PNIPAAm released higher amounts of MTX at 40 °C than at 37 °C; the increase in release rate being greater in the case of dextran–PNIPAAm–AEM. It is known that PNIPAAm chains dehydrate above the LCST61 and micelles diminish their size as temperature increases (as observed by means of DLS). Consequently, the collapse of the micelle may have promoted the release of the drug owing to diminution of the micellar core volume. On the other hand, polymers bearing PEGMA showed different behavior depending on the presence or absence of AEM. Dextran–PEGMA micelles released slightly higher amounts of MTX at 40 °C than at 37 °C. DLS data did not show a micelle size reduction with the increase of the temperature, but this finding may have been due to aggregation of small micelles.56 In contrast, dextran–PEGMA–AEM micelles released higher amounts of MTX at 37 °C, which may have been associated with the more open and less micellar conformation of this polymer below its cloud point of 38 °C.
Cytotoxicity of methotrexate-loaded and non-loaded micelles
The bioactivity of MTX-loaded (by dialysis and solvent evaporation methods) and non-loaded micelles was tested against two cancer cell lines: an ovarian cancer cell line from hamster (CHOK-1) and adenocarcinoma cells from human cervix (HeLa), using polymer concentrations equal to two-fold CMC (Fig. 7). Dextran was tested at the highest concentration used (400 μg mL−1) and the results confirmed its excellent cytocompatibility. | ||
| Fig. 7 Cytotoxicity of the MTX-loaded and empty micelles against tumour cells. Viability of CHO-K1 and HeLa cells after incubation with placebo dextran (400 μg mL−1), dextran–PNIPAAm (320 μg mL−1), dextran–PNIPAAm–AEM (400 μg mL−1), dextran–PEGMA (32 μg mL−1) and dextran–PEGMA–AEM (26 μg mL−1) micelles and the corresponding MTX-loaded micelles prepared applying dialysis (DM) and solvent evaporation (EM) methods. | ||
All non-loaded micelles, except those prepared with dextran–PNIPAAm-AEM, were highly cytocompatible with the tumour cells. In the case of MTX-loaded micelles, dextran–PNIPAAm, dextran–PEGMA and dextran–PEGMA–AEM, the presence of MTX slightly increased the cytotoxicity of the micelles particularly after 48 h of incubation with both cell lines. In addition, they were more cytotoxic than a solution of free MTX prepared with a drug concentration higher than that provided by any of the micelle formulations. In the case of CHO-K1 and for a given copolymer, the performance of the drug-loaded polymeric micelles was similar independent of the loading method. However, in HeLa cells, dextran–PEGMA and dextran–PEGMA–AEM micelles, loaded via the dialysis method, were more cytotoxic due to the higher amount of MTX encapsulated. By contrast, dextran–PNIPAAm micelles were more cytotoxic when they were loaded applying the evaporation method. In general, HeLa cells were less sensitive to MTX than CHO-K1 cells, as observed for the control drug solution.
Dextran–PNIPAAM–AEM micelles were cytotoxic in the absence of the cytostatic agent. After MTX encapsulation, the toxicity of the micelles diminished. This behavior could be due to the interaction of MTX with the AEM groups, which in turn diminished the number of free cationic amine groups responsible for the cytotoxicity of the copolymer. A similar finding has been previously reported for complexes of poly(2-dimethyl amino ethyl methacrylate) with DNA, which were less cytotoxic than the polymer itself.46
Regarding dextran controls, after 48 h of incubation, MTX-loaded dextran system caused higher cytotoxicity than free MTX, which may suggest a possible cellular uptake of MTX promoted by the free dextran. Overall, both loading methods proved suitable for the preparation of MTX-loaded micelles as cytotoxicity effects were dependent on the copolymer type and the cell susceptibility.
Cellular uptake
The uptake of the polymeric micelles by CHO-K1 cells was monitored by means of confocal microscopy (Fig. 8 and 9). Cells were incubated with non-loaded FITC-labelled micelles over 2 and 24 h and confocal images were recorded. The cells did not modify their morphology, as compared with the negative control (Fig. 9.1). The highest cellular uptake was observed for dextran–PNIPAAm–AEM (Fig. 8C and D and 9.3) at both concentrations tested (400 μg mL−1 and 500 μg mL−1), which was likely due to the high AEM content in the copolymer which enhanced cell membrane interaction and internalization.38 Moreover, this fast internalization could destabilize the ionic balance and the structure of the membrane, which in turn may have been a reason for the high cytotoxicity of these micelles. Dextran–PNIPAAm (Fig. 8A), dextran–PEGMA (Fig. 8E) and dextran–PEGMA–AEM (Fig. 8G) were almost not internalized by the CHO-K1 cells at the lower concentration tested (2-fold CMC) probably due to the short time in contact with cells and the low copolymer concentration used (26–160 μg mL−1). | ||
| Fig. 8 Cellular internalization of FITC-labeled micelles by CHO-K1 after 2 h of incubation with cells by confocal microscopy. (A) Dextran–PNIPAAm at 160 μg mL−1 and (B) 500 μg mL−1, (C) dextran–PNIPAAm–AEM at 400 μg mL−1 and (D) 500 μg mL−1, (E) dextran–PEGMA at 32 μg mL−1 and (F) 500 μg mL−1 and (G) dextran–PEGMA–AEM at 26 μg mL−1 and (H) 500 μg mL−1. Blue channel corresponds to the nucleus dyed with DAPI, red channel the cytoskeleton stained with phalloidin and green channel the micelles labeled with FITC. Scale bar 25 μm. | ||
 | ||
| Fig. 9 Cellular internalization of the micelles by CHO-K1 after 24 h of incubation with cells by confocal microscopy. (1) Fresh medium and (2–5) FTIC-labeled polymeric micelles (500 μg mL−1) of dextran–PNIPAAm (2), dextran–PNIPAAm–AEM (3), dextran–PEGMA (4) and dextran–PEGMA–AEM (5). (A) Blue channel, the nucleus dyed with DAPI; (B) red channel, the cytoskeleton stained with phalloidin; (C) green channel, the micelles labeled with FITC and (D) all merged. Scale bar 20 μm. | ||
The experiments were repeated testing all copolymers at the same concentration (500 μg mL−1). At this higher concentration, micelles of dextran–PNIPAAm, dextran–PEGMA and dextran–PEGMA–AEM were internalized to a low extent again (Fig. 8B, F and H). This finding is in agreement with previous reports that demonstrate that PEGMA-based copolymers are barely internalized in HeLa cells after 2 h of incubation if no internalising ligand is used to enhance the uptake.60 The low AEM proportion present in dextran–PEGMA–AEM seemed to be not enough to enhance the cellular uptake.
To evaluate the cellular internalization, confocal images were taken in two planes of the cells containing micelles. Micelles, that are represented in the green channel due to the conjugation of the polymers with FITC, could be detected between the cytoplasm (red channel) and the nucleus of cells (blue channel). MTX-loaded dextran–PNIPAAm, dextran–PEGMA and dextran–PEGMA–AEM demonstrated their cytotoxic effects after 24 and 48 h of incubation with CHO-K1 cells when a concentration of twice the CMC was used. We hypothesised that the short time of the incubation for confocal studies was the reason for the low internalization observed, and further studies after 24 h of incubation with cells were carried out (Fig. 9). More micelles were internalized with the increase of the incubation time. A higher internalization was observed with the incorporation of AEM into the copolymers, especially in the case of dextran–PNIPAAm–AEM. Interestingly, in the case of PEGMA copolymers the micelles were colocalized in the cytoplasm as the green colour from the micelles turned to yellow when it is colocalized with the red colour from the cytoskeleton dyed with phalloidin.
Conclusions
Copolymers of dextran and PNIPAAm or PEGMA were successfully prepared by conducting free radical polymerisations from initiator sites on the dextran backbone, without the requirement of protecting the hydroxyl groups. To overcome the common limitation of micelles for achieving high MTX loading capacities, a monomer with amine groups (AEM) was also successfully incorporated on the polymer chains. Micelles obtained showed an appropriate size for being internalized by cells and were able to encapsulate a hydrophobic probe in their nuclei. In addition, except for the case of dextran–PNIPAAm–AEM, the copolymers were very cytocompatible in non-tumoural mammalian cells at concentrations up to 0.1 mg mL−1. MTX-loaded polymeric micelles showed pH- and temperature-responsive release and were slowly internalized into tumour cells, being more cytotoxic than solutions of the free drug. AEM in the chains grafted to the dextran enhanced cellular uptake. Overall the results indicate that an adequate balance in the proportion of temperature-sensitive grafted chains and of AEM moieties may lead to dextran-based polymeric micelles suitable as MTX nanocarriers.Acknowledgements
Work supported by MICINN (SAF2014-52632-R) and FEDER. B. Blanco-Fernandez is grateful to the Spanish Ministry of Education for a FPU grant. The Instituto de Ortopedia y Banco de Tejido Musculoesquelético (USC) is acknowledged for help with the cell cultures. CA thanks the UK Engineering and Physical Sciences Research Council (EPSRC) for a Leadership Fellowship (EP/H005625/1).References
- S. S. Abolmaali, A. M. Tamaddonm and R. Dinarvand, Cancer Chemother. Pharmacol., 2013, 71, 1115–1130 CrossRef CAS PubMed.
- C. M. Paulos, J. A. Reddy, C. P. Leamon, M. J. Turk and P. S. Low, Mol. Pharm., 2006, 66, 1406–1414 CrossRef PubMed.
- J. A. Moura, C. J. Valduga, E. R. Tavares, I. F. Kretzer, D. A. Maria and R. C. Maranhao, Int. J. Nanomed., 2011, 6, 2285–2295 CAS.
- S. Jain, R. Mathur, M. Das, N. K. Swarnakar and A. K. Mishra, Nanomedicine, 2011, 6, 1733–1754 CrossRef CAS PubMed.
- P. Srisuk, P. Thongnopnua, U. Raktanonchai and S. Kanokpanont, Int. J. Pharm., 2012, 427, 426–434 CrossRef CAS PubMed.
- Y. Zhang, T. Jin and R. X. Zhuo, Colloids Surf., 2005, 44, 104–109 CrossRef CAS PubMed.
- Y. I. Jeong, D. H. Seo, D. G. Kim, C. Choi, M. K. Jang and J. W. Nah, Macromol. Res., 2009, 17, 538–543 CrossRef CAS.
- C. F. v. Nostrum, Soft Matter, 2011, 7, 3246–3259 RSC.
- Y. Y. J. Zou, Y. K. Li, W. Ji, C. K. Chen, W. C. Law, P. N. Prasad and C. Cheng, Biomater. Sci., 2015, 3, 1078–1084 RSC.
- T. C. Lai, H. Cho and G. S. Kwon, Polym. Chem., 2014, 5, 1650–1661 RSC.
- A. R. Khan, J. P. Magnusson, S. Watson, A. M. Grabowska, R. W. Wilkinson, C. Alexander and D. Pritchard, Polym. Chem., 2014, 5, 5320–5329 RSC.
- N. Chan, S. Y. An and J. K. Oh, Polym. Chem., 2014, 5, 1637–1649 RSC.
- Y. Kim, M. H. Pourgholami, D. L. Morris, H. Lu and M. H. Stenzel, Biomater. Sci., 2013, 1, 265–275 RSC.
- F. Z. J. Chen, J. Ouyang, J. Kong, W. Zhong and M. M. Q. Xing, J. Mater. Chem., 2012, 22, 7121–7129 RSC.
- X. Zhang, Z. Zhang, Z. Zhong and R. Zhuo, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2687–2696 CrossRef CAS.
- M. Yokoyama, J. Exp. Clin. Med., 2011, 3, 151–158 CrossRef CAS.
- H. Maeda, J. Controlled Release, 2012, 164, 138–144 CrossRef CAS PubMed.
- A. J. Clark, D. T. Wiley, J. E. Zuckerman, P. Webster, J. Chao, J. Lin, Y. Yen and M. E. Davis, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 3850–3854 CrossRef CAS PubMed.
- C. Alvarez-Lorenzo and A. Concheiro, Chem. Commun., 2014, 50, 7743–7765 RSC.
- L. M. Randolph, M. P. Chien and N. C. Gianneschi, Chem. Sci., 2012, 3, 1363–1380 RSC.
- S. J. T. Rezaeia, M. R. Nabida, H. Niknejadb and A. A. Entezamic, Int. J. Pharm., 2012, 437, 70–79 CrossRef PubMed.
- Y. P. Li, K. Xiao, W. Zhu, W. B. Deng and K. S. Lam, Adv. Drug Delivery Rev., 2014, 66, 58–73 CrossRef CAS PubMed.
- C. Alvarez-Lorenzo, B. Blanco-Fernandez, A. M. Puga and A. Concheiro, Adv. Drug Delivery Rev., 2013, 65, 1148–1171 CrossRef CAS PubMed.
- V. Vázquez-Dorbatt, J. Lee, E. W. Lin and H. D. Maynard, ChemBioChem, 2012, 13, 2478–2487 CrossRef PubMed.
- C. Houga, J. F. L. Meins, R. Borsali, D. Taton and Y. Gnanou, Chem. Commun., 2007, 29, 3063–3065 RSC.
- Y. I. Jeong, D. H. Kim, C. W. Chung, J. J. Yoo, K. H. Choi, C. H. Kim, S. H. Ha and D. H. Kang, Int. J. Nanomed., 2011, 6, 1415–1427 CrossRef CAS PubMed.
- Z. Zhao, Z. Zhang, L. Chen, Y. Cao, C. He and X. Chen, Langmuir, 2013, 29, 13072–13080 CrossRef CAS PubMed.
- S. Tan, D. Zhao, D. Yuan, H. Wang, K. Tu and L. Q. Wang, React. Funct. Polym., 2011, 71, 820–827 CrossRef CAS.
- H. T. Duong, F. Hughes, S. Sagnella, M. Kavallaris, A. Macmillan, R. Whan, J. Hook, T. P. Davis and C. Boyer, Mol. Pharm., 2012, 9, 3046–3061 CrossRef CAS PubMed.
- G. Y. Liu, C. J. Chen, D. D. Li, S. S. Wang and J. Ji, J. Mater. Chem., 2012, 22, 16865–16871 RSC.
- A. Zhang, Z. Zhang, F. Shi, C. Xiao, J. Ding, X. Zhuang, C. He, L. Chen and X. Chen, Macromol. Biosci., 2013, 13, 1249–1258 CrossRef CAS PubMed.
- S. M. Sagnella, H. Duong, A. MacMillan, C. Boyer, R. Whan, J. A. McCarroll, T. P. Davis and M. Kavallaris, Biomacromolecules, 2014, 15, 262–275 CrossRef CAS PubMed.
- D. Bontempo, G. Masci, P. Leonardis, L. Mannina, D. Capitani and V. Crescenzi, Biomacromolecules, 2006, 7, 2154–2161 CrossRef CAS PubMed.
- Z. H. Wang, W. B. Li, J. Ma, G. P. Tang, W. T. Yang and F. J. Xu, Macromolecules, 2011, 44, 230–239 CrossRef CAS.
- S. Çakmak, A. S. Çakmak and M. Gümüşderelioğlu, Mater. Sci. Eng., C, 2013, 33, 3033–3040 CrossRef PubMed.
- M. L. Patrizi, G. Piantanida, C. Coluzza and G. Masci, Eur. Polym. J., 2009, 45, 2779–2787 CrossRef CAS.
- F. Li, D. F. Pei, Q. R. Huang, T. F. Shi and G. Zhang, Carbohydr. Polym., 2014, 99, 728–735 CrossRef CAS PubMed.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC.
- A. L. H. Vihola, L. Valtola, H. Tenhu and J. Hirvonen, Biomaterials, 2005, 26, 3055–3064 CrossRef PubMed.
- J. P. Magnusson, A. Khan, G. Pasparakis, A. O. Saeed, W. Wang and C. Alexander, J. Am. Chem. Soc., 2008, 130, 10852–10853 CrossRef CAS PubMed.
- X. Lu, S. Gong, L. Meng, C. Li, F. Liang, Z. Wu and L. Zhang, Eur. Polym. J., 2007, 43, 2891–2900 CrossRef CAS.
- Y. Chen, X. Sha, W. Zhang, W. Zhong, Z. Fan, Q. Ren, L. Chen and X. Fang, Int. J. Nanomed., 2013, 8, 1463–1476 Search PubMed.
- Z. Tyeklar, R. R. Jacobson, N. Wei, N. N. Murthy, J. Zubieta and K. D. Karlin, J. Am. Chem. Soc., 1993, 115, 2677–2689 CrossRef CAS.
- A. J. Graaf, E. Mastrobattista, C. F. van Nostrum, D. T. S. Rijkers, W. E. Hennink and T. Vermonden, Chem. Commun., 2011, 47, 6972–6974 RSC.
- Y. Z. Du, Q. Weng, H. Yuan and F. Q. Hu, ACS Nano, 2010, 4, 6894–6902 CrossRef CAS PubMed.
- Z. H. Wang, Y. Zhu, M. Y. Cahi, W. T. Yang and F. J. Xu, Biomaterials, 2012, 33, 1873–1883 CrossRef CAS PubMed.
- C. Porsch, S. Hansson, N. Nordgren and E. Malmstroem, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 2011, 52, 434–435 CAS.
- W. Ji, D. Panus, R. N. Palumbo, R. Tang and C. Wang, Biomacromolecules, 2011, 12, 4373–4385 CrossRef CAS PubMed.
- W. Li, J. Li, J. Gao, B. Li, Y. Xia, Y. Meng, Y. Yu, H. Chen, J. Dai, H. Wang and Y. Guo, Biomaterials, 2011, 32, 3832–3844 CrossRef CAS PubMed.
- R. Nagarajan and K. Ganesh, Macromolecules, 1989, 22, 4312–4325 CrossRef CAS.
- L. Y. Qiu, R. J. Wang, C. Zheng, Y. Jin and L. Q. Jin, Nanomedicine, 2010, 5, 193–208 CrossRef CAS PubMed.
- G. Gaucher, P. Satturwar, M. C. Jones, A. Furtos and J. C. Leroux, Eur. J. Pharm. Biopharm., 2010, 76, 147–158 CrossRef CAS PubMed.
- M. F. Francis, L. Lavoie, F. M. Winnik and J. C. Leroux, Eur. J. Pharm. Biopharm., 2003, 337–346 CrossRef CAS.
- C. Porsch, S. Hansson, N. Nordgren and E. Malmstroem, Polym. Chem., 2011, 2, 1114–1123 RSC.
- W. Yuan, J. Zhang, H. Zou, T. Shen and J. Ren, Polymer, 2012, 53, 956–966 CrossRef CAS.
- B. Trzebicka, D. Szweda, S. Rangelov, A. Kowalczuk, B. Mendrek, A. Utrata-Wesolek and A. Dworak, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 614–623 CrossRef CAS.
- K. E. Schmalenberg, L. Frauchiger, L. Nikkhouy-Albers and K. E. Uhrich, Biomacromolecules, 2001, 2, 851–855 CrossRef CAS PubMed.
- J. H. Hwang, C. W. Choi, H. W. Kim, D. H. Kim, T. W. Kwak, H. M. Lee, C. H. Kim, C. W. Chung, Y. I. Jeong and D. H. Kang, Int. J. Nanomed., 2013, 8, 3197–3207 Search PubMed.
- H. Gao, M. Li and Y. Wu, J. Appl. Polym. Sci., 2011, 120, 2448–2458 CrossRef CAS.
- K. L. Thompson, E. S. Read and S. P. Armes, Polym. Degrad. Stab., 2008, 93, 1460–1466 CrossRef CAS.
- H. Wei, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, Prog. Polym. Sci., 2009, 34, 893–910 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Hydrodynamic radius of dextBr0.4 eq., dextran–PNIPAAm, dextran–PNIPAAm–AEM, dextran–PEGMA and dextran–PEGMA–AEM solutions in PBS (0.5 mg mL−1) at 20, 37 and 55 °C determined by DLS; and AFM micrographs of dextran–PNIPAAm–AEM at 20 °C (A) and 37 °C (B). See DOI: 10.1039/c7ra00696a |
| ‡ Current Address: Department of Radiology, Michigan State University, East Lansing, 48823,USA. |
| § Current address: School of Chemistry, University of Birmingham, Birmingham B15 2TT, UK. |
| This journal is © The Royal Society of Chemistry 2017 |