
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent developments in chemical reactivity of N,N-dimethylenamino ketones as synthons for various heterocycles
Hatem M. Gaber
a,
Mark C. Bagley
b,
Zeinab A. Muhammad
a and
Sobhi M. Gomha
 *c
*c
aNational Organization for Drug Control and Research (NODCAR), P.O. Box 29, Cairo, Egypt. E-mail: hatem.gaber@yahoo.com; zeinab.a.muhammad@gmail.com
bDepartment of Chemistry, University of Sussex, School of Life Sciences, Falmer, Brighton, East Sussex BN19QJ, UK. E-mail: M.C.Bagley@sussex.ac.uk
cDepartment of Chemistry, Faculty of Science, Cairo University, Giza, 12613, Egypt. E-mail: s.m.gomha@gmail.com
First published on 6th March 2017
Abstract
The current review presents recent progress in the utility of N,N-dimethyl enaminones as building blocks for a diverse range of acyclic, carbocyclic, five- and six-membered heterocyclic a broad range of heterocyclic and fused heterocyclic derivatives. Most importantly, these N,N-dimethyl analogues have proven to be of biological interest and provide an access to new class of biologically active heterocyclic compounds for biomedical applications. All of these topics are drawn from the recent literature till 2016.
 Hatem M. Gaber | Hatem Moustafa Gaber was born in November 1969 in Menoufia and obtained his education in Cairo, Egypt. He graduated with B.Sc. degree in chemistry from Helwan University, Cairo in 1991 with general grade of “Very Good”. After getting his M.Sc. degree in organic chemistry in 1995, he had a permanent position as a Senior Research Assistant at the National Organization for Drug Control and Research (NODCAR) in Cairo. He earned a Ph.D. degree in organic chemistry from Cairo University in 1999. He was promoted to the rank of a Researcher in organic chemistry in 1999 and an Associate Professor in 2006, before taking up his current position as a Professor of organic chemistry at NODCAR in 2011. Also, he was awarded three Postdoctoral Research Fellowships from several institutions, including an Egyptian Government Scholarship in the USA, working as a Visiting Fellow with Professor Dr C. Parkanyi at Florida Atlantic University in Florida to do research in the area of open-chain nucleoside chemistry with a grant from the General Egyptian Administration for Scholarships in Egypt (2001–2002); then a World Laboratory Scholarship in Denmark taken at Nucleic Acid Center at the University of Southern Denmark in Odense for the period of one year (2003–2004) under the direction of Professor Dr E. Pedersen in the field of nucleotide/oligonucleotide chemistry with support from ICSC–World Laboratory grant in Switzerland; moreover, a Research Fellowship in the UK at Cardiff University in Cardiff for the period of three years (2009–2009), working with Professor Dr M. Bagley on synthetic approaches towards new anti-cancer and antibiotic agents in addition to the synthesis and properties of anticonvulsant enaminones. In 2012, Dr Gaber was appointed Head of the Organic Chemistry Department at NODCAR, then Head of the General Pharmaceutical Chemistry Division (Dean) in 2014. His research interests include heterocyclic and medicinal chemistry with emphasis on the design and synthesis of novel bioactive heterocycles and the study of their biomedical applications with the aim of developing new therapeutic agents. |
 Mark C. Bagley | Mark C. Bagley was educated at the University of Oxford (BA 1991; DPhil 1994 with Prof. L M Harwood), with post-doctoral studies at the Université de Genève (with Prof. W Oppolzer), Loughborough and Exeter (with Prof. C J Moody), before being appointed as a Lecturer in Organic Chemistry at Cardiff University in 1999. He stayed at Cardiff for 13 years, being promoted to a Senior lectureship in Organic Chemistry in 2004 and a Reader in 2006, before taking up his current position as Professor of Organic Chemistry at the University of Sussex in February 2012. |
 Zeinab A. Muhammad | Zeinab A. Muhammad was born in Cairo, Egypt. She graduated from B.Sc, then she carried out his M.Sc. and Ph.D. studies in 2012 and 2015, respectively in the field of organic synthesis. Work at National Organization for Drug Control and Research (NODCAR), P.O. Box 29, Cairo, Egypt. She joined the scientific school of Prof. A. S. Shawali in 2009 and she has published scientific papers and reviews all in international journals in the fields of physical organic chemistry, chemistry of hydrazonoyl halides and bioactive heterocyclic chemistry. |
 Sobhi M. Gomha | Sobhi Mohamed Gomha was born in Fayoum, Egypt. He graduated from Cairo University, Egypt in 1995 then he carried out his M.Sc. and Ph.D. studies in 2002 and 2006 respectively, at Cairo University in the field of organic synthesis. In 2011 he promoted to Associate Professor and in 2016 he was appointed as a full Professor of Organic chemistry at Cairo University. He joined the scientific school of Prof. A. S. Shawali in 1996 and he has published 106 scientific papers and reviews all in international journals in the fields of physical organic chemistry, chemistry of hydrazonoyl halides and bioactive heterocyclic chemistry (there are about 755 citations of his work from 2000 until February 2017 (h-index 16). |
1. Introduction
β-Aminovinyl ketones are versatile synthetic intermediates that combine the ambident nucleophilicity of enamines with the ambident electrophilicity of enones. As can be rationalized, these systems have “enamine” character, and can act as building blocks for the synthesis of various heterocycles such as pyridine, pyrimidine, and pyrrole derivatives.1 In addition, they also have “enone” character, and may act as acceptors in both 1,2- and 1,4-additions. In this way, β-aminovinyl ketones serve as scaffolds for annulation, and can provide access to systems such as pyrroles, indolizidines, quinolizidines, and perhydroindoles, all of which are common motifs in alkaloid structures.1,2N,N-Dimethyl derivatives of β-aminovinyl ketones are chemical compounds consisting of an amino group linked through a carbon–carbon double bond to a keto group.6 They are typical push–pull ethylenes in which the amine group pushes and the carbonyl pulls electron density. The chemistry of the enamino carbonyl group (1) is potentially an area of considerable scope when one considers that there are present in this moiety two electron-deficient centres (i.e. two electrophilic sites) at C-1 and C-3, while the C-2, carbonyl oxygen and amino functions are electron rich (i.e. three nucleophilic sites) (Fig. 1).3,4 They can thus function both as nucleophiles and as electrophiles, their versatility in either case being extended by their ability to show ambident reactivity.2 Accordingly, these β-acyl enamines proved to be suitable for a number of further synthetic transformations, mainly; hydrolysis to diones, reduction and condensation to fused heterocycles.
 | ||
| Fig. 1 General formula of N,N-dimethyl enaminone derivatives. | ||
A few review articles, describing the chemistry of β-aminovinyl ketones, have appeared in the literature,1,3,5,6 although hitherto with little emphasis on the reactivity of the corresponding N,N-dimethyl derivatives of β-aminovinyl ketones. Since a large number of developments in the use of these N,N-dimethyl derivatives in heterocyclic and medicinal chemistry have been reported recently, and in conjunction with our long-term continuing interest in exploring the synthetic applications of biologically active enamino compounds,7–19 the current review presents firstly recent progress in the utility of N,N-dimethyl enaminones as building blocks for a diverse range of acyclic, carbocyclic, five- and six-membered heterocyclic as well as condensed heterocyclic compounds, with particular emphasis on the useful chemical transformations of this class of N,N-disubstituted enaminones to the structurally related α-(arylhydrazono)-β-ketoaldehydes. Structural investigation for these aldehydes and some of other products has also been made. This is finally followed by a consideration of the ever-increasing chemotherapeutic potentials of these N,N-dimethyl derivatives and their biomedical applications as valuable synthons on the way to a variety of bioactive heterocyclic compounds. All of these topics are drawn from the recent literature till 2016.
2. Synthetic applications of N,N-dimethylenamino ketones
2.1. Preparation of acyclic compounds
Coupling21,22,32–34 of diazotized aromatic amines with the aminovinyl ketones 1d,f,g,h and 1q resulted in products of coupling and hydrolysis of the dimethylamine moiety. These products were shown, based on 1H NMR and 13C NMR, to exist as a mixture of anti and syn hydrazones with the anti form always prevailing.22 It is assumed that initially formed arylazo intermediates 2a–k are hydrolyzed, by the action of the aqueous base existing in the medium, into the arylhydrazonoaldehydes 3a–k (Scheme 1).
 | ||
| Scheme 1 | ||
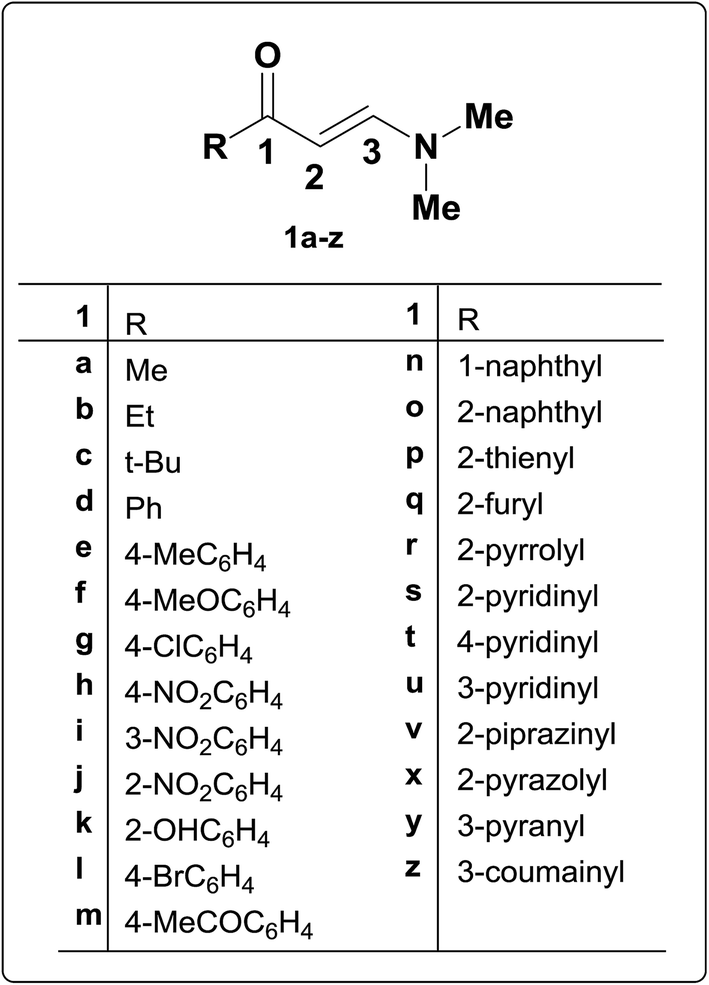
In addition, 3-(thiophen-2-yl)- or 3-aryl-propionaldehydes 4a–k were prepared via coupling of aminovinyl ketones 1p and 1e,g,h with aromatic diazonium salts.28 It is believed that assumed fixation of hydrazones by hydrogen bonding in DMSO solution is least likely although it was observed in almost every case of aroyl derivatives the presence of two NH signals integrating for a total of one proton with varying intensity in each case depending upon the nature of the substituent at the carbonyl moiety (Scheme 2). This may point to the existence of an equilibrium between the two forms 4C and 4D in DMSO solution.28
 | ||
| Scheme 2 | ||
Furthermore, it was found that diazotized anthranilonitrile (5a) or diazotized methyl anthranilate (5b) couples readily with the aminovinyl ketones 1a, 1d, 1p–s to yield products of coupling and hydrolysis of dimethylamino moiety. The coupling products can thus be formulated as the hydrazone forms 8A,B or potential tautomeric enol azo forms 7,8C or a mixture of one or more of these forms. The hydrazone structure 8A,B was established for all these products and the E-form 8A is believed to be the predominant form except for the product of coupling 1-methyl-3-dimethylaminoprop-2-enone (1a) with diazotized anthranilonitrile (5a) (Scheme 3). 1H NMR of this product show that it exists in DMSO as an equilibrium mixture of E-form 8A, Z-form 8B and the enol azo form 8C, as 1H NMR revealed three signals for a total of one proton, and it is interesting to note that the E-form was the major constituent in this equilibrium mixture (70%). All these forms are stabilized by hydrogen bonding.27,35
 | ||
| Scheme 3 | ||
Similarly, coupling of aminovinyl ketones either 1d,p with benzonitrile diazonium chloride (9), following recently reported procedure,21 produced the corresponding acyclic β-ketoaldehydes 10a,b (Scheme 4).36
 | ||
| Scheme 4 | ||
Also, 3-oxo-2-(arylhydrazono)pentanal 11a–h were obtained in 50–80% yields via coupling N,N-dimethyl enaminones either 1b or 1m with aromatic diazonium salts. 1H NMR of the products indicated that they exist at least in DMSO solution as mixtures of the anti-form 11 and syn-form 12 (Scheme 5). The anti-from generally predominated.29
 | ||
| Scheme 5 | ||
The diazotized 2-aminocyclohexenethiophene (13) coupled readily with N,N-dimethylenamino ketones 1a,d,p–r to yield the corresponding hydrazonopropanals 14a–e, respectively (Scheme 6).27
 | ||
| Scheme 6 | ||
It has been found that 5-methylisoxazole-3-diazonium chloride (15a) coupled readily with β-acyl enamines 1q,u to produce the expected β-ketoaldehydes 16a,b.33 However, a similar treatment of ketoenamines 1d and 1q resulted in the cyclization into pyrazolo[5,1-c][1,2,4]triazines 18a,b via the assumed intermediacy of acyclic aldehydes 17a,b (Scheme 7).33
 | ||
| Scheme 7 | ||
Elnagdi et al.37 reported on the reaction of 1-dimethylamino-5-arylpenta-1,4-diene-3-one 19a–c with diazonium ions in aqueous ethanol, where the dimethylamino group was substituted by the hydroxyl group and thus the corresponding 5-aryl-2-arylhydrazono-3-oxopent-4-enals 20a–c were isolated. Compounds 20a–c were presumably formed by hydrolysis of the enamine primary products. The 1H NMR indicated the presence of a mixture of both E and Z forms of these pentenals in approximately equivalent ratios (Scheme 8).37
 | ||
| Scheme 8 | ||
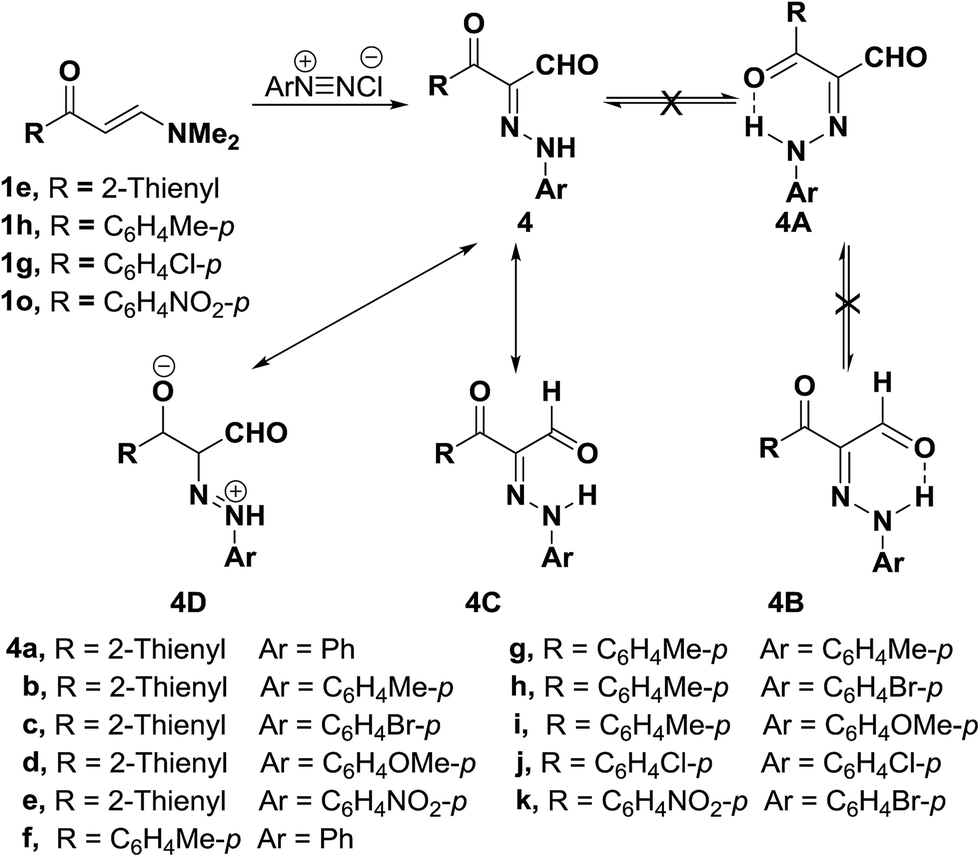
Also, enaminophthalimidoacetone derivative 21 readily underwent coupling with aromatic diazonium salts in the presence of sodium hydroxide to yield the analogous aldehydes 22a–d in acceptable yields (Scheme 9).38
 | ||
| Scheme 9 | ||
Compounds 24a,b were obtained in the same way as aldehydes 22 where the N,N-dimethylenamino ketone 21 coupled with heterocyclic diazonium salts 23a,b to give rise to the corresponding hydrazones 24a,b in good yields (Scheme 10).38
 | ||
| Scheme 10 | ||
As recently reported by our research group,7 pyrazole enaminone derivative 1v also coupled with p-chlorobenzenediazonium chloride to provide the pyrazoloylhydrazone derivative 25, in 82% yield (Scheme 11).
 | ||
| Scheme 11 | ||
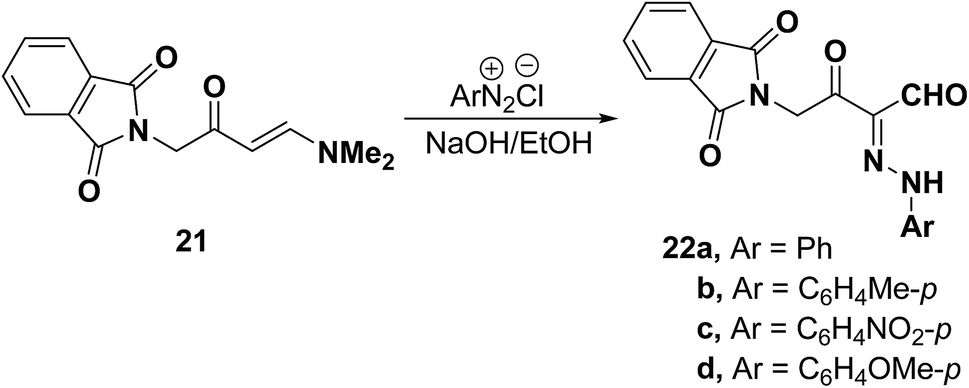
Trials to couple enaminone of the coumarin 1z with benzenediazonium chloride failed. However, p-nitrobenzenediazonium chloride coupled readily with 1z to yield the respective arylhydrazonopropanals that proved to exist as an equilibrium mixture of the anti-form 26A and the syn-form 26B (Scheme 12).25
 | ||
| Scheme 12 | ||
An interesting synthesis of arylhydrazonals is the formation of 27a–e39 on coupling of enaminal 27a or enaminoester 27b with various aromatic diazonium salts. Compounds 28a–e were assigned the indicated hydrazone structure in preference to a potentially tautomeric enolazo structure. This assignment is based on 1H NMR and 13C NMR spectra which revealed signals for two formyl protons and carbons in 28a–d and one such signal in the spectra of 28e (Scheme 13).40
 | ||
| Scheme 13 | ||
On the other hand, when dimethylaminomethylene derivative 29 was coupled with benzenediazonium chloride, the phenylhydrazone 32 was formed. It is believed that, as a result of nitrogen lone pair donation to enamine β-carbon, the latter becomes sufficiently nucleophilic and intermediate diazonium salt 30 is initially formed. This then readily hydrolyses into the azo derivative 31 that then undergoes Japp–Klingemann cleavage to yield the final isolated product 32 (Scheme 14).41
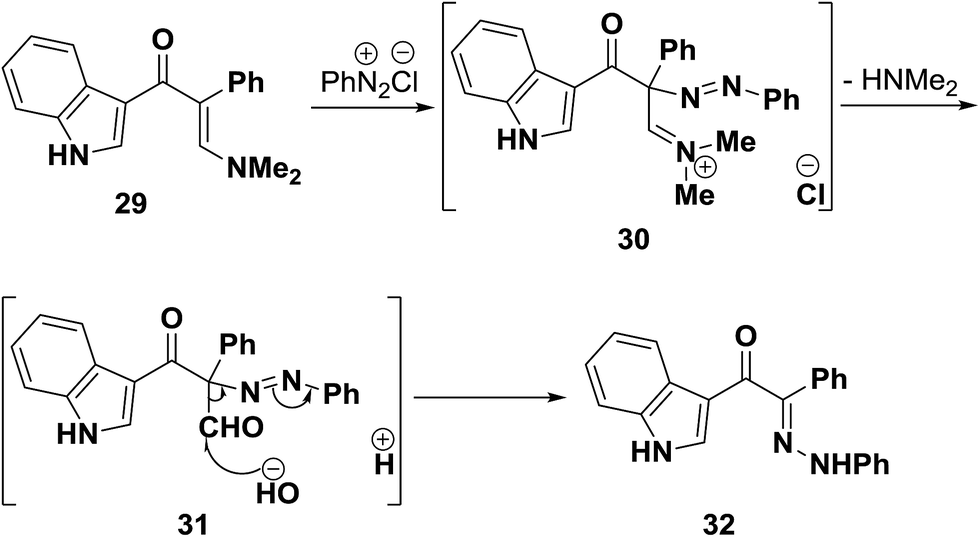 | ||
| Scheme 14 | ||
 | ||
| Scheme 15 | ||
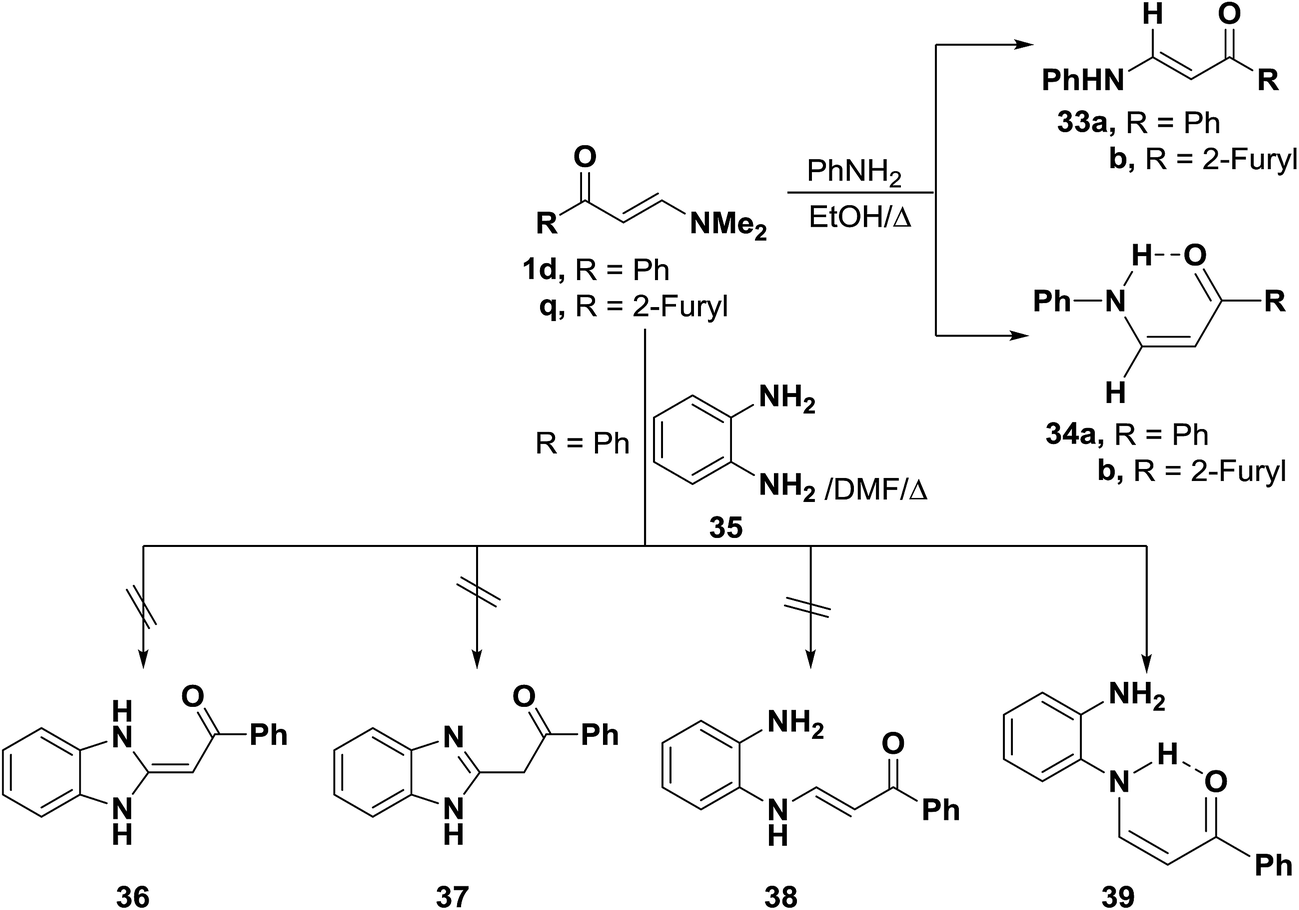
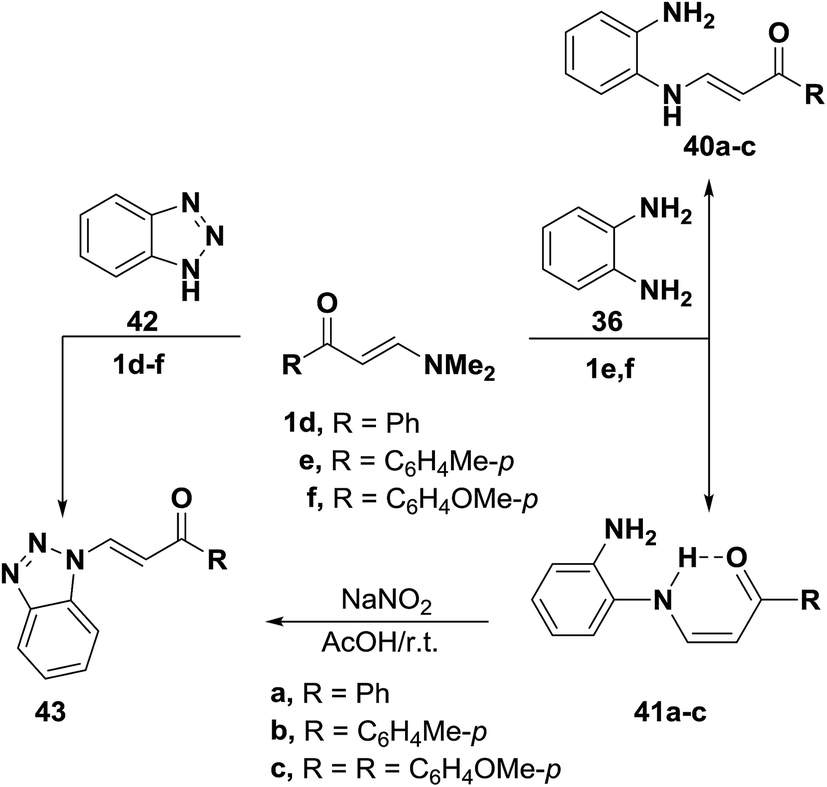
In accordance with the previous observation,42 β-aminovinyl ketones 1e,f reacted with o-phenylenediamine (36) to afford products of condensation via dimethylamine elimination. These were assigned cis structure 41 rather than trans structure 40 based on 1H NMR, which revealed signals for cis olefinic protons in their proper positions with J = 9 Hz. The predominance of this form may be due to fixation by hydrogen bonding.43 In contrast to this observation, it has been found that a similar treatment of ketones 1d–f with benzotriazole (42), instead of o-phenylenediamine (36), led to the formation of trans enaminones 43, which were also obtained from the diazotization of cis enaminones 41 with sodium nitrite in acetic acid under stirring at room temperature (Scheme 16).43
 | ||
| Scheme 16 | ||
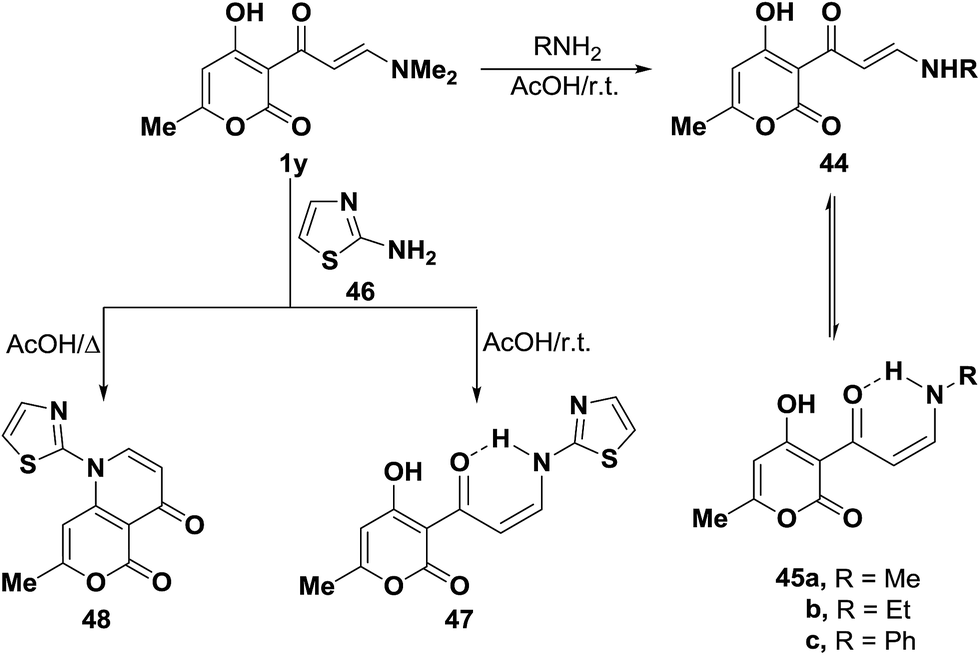
On the other hand, tertiary enaminone 1y reacted with methylamine, ethylamine and aniline in acetic acid under stirring at room temperature to yield the corresponding secondary enaminones, which were found to exist, in each case, as a mixture of the E- and Z-forms 44a–c and 45a–c. The existence of Z-form for these compounds is in contrast to the observed predominance of E-form for the starting 1y. This is attributed to the stabilization of Z-form for those compounds through hydrogen bonding.44 Interestingly, when compound 1y was similarly treated with 2-aminothiazole (46) in acetic acid under stirring for three hours at room temperature, only the acyclic Z-enaminone 47 was formed in exclusively based on considering J value for olefinic protons (J = 9 Hz). While bicyclic pyridine derivative 48 was isolated upon heating of 1y with 46 under reflux in acetic acid for two hours (Scheme 17).44
 | ||
| Scheme 17 | ||
Tertiary amines 49a,b were converted into the corresponding secondary amines 50a,b when heated at reflux with equivalent amount of aniline in ethanol for two hours in yields of 53% and 37%, respectively.45 Also, it has been found that a much better yield (71%) of product 50a could be obtained on microwave heating of 49 with equimolar amount of aniline in a domestic microwave oven at full power for two minutes.45 On the other hand, microwave heating of 49 with ammonium acetate for two minutes in a domestic microwave oven at full power produced the primary amine 51 in yield of 91%. The authors46 failed to obtain 51 by conventional heating with ammonium acetate in acetic acid (Scheme 18).
 | ||
| Scheme 18 | ||
Dimethylaminomethylene thiazolones 52a,b reacted with equivalent amounts of aromatic amines with reflux in acetic acid for one hour to yield the corresponding secondary amines 53a–e, respectively.47 Similar treatment with piperidine in refluxing ethanol for seven hours afforded the piperidino derivatives 54a,b, which were also obtained on reacting thiazolinones 55a,b directly with triethyl orthoformate and piperidine in DMF.47 Utilization of triethyl orthoformate and piperidine in DMF solution was found more economic and safer than using DMFDMA. It is believed that piperidine reacts with acetal forming non-isolable intermediate 56 which then condensed with 52 to give the final isolable products as depicted in Scheme 19.47,48
 | ||
| Scheme 19 | ||
Dimethylaminomethylene thiazolone 57 could be converted into the enamine 58 on treatment with p-toluidine in refluxing acetic acid for one hour.47 Nucleophilic displacement of the active dimethylamino group of 57 by cycloaliphatic amines, in refluxing ethanol for seven hours, resulted in the formation of the corresponding derivatives of piperidino and morpholino 59a,b, respectively. Structure 59b was confirmed by X-ray crystal determination (Scheme 20).47
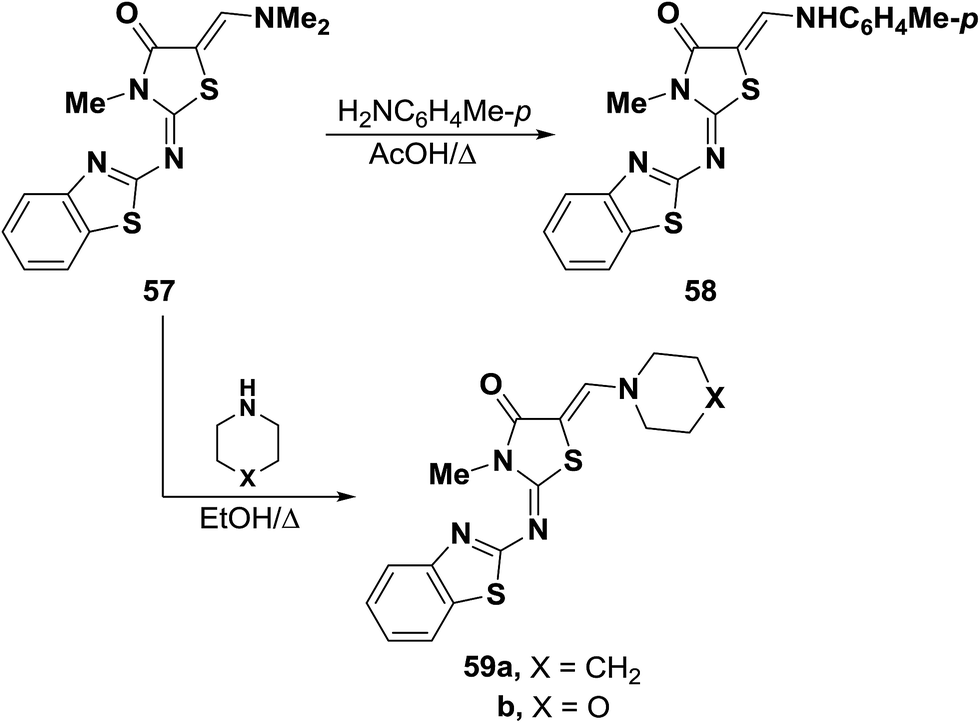 | ||
| Scheme 20 | ||
In addition, interaction of dimethylaminomethylidenes 1d,g,j and 1p with anthranilonitrile (60a) or methylanthranilate (60b) yielded the cis enaminones 61a–f, respectively (Scheme 21). Attempted cyclization of 61a–c into the corresponding quinolines failed. Successful cyclization of 61d into the quinolinone 63 could be affected, on heating for five minutes in domestic microwave oven at full power. When compounds 1d,g,j and 1q were similarly treated with 2-amino-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carbonitrile (64a) or its 3-carboxylate derivative 64b, the corresponding cis enaminones 65a–e were obtained. Again, 1H NMR indicated the predominance of the cis-form (c.f. Table 1).49
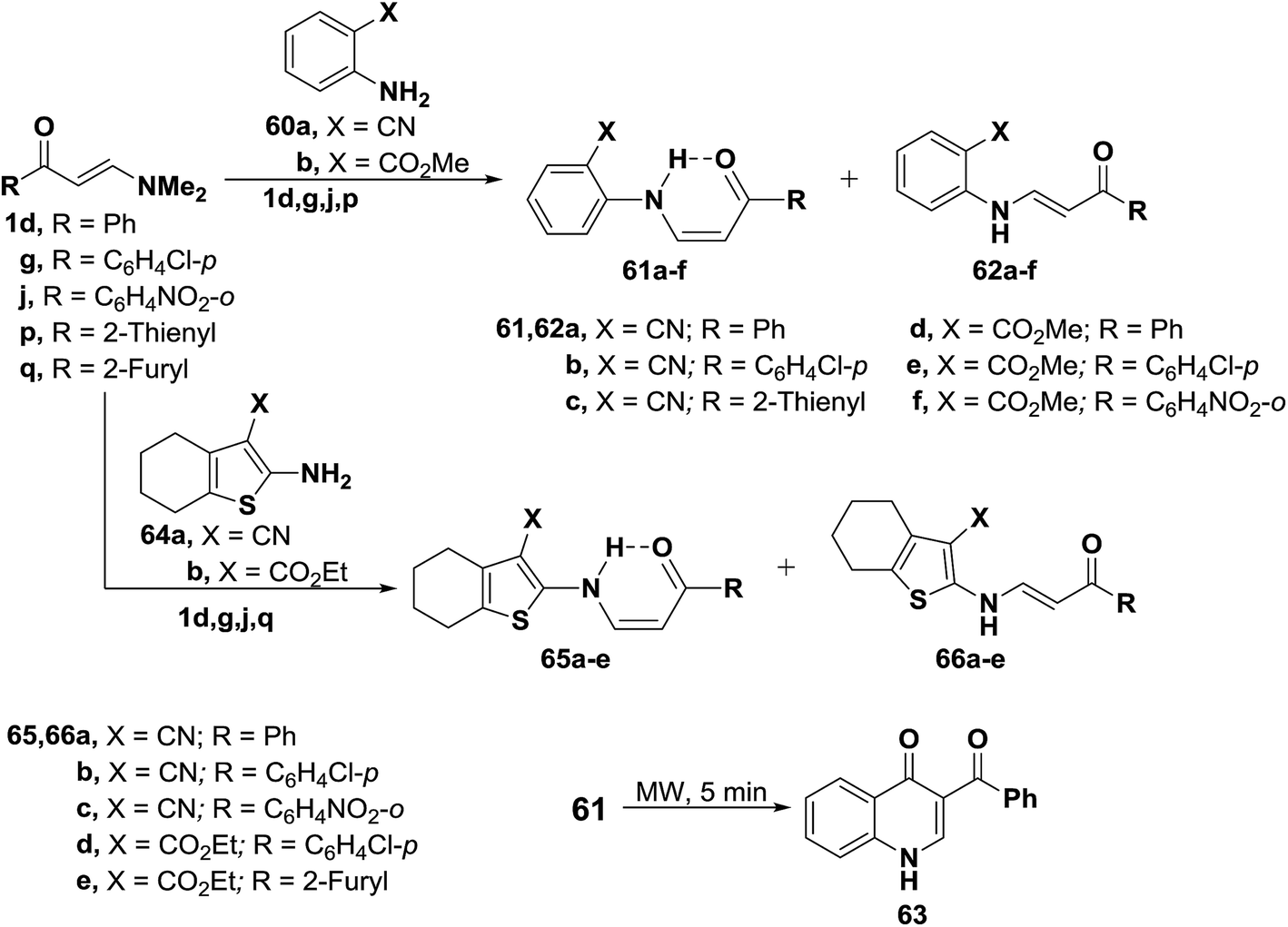 | ||
| Scheme 21 | ||
| Compounds | cis-Form | trans-Form |
|---|---|---|
| 61b | 7 | 1 |
| 61e | 4 | 1 |
| 61f | 4.2 | 1 |
| 65a | 6.5 | 1 |
| 65c | 6 | 1 |
| 65 | 4 | 1 |
Reacting enaminonitrile 67 with ethyl glycinate 68 in ethanol/potassium carbonate solution yielded the analogous ethyl ester 69, which could not be cyclized into the pyrrole derivative 70 under a variety of conditions.41 Also, compound 67 reacted with aminothiazole derivative 71 and with aminothiophene derivative 64b to afford the secondary amines 72B and 73B, respectively. Although 72B, 73B may also exist as 72A or 73A, the hydrogen bonded form 72B and 73B seems more stable (Scheme 22).50
 | ||
| Scheme 22 | ||
On the other hand, when compounds 1d,e,g,j,x were similarly treated with piperidine (74a), only the E-enaminones 75a–d, respectively, were formed in exclusively as indicated from the coupling constant values for olefinic doublets. No trace of Z-form was observed in this reaction. Analogously, compounds 75f,g were formed from the reaction of 1d,x with morpholine (74b) (Scheme 23).49,51–53
 | ||
| Scheme 23 | ||
Interaction of 2-aminobenzothiazole (76) with aminovinyl ketones 1d and 1q provided the heteroaromatic aminoenones 77a,b. Similarly, compounds 1d and 1q,s were transformed to the corresponding acyclic aminoenones 80a–c by treatment with 3-amino-5-methylisoxazole (79) (Scheme 24). These products 77a,b and 80a–c are believed to exist in equilibrium with enols 78a,b and 81a–c, which are stabilized through hydrogen bonding.54
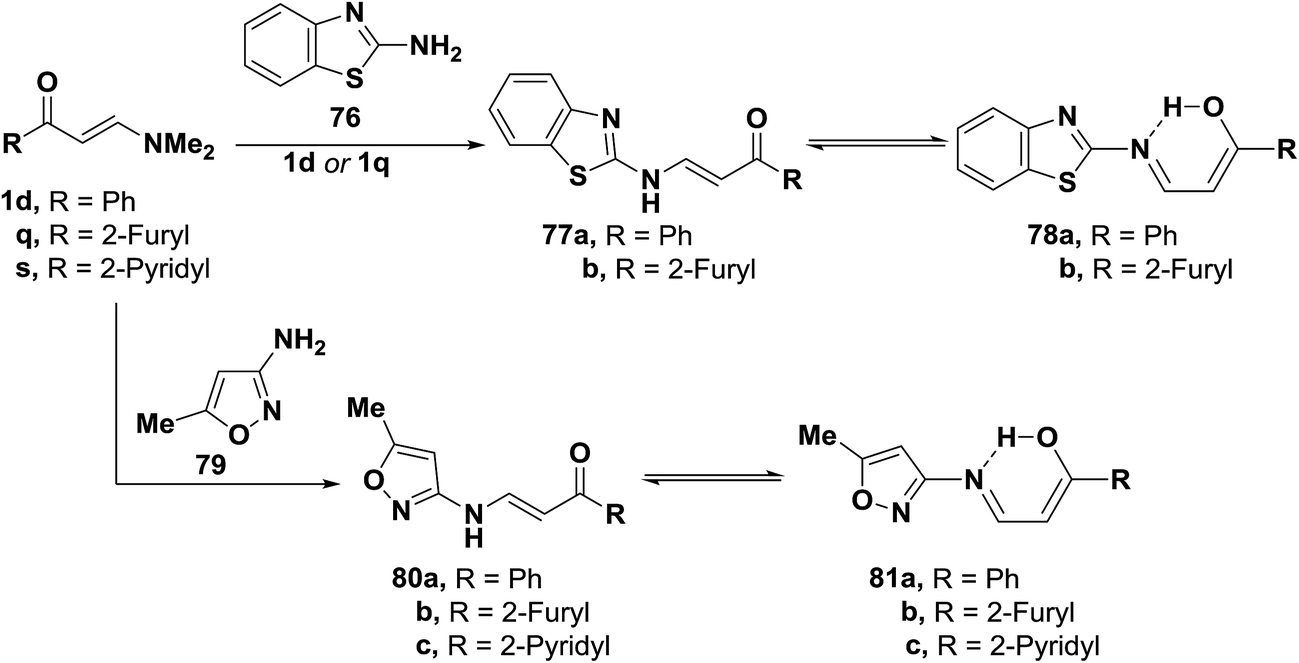 | ||
| Scheme 24 | ||
N,N-Dimethylenamino ketones 1e,g and 1p reacted with aminopyrazoles 82a,b to yield the acyclic enaminones 83a–f. The structure of which proved to exist in the cis-form based on considering J values for olefinic protons (J = 9 Hz).49 Compound 83a–f readily cyclized into the bicyclic pyrimidines 84a–f on reflux in pyridine solution in the presence of concentrated hydrochloric acid (Scheme 25).49
 | ||
| Scheme 25 | ||
Treatment of N,N-dimethylamino derivatives 1d–f with thienocoumarin 85 in acetic acid at room temperature furnished products of addition and dimethylamine elimination in 80–85% yields. IR and 1H MNR spectra of these products indicated involvement of the amino function in this reaction. Therefore, structure 86a–c was suggested for those products. On the other hand, when the reaction of 1d–f with 85 was conducted in refluxing acetic acid, solid products 86a–c were isolated by filtering the hot solution in 33–43% yields. When the mother liquor was left to stand at room temperature, the C-1 alkylation products 87a–c were isolated in 32–36% yields. These products were found to be isomeric with 86a–c. Both IR and 1H NMR indicated that the amino function was not involved into the reaction.53 Compounds 86a–c rearranged into the corresponding α,β-unsaturated ketones 87a–c on prolonged boiling under reflux in dioxane solution in the presence of diethylamine or in pyridine in 60–70% yields. Better conversion yields could not be achieved under a variety of conditions. It is thus believed that products 266 are kinetic products, while 71 are the thermodynamic ones. Conversion of 86 into 87 in basic media is believed to proceed via base addition across the double bond and elimination of 85, thus allowing more of the thermodynamic product to be formed (Scheme 26).55
 | ||
| Scheme 26 | ||
An interesting reaction leading to cis-enaminone is the condensation of aminothienopyridazines (88a,b) with aminovinyl ketone 23 to yield products of condensation via dimethylamine elimination for which structures 89 or 90 seemed possible. Structure 90 was established for those products based on the 1H NMR and IR spectral data that revealed involvement of amino function in the reaction. Typical for secondary enaminone, compounds 90a,b existed solely in Z-form as this form is fixed by hydrogen bond and this preferred over sterically and stereoelectronically fixed E-form.54 Further confirmation of this structure assignment was obtained via successful conversion of 90b into the thiophene 91 through hydrolysis of the alkylated amino moiety (Scheme 27).56
 | ||
| Scheme 27 | ||
Dienone 19 underwent analogous reactions with aminothienopyridazines 88a,b in a microwave oven at 560 W for 90 seconds to give rise to the expected N-alkylated products 92a,b.56 A similar treatment of 6a with benzothienocoumarin 93 led to the corresponding enaminone 94 (Scheme 28).56
 | ||
| Scheme 28 | ||
Aminothiophene derivatives 85, 88a,b and 93 reacted with the N,N-dimethylenamino ketones 95a,b in a similar fashion yielding the corresponding secondary enaminones 95a,b, 96a–d and 97a,b (Scheme 29).56
 | ||
| Scheme 29 | ||
In contrast to the behavior of aminothiophene toward N,N-dimethylenamino ketones, interaction55 of compounds 1d,e with 88a in acetic acid at reflux for three hours gave C-1 alkylation products 99a,b instead of the N-alkylated derivatives 98a,b that would be expected by analogy with the other reports42,56 on the reactivity of nitrogen nucleophiles toward electron poor olefins (Scheme 30).
 | ||
| Scheme 30 | ||
Similar to the behavior of 73a toward 1d,e enaminoketone 23 reacted with thienocoumarin 85 under microwave heating in the presence of few drops of acetic acid to produce only the C-1 alkylation product 100. This approach was also suitable for the preparation of another C-1 alkylation product 81, where reaction of 23 with benzothienocoumarin 92 under the same reaction conditions led to the corresponding α,β-unsaturated ketone 101 (Scheme 31).56
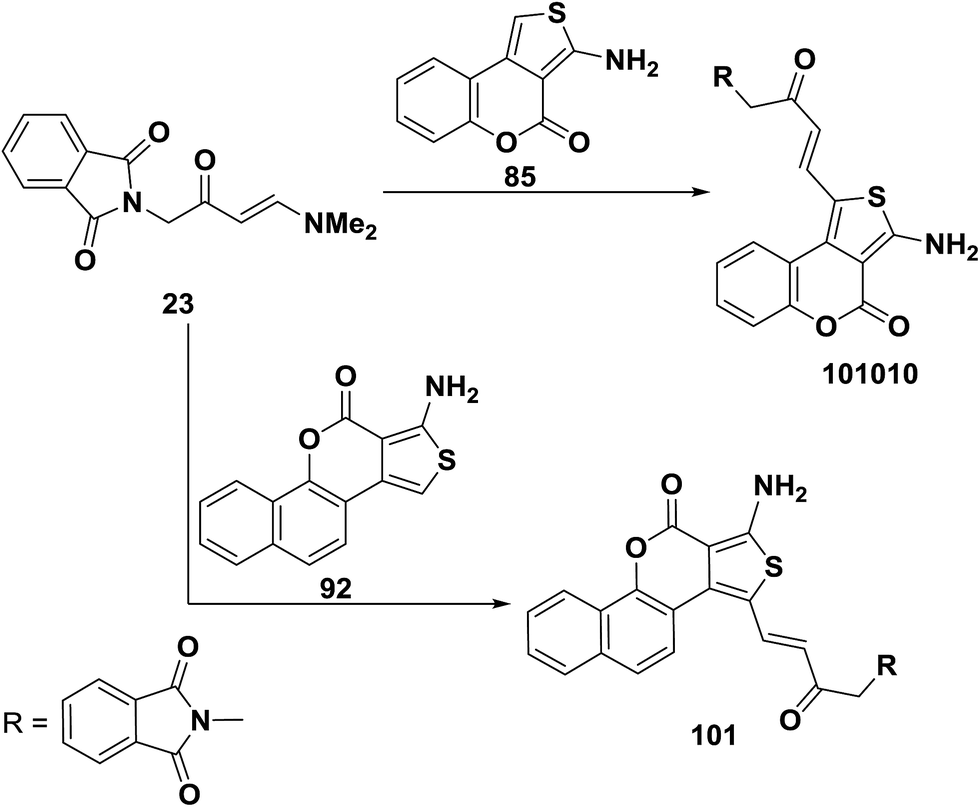 | ||
| Scheme 31 | ||
Also, compound 85 reacted with aryl vinyl ketones 103a,b, generated in situ from corresponding dimethylaminopropanone hydrochlorides 102a,b, in acetic acid/ethanol mixture at reflux for three hours to yield 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 adducts. These were stable on reflux in protic solvents and thus possible formation of cycloadducts similar to 104 was ruled out (Scheme 32). Moreover, 13C NMR of the reaction products indicated presence of only two sp3 carbons while in cycloadducts 104 four such carbons should have been observed. Therefore, the products for this reaction could be assigned structure 105.55
:1 adducts. These were stable on reflux in protic solvents and thus possible formation of cycloadducts similar to 104 was ruled out (Scheme 32). Moreover, 13C NMR of the reaction products indicated presence of only two sp3 carbons while in cycloadducts 104 four such carbons should have been observed. Therefore, the products for this reaction could be assigned structure 105.55
 | ||
| Scheme 32 | ||
It can thus be concluded that outcome of reactions of amines with electron poor olefins are dependent on nature of reagents used. Amino function, C-1 as well as the diene system are all possible sites of attack.56
:1 adducts. These products could be formulated as pyran structure 107 or enamine structure 109. However, structure 109 was assigned on the basis of 1H NMR spectra, in which two olefinic doublets with a J value of 13 Hz were observed indicating that the trans olefinic moiety has not been involved in the reaction and hence this excluded completely the possibility of pyran structure 107 for this reaction products. Consequently, formation of 109 is assumed to occur via the addition of the active methylene reagents to carbonyl groups in 1a, 1d or 1p,q and subsequent water elimination to furnish α,β-unsaturated nitriles 108. Water eliminated in this process then hydrolyses the cyano group in the intermediates 108 affording the final isolated products 109.57,58 This structure was also confirmed by preparing the same reaction products 109c–e via condensation of cyanoacetamide with 1d or 1p,q under the same reaction conditions (Scheme 33).58
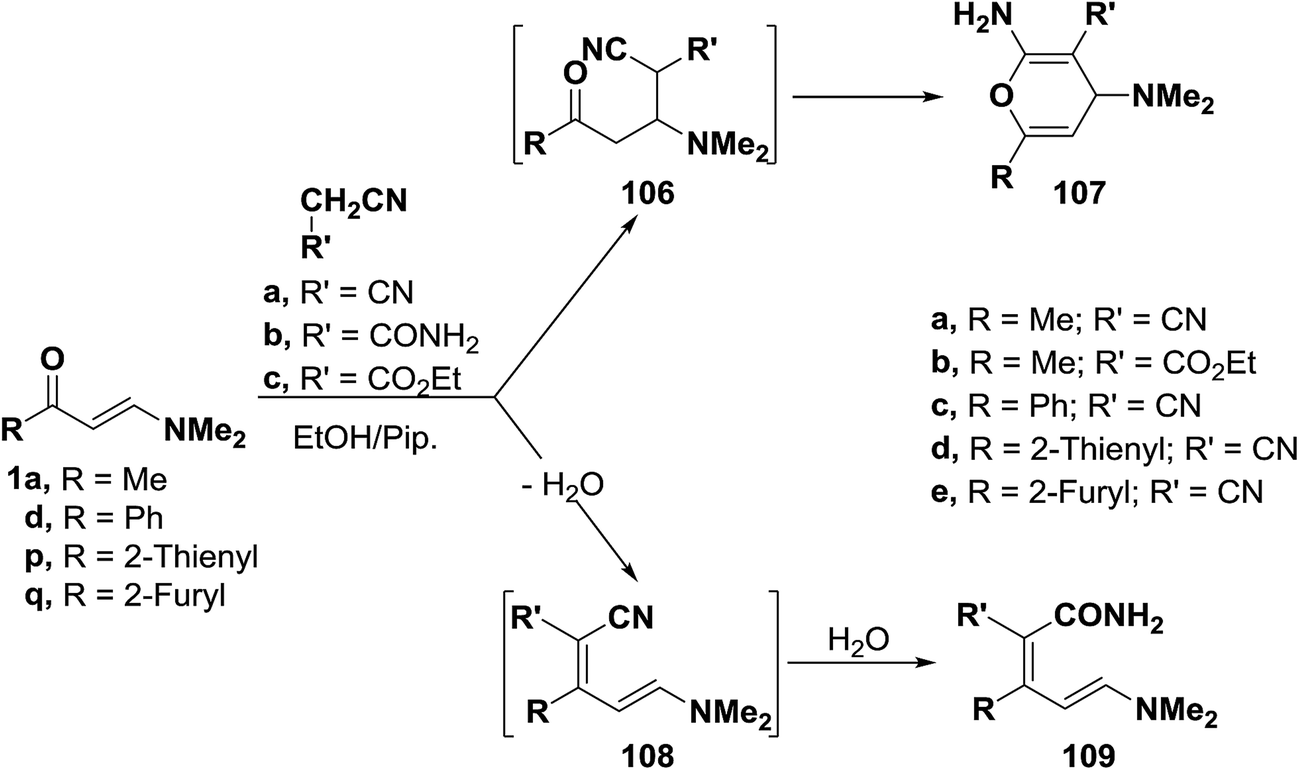 | ||
| Scheme 33 | ||
Interaction of enaminonitrile 67b with ethyl thioglycolate in ethanol/potassium carbonate solution yielded the ethyl ester 110. Trials to effect cyclization of 110 into 111 under a variety of conditions failed. In protic medium 110 decomposed and in aprotic medium 100 was recovered unreacted (Scheme 34).41 Moreover, compound 109 reacted also with 1-methyl-6,7-dimethoxy-3,4-dihydroisoquinoline (112) in refluxing acetic acid to provide the respective dihydroisoquinolinylbutenenitrile derivative 113. The authors50 indicated that free donation around the single bond would allow for a trans-form that should in theory experience less steric interaction.
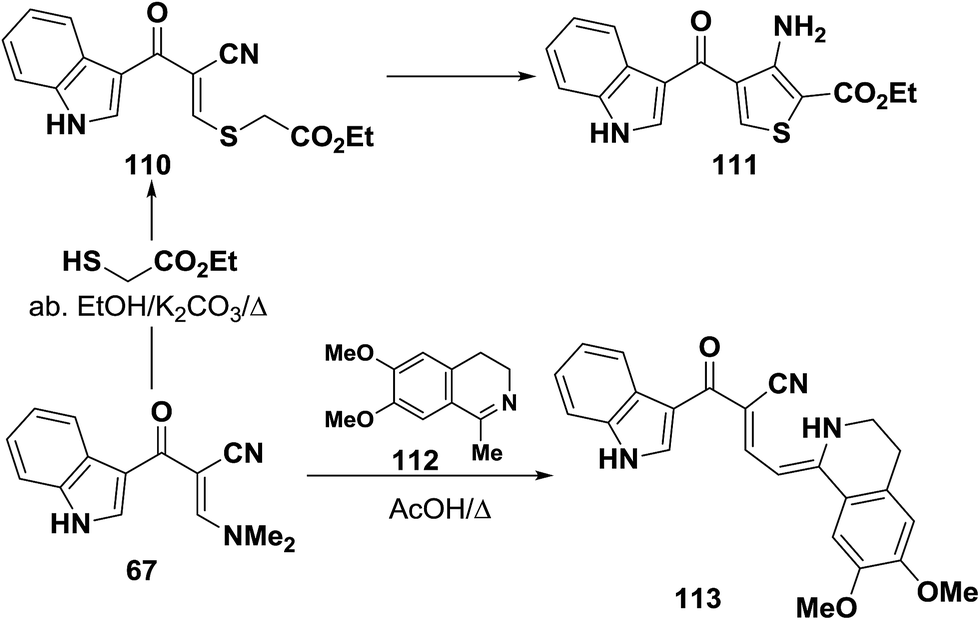 | ||
| Scheme 34 | ||
Also, interaction of enaminocarbonyl compound 1p with pyridazine-3-imine derivative 114 in refluxing ethanol containing a few drops of triethylamine led to the acyclic compound 115 via loss of a dimethylamine molecule, while on being heated in a mixture of aqueous acetic acid and hydrochloric acid these afforded the tricyclic product 116 (Scheme 35). The latter could be also obtained via cyclization of α,β-unsaturated compound 115 into 116 on boiling in aqueous acetic acid/hydrochloric acid mixture.59
 | ||
| Scheme 35 | ||
Interestingly, reaction of N,N-dimethylamino derivatives 1d and 1p with phenylisothiocyanate in DMF in the presence of potassium hydroxide afforded after acidification with hydrochloric acid two products, the major of which were found to be of molecular formulae corresponding to structure 120 or its tautomeric forms 117–119. However, thiol structure 120 better agreed with the obtained spectral data for these compounds, thus thione structures 117 and 118 could be ruled out. Structure 119 was also excluded on the basis of 13C NMR spectra of the isolated products, which revealed the absence of any sp3-hybridized carbon atoms. These results strongly support structure 120 for the major products. The minor products for this reaction were assigned structure 34 based on the elemental analyses and spectral data of the isolated reaction products (Scheme 36).60
 | ||
| Scheme 36 | ||
Unexpectedly, α-(dimethylamino)methylene-β-oxo nitriles 121a,b reacted with hydroxylamine hydrochloride in absolute ethanol in the presence of anhydrous sodium carbonate under reflux to give the acyclic hydroxylaminopropenonitriles 123a,b, in excellent yields, based on elemental analyses and spectral data of the isolated reaction products. It is of value to report here that all trials to convert compounds 123a,b into the corresponding isoxazoles 124 were unsuccessful. This can be attributed to the fact that compounds 123a,b are mainly existing in the anti-form as indicated from their 1H NMR spectra.61 On the other hand, reaction of 3-dimethylamino-2-benzoylpropenenitrile (121a) with N-substituted ureas or thioureas 125a–l in acidic medium yielded ureidopropenenitriles 126a–l, respectively (Scheme 37).62
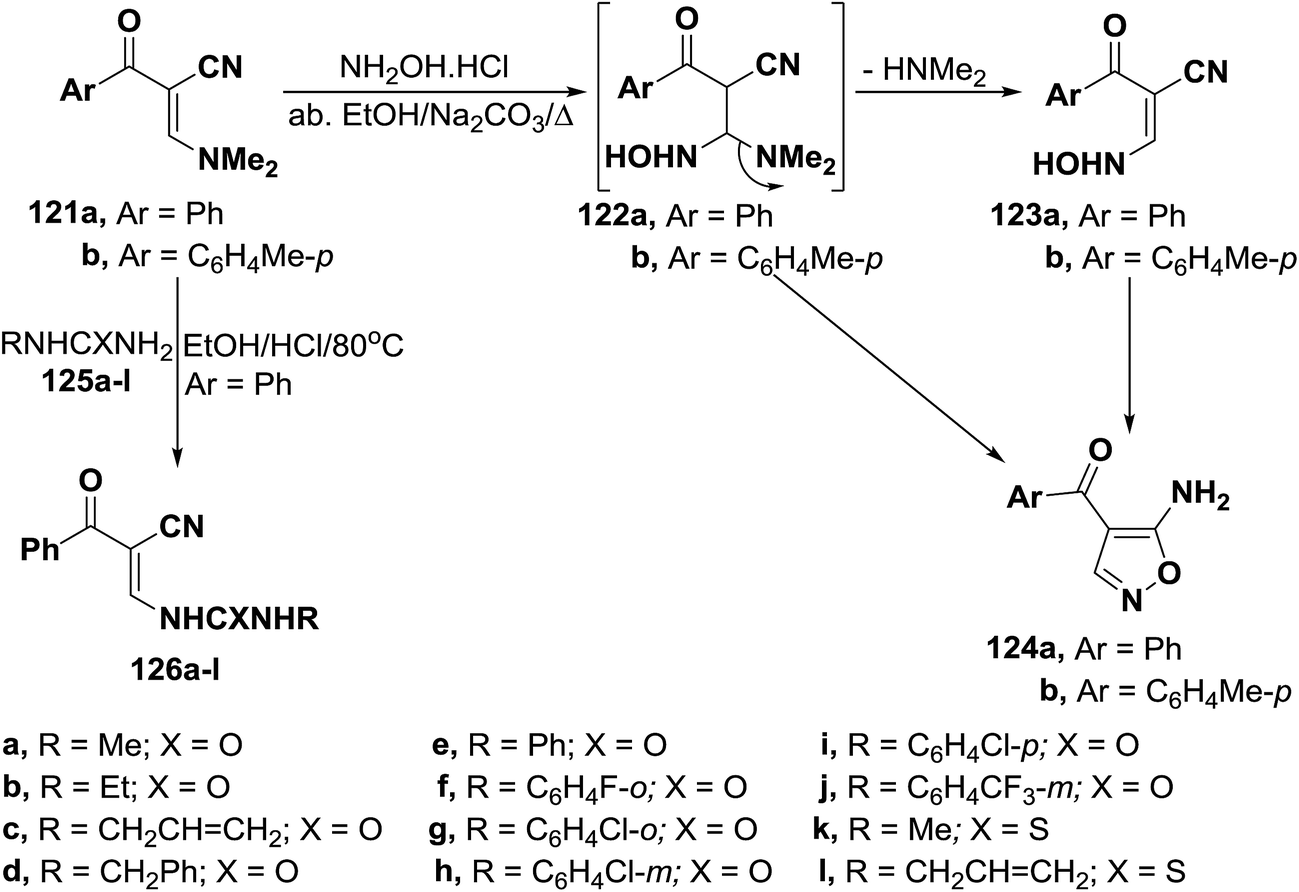 | ||
| Scheme 37 | ||
Reacting enaminoketones 1d,g and 1p,q with hydroxylamine hydrochloride in ethanolic sodium acetate at room temperature resulted in the formation of products of condensation via dimethylamine elimination. These products were found to exist, in each case, as an equilibrium mixture of the hydroxylamino E- and Z-forms (127 or 128). Assignment of these forms were based on their 1H NMR spectra, where two doublets for CH2 protons and other two triplets for oxime CH were detected (Scheme 38).63
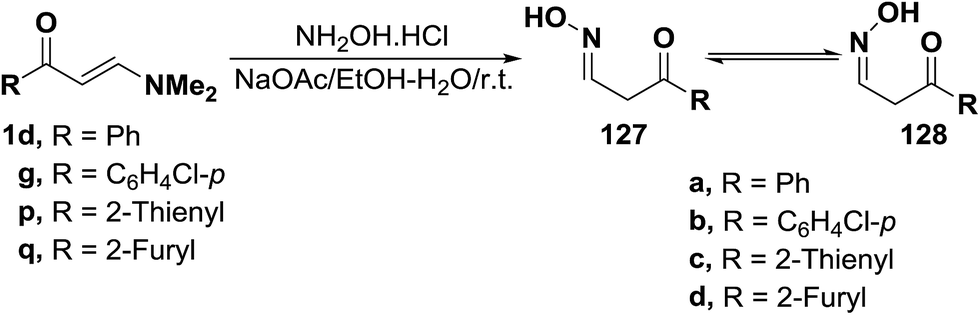 | ||
| Scheme 38 | ||
When N,N-dimethyl enaminone 1h was allowed to react with 2,4-bis(4-methoxyphenyl)-1,3,2,4-dithiadiphosphetane 2,4-disulfide, Lawesson's Reagent (LR), in benzene at room temperature, the enaminothione 129 was produced in a high yield (Scheme 39).64
 | ||
| Scheme 39 | ||
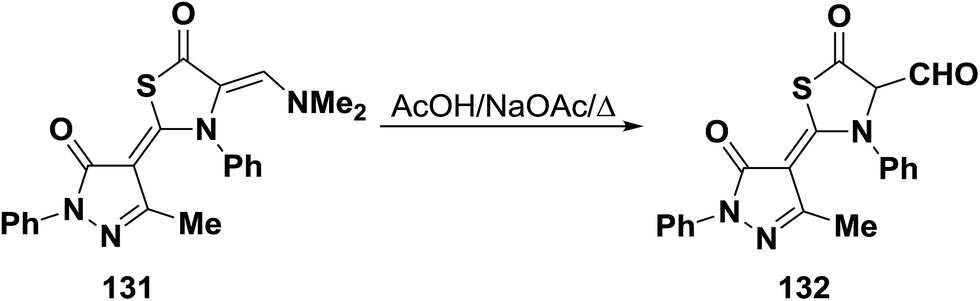
N,N-Dimethyl enamine 131 underwent hydrolysis upon boiling in acetic acid containing sodium acetate to give the bifunctional thiazolidinone-α-carboxaldehyde derivative 132 (Scheme 40).65
 | ||
| Scheme 40 | ||
2.2. Preparation of carbocyclic compounds
As mentioned earlier in this review, β-aminovinyl ketones 1d–f underwent self-condensation on reflux in acetic acid yielding the 1,3,5-trisubstituted benzene derivatives 135a–c.43,66,67 The electron rich C-2 in one molecule of 1d, 1e or 1f adds to the electron deficient C-3 in another molecule, forming the intermediates 133a–c. It is most likely that these intermediates 133 reacted, in each case, swiftly with a third molecule of 1, yielding the intermediates 134 that lose three molecules of dimethylamine, leading eventually to the triaroylbenzene derivatives 135. It is believed that acidity of the reaction mixture has prompted such trimerization of enaminones into triaroylbenzenes (Scheme 41). | ||
| Scheme 41 | ||
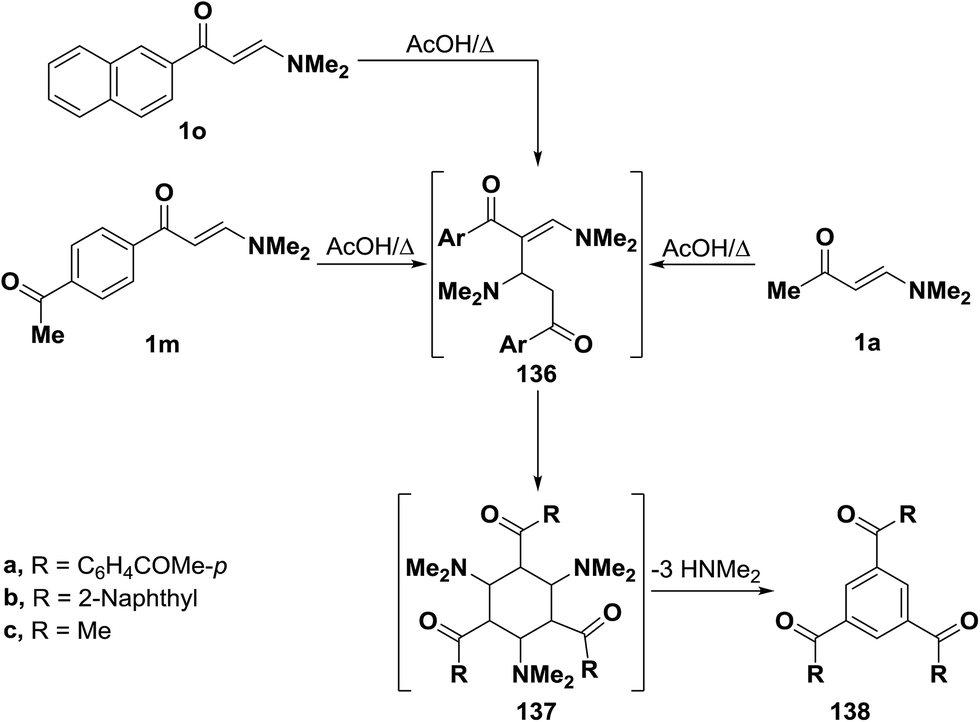
By using the synthetic sequence as was suggested for the synthesis of 135a–c, the trisubstituted benzene derivatives 138a–c were formed from aminovinyl ketones 1m, 1o and 1a, respectively.66–68 Formation of intermediates 136 and 137 is suggested although direct concerted 2 + 2 + 2 cycloaddition leading directly to 137 can not be overlooked (Scheme 42).49
 | ||
| Scheme 42 | ||
This approach was also suitable for the preparation of the 1,3,5-trisubstituted benzenes 140a–c, where reaction of the enaminone-derived heterocycles, such as thieno, furo and pyrido enaminones 1p,q,s in refluxing acetic acid led to the corresponding trisubstituted derivatives 140a–c. The reaction takes place by condensation of three moles of each enaminone 1p,q or 1r to form the corresponding intermediate 139 which loses, in each case, 3 moles of dimethylamine, aromatizes and affords the final isolated product 140a, 140b or 140c (Scheme 43).49,67
 | ||
| Scheme 43 | ||
Furthermore, it has been found that heating a mixture of 141 and an excess of an aryl ethynyl ketone 142 (∼8 equivalents) in toluene resulted in smooth trimerization to afford the linked 1,3,5-triaroylbenzenes 143a–d in good isolated yields, especially given the fact that six new C–C bonds are formed during the course of the reaction. Both electron rich (X = OMe) and electron deficient (R = NO2) aryl ethynyl ketones proved to be suitable reactants, although nitro-substituted linked triaroylbenzenes were isolated in slightly lower yields (Scheme 44).69
 | ||
| Scheme 44 | ||
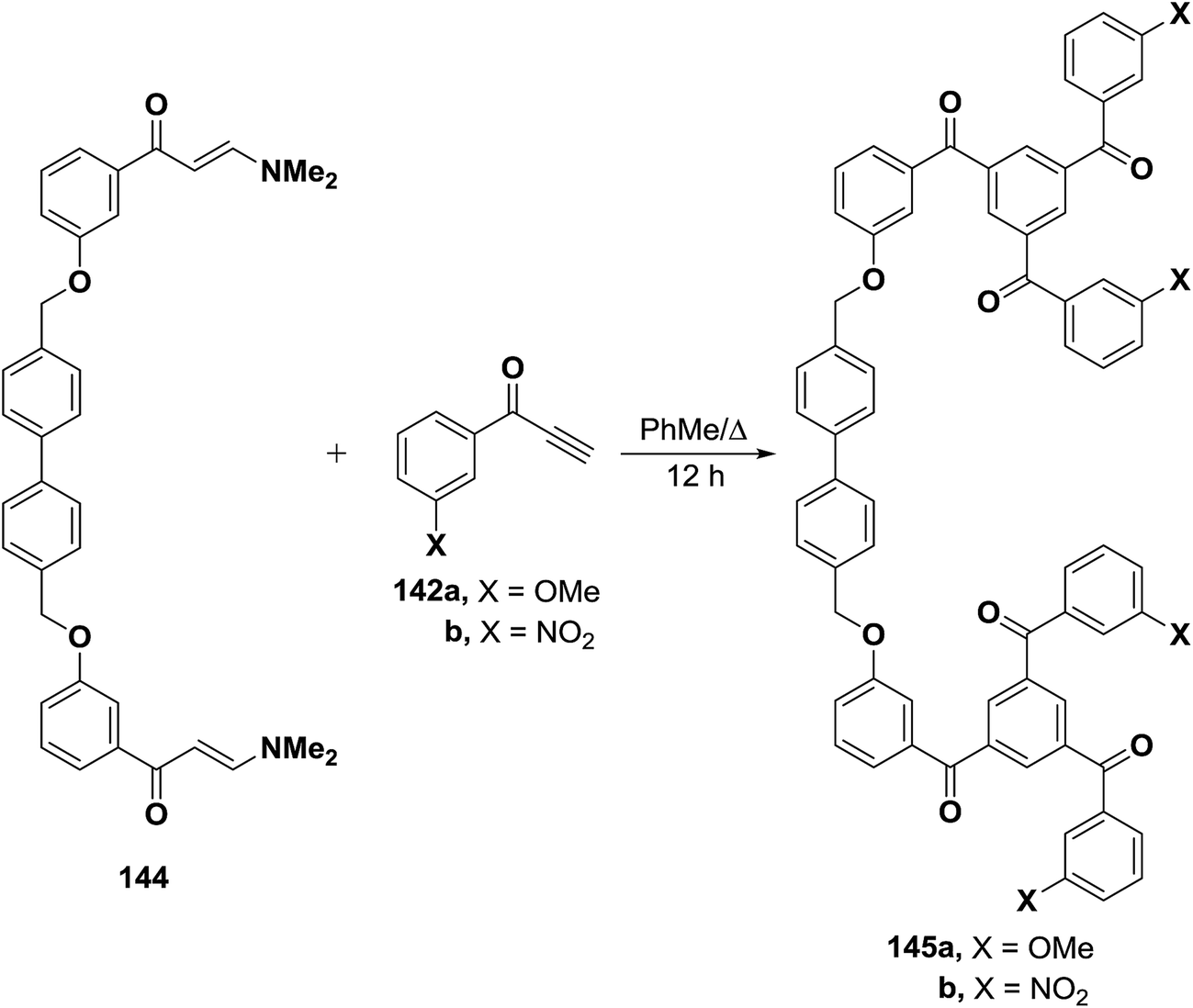
A similar treatment of the bis(enaminone) 144 with aryl ethynyl ketones 142a,b resulted in the formation of the corresponding 1,3,5-triaroylbenzenes 145a,b (Scheme 45).69
 | ||
| Scheme 45 | ||
This protocol was also suitable for the preparation of tris(1,3,5-triaroylbenzene) derivative 147 in which the individual cyclotrimers are connected via a 1,3,5-trisubstituted phenyl ring (Scheme 46).69
 | ||
| Scheme 46 | ||
Reaction of 3-dimethylaminoacrylaldehyde (28a) with aminothienocoumarin 85 in DMF at reflux for four hours gave a product of cycloaddition and dimethylamine elimination for which structure 148 was established. Similarly, condensed aminothiophene 91 reacted with 3-dimethylaminoacrylaldehyde (28a) under the same applied reaction conditions to afford the analogous cycloaddition product 149.56 Structures 138 and 149 were confirmed by preparing the same reaction products via alternative synthetic routes involving the reaction of acrylaldehyde (150) with condensed aminothiophenes 85 and 92, respectively, under the same reaction conditions (Scheme 47).56
 | ||
| Scheme 47 | ||
Lewis acid-catalyzed [3 + 2] cycloaddition of donor–acceptor cyclopropanes 151 and enamines 1d,o in methylene chloride in presence of MgI2 yielded nitrogen-functionalized cyclopentane derivatives 152 in good yields (Scheme 48).70
 | ||
| Scheme 48 | ||
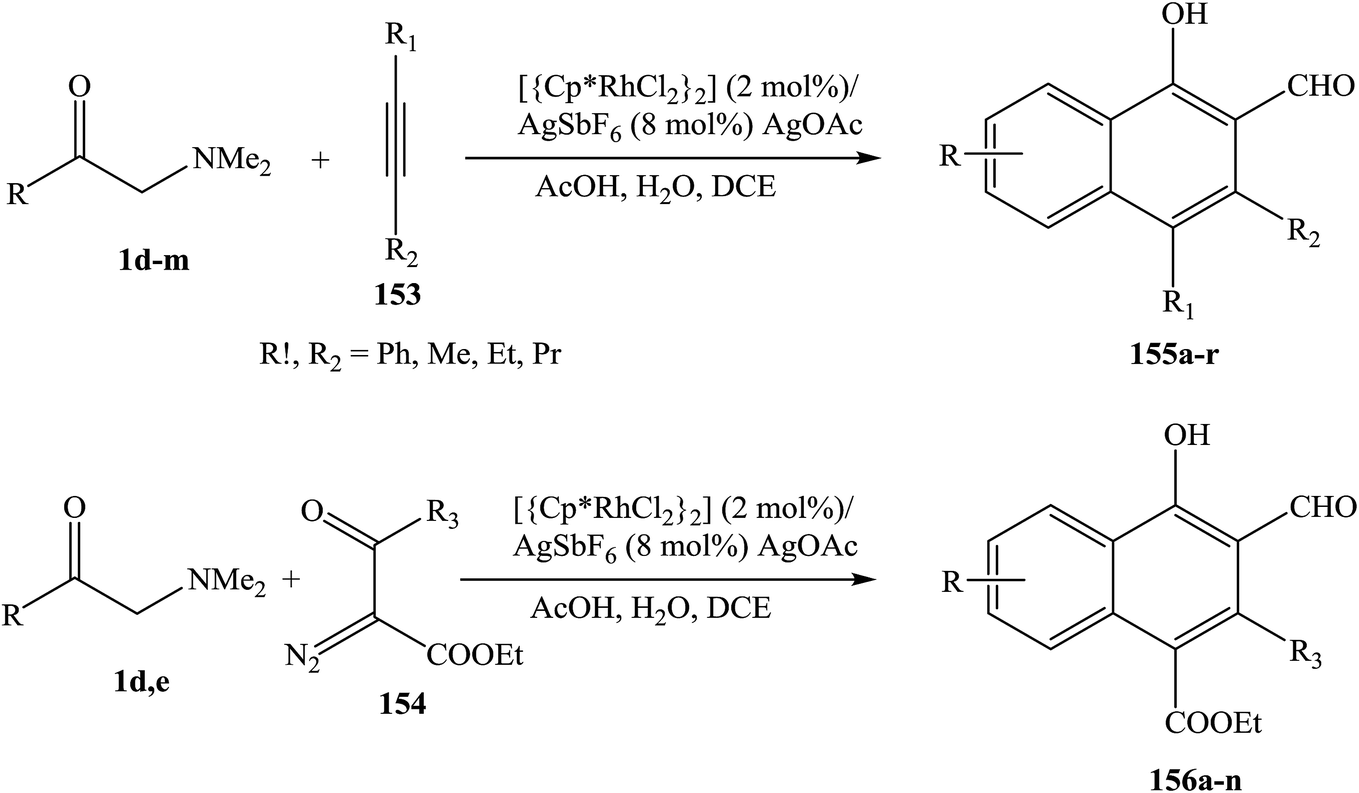
The use of enaminones as effective synthons for a directed C–H functionalization is reported. Proof-of-concept protocols have been developed for the Rh III-catalyzed synthesis of naphthalenes, based on the coupling of enaminones 1 with either alkynes 153 or α-diazo-β-ketoesters 154. Two inherently reactive functionalities (hydroxy and aldehyde groups) are integrated into the newly formed cyclic framework and a broad range of substituents are tolerated, rendering target products 155, 156 readily available for further elaboration (Scheme 49).71
 | ||
| Scheme 49 | ||
2.3. Preparation of heteroaromatic compounds
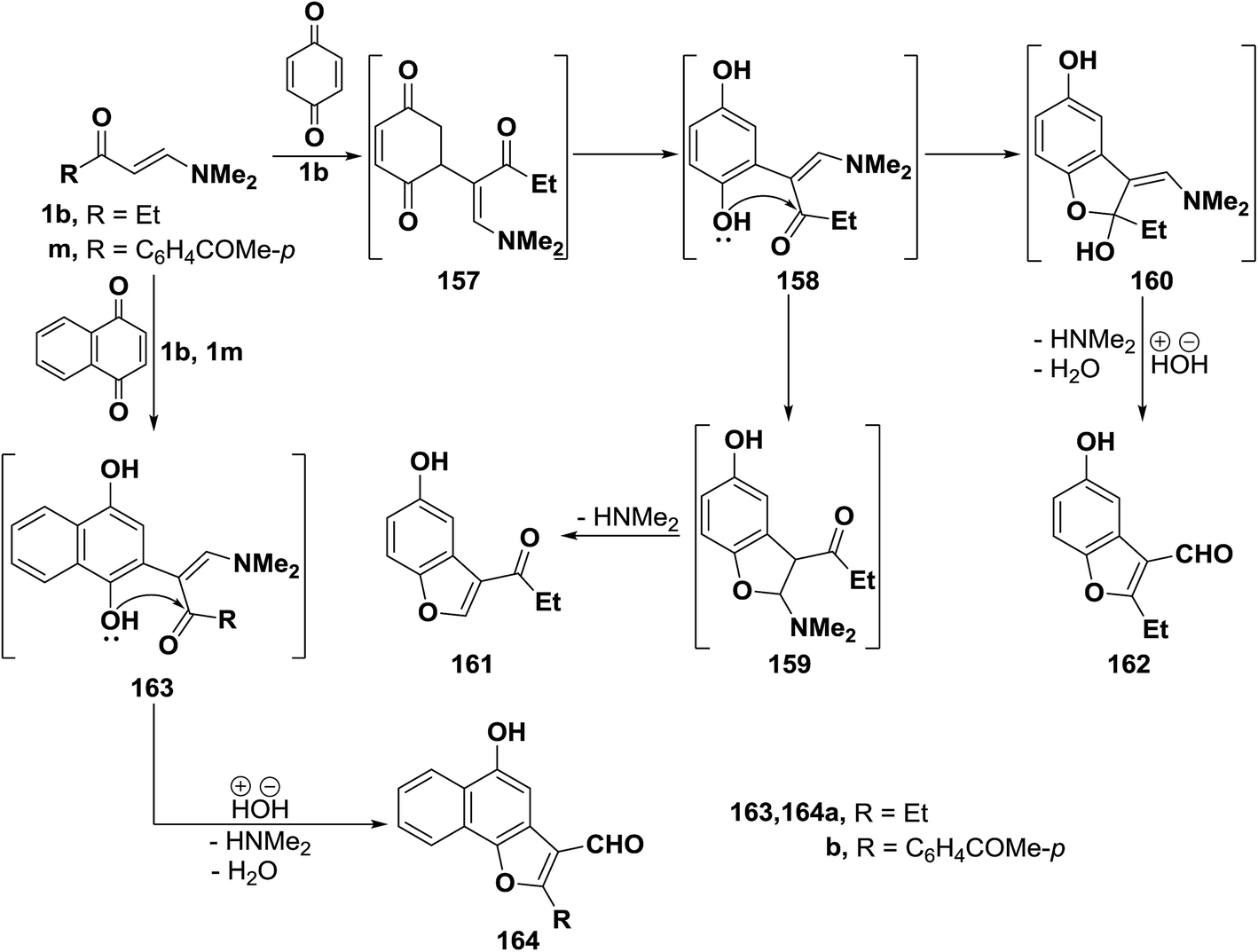
β-Aminovinyl ketones are important synthetic intermediates, particularly in heterocyclic chemistry.6,72 Heterocycles prepared from those ketones include carbazolequinone alkaloids,73 tricyclic benzo[a]quinolizines,74 pyrroles,75 benzodizepines,76 pyrimidines,77 pyridines,78 isoxazoles,79 quinolines,80,81 1,3-thiazines,82 furans,83 benzothiazoles,84 pyrazoles,14,85–87 triazole,88 isochromanes,89 and 1,4-diazepines.90 In this regard, many other recent examples are herewith provided including the preparation of five and six membered heterocycles and some of their condensed derivatives.2.3.1.1. Preparation of furans. Interaction of β-aminovinyl ketones with quinones represents an interesting approach to furan ring system. Many scientific publications in this area of investigation have been found. Thus, e.g., treating vinyl ketone 1b with p-benzoquinone, in acetic acid with stirring at room temperature, yielded a product of addition via dimethylamine and water elimination, this product can thus be formulated as 161 or isomeric 162. However, the benzofuran-3-al structure 162 was assigned for that product on the basis of its elemental analysis and spectra data. Formation of 162, as illustrated in Scheme 111, may be explained to occur via initial addition of electron rich C-2 in the enaminone 1b to the electron-deficient quinone carbon, forming the non-isolable acyclic adduct 157, followed by subsequent enolization would take place, affording the dihydroxy intermediate 158. The latter underwent intramolecular ionic heterocyclization at room temperature via nucleophilic attack by the OH function on the carbonyl carbon rather than the methylene CH, yielding the N,N-dimethylaminomethylidene intermediate 160, accompanied by hydrolysis with the release of dimethylamine and water, leading eventually to the final isolable benzofuranal derivative 162.68
Similar to the behavior of β-aminovinyl ketone 1b toward p-benzoquinone, compounds 1b and 1p also reacted with 1,4-naphthoquinone to produce the corresponding furanals 164a,b through the intermediate Michael adducts 163a,b (Scheme 50).68
 | ||
| Scheme 50 | ||
In addition, dienones 19a–c reacted with p-benzoquinone, in acetic acid with stirring at room temperature, to yield products of addition via dimethylamine and water elimination, for which structure 167a–c was considered as indicated from 1H NMR spectra where a formyl-H was observed, in each case, at δ 9.0 (1H), δ 9.21 (1H) or δ 9.17 (1H) ppm. It is thus believed that p-benzoquinone initially adds to electron rich C-2, yielding acyclic dihydroxy intermediates 165a–c which then cyclize exclusively into benzofuranals 167a–c rather than the isomeric 160a–c. Although cyclization into 160 seems to be kinetically more favored, products 167 are apparently thermodynamically more stable because of their extended conjugated double bond system.37 By using the synthetic sequence as was suggested for the synthesis of 167a–c, the naphthofuran-3-al derivatives 168a–d were formed from 1,4-naphthoquinone (Scheme 51).37,60
 | ||
| Scheme 51 | ||
In contrast to the observed formation of 3-formyl derivatives on reacting β-aminovinyl ketones with quinones, our research group reported8 that pyrazolo enaminone 1x reacted with 1,4-naphthoquinone to yield a product of addition and dimethylamine elimination. The naphthofuranoylpyrazole structure 173 was established for that product on the basis of its elemental analysis and spectra data. Formation of 173, as illustrated in Scheme 50, may be rationalized via initial addition of electron rich C-2 in the enaminone 1x to the active double bond of 1,4-naphthoquinone, followed by subsequent enolization would take place, forming the non-isolable acyclic dihydroxy intermediate 169. The latter underwent intramolecular ionic heterocyclization at room temperature via nucleophilic attack by the OH function on the methylene CH rather than the carbonyl carbon, yielding the dihydronaphthofuran intermediate 171, accompanied by 1,2-elimination with the release of dimethylamine, leading eventually to the final isolable 2-unsubstituted naphthofuran derivative 173 as described in our earlier report.7 It seemed that our approach was not suitable for the synthesis of the naphthofuranal derivative 172 that would be expected by analogy with the aforesaid reports37,60,68 on the reactivity of β-aminovinyl ketones toward quinones (Scheme 52).
 | ||
| Scheme 52 | ||
In support of this view, pyrano enaminone 1y also reacted with p-benzoquinone to give the corresponding pyranyl benzofuryl ketone 175 and not the pyranylbenzofuran-3-carboxaldehyde derivative 148. Furthermore, reacting 1y with 1,4-naphthoquinone afforded naphthofuryl ketone 177. The formation of 175 and 177 from 1y, is assumed to occur via initial addition of the aminovinyl ketone, of electron rich C-2, to the active double bond in quinones, forming the intermediate phenolic adducts 175 and 176, that cyclize via loss of dimethylamine to yield the final isolable products (Scheme 53).44
 | ||
| Scheme 53 | ||
In accordance with the previous observations reported by us7 and by others,44 β-ketoenamines 1z, 23 and 179 reacted with p-benzoquinone to furnish the corresponding benzofuryl ketones 181a–c, via the intermediacy of dihydroxy compounds 180a–c.90,91 The exact structure of products in these reactions has been firmly established on the basis of Heteronuclear Multiple Quantum Coherence spectroscopy (HMQC). Thus, HMQC of compound 181b, as a representative example, indicated that carbonyl group at δ 188.9 ppm is not bonded to any hydrogen atoms. This fully supported the proposed structure 181. A similar treatment of the N,N-dimethylamino derivatives of coumarin 1z and of benzene 1d with 1,4-naphthoquinone led to the naphthofuryl ketones 182a90 and 182b,42 respectively (Scheme 54).
 | ||
| Scheme 54 | ||
2.3.1.2. Preparation of pyrazoles and isoxazoles. One of the common route to pyrazole and isoxazole ring systems is the interaction of β-aminovinyl ketones with N-nucleophiles. Several contributions have been made to this area of research. Thus, e.g., treatment of vinyl ketone 183b with hydrazine hydrate and with phenylhydrazine in absolute ethanol at reflux temperature for four hours gave rise to pyrazole products for which two possible structures 188 or 189 can be formulated. However, structure 189a,b could be established for these products based on the non-identity of reaction product, obtained by reacting 183b with phenylhyrazine, with a sample of 188b prepared via initial condensation of acetophenone derivative 182b with phenylhydrazine in refluxing absolute ethanol in the presence of acetic acid and subsequent treatment of the formed phenylhydrazone 190a with DMFDMA (Scheme 55).92 Compound 183b also reacted with hydroxylamine hydrochloride, in absolute ethanol in the presence of fused sodium acetate under reflux for five hours, to yield a product that may be also formulated as 188c or 189c. Attempt to prepare a sample of 188c from reaction of oxime 190b with DMFDMA failed. However, structure 189c could be established for the reaction product based on the fact that this reaction product readily converted, under basic reaction conditions, into the nitrile 191, which was also prepared via direct cyanation of 192b with N-cyanobenzotriazole 193 as an efficient C-cyanating reagent.92,93
 | ||
| Scheme 55 | ||
A similar treatment of β-acylated enamine 194 with hydrazines led to 3-unsubstituted derivatives of pyrazole and isoxazole 196a–c on treatment with hydrazines and hydroxylamine. The isomeric 5-unsubstituted pyrazole 195b could be prepared by condensing active methylene compound 199 with phenylhydrazine and subsequent condensation of the formed phenylhyrazone 197 with DMFDMA. On the other hand, the 5-unsubstituted isoxazole 196c was converted into the α-cyanoketone 198 on reacting with a little excess of sodium hydride in refluxing dioxane. Alternatively, compound 198 was directly prepared from reaction of 199 with 193 (Scheme 56).92
 | ||
| Scheme 56 | ||
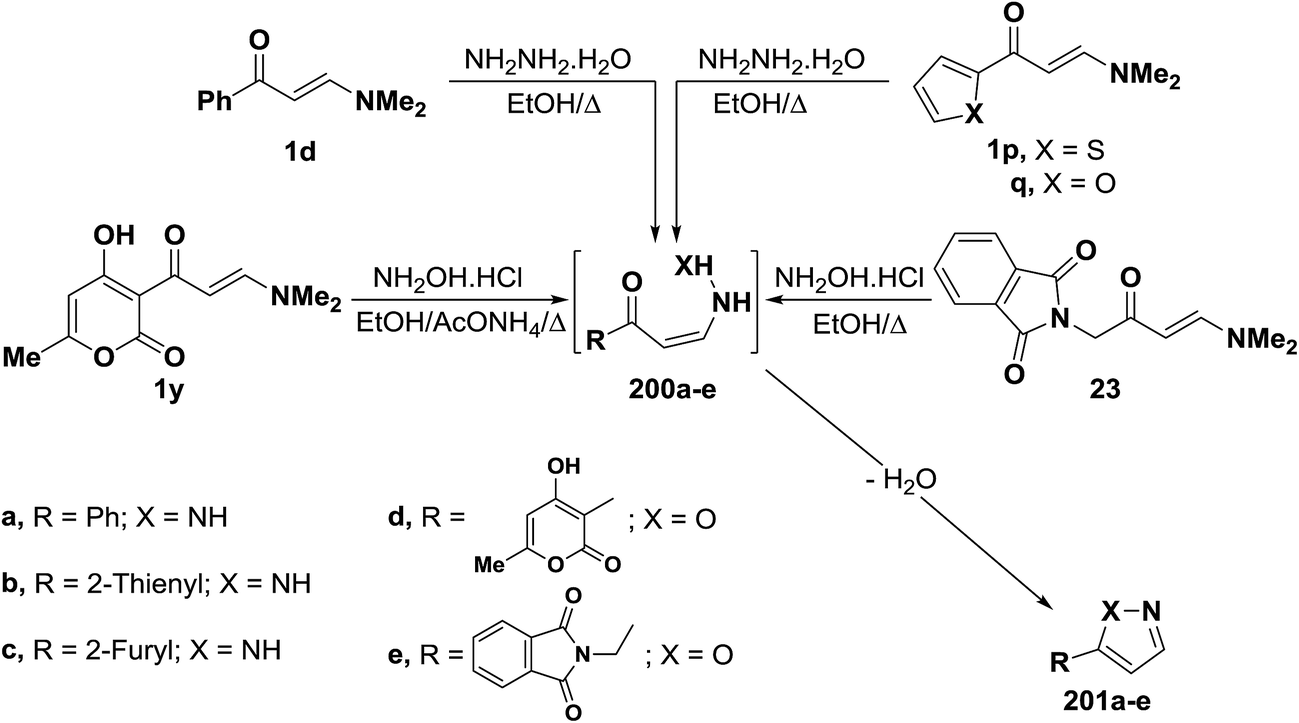
In accordance with the observed formation of 3-unsubstituted heterocyclic compounds, enaminoketones 1d and 1p,q reacted with hydrazine hydrate in refluxing ethanol to give the corresponding pyrazoles 201a–c, respectively. It is believed that hydrazine adds, in each case, reversibly across the α,β-unsaturated moiety in 1d or 1p,q affording non-isolable intermediates 200a–c. This is followed by deamination and dehydration, leading eventually to the final isolable pyrazoles 201a–c.55,94 In a similar manner, treatment of N,N-dimethylaminomethylene derivatives 1y and 23 with hydroxylamine hydrochloride led to the isoxazoles 201d,e, respectively (Scheme 57).44,95,96
 | ||
| Scheme 57 | ||
In addition, heterocyclic enaminone 202 also reacted with each of hyrazine hydrate and methylhydrazine in refluxing ethanol to furnish the anticipated pyrazoles 204a,b, via the intermediacy of 203a,b. On the other hand, when the reaction of compound 202 with acetyl-, phenyl- and benzoyl-derivatives of hydrazine, was conducted in refluxing glacial acetic acid, in all cases, a single tetracyclic product was isolate for which structure 205 was established as indicated from spectral data of that reaction product. Formation of the tetracyclic azepine 205 would involve an initial formation of intermediates 203c–e which underwent nucleophilic cyclization with loss of hydrazine molecules to afford the final single product 205. The same compound 205 was also obtained by refluxing enaminone 202 in a mixture of glacial acetic acid and hydrochloric acid (1:1) for four hours (Scheme 58).97
 | ||
| Scheme 58 | ||
In contrast to the observed formation of 3-unsubstituted heterocycles, 5-unsubstituted pyrazoles 207a,b were obtained upon heating of N,N-dimethylamino derivatives of pyridine 1t,u to 60–65 °C under stirring, with hydrazine hydrate through the intermediate formation of acyclic condensation products 206a,b.98 N,N-Dimethylamino derivative of pyrazole 1x underwent analogous reaction in refluxing DMF to yield the respective pyrazolylpyrazole derivative 207c as described by us in a previous communication.7 Compounds 1y also reacted with hydrazine and with phenylhydrazine in acetic acid at room temperature to afford the pyranylpyrazoles 207d,e.44 When compound 1z reacted with hydrazines in refluxing ethanol, the pyrazoles 207f,g were isolated, while the isoxazole 207h was obtained upon heating of 1z with hydroxylamine hydrochloride in ethanol in the presence of anhydrous sodium acetate at reflux for six hours.90 Compound 207f was also obtained in yield of 60%, when the reaction of 1z with hydrazine was carried out under microwave heating for five minutes (Scheme 59).99
 | ||
| Scheme 59 | ||
An interesting reaction leading to 3-unsubstituted pyrazoles is the interaction of 3-(N,N-dimethylamino)-methylene derivatives 49a with hydrazines in refluxing ethanol to give the pyrazoles 208a–d.100,101 On the contrary, compound 49a reacted with hydroxylamine hydrochloride in refluxing ethanol to yield a product that was assigned 5-unsubstituted isoxazole structure 209 based on 1H and 13C NMR spectra.101 Interestingly, it has been found that interaction of 49a with an excess of hydroxylamine hydrochloride, in pyridine at reflux temperature for two hours, led to the 5-aminoisoxazole derivative 212, which is assumed to proceed by reaction of the vinyl ketone 49a with two molecules of hydroxylamine, affording the dioxime 210, that then loses a molecule of water, yielding the α-cyano oxime 211, followed by spontaneous cyclization to the final isolable aminoisoxazole 212 (Scheme 60).100,102
 | ||
| Scheme 60 | ||
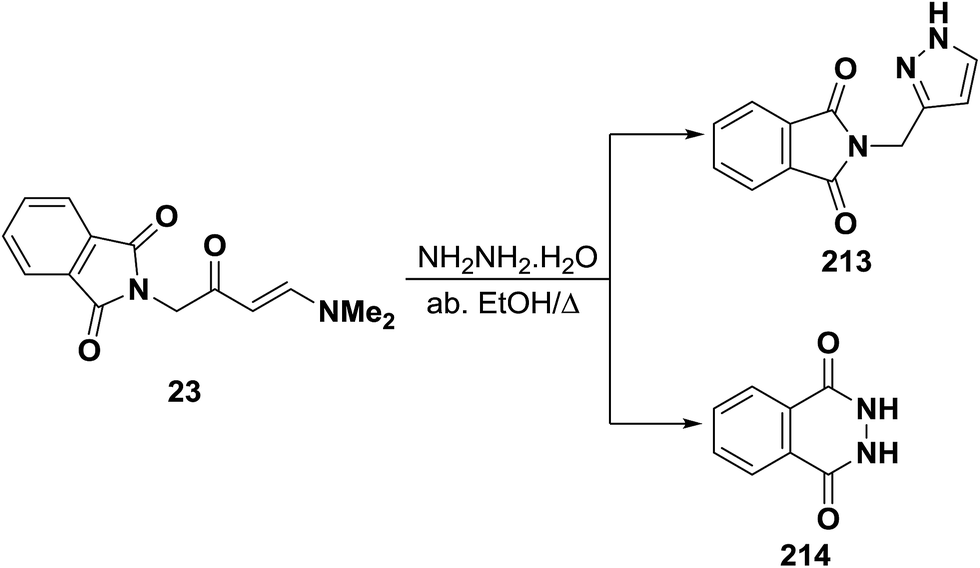
Unexpectedly, treatment of aminovinyl ketone 23 with hydrazine hydrate in refluxing absolute ethanol in an attempted to obtain the pyrazole derivative 213 was unsuccessful (Scheme 61). The isolated product was identified as 2,3-dihydrophthalazine-1,4-dione (214) identical to that reported in the literature.95,103
 | ||
| Scheme 61 | ||
Interestingly, it has been found that enaminonitriles underwent somewhat different reactions with N-nucleophiles, providing access to cyano or amino heterocyclic compounds, depending on the applied reaction conditions. In the first step of those reactions, the dimethylamino group was substituted by hydrazines or by hydroxylamine. The final products were obtained by ring closure to the carbon atoms either of the carbonyl group, affording cyano compounds, or of the nitrile group, yielding amino analogues. Several data have been reported on this area of study (Scheme 62).
 | ||
| Scheme 62 | ||
As previously indicated in this review, interaction of enaminonitriles 127a,b with hydroxylamine hydrochloride did not give expected isoxazoles 130a,b, but instead afforded the acyclic hydroxylaminomethylenes 215a,b, that could not be cyclized into 130a,b under a variety of reaction conditions.61 In contrast, compounds 126a,b reacted, on one hand, with hydrazine hydrate in refluxing absolute ethanol for six hours to yield the cyanopyrazoles 217a,b and, on the other, with phenylhydrazine under the same experimental conditions to produce the aminopyrazoles 219a,b. It is believed that compounds 127a,b react initially with both hydrazines to provide the non-isolable acyclic hydrazino derivatives 216a–d. The intermediates 216a,b cyclize readily via water elimination, leading to pyrazoles 217a,b.61 No traces of the other possible regioisomers 218a,b could be isolated, since the 1H NMR spectra of the obtained products showed, in each case, the presence of pyrazole H-3 proton as a singlet at δ 7.9 ppm and this disagree with the tautomeric 218a,b in which the corresponding pyrazole H-5 protons would appear as doublets.104,105 Similar cyclization of 216c,d into 219a,b is sterically hindered as it could produce pyrazoles with two adjacent bulky substituents. Consequently, intermediates 216c,d cyclize via nucleophilic addition to cyano group, yielding the aminopyrazole derivatives 218a,b as confirmed by elemental analyses and spectral data (Scheme 63).
 | ||
| Scheme 63 | ||
In contrast to the observed formation of acyclic products 129a,b, enaminonitrile 220 reacted with hydroxylamine hydrochloride in absolute ethanol in the presence of potassium carbonate anhydrous at reflux temperature to give the aminoisoxazole derivative 223 as confirmed by elemental and spectral data. Compound 223 is assumed to be formed via a Michael type addition of the amino group of hydroxylamine to the enamine double bond in 220 with loss of dimethylamine molecule, forming acyclic intermediate 221a, which cyclized into the final amino derivative 223 via nucleophilic addition to cyano group.106 On the other hand, it has been found that reacting enaminonitriles 67 and 220 with hyrazines in refluxing ethanol led to the cyanopyrazoles 223a–c, respectively, which is a contradiction to the behavior of compound 220 toward hydroxylamine. The spectral data of the isolated products were in complete agreement with structure 222a–c. Consequently, the acyclic hydrazino intermediates 221b–d, formed in this process, are not subjected to nucleophilic addition, but are stabilized by the evolution of water molecules (Scheme 64).50,106
 | ||
| Scheme 64 | ||
An interesting reaction leading to aminopyrazoles is the interaction of enaminonitriles 224a–c with hydrazine hydrate in refluxing dioxane for three hours to furnish the aminopyrazoles 226 and not the cyanopyrazoles 225. Inspection of 1H NMR spectrum enabled establishing structure 226 for these pyrazole derivatives since the pyrazole H-3 appeared as a singlet at δ 8.0–8.3 ppm, the authors could not trace in the 1H NMR any signals for the tautomeric 227 as this would reveal pyrazole H-5 as doublet (Scheme 64).104
It is worthwhile to report here that interaction of β-aminovinyl ketones with hydrazonoyl halides in the presence of bases also provides access to pyrazole ring system. Many examples have been reported on this interesting area of research.
Al-Zaydi and co-workers107 reported that treatment of vinyl ketones 1d and 1p–s with nitrilimines 229a,b (liberated in situ from the corresponding hydrazonoyl halides 228a,b by the action of triethylamine in dry benzene solutions at room temperature) led to, in each case, the formation of only one isolable product as tested by TLC analysis. The reaction products were identified as the 5-unsubstituted pyrazole structure 233a–j (Scheme 65) that are assumed to be formed via 1,3-dipolar cycloaddition of the nitrilimines 229a,b to the activated double bond in the vinyl ketones 1d and 1p–s to afford the non-isolable dihydropyrazole intermediates 232a–j followed by elimination of dimethylamine yielding the final pyrazole derivatives 233a–j.94,107–109 The other possible regioisomers 231a–j are not observed through out the reaction course and were excluded on the basis of the spectral data of the isolated products. This conclusion was further confirmed chemically via reacting some examples of the isolated products 233a,b,e,f with hydrazine hydrate in ethanol under reflux to afford the pyrazolo[3,4-d]pyridazine derivatives 234a–d in almost quantitative yields. It is of importance to report here that compounds 234a–d can not be prepared by the action of hydrazine on the other possible regioisomers 234a–j under the same experimental conditions as shown in (Scheme 65).
 | ||
| Scheme 65 | ||
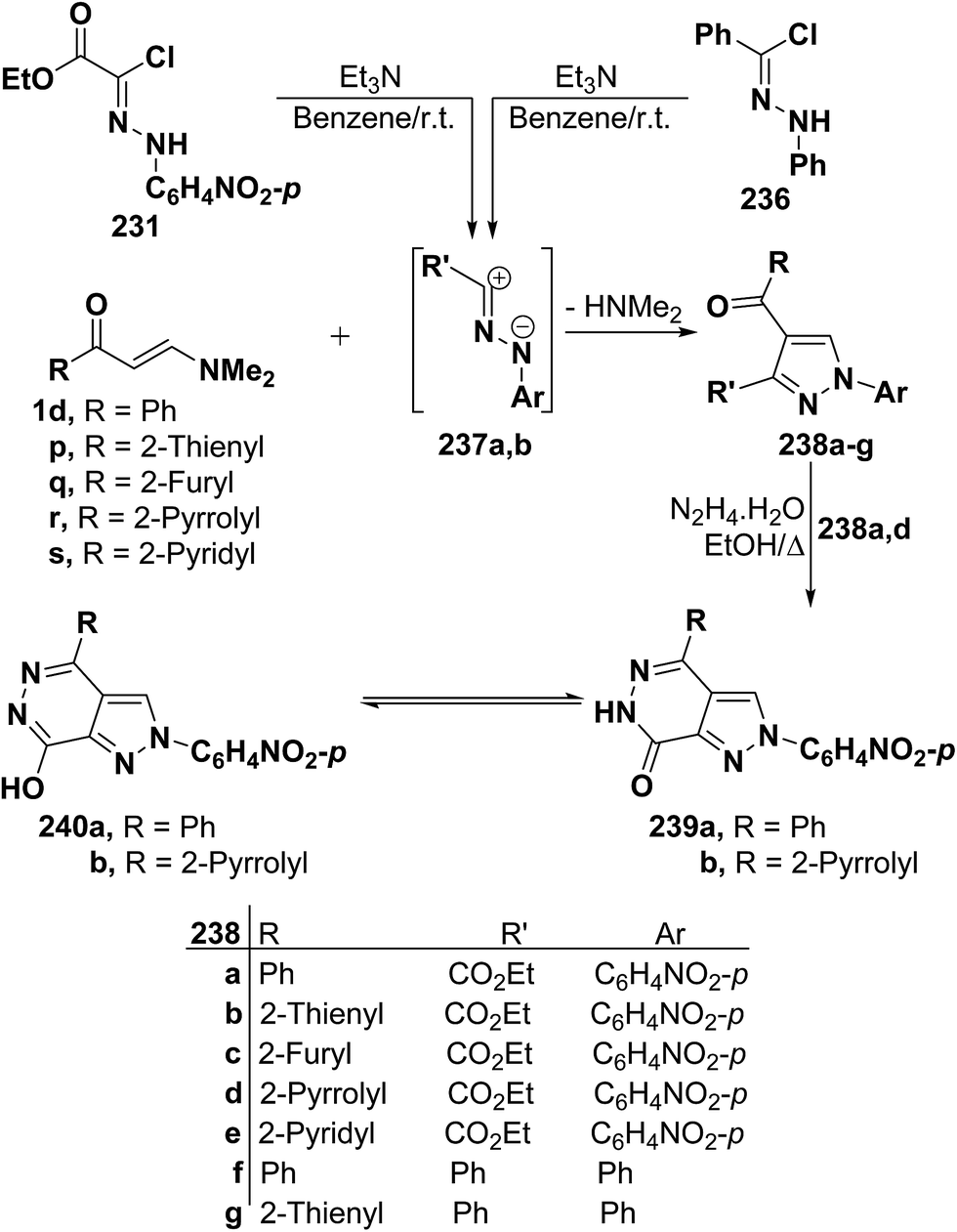
By using the synthetic sequence as was suggested for the synthesis of 233a–j, the 5-unsubstituted pyrazole derivatives 237a,b were formed from nitrilimines 237a,b that generated by the action of triethylamine on hydrazonoyl halides 235 and 236, respectively, under the same experimental conditions.107 Further confirmation of the proposed structure 238 comes from reacting the isolated products 237a,d with hydrazine hydrate in ethanol under reflux to give products that were formulated as the pyrazolo[3,4-d]pyridazinones 239a,b rather than the corresponding hydroxy derivatives 240a,b as confirmed by the spectroscopic data of the isolated products (Scheme 66).107
 | ||
| Scheme 66 | ||
In accordance with the observed formation of 5-unsubstituted pyrazoles, aminomethylene derivatives 241, 242 and 243 reacted with nitrilimines 245a–h (generated from hydrazonoyl halides 244a–k and triethylamine) to furnish the corresponding pyrazole derivatives 246a–k by cycloaddition and dimethylamine elimination.4,99,110,111 Structure 246 was further established based on conversion of the isolated products 246a–e with hydrazine hydrate into the corresponding pyrazolo[3,4-d]pyridazines 247 and 249a–e, respectively. Clearly, these products can only be obtained from 5-unsubstituted isomer. It is worth mentioning herein that treatment of pyrazoles 246d–h with hydrazine hydrate under reflux in ethanol led to the expected pyrazolopyridazines 249a–e, respectively.4,99,110 Similar hyrazinolysis of pyrazoles 246a–c, in refluxing ethanol for two hours, resulted in the formation of only one isolable product. This product was identified as the 7-hydroxypyrazolopyridazine structure 247 that is formed via loss of a molecule of methanol, ethanol or aniline (Scheme 67).110
 | ||
| Scheme 67 | ||
Also, it has been found that reaction of enaminone 23 with nitrilimines 250a–c gave rise to the pyrazoles 251a–c. Refluxing compound 251a,b with hydrazine hydrate for five minutes led to the formation of 4-(aminomethyl)pyrazolopyridazines 253 and 254, respectively, that are assumed to be formed as a result of hydrolysis accompanied by the release of phthalic acid (Scheme 68).112
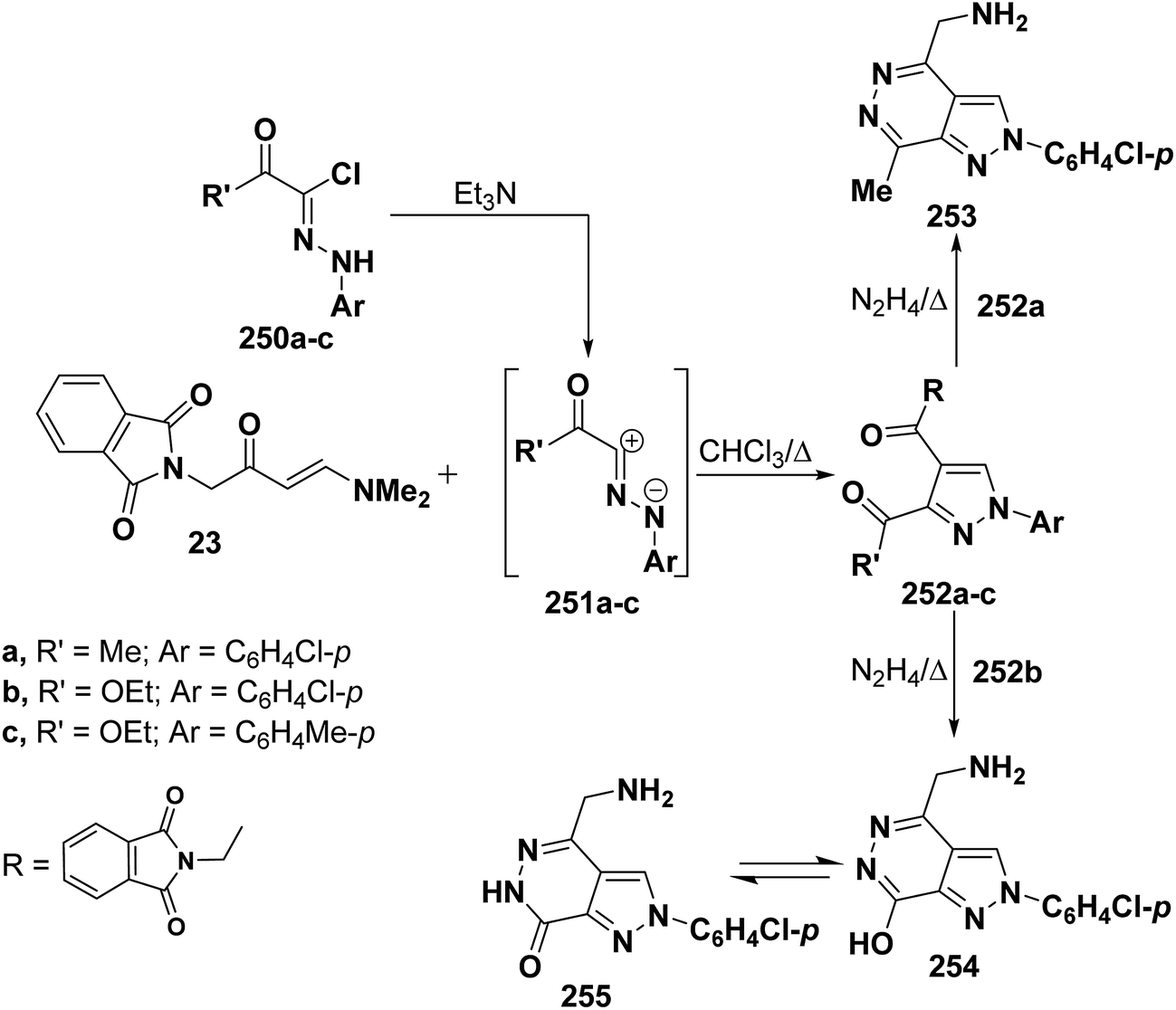 | ||
| Scheme 68 | ||
By analogy with hydrazonoyl chlorides, hydroximoyl chlorides 256 also reacted with vinyl ketones 1d and 1p–s in the presence of triethylamine to produce products of condensation via dimethylamine hydrochloride elimination for which three isomeric structures are possible. However, the structure of isolated products was considered to be 5-unsubstituted isoxazole structure 257 rather than other potential isomeric structures 259 and 261 based on spectral data and on the fact that the reaction products 263a,b,d,f were readily converted into the corresponding isoxazolo[3,4-d]pyridazines 264a–d, respectively, upon treating with hyrazine hydrate in refluxing absolute ethanol for 3–4 hours. It is believed that 256a,b initially generate nitrile oxides 257a,b in situ and these then undergo 1,3-dipolar cycloaddition to vinyl ketones 1d and 1p–s yielding intermediate cycloadducts 262a–g that aromatize via dimethylamine elimination (Scheme 69).107,108
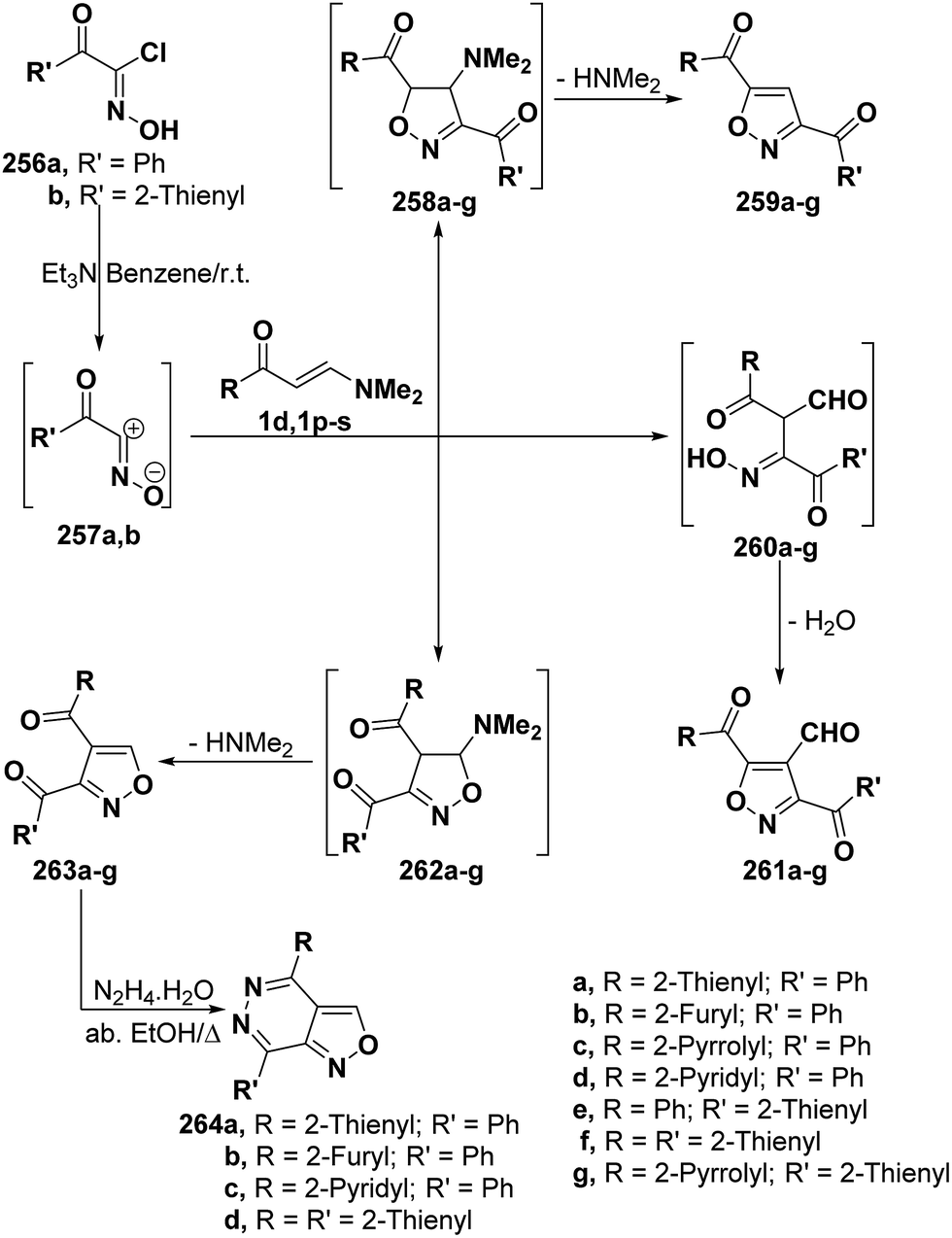 | ||
| Scheme 69 | ||
In a similar way, aminomethylene derivatives 1p, 239 and 1z underwent analogous reactions with a variety of nitrile oxides 266a–f (generated from hydroximoyl chlorides 265a–f and triethylamine) to give the expected isoxazole derivatives 267a–h by cycloaddition and dimethylamine elimination in a straightforward manner (Scheme 70).4,99,108 Spectral data as well as chemical behavior of the formed isoxazoles indicated that they are the 5-unsubstituted isomeric structure 267a–h and this agrees with an already established trend in the behavior of β-aminovinyl ketones toward both nitrilimines and nitrile oxides.
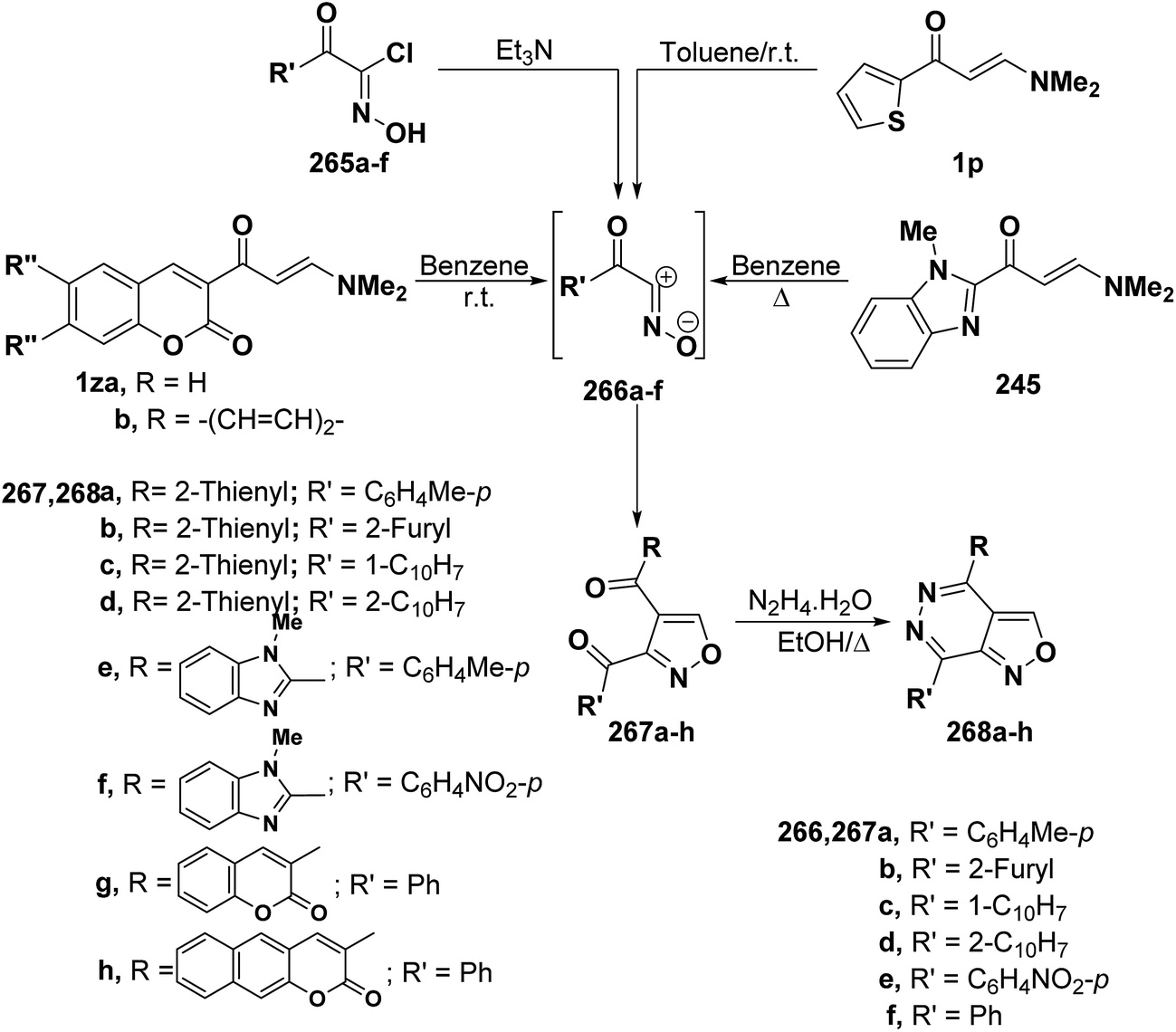 | ||
| Scheme 70 | ||
An interesting synthesis of 5-unsubstituted pyrazole is the formation of 270 upon fusion of aminomethylene derivative 1d with phenylhydrazonylpyridinium bromide 269 (Scheme 71).113
 | ||
| Scheme 71 | ||
The synthesis of 1,3-disubstituted pyrazole derivative 272 was initiated with enaminone 1d–f,o, hydrazine hydrate, and aryl halide derivatives 271 in the presence of presence of Cs2CO3 and Cu catalyst in DMF at elevated temperature (Scheme 72).114
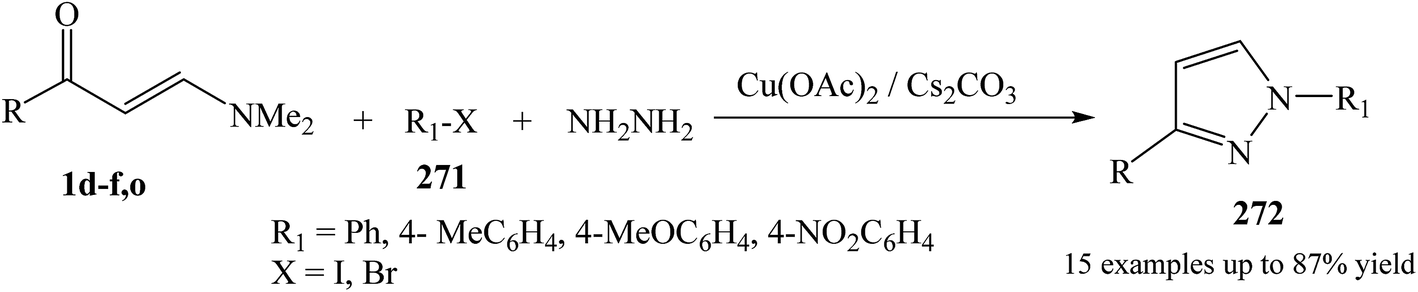 | ||
| Scheme 72 | ||
4-Hydrazinothienopyrimidine 273A or 273B and the appropriate (E)-3-(dimethylamino)-1-phenylprop-2-en-1-one (1) were mixed in ethanol containing HCl, and the mixture was heated to 70 °C for 6 h, yielded 4-(5-aryl-1H-pyrazol-1-yl)thieno[2,3-d]pyrimidine 274A and 4-(5-aryl-1H-pyrazol-1-yl)thieno[3,2-d]pyrimidine 274B, respectively (Scheme 73).115
 | ||
| Scheme 73 | ||
2.3.1.3. Miscellaneous. It is also of value to report here that reactions with β-aminovinyl ketones allow an easy access to some other five membered heterocycles such as thiophenes and pyrroles. Some recent reports on this area have been published.
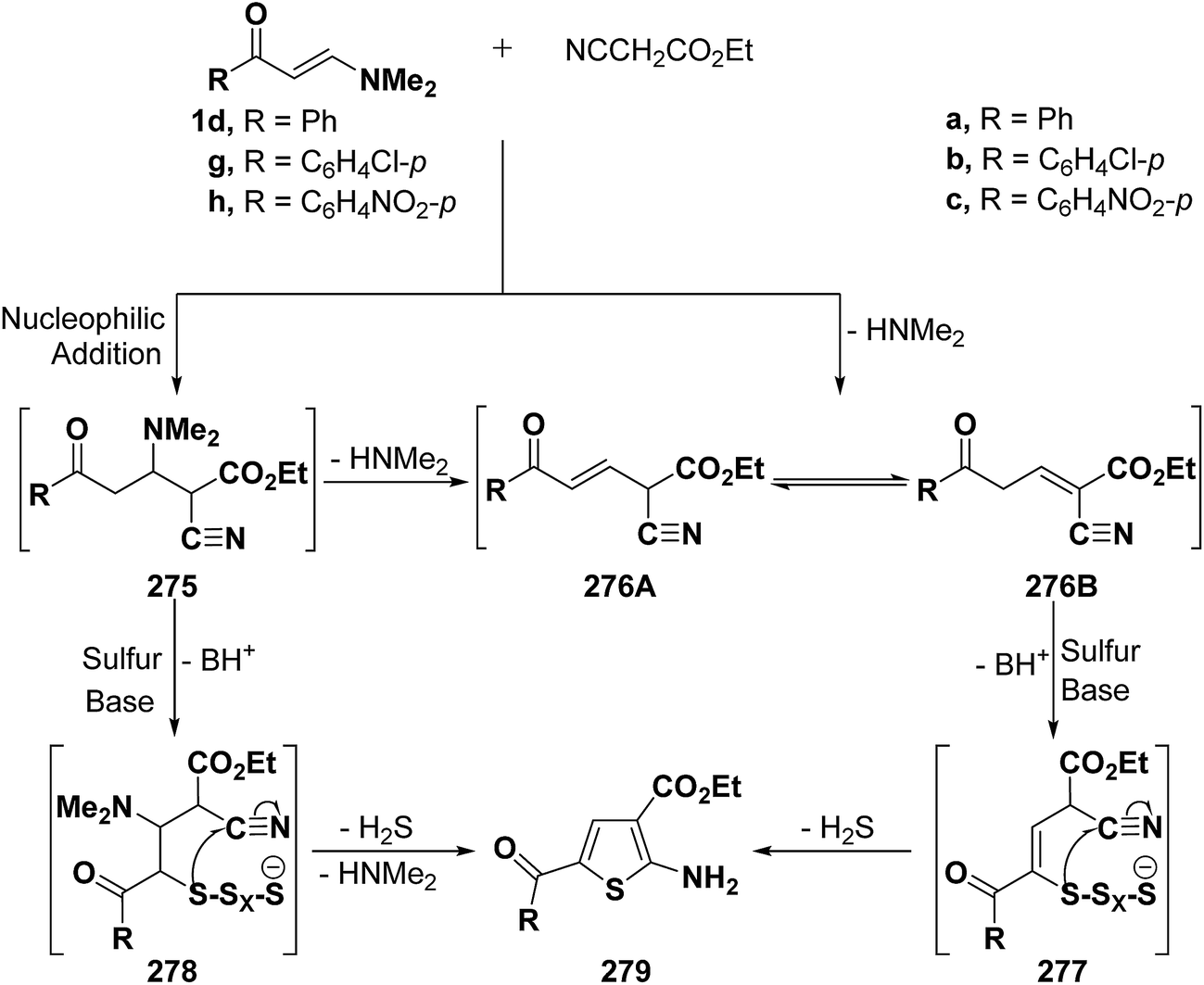
Elnagdi et al.116 reported on the utility of vinyl ketones as aldehyde equivalents in Gewald reactions. It has been found that ketones 1d,g,h reacted smoothly with elemental sulfur and ethyl cyanoacetate in the presence of equivalent amounts of piperidine, under reflux in dry DMF for 6–8 hours, to yield 4-unsubstituted-2-aminothiophenes 279a–c in good yields. It is believed that the initial step in the reaction sequence would be the nucleophilic addition of an active methylene nitrile to the α,β-unsaturated moiety in ketones 1d,g,h with subsequent elimination of dimethylamine to yield 276A or 276B. Alternatively, reaction of adducts 275 with sulfur in the presence of an equivalent amount of piperidine would yield 278 that would cyclize and aromatize to yield 279 via loss of hydrogen sulfide and dimethylamine molecules during the reaction course. This sequence is quite similar to the general mechanism of the Gewald reaction.117 In either case, the reactivity of methylene moiety in either 275 or 276 is essential for the success of reaction (Scheme 74). Consequently, vinyl ketones 1d,g,h proved to fulfill this prerequisite as they contain electron attracting substituents.116
 | ||
| Scheme 74 | ||
In addition, Al-Zaydi et al.99 reported on the formation of pyrrole derivatives 281 upon treatment with acetic acid and ammonium acetate in a domestic microwave oven at full power. It is assumed that initially formed 2,4-dicoumarinoylpyrroles 280 undergo a Nenitzescu like cyclization118 and decarbonylation; thus yielding the final products 281, respectively (Scheme 75).
 | ||
| Scheme 75 | ||
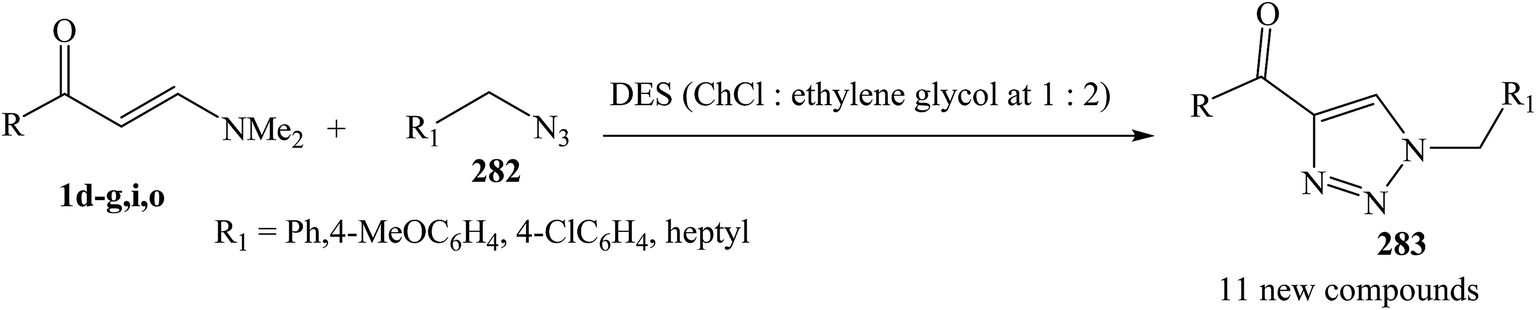
Martins and et al. evaluated the effect of type III DES in a reaction between β-enaminones 1d–g,i,o and organic azide 282 for the synthesis of 4-acyl-1-substituted-1,2,3-triazoles 283 via 1,3-dipolar cycloaddition. The products were obtained in high selectivity and good yields (70–84%). The advantages of the method include easy work-up, metal-free conditions, inexpensiveness, and the ability to be used four times without a loss in yield (Scheme 76).119
 | ||
| Scheme 76 | ||
1,5-Disubstituted 1,2,3-triazole derivatives 286 were prepared via the three-component reactions enaminone 1, amine 284, and tosylhydrazine 285. This metal- and azide-free, regioselective synthetic method proceeds in the presence of only molecular iodine (Scheme 77).120
 | ||
| Scheme 77 | ||
2.3.2.1. Preparation of pyridines and related compounds. As mentioned earlier in this review, refluxing β-aminovinyl ketones 1d–f in acetic acid alone resulted in the formation of 1,3,5-triaroylbenzene derivatives 135a–c by a self condensation-elimination route.43,66,67 In contrast, on being heated in acetic acid in the presence of ammonium acetate, those ketones 1d–g afforded 6-substituted-3-aroylpyridines 289a–d, respectively.66,67 The electron rich C-2 in one molecule of 1 adds to the electron deficient C-3 of another molecule, followed by dimethylamine elimination, forming the intermediates 21 and 287, respectively. The latter intermediates 287 cyclizes by the action of ammonia into the final isolable products 289, through the intermediacy of 288 (Scheme 78).
 | ||
| Scheme 78 | ||
By using the synthetic sequence as was suggested for the synthesis of 289, the 6-substituted-3-aroylpyridines 291a–f were formed from dimethylaminomethylene compounds 1a, 1m, 1o and 1p,q,s respectively, by self cyclocondensation through the intermediate formation of 290a–f (Scheme 79).66–68
 | ||
| Scheme 79 | ||
Also, treating vinyl ketones 1z in acetic acid/ammonium acetate afforded the corresponding pyridines 277 (ref. 78 and 99) while interaction of 1z with enamino carbonyl compound 1a in a microwave oven in the presence of ammonium acetate and acetic acid for two minutes gave a mixture of 292 and methylpyridine derivative 293 (Scheme 80).99
 | ||
| Scheme 80 | ||
It is of value to report here that reaction of β-aminovinyl ketones with acetyl derivatives also provide access to pyridines. Lots of recent reports on this area have been found. Thus, e.g., reacting aminovinyl ketone 1d with acetophenone in refluxing acetic acid in the presence of ammonium acetate yielded a product that may be formulated as 294 or isomeric 295. The cyclization reaction may proceed by two possible mechanisms, which differ in their sequential nucleophilic attack/amine exchange reaction. While initial condensation of the methyl function with the carbonyl group of the vinyl ketone 1d and subsequent cyclization could lead to structure 294, initial Michael addition of the methyl ketone across the activated double bond in 1d and subsequent cyclization might afford compound 295 (Scheme 81).121
 | ||
| Scheme 81 | ||
In a similar way, pyridine enaminones 1t,u reacted with acetylpyridines in anhydrous tetrahydrofuran in the presence of potassium t-butoxide under stirring followed by refluxing in glacial acetic acid in the presence of ammonium acetate to afford the terpyridines 297a–e in 25.8–29.7% yields through the intermediate formation of Michael adducts 296a–e (Scheme 82).122
 | ||
| Scheme 82 | ||
Analogously, aminovinyl ketone 1z reacted with acetyl derivative 298 in refluxing acetic acid in the presence of ammonium acetate to produce the corresponding pyridine derivative 299 (Scheme 83).78
 | ||
| Scheme 83 | ||
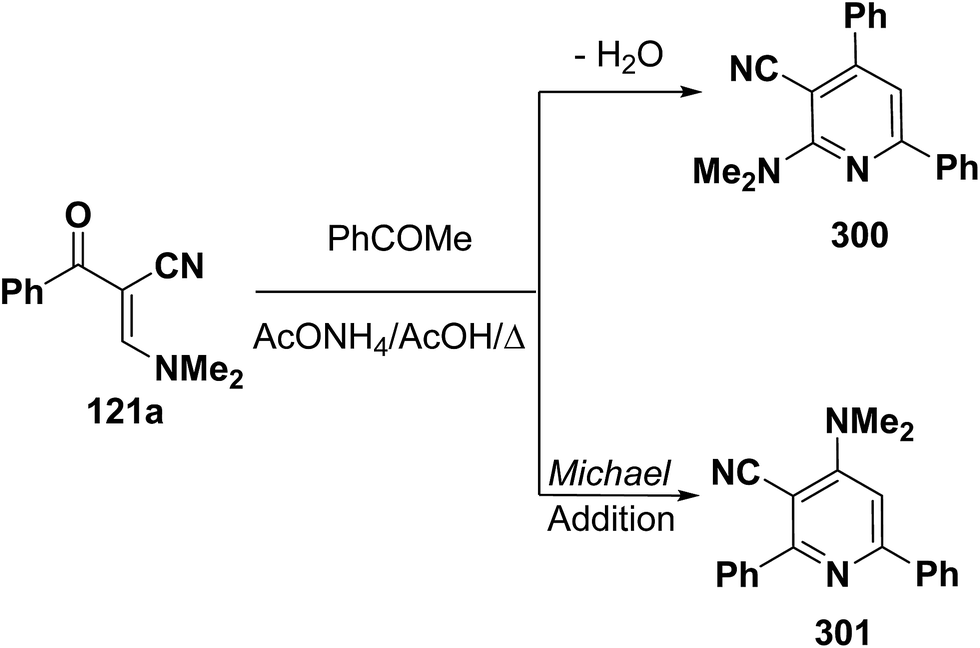
An interesting reaction leading to pyridine ring system is the interaction of enaminonitrile 121a with acetophenone to give a sole isolable reaction product that was assigned structure 301 rather than isomeric 300 based on NOE difference NMR experiments where irradiation of pyridine 3-H at δ 7.37 ppm enhanced the dimethylamino signal (Scheme 84).121
 | ||
| Scheme 84 | ||
It is also worthwhile to report here that interaction of β-aminovinyl ketones with active methylenes allows an interesting access to pyridine ring system. Intensive research work on this area of studies have been described. Elnagdi et al.42,66,68,121 reported that reaction of aryl enaminones 1d,g, 1m with acetylacetone in refluxing acetic acid in the presence of ammonium acetate afforded pyridine derivatives. Two pathways can be envisioned for formation of these final products (Scheme 85). Thus, initial addition of active methylene moiety to α,β-unsaturated double bond in enaminones would afford Michael adducts 304a,d–f that in the presence of ammonium ion condensed, in each case, with acetylcarbonyl group yielding an enamine that cyclizes into 6-arylpyridines 305a,d–f. In contrast, initial condensation of active methylene moiety with carbonyl function would yield 302a,d–f that in the presence of ammonium acetate cyclizes into the alternative 4-aryl analogues 303. Structure 305 was preferred over possible isomeric structure 303 on the basis of 1H NMR spectra.7,42,66 A similar treatment of the aromatic enaminones 1e–g and 1o with ethyl acetoacetate led to the corresponding 4,5-unsubstituted pyridines 306b–d,f as confirmed by the 1H NMR spectra, which indicated pyridine protons with J = 8.0–8.2 Hz, that is characteristic for pyridine H-5 and H-4.66
 | ||
| Scheme 85 | ||
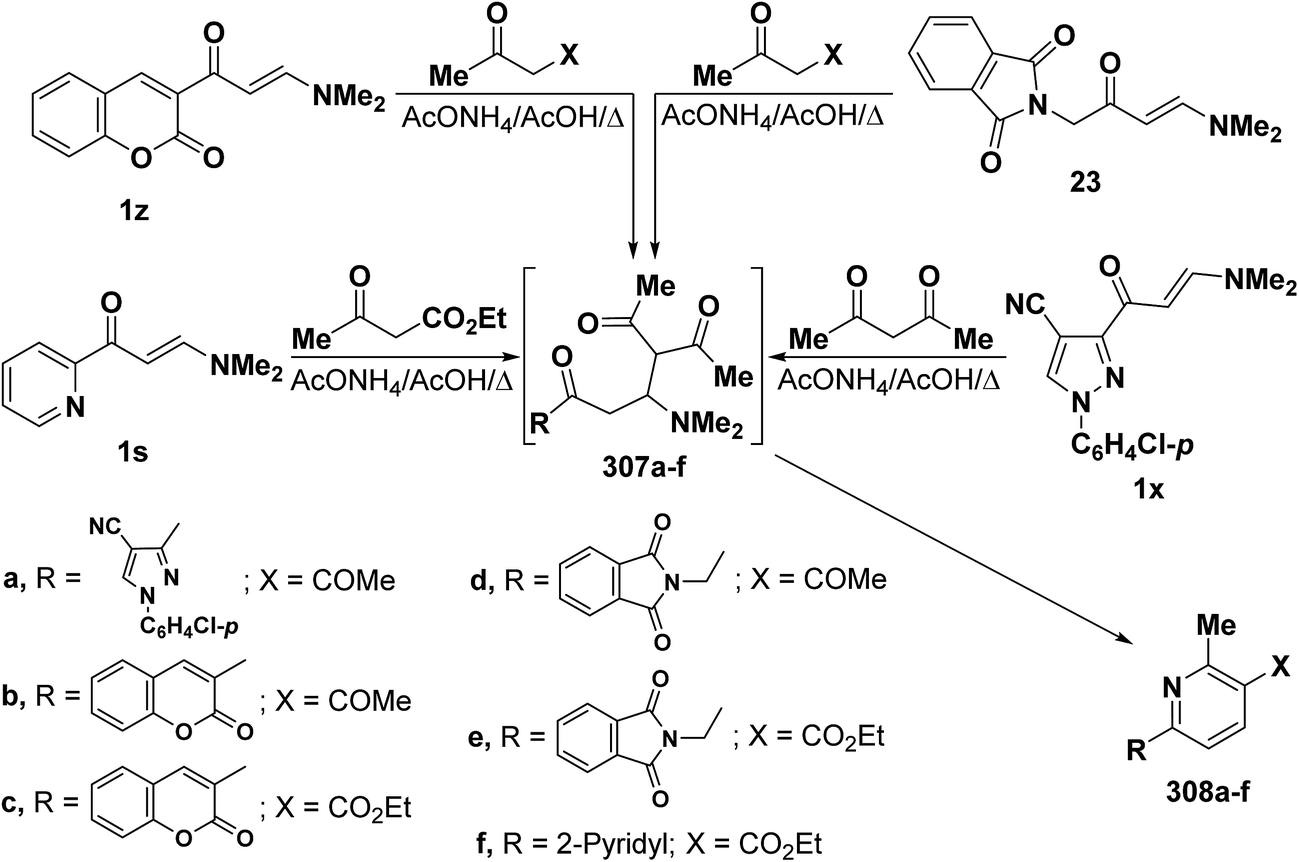
In addition, heterocyclic enaminones 1s, 1x, 1z and 23 also underwent analogous reactions with either acetylacetone or ethyl acetoacetate to yield the expected 6-heterocyclylpyridine derivatives 308a–f, respectively, as the sole isolable products (Scheme 86). Formation of these compounds would involve an initial formation of Michael adducts 307a–f, followed by intramolecular cyclization to give the final products 306a–f.7,25,91,121 6-Substituted pyridine structure 308a–f was assigned for those products based on the presence of pyridyl ring protons, in each case, as doublets with J = 8 Hz as reported by us7 and by others.25,79,121
 | ||
| Scheme 86 | ||
In accordance with the observed formation of 6-heterocyclylpyridine derivatives 308, enaminonitrile 67 reacted with ethyl acetoacetate in refluxing acetic acid in the presence of ammonium acetate to provide the expected pyridine derivative 310. In contrast, treating compound 67 with acetylacetone under the same experimental conditions resulted in the formation of an acyclic product 310 that could not be cyclized into the anticipated pyridine derivative 312 (Scheme 87).50
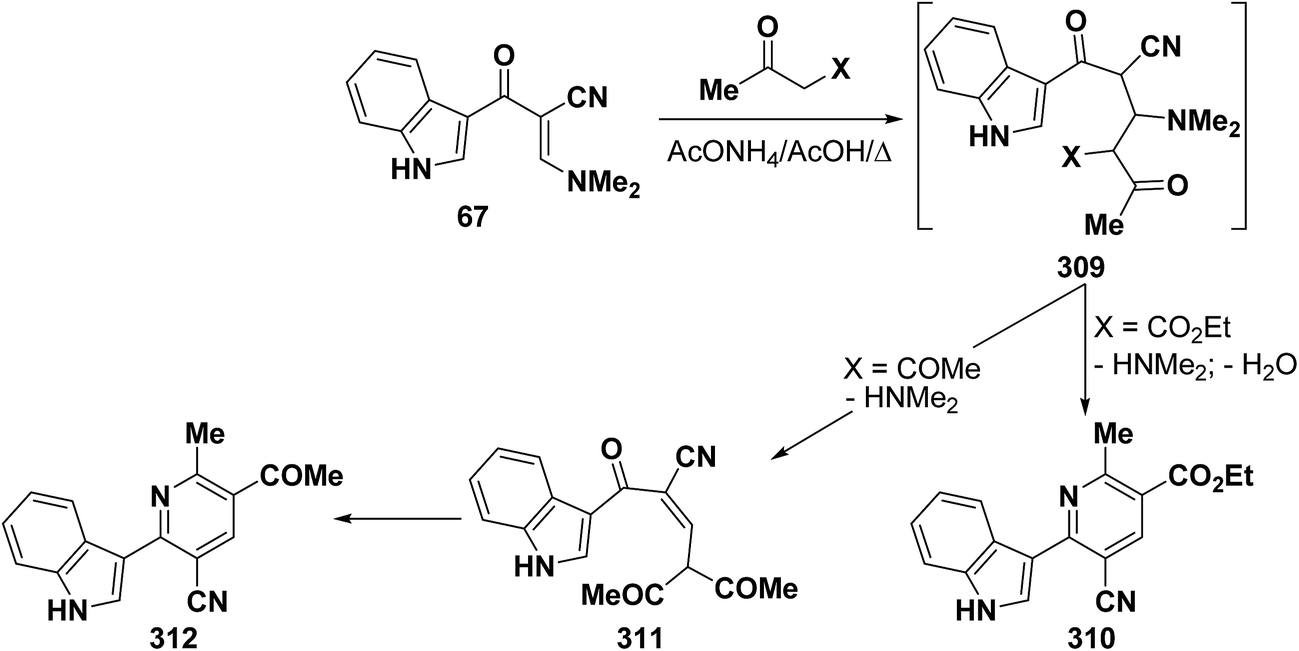 | ||
| Scheme 87 | ||
Similar to the behavior of β-aminovinyl ketones toward acetylmethylene compounds, those vinyl ketones also reacted with cyanomethylene compounds to furnish pyridines. Thus, reaction of vinyl ketones 95a,b with malononitrile in refluxing acetic acid and in presence of ammonium acetate yielded products of condensation via dimethylamine elimination. These may thus be formulated as 313–316. Initial Michael addition to the α,β-unsaturated linkage can afford 314 that would then lose dimethylamine and cyclize into 315 or isomerize into 317 (Scheme 88). Alternatively, condensation of the carbonyl group in 95a,b with the active methylene moiety would afford 313 that can then cyclize into 316.25
 | ||
| Scheme 88 | ||
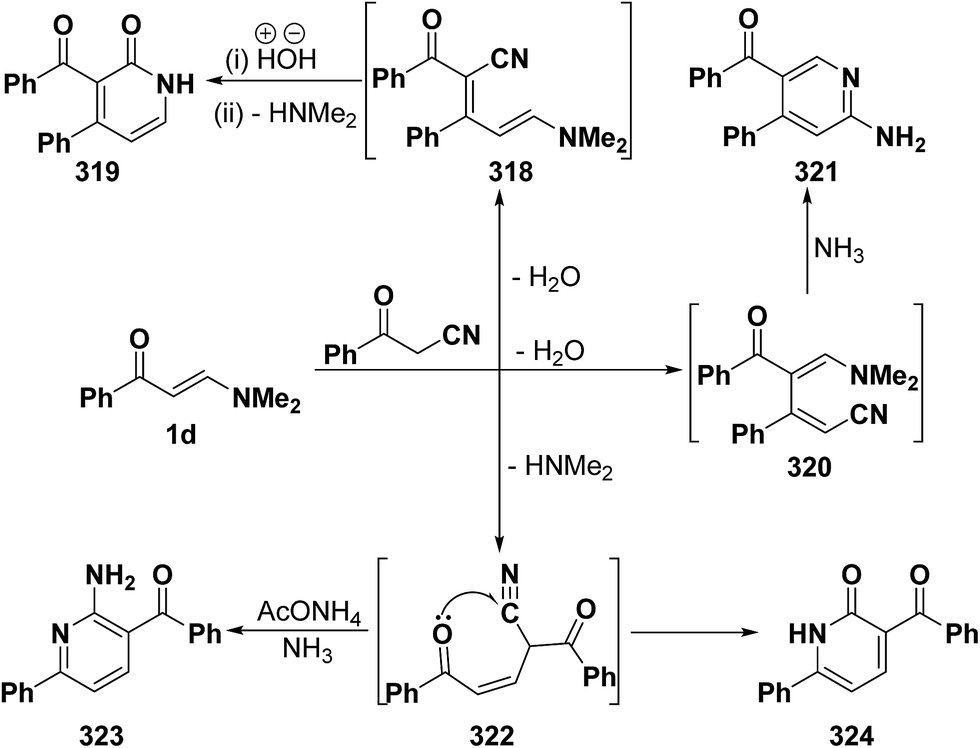
An interesting reaction leading also to pyridine ring system is the interaction of phenyl enaminone 1a with benzoylacetonitrile in refluxing acetic acid in the presence of ammonium acetate to furnish a mixture of two products. One of the products was assigned structure 323 and is assumed to be formed via initial addition of the active methylene moiety in benzoylacetonitrile to the enaminone C-3 and subsequent elimination of the dimethylamine yielding the acyclic Michael adduct 322, that would react with ammonia to yield aminopyridine derivative 323 (Scheme 89). Several isomeric structures seemed possible for the other product, depending on the reaction route, namely; the pyridone structure 319 resulting from initial condensation of the active methylene moiety with the enaminone carbonyl function, forming 318, followed by hydrolysis and subsequent cyclization, or an isomeric pyridone structure 321 resulting from initial condensation of the benzoylacetonitrile carbonyl function with the enaminone C-2, affording 320 and subsequent cyclization. Both structures 319 and 321 were excluded based on the absence of a low field (δ > 8.5 ppm) pyridine 6-H signal. Consequently, the pyridone structure 324 was considered for the second reaction product that showed its stability on reflux in acetic acid or mineral acid, confirming structure 324.121
 | ||
| Scheme 89 | ||
In a similar way, β-aminovinyl ketones 1d and 1p,q,s reacted with malononitrile in refluxing ethanolic sodium ethoxide to yield the ethoxypyridines 327a–d. Initial addition of carbanion of malononitrile across the activated double bond in vinyl ketones would yield the Michael adducts 325a–d. Subsequent addition of ethoxide anion to one of the cyano groups would give the iminoethers 326a–d, which would be cyclized via a nucleophilic attack of an NH group on a cyano carbon, affording the intermediates 327a–d. This would be finally followed by deamination and dehydration, leading to the isolated ethoxypyridine products 328a–d (Scheme 90).94
 | ||
| Scheme 90 | ||
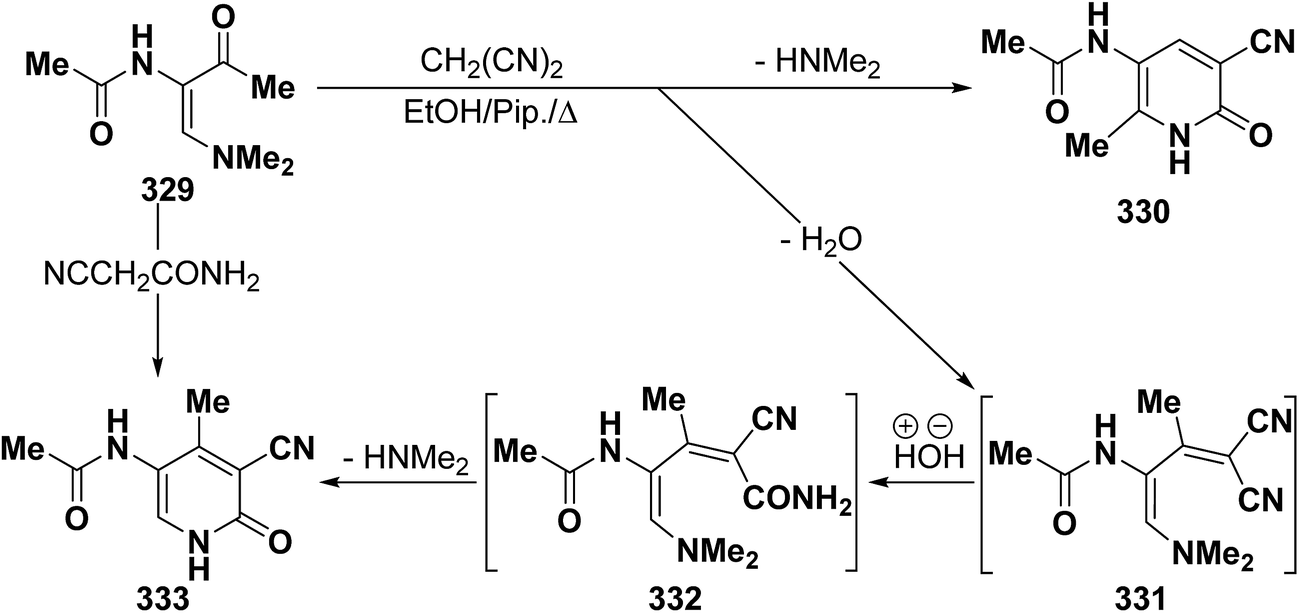
In contrast to the behavior of β-aminovinyl ketones toward cyanomethylene reagents, vinyl ketone 329 was treated with malononitrile, in ethanol in the presence of a catalytic amount of piperidine at reflux temperature to give a condensation product for which 6-unsubstituted pyridone structure 333 was considered rather than the alternative 4-unsubstituted structure 330 as indicated from spectral and chemical evidence.116 In support of the proposed structure, compound 333 was also formed on treating 329 with cyanoacetamide in refluxing pyridine. Based on the above findings, the initial step in the reaction involves condensation of the carbonyl function in 329 with malononitrile, yielding the diene 331 which is hydrolysed to an intermediate amide 332 and then cyclizes to 333 with the loss of dimethylamine molecule (Scheme 91).123
 | ||
| Scheme 91 | ||
In a similar manner, vinyl ketone 49a reacted with ethyl cyanoacetate in refluxing dioxane in the presence of sodium hydride to yield 6-unsubstituted pyridone derivative 335 via the intermediacy of the dienes 334a,b (Scheme 92).45
 | ||
| Scheme 92 | ||
One of the main route to 3-cyanopyridine-2-thiones and -2-ones is the interaction of β-aminovinyl ketones with cyanothioacetamide and with cyanoacetamide. Several data on this research area have been published. For instance, 3-cyanopyridines 337a–d were prepared by treating enaminone 1d,p with cyanothioacetamide in refluxing acetic acid in the presence of ammonium acetate for 1.5 hours, or with cyanoacetamide in sodium ethoxide solution at reflux, yielding the target molecule via the intermediacy of Michael adduct 336 (Scheme 93).42,58,109,124,125
 | ||
| Scheme 93 | ||
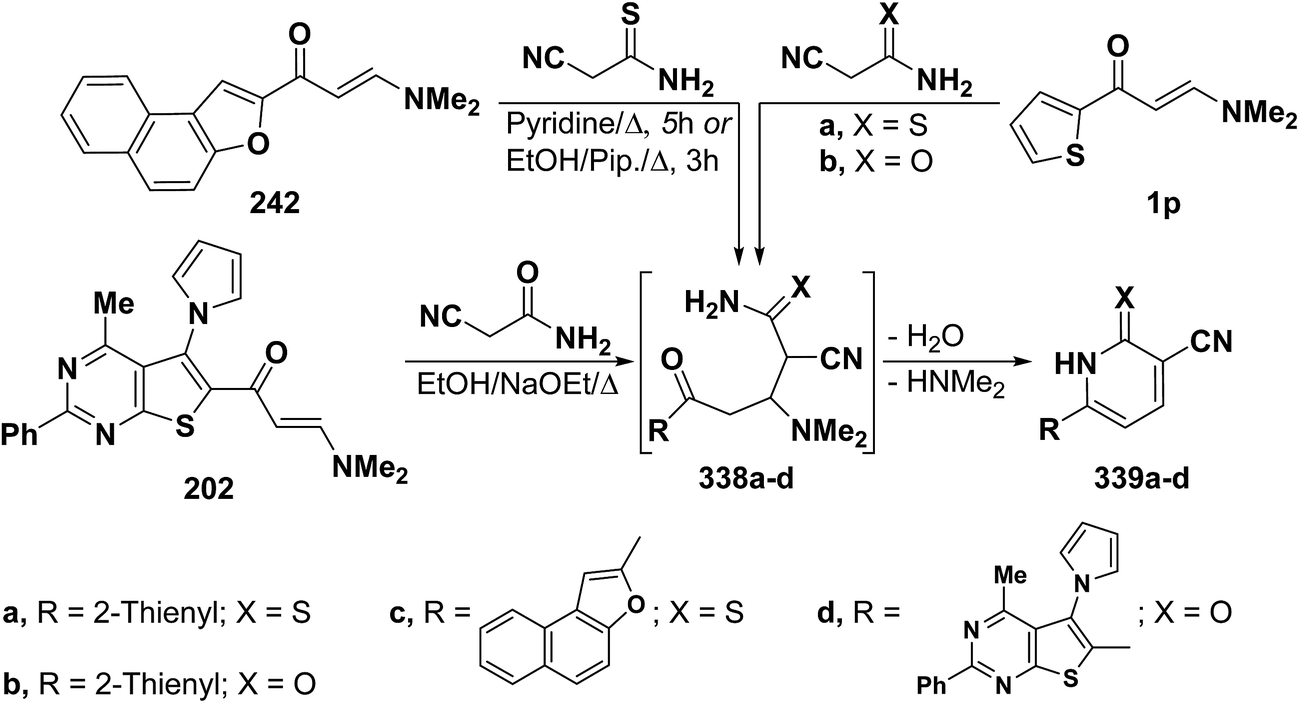
In accordance with the observed formation of 4,5-unsubstituted pyridines, heterocyclic enaminones 1p, 242 and 202 reacted with cyanothioacetamide or cyanoacetamide to give the corresponding pyridines 339a–d, respectively, via the intermediacy of Michael adducts 338a–d (Scheme 94).67,108,110,126
 | ||
| Scheme 94 | ||
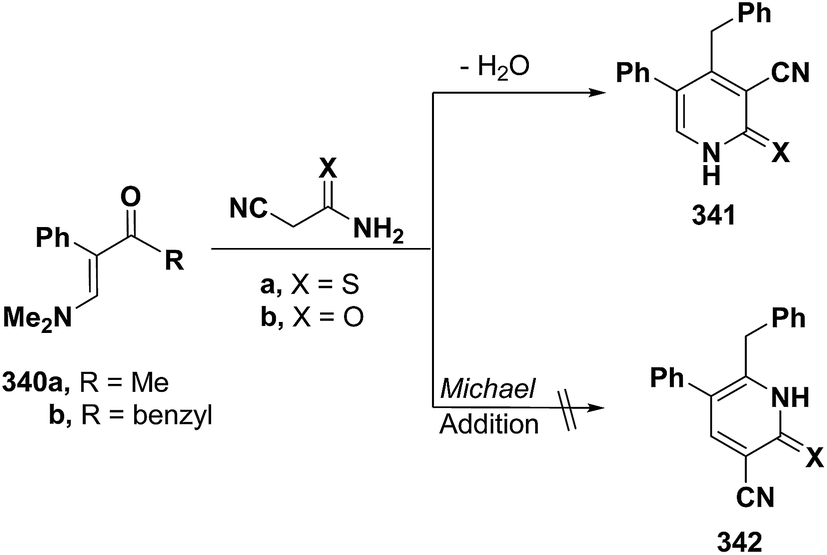
Also, aminovinyl ketone 340b condensed with cyanothioacetamide and with cyanoacetamide via dimethylamine elimination to yield the 3-cyanopyridines that were formulated as 341a,b rather than isomeric 342a,b as indicated from the 1H NMR spectra for the reaction products. In case of X = S, 1H NMR spectrum showed the pyridine ring CH as singlet at δ 7.76 ppm. This may suggest a favorable reaction product 342a formed via initial addition of the active methylene moiety in cyanothioacetamide across the activated double bond in 340b and subsequent cyclization and aromatization via dimethylamine elimination (Scheme 95). The alternative structure 341a which could have resulted from initial condensation of the active methylene with the carbonyl function, should display a doublet for the pyridine ring CH.127
 | ||
| Scheme 95 | ||
Treatment of dimethylaminomethylene compound 228a with cyanothioacetamide, cyanoacetamide or anion of malononitrile dimer in dry DMF and sodium hydride led to 5-benzoylpyridines 345a–c rather than isomeric 344a–c based on the mass spectra (MS) of the isolated products. Additionally, the IR spectrum showed the disappearance of the cyano group so that structure 345b became 346 upon hydrolysis (Scheme 96).128
 | ||
| Scheme 96 | ||
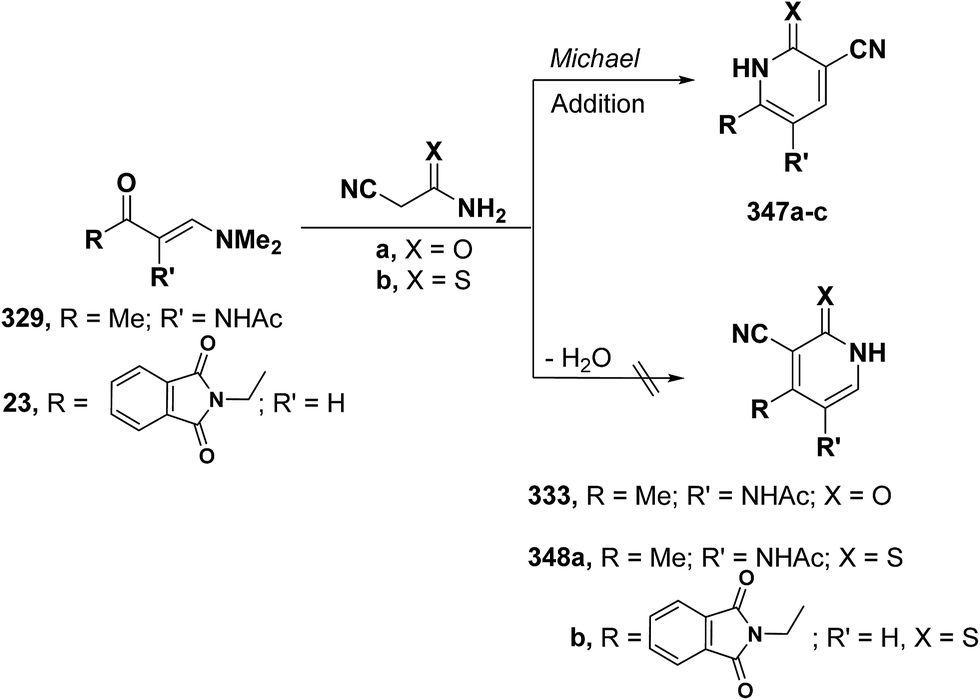
Quite surprisingly, interaction of vinyl ketones 329 and 23 with cyanoacetamide or cyanothioacetamide gave, in each case, a sole isolable reaction product that was assigned 4-substituted pyridine structure 333 or 348a,b rather than isomeric 347a–c. Structure 333 or 348, in this case, resulted from initial condensation of the active methylene in cyanoacetamide or cyanothioacetamide with carbonyl function in 329 or 23 and subsequent cyclization and aromatization via dimethylamine elimination (Scheme 97).95,123
 | ||
| Scheme 97 | ||
On the other hand, it has been found that interaction of vinyl ketones 1d–f with 3-aminocrotononitrile 349 afforded the 5,6-unsubstituted pyridine derivative 353 via the intermediates 351, but not 350 (Scheme 98).43 Although this reaction can afford 4,5-unsubstituted analogue 352 as well, structure 353 is established based on H-5,6 coupling of pyridine which showed a value of 4 Hz, if the reaction product is 352 those protons would be expected to have a much higher value (J ∼ 9 Hz).7,42,43,66
 | ||
| Scheme 98 | ||
An interesting synthesis of quinolones is the formation of 354 on refluxing aminovinyl ketone 1j in ethanol for 60 minutes under catalytic transfer hydrogenation conditions (CTH) using cyclohexene as hydrogen source and 10% Pd–C as catalyst (Scheme 99).129
 | ||
| Scheme 99 | ||
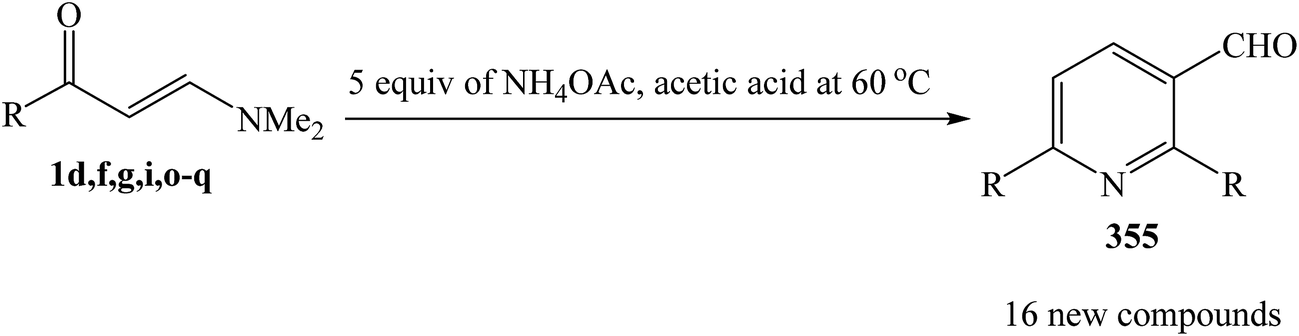
Shankaraiah and et al. was reported a clean, convenient, and highly efficient method for the rapid construction of pyridine 2,6-diarylnicotinaldehydes 355 by an unexpected self condensation of enaminoketones 1 in the presence of NH4OAc in acetic acid under conventional conditions and also under microwave irradiation (Scheme 100).130
 | ||
| Scheme 100 | ||
The three-component sequential reaction of enaminones 1d,f–h, cinnamaldehyde 356 and the appropriate amines 357, proceeded smoothly to give 1,3,4-trisubstituted 1,4-dihydropyridines 358a–t in aqueous DMF (Scheme 101).131,132
 | ||
| Scheme 101 | ||
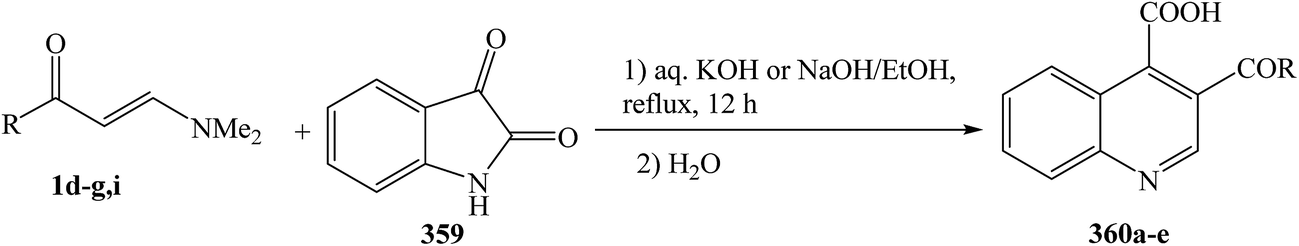
Enaminones 1d–g,i was heated at reflux with isatin 359 in the presence of an aqueous solution of KOH or NaOH, followed by subsequent acidification with dilute hydrochloric acid to give the quinoline-4-carboxylic acids 360a–e in good to excellent yields (75–90%) (Scheme 102).133
 | ||
| Scheme 102 | ||
2.3.2.2. Preparation of pyrimidines. One of the major routes to pyrimidines is the ring closure of β-aminovinyl ketones by bifunctional reagents. This one-pot cyclization involves both the carbonyl and the dimethylamino groups, leading eventually to the target pyrimidines. Some representative examples have been provided herewith. Thus, treatment of vinyl ketones 1d and 1p,q with thiourea in refluxing sodium ethoxide yielded the pyrimidine-2-thiones 363a–c, respectively. Formation of these products would involve, in each case, an initial Michael addition of the amino group in thiourea to the activated double bond in 1d or 1p,q yielding Michael adduct 361a, 361b or 361c. This is followed by deamination to form the acyclic non-isolable intermediates 362a–c, which then undergo enolization and subsequent nucleophilic cyclization via loss of water, affording the final products 363a–c. A plausible mechanism for the formation of pyrimidines 363a–c is depicted in scheme 163 (Scheme 103).94
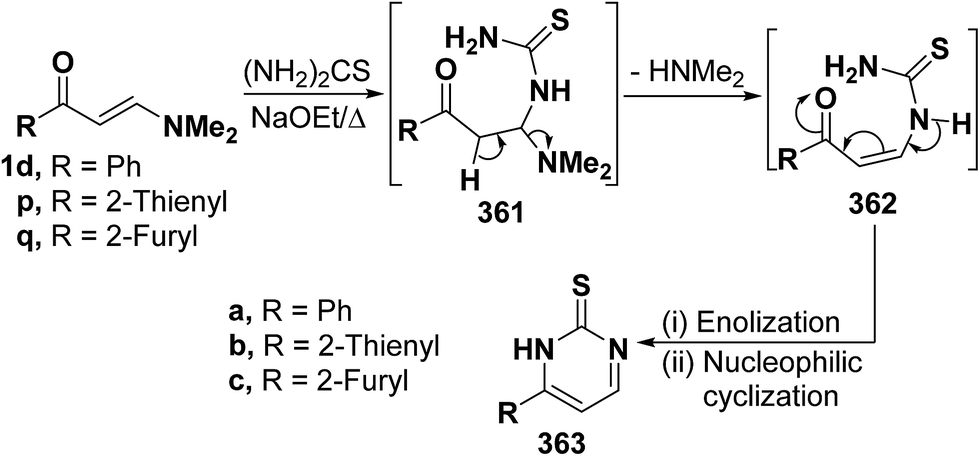 | ||
| Scheme 103 | ||
In a similar manner, heterocyclic enaminone 202 reacted with thiourea in refluxing DMF/ethanol in the presence of excess anhydrous potassium carbonate to afford the pyrimidine 364, while the S-alkylated product 365 was obtained under similar reaction conditions, except that DMF/ethanol was replaced by methyl glycol.97 On the other hand, treating 202 with guanidine compounds 366a–c in refluxing methyl glycol in the presence of excess anhydrous potassium carbonate resulted in the formation of the corresponding pyrimidines 368a–c, respectively, via the intermediate formation of acyclic secondary enaminones 367a–c (Scheme 104).97
 | ||
| Scheme 104 | ||
In addition, reaction of aryl enaminone 1m with guanidine carbonate (369) in refluxing ethanolic sodium ethoxide solution for sixteen hours resulted in the formation of 2-aminopyrimidine derivative 370,68 whereas pyrimidine derivative 372 was isolated on treatment of 3-pyridyl enaminone 1u with guanidine derivative 371 via loss of dimethylamine and water molecules (Scheme 105).134
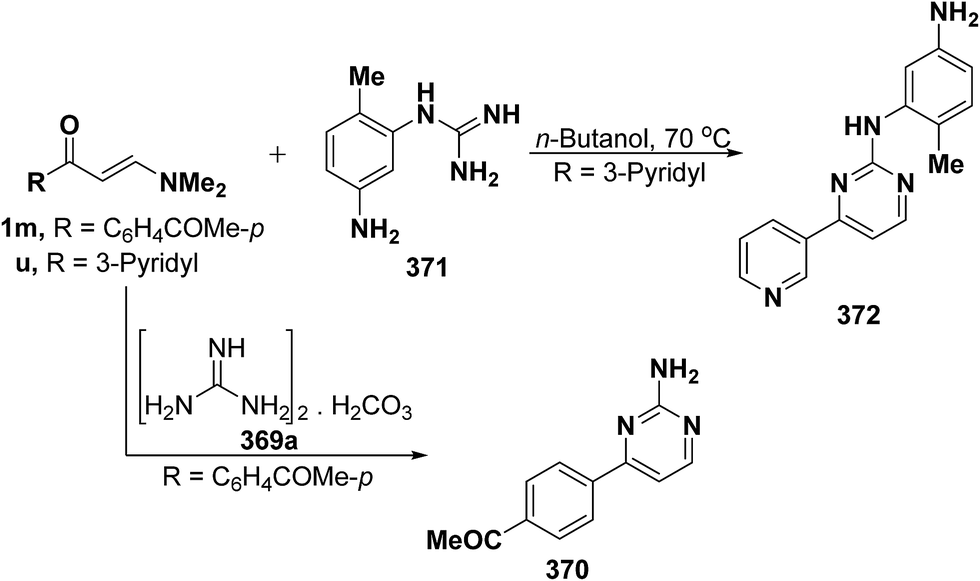 | ||
| Scheme 105 | ||
Interestingly, reaction of enaminonitriles 121a,b with guanidine nitrate (369) in absolute ethanol in the presence of excess anhydrous potassium carbonate at reflux temperature furnished the pyrimidine-5-carbonitriles 375a,b, via the intermediacy of Michael adducts 373a,b, rather than the 5-aroyl analogues 374a,b as proven from spectral data of the isolated products (Scheme 106).61
 | ||
| Scheme 106 | ||
Recently, Alinezhad and et al. reported that phenylphosphinic acid or 2-pyrrolidonium bisulphate are found to catalyze the three-component condensation of an enaminone 1d,h, aldehyde 376, and urea or thiourea to afford the corresponding 6-unsubstituted dihydropyrimidinones 377 in high to excellent yields. This methodology is simple and fast synthetic route for the preparation of interesting class of heterocycles (Scheme 107).135,136
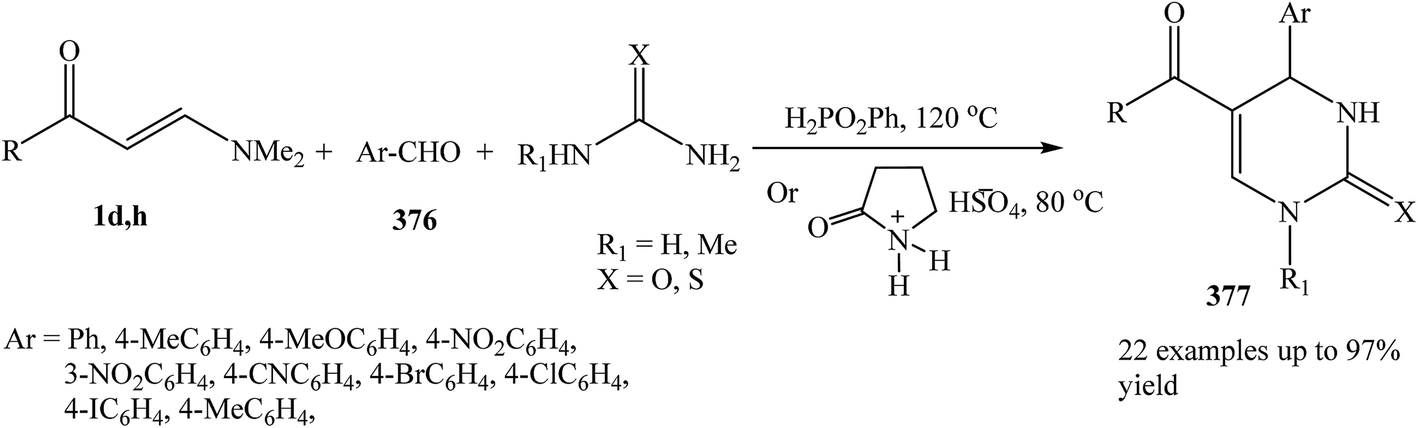 | ||
| Scheme 107 | ||
Ingham and et al.137 was reported the development of a monolith-supported synthetic procedure of enaminone 1 in the presence of Huunig's base, taking advantage of flow processing and the superior flow characteristics of monolithic reagents over gel-phase beads, to allow facile access to an important family of 2-aminopyrimidine derivatives 379 through formation of thioether derivative 378. The process has been successfully applied to a key precursor on route to Imatinib (Ar = 3-pyridyl, R1 = 2-methyl-5-nitrobenzyl, R2 = H) (Scheme 108).
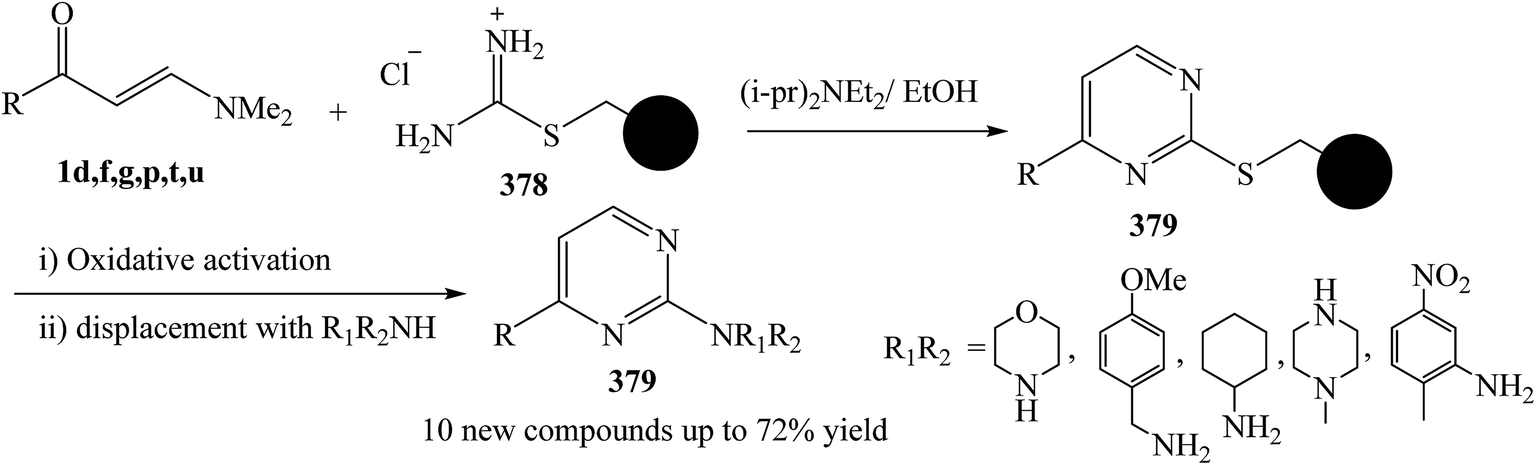 | ||
| Scheme 108 | ||
The refluxing reaction of (E)-3-(dimethylamino)-1-(pyridin-3-yl)prop-2-en-1-one (1u) with 1-(2-methyl-5-nitrophenyl)guanidine nitrate 380 was processed in isopropyl alcohol for 12 h, to give N-(2-methyl-5-nitrophenyl)-4-(pyridin-3-yl)pyrimidin-2-amine 381 in 82% (Scheme 109).138
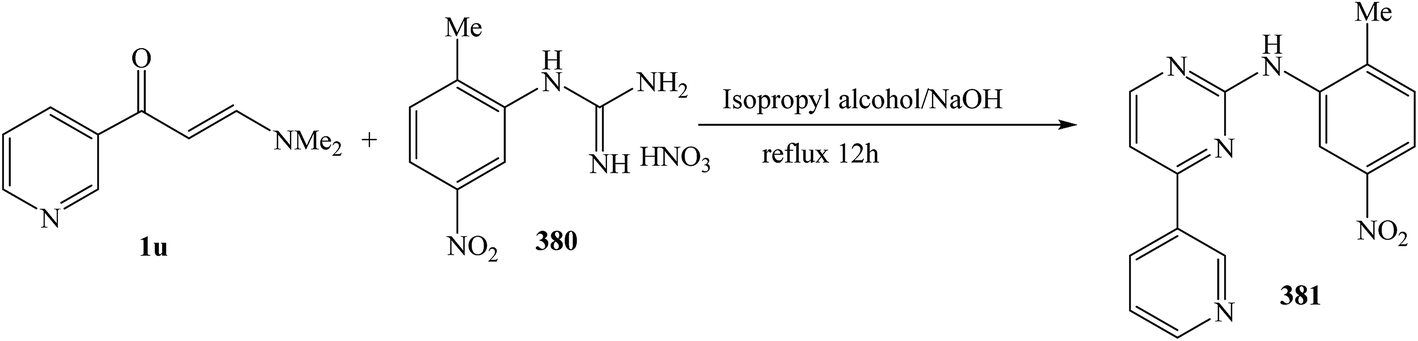 | ||
| Scheme 109 | ||
2.3.2.3. Preparation of pyrans. Recently, Kepe et al.139,140 reported on an one-pot synthesis of different 2H-pyran-2-one derivatives via interaction of either monoactivated methyl ketones or 1,3-dicarbonyl compounds with DMFDMA and N-acylglycines in acetic anhydride. More recently, the general nature of Kepe's pyranone synthesis was extended to reaction of N-substituted glycines with various β-aminovinyl ketones, with especial emphasis on ketones incorporating heterocyclic moieties. Thus, treating aliphatic enaminones 1a–c with N-acetylglycine (382a), hippuric acid (382b) or N-pyrazinylcarbonylglycine (382c) in acetic anhydride led to the corresponding pyranones 386a–g, respectively. It is believed that N-acylglycines 382a–c are first cyclized to the corresponding oxazolones 383a–g and subsequent reaction of the latter with enaminones 1a–c yielded initially Michael adducts 384a–g. These adducts undergo deamination and enolization to afford the non-isolable enols 385a–g that rearrange into the final isolable products 386a–g, respectively, via an attack of hydroxyl function on the oxazolone ring (Scheme 110).68,141
 | ||
| Scheme 110 | ||
As an extension to the Kepe acylaminopyranone synthesis,141,142 dienones 19a–c reacted with N-acetyl- and N-benzoyl-glycines 382a,b in acetic anhydride to yield products assumed to be the pyranones 388a–f rather than the isomeric 389a–f. It is thus assumed that in situ generated oxazolones 383a,b react with 19a–c via initial addition of the active methylene in 383a,b to the enaminone moiety at C-3, yielding intermediates 387a–g that further rearrange into the final isolable products 388a–f (Scheme 111).37
 | ||
| Scheme 111 | ||
Similar reactions of 382a,b with either aromatic enaminones 1d and 1o or with simple heterocyclic enaminones 1p–s in acetic anhydride resulted in the formation of the corresponding acylaminopyranones 389a–j, respectively (Scheme 112).58
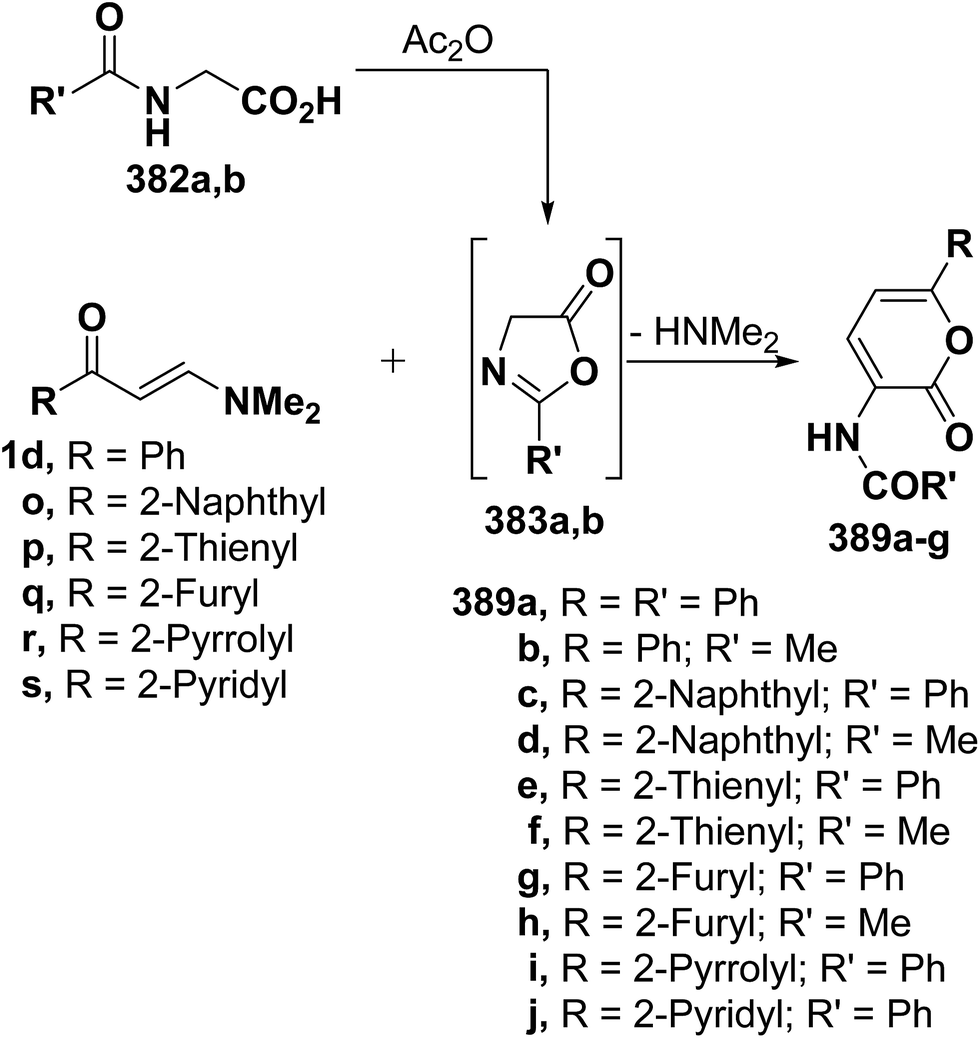 | ||
| Scheme 112 | ||
In accordance with the observed formation of pyranones, heterocyclic enaminones 1y, 1z and 196 reacted with hippuric acid (382b) to give the desired pyranones 390a–d, respectively (Scheme 113).44,77,99,126 The formation of compounds 390a–d starting from the corresponding heterocyclic enaminones and 382b can thus be considered as an extension to the Kepe pyranone synthesis141,142 to enable the synthesis of 6-heteroarylpyran-2-ones.
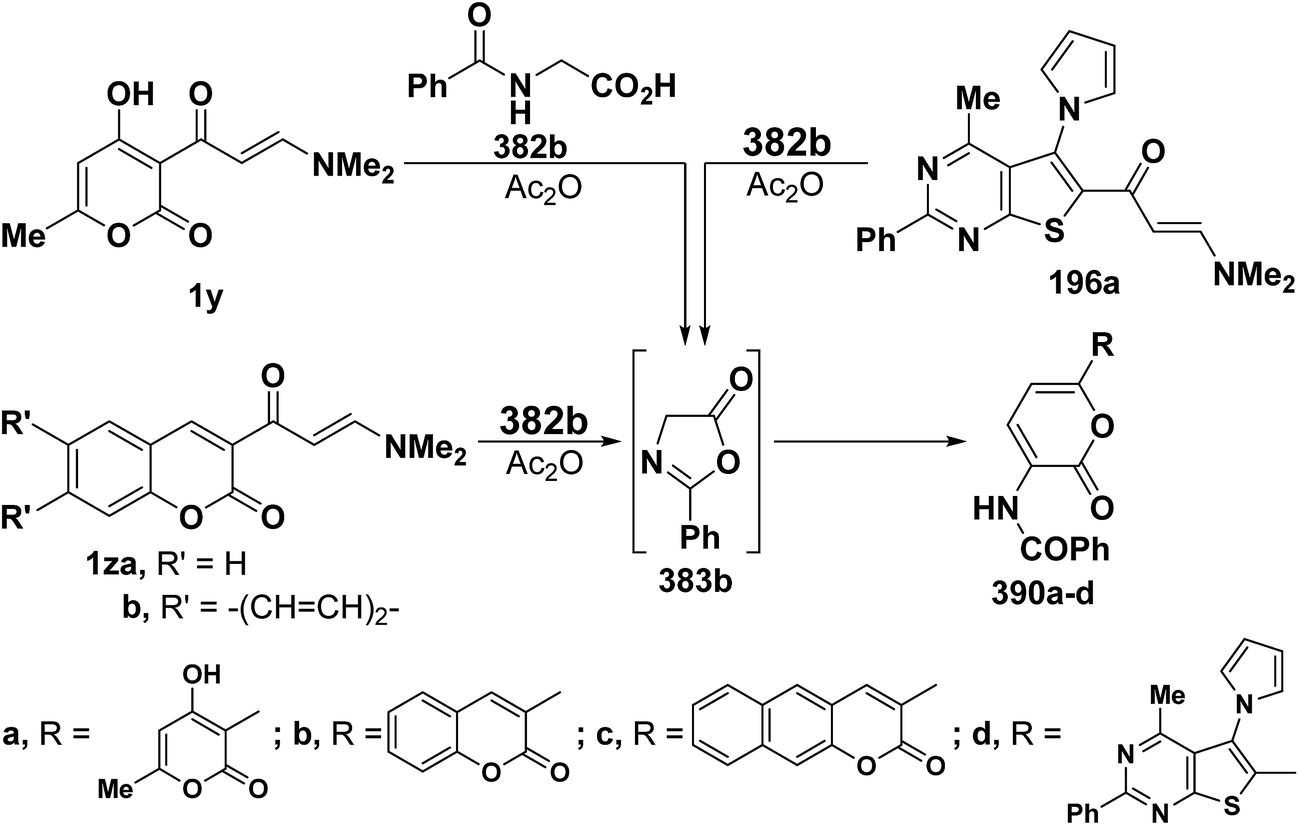 | ||
| Scheme 113 | ||
An interesting reaction leading to the formation of a pyranone ring system78 is the interaction of aminovinyl ketone 1z with dithiocarboxylic acid 391, affording the pyranone derivative 394 via the intermediate formation of 392 and 393, respectively, in a manner similar to that suggested to account for formation of pyranone 390b from reaction of hippuric acid (382b) with the same vinyl ketone 1z.78 This is also a new extension to the Kepe acylaminopyranone synthesis (Scheme 114).141,142
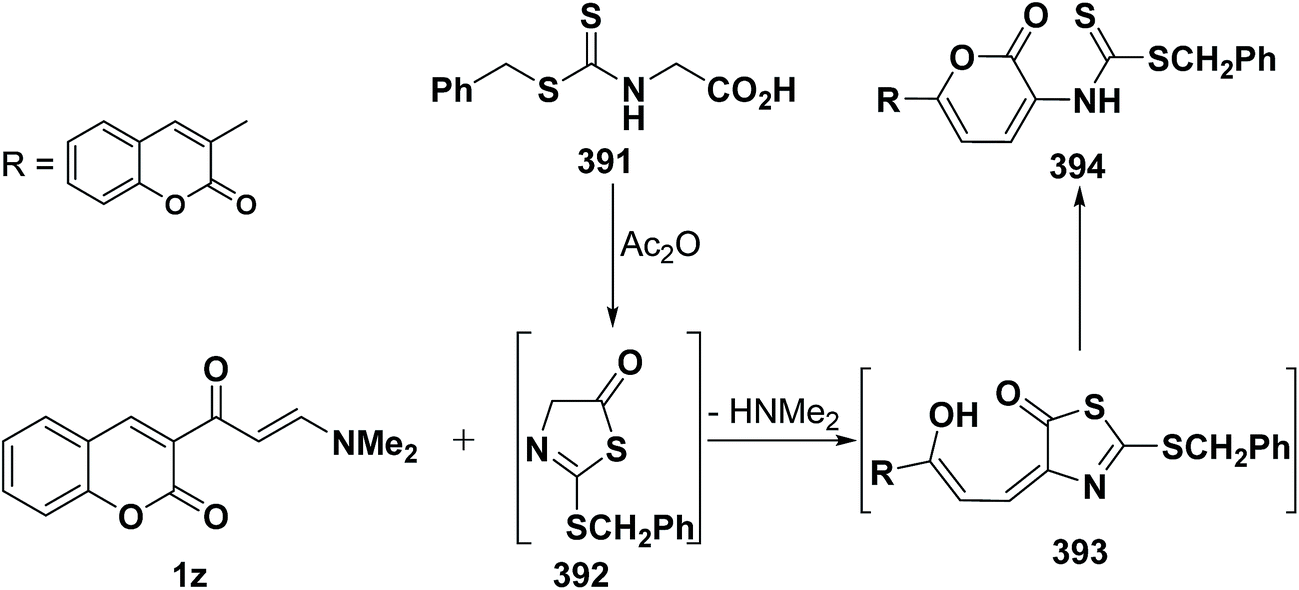 | ||
| Scheme 114 | ||
Unexpectedly, interaction of aminovinyl ketone 1k with aromatic diazonium salts did not give the anticipated arylhydrazonals 396a–c, instead cyclic N,O-acetals 398a–c were formed in good yields. It is believed that the formed enazo compounds 395a–c were initially cyclized into 397a–c that reacted with ethanol to yield the final isolable chromones (4H-benzopyran-4-ones, 398a–c). Both 1H and 13C NMR spectra of the reaction products fit completely with the proposed structure. Additionally, the X-ray crystal structure determination of 398a confirmed the existence of the hydrazono-chromone form 398 rather than the alternative possible enol azo form 399.141 Furthermore, the o-hydroxy substituted derivative 1k showed an additional reactivity when treated with acids: it underwent intramolecular ring closure wherein the aromatic hydroxy group attacked the aminopropenone side chain, leading to chromone 400 in quantitative yield (Scheme 115).142
 | ||
| Scheme 115 | ||
A facile and efficient synthetic strategy to 3-((trifluoromethyl)thio)-4H-chromen-4-one 401 was developed. AgSCF3 and trichloroisocyanuric acid (TCCA) were employed here to generate active electrophilic trifluoromethylthio species in situ.143 Or by using iodine in toluene.144 This reactions could proceed under mild conditions in a short reaction time and be insensitive to air and moisture (Scheme 116).
 | ||
| Scheme 116 | ||
 | ||
| Scheme 117 | ||
On the other hand, reaction of vinyl ketones 1d and 121a with 4-unsubstituted aminopyrazole 305 in the presence of zinc chloride and pyridine, respectively, yielded addition products with elimination of dimethylamine and water for which structure 407 was established. Additionally, when compounds 1d and 405 were refluxed in ethanol, compound 406a was isolated in a good yield. It could then be cyclized into 407a on fusion with zinc chloride.146 Likewise, treating vinyl ketones 1d and 121a with aminopyrazolone 408 in refluxing pyridine for three hours led to the corresponding pyrazolo[3,4-b]pyridines 409a,b (Scheme 105).144 In contrast to the observed formation of pyrazolopyridines,146,147 the same ketones 1d and 121a reacted with 4-unsubstituted aminopyrazole 410 in refluxing pyridine to give the pyrazolo[1,5-a]pyrimidines 412a,b as confirmed by the spectral data of the reaction products. Accordingly, formation of compounds 412a,b, in this case, may be rationalized via initial nucleophilic attack by the exocyclic nitrogen on the electron deficient C-3 of enaminone moiety in 1d and 121a with subsequent elimination of dimethylamine, yielding intermediates 411a,b. These undergo intramolecular cyclization via loss of water, affording the final products 412a,b (Scheme 105).54,148
Similar to the previously described52 behavior of vinyl ketone 1d toward aminopyrazole 410, compound 1d reacted with 3-aminopyrazolo[3,4-d]pyrimidine derivative 413 in refluxing glacial acetic/hydrochloric (1:1) mixture for ten hours to give the respective pyrimido[2,3:4,3]pyrazolo[1,5-a]pyrimidine derivative 414 (Scheme 105).149 In contrast to the reported146 behavior of enaminonitrile 121a toward aminopyrazole 410, compound 121a reacted with 3-amino-1H-pyrazolo[3,4-b]pyridine derivative 415 in glacial acetic acid at reflux temperature for three hours to furnish the tricyclic pyrimidine derivative 416 (Scheme 118).150
 | ||
| Scheme 118 | ||
Refluxing vinyl ketone 1d or 1d with 2-aminobenzimidazole (417) in pyridine solution afforded the 4-substituted pyrimido[1,2-a]benzimidazoles 419a,b,54 while 1-substituted 4-cyanobenzimidazo[1,2-a]pyridines 422a–c were obtained on treatment of vinyl ketones 1d,g or 1p with 2-cyanomethylbenzimidazole (420) in refluxing ethanol containing a catalytic amount of piperidine.23 Formation of compounds 419a,b is thus formed via addition of the ring nitrogen to the activated double bond in 1d and 1s yielding Michael adducts 418a,b which then cyclize by elimination of water and aromatize via loss of dimethylamine, affording the isolable products 419a,b.54 On the contrary, compounds 422a–c are formed via addition of active methylene to the activated double bond, leading eventually to the final products through the intermediate formation of 421a–c (Scheme 119).23
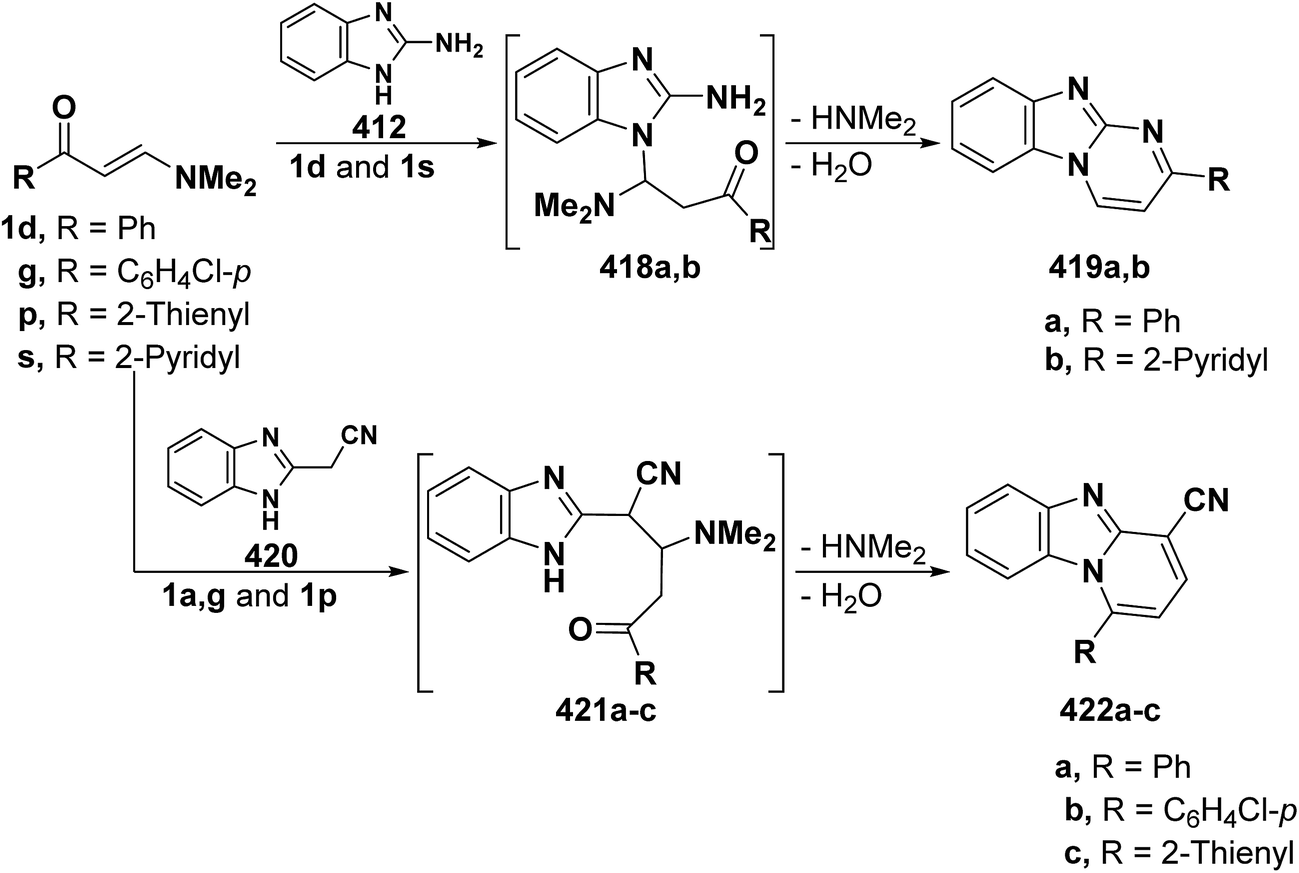 | ||
| Scheme 119 | ||
Moreover, heterocyclic enaminones 1z and 49a reacted with 3-amino-1,2,4-triazole (405) to yield products that may be formulated as 424 or the isomeric 425.45,90 However, structure 425 was established for the reaction products based on 1H NMR data and NOE experiments (Scheme 120).
 | ||
| Scheme 120 | ||
It has been reported68 that vinyl ketones 1b and 1m coupled with the diazotized 5(3)-amino-3(5)-phenylpyrazole (426a), which exists in equilibrium with an isolable diazobetaine 426b, to give azolotriazines 429a,b rather than the expected acyclic α-hydrazono-β-ketoaldehydes 428a,b. Thus, the inability to isolate the target hydrazones 428a,b from this reaction may indicate that the reagents react via a direct 4 + 2 cycloaddition mechanism to the activated double bond in 1b and 1m leading to the isolated products 429a,b (Scheme 121). Likewise, coupling compound 1m with diazotized 2-aminobenzimidazole 430 and with diazotized 3-amino-1H-1,2,4-triazole 431 afforded the analogous [1,2,4]triazino[4,3-a]benzimidazole derivative 432 and triazolo[3,4-c][1,2,4]triazine derivative 433, respectively, via direct 4 + 2 cycloaddition.68
 | ||
| Scheme 121 | ||
Interestingly, the behavior of cyclic enaminone 95b toward diazotized heterocyclic amine 444 differs also from the well established behavior of acyclic enaminones toward arenediazonium salts, where formation of 2-heteroylhydrazonopropanal 445 has not been observed, but instead a biscoupling product 448 has been obtained. This product is assumed to be formed via initial Japp Klingemann type cleavage of the dimethylaminomethylene moiety in 95b, yielding 446 that cyclizes into 447 and then couples further with 444 to yield a hydrazone that cyclizes into the final isolable product 448 (Scheme 122).25
 | ||
| Scheme 122 | ||
Multi-component reaction of sulphone 449, 3-aminotriazole 405 and dimethylformamide–dimethylacetal (DMF–DMA) in DMF under microwave irradiation at 150 °C for 10 min. Afforded compound 450 rather than its isomeric structure 451 (Scheme 110). The conformation of compound 450 was established on the bases of spectral data (MS, IR, 1H NMR) and elemental analyses. Furthermore, alternative synthesis of compound 452 was achieved via condensation of sulphone 1 with dimethylformamide dimethylacetal (DMF–DMF) to give compound 452, and treatment of the product 5 with 3-aminotriazole 305 under the same reaction condition to yield authentic product 450 (Scheme 123).151
 | ||
| Scheme 123 | ||
With this result in hand, the scope of such multi-component protocol were expanded and derivatives 453–457 were synthesized; sulphone 452 and dimethylformamide–dimethylacetal (DMF–DMA), under the same reaction conditions, afforded, in each case, the corresponding heterocyclic ring systems 458–462, respectively (Scheme 124).151
 | ||
| Scheme 124 | ||
To account for the formation of the products 450 and 458–462, it was suggested that the studied reactions started with Michael-type addition of the exocyclic amino group of each the amines used to the activated double bond of 5 followed by in situ tandem elimination of dimethylamine and dehydrative cyclization (Scheme 125).151
 | ||
| Scheme 125 | ||
Also, the reaction of enaminone 465 with active methylene under microwave irradiation. Reaction of enaminone 465 with active methylene derivatives 466–469 gave in glacial acetic acid in the presence of ammonium acetate gave poly-heterocyclic ring systems 470–473, respectively (Scheme 126). The structure of the products was assigned based on the spectral data and elemental analyses.152
 | ||
| Scheme 126 | ||
The reactivity of the enaminone 474a or the morpholinyl derivative 474b towards some heterocyclic amines were also examined. Thus, reaction of 474a or 474b with 5-amino-1H-pyrazole-4-carbonitrile 453 in acetic acid under reflux yielded the respective pyrazolo[1,5-a]pyrimidine derivative 475 (Scheme 114). Similar treatment of 474a or 474b with 5-amino-1,2,4-triazole (405), 5-amino-1H-tetrazole (455), 2-aminobenzimidazole (451) and 6-amino-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (479) under the same reaction conditions afforded the respective 1,2,4-triazolo[1,5-a]pyrimidine 441, tetrazolo[1,5-a]pyrimidine 477, benzimidazo[1,2-a]pyrimidine 478 and 2,3-dihydropyrido[2,3-d]pyrimidinone 480 derivatives, respectively (Scheme 127).51
 | ||
| Scheme 127 | ||
The catalytic activity of the as-synthesized and activated hydrotalcites towards the aza-Michael addition reaction was evaluated. Thus, the reaction of an enaminone 1d with the 5-amino-1H-pyrazole derivatives 481a–c in the presence hydrotalcite catalysts was carried out without solvent under microwave irradiation, to obtain only one isolable product in each case, (as examined by TLC) which were identified as pyrazolo[1,5-a]pyrimidine derivatives 482a–c (Scheme 128).153
 | ||
| Scheme 128 | ||
An environmentally benign multicomponent synthetic method has been realized for the diastereoselective construction of fused tetrahydropyridines 485 from reaction of enaminone 1b,d,f,g, o-aminophenol 483, and cinnamaldehyde 484 in the presence of lactic acid in water–ethanol media (Scheme 129).154
 | ||
| Scheme 129 | ||
A mixture of enaminone 1d,g,p,q and 3-amino-2-cyanopent-2-enedinitrile (486) in AcOH/NH4OAc was heated under reflux for 2 h to give 7-amino-5-oxo-2-(thienyl)-5,6-dihydro-1,6-naphthyridine-8-carbonitriles 487a–d (Scheme 130).155
 | ||
| Scheme 130 | ||
The reaction of enaminone 1t with 6-amino-2-thioxo-(1H)-pyrimidin-4-one (489) produced 5-(pyridin-4-yl)-2-thioxo-2,3-dihydropyrido[2,3-d]pyrimidin-4(1H)-one (490) (Scheme 2). Treatment of thione 490 with hydrazonoyl halides 491a–h in dioxane, in the presence of triethylamine under reflux gave in each case a single product consistent with structure 492 (Scheme 2) based on spectroscopic data (IR, 1H NMR and MS) and elemental analyses (Scheme 131).16
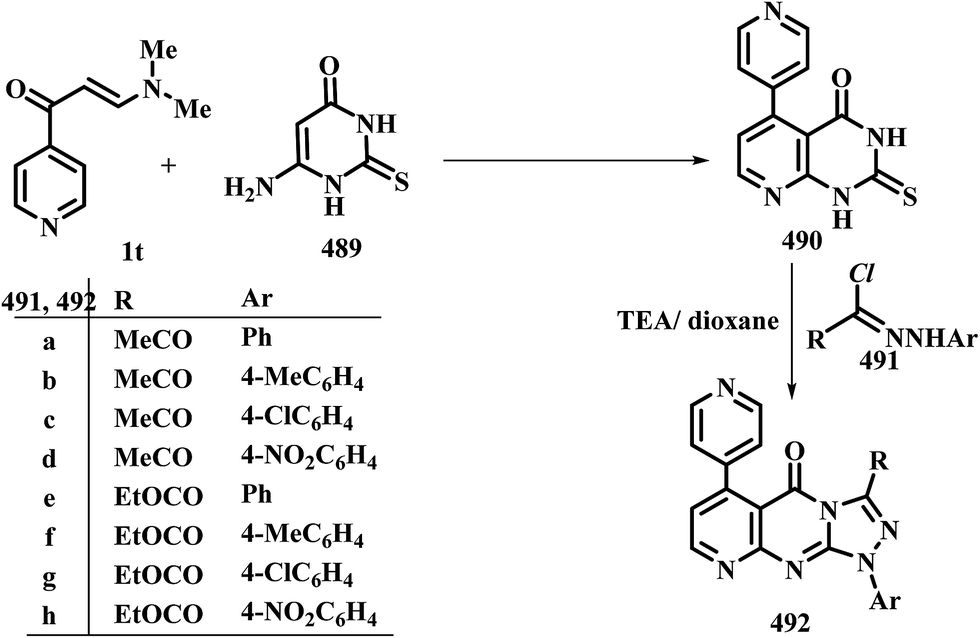 | ||
| Scheme 131 | ||
3. Conclusions and future directions
The data considered in this review clearly demonstrate the high synthetic applications of N,N-dimethylenamino ketones in the preparation of acyclic and carbocyclic compounds as well as a broad range of heterocyclic and fused heterocyclic derivatives. Most importantly, these N,N-dimethyl analogues have proven to be of biological interest and provide an access to new class of biologically active heterocyclic compounds for biomedical screening. Presently, the great interest of researchers worldwide in such ketones is confirmed by the fact that most of the articles cited in this review are dated in the last two decades. We are sure that they will generate new and innovative applications with future generations of compounds, and thus providing a valuable adjunct to therapy. Further studies in this direction are currently underway and the results of this research will be reported elsewhere.Acknowledgements
H. Gaber is greatly indebted to School of Chemistry, Cardiff University, Cardiff, U.K., for granting him a Post-Doctoral Research Fellowship and for all laboratory facilities provided to this fellowship. Also, the authors wish to acknowledge the generous financial support of this research by grant from Cardiff University, the Engineering and Physical Sciences Research Council (EPSRC) and the Biotechnology and Biological Sciences Research Council (BBSRC), in collaboration with the University of Wales College of Medicine, U.K.References
- G. Negri, C. Kascheres and A. J. Kascheres, J. Heterocycl. Chem., 2004, 41, 461 CrossRef CAS.
- J. P. Michael, C. B. de Koning, D. Gravestock, G. D. Hosken, A. S. Howard, C. M. Jungmann, R. W. M. Krause, A. S. Parsons, S. C. Pelly and T. V. Stanbury, Pure Appl. Chem., 1999, 71, 979 CrossRef CAS.
- P. Lue and J. V. Greenhill, Adv. Heterocycl. Chem., 1997, 67, 207 CrossRef.
- M. R. Shaaban, T. S. Saleh, F. H. Osman and A. M. Farag, J. Heterocycl. Chem., 2007, 44, 177 CrossRef CAS.
- C. M. Kascheres, J. Braz. Chem. Soc., 2003, 14, 945 CrossRef CAS.
- J. V. Greenhill, Chem. Soc. Rev., 1977, 6, 277 RSC.
- S. A. S. Ghozlan, I. A. Abdelhamid, H. M. Gaber and M. H. Elnagdi, J. Heterocycl. Chem., 2005, 42, 1185 CrossRef CAS.
- S. A. S. Ghozlan, I. A. Abdelhamid, H. Gaber and M. H. Elnagdi, J. Chem. Res., 2004, 789 CrossRef CAS.
- H. F. Anwer, N. M. Hanafi, H. M. Gaber and M. H. Elnagdi, J. Chem. Res., 2005, 29 Search PubMed.
- H. M. Gaber, A. W. Erian, S. A. Ouf and S. M. Sherif, Afinidad, 2005, 62, 143 CAS.
- H. M. Gaber, F. A. Abu-Shanab, S. A. Ouf and S. M. Sherif, Afinidad, 2005, 62, 57 CAS.
- H. M. Gaber and S. A. Ouf, Afinidad, 2005, 62, 337 CAS.
- T. A. Farghaly, M. A. Abdallah and Z. A. Muhammad, Molecules, 2011, 16, 10420 CrossRef CAS PubMed.
- F. A. Abu-Shanab, A. Hessen, H. M. Gaber and S. A. S. Mousa, J. Sulfur Chem., 2006, 27, 293 CrossRef CAS.
- H. M. Gaber, S. M. Sherif and F. A. Abu-Shanab, J. Sulfur Chem., 2005, 26, 393 CrossRef CAS.
- S. M. Gomha, F. M. Abdelrazek, A. H. Abdelrahman, P. Metz and M. A. Sayed, Lett. Drug Des. Discovery, 2017, 14 DOI:10.2174/15701808146661611 28120240.
- S. M. Gomha and H. A. Abdel-Aziz, Heterocycles, 2012, 85(9), 2291 CrossRef CAS.
- S. M. Sherif, R. M. Mohareb, H. Z. Shams and H. M. M. Gaber, J. Chem. Res., 1995, 434, 2658 Search PubMed.
- S. M. Gomha, Y. H. Zaki and A. O. Abdelhamid, Molecules, 2015, 20, 21826 CrossRef CAS PubMed.
- L. J. O. Figueiredo and C. Kascheres, J. Org. Chem., 1997, 62, 1164–1167 CrossRef CAS.
- F. Al-Omran, M. M. Abdel Khalik, A. Abou-Elkhair and M. H. Elnagdi, Synthesis, 1997, 91 CrossRef CAS.
- N. A. Al-Awadi, M. H. Elnagdi, Y. A. Ibrahim, K. Kaul and A. Kumar, Tetrahedron, 2001, 57, 1609 CrossRef CAS.
- A. Z. A. Hassanien, Afinidad, 2003, 60, 468 CAS.
- P. Šimůnek, A. Lyčka and V. Macháček, Eur. J. Org. Chem., 2002, 2764 CrossRef.
- S. Al-Mousawi, M. M. AbdelKhalik, E. John and M. H. Elnagdi, J. Heterocycl. Chem., 2003, 40, 689 CrossRef CAS.
- N. Al-Awadi, M. R. Ibrahim, I. A. Abdelhamid and M. H. Elnagdi, Tetrahedron, 2008, 64, 8202 CrossRef CAS.
- K. M. Al-Zaydi, E. A. Hafez and M. H. Elnagdi, J. Chem. Res., 2000, 154, 510 Search PubMed.
- M. A. Al-Shiekh, H. Y. Medrassi, M. H. Elnagdi and E. A. Hafez, J. Chem. Res., 2007, 432 CrossRef CAS.
- M. A. Al-Shiekh, A. M. Salaheldin, E. A. Hafez and M. H. Elnagdi, J. Heterocycl. Chem., 2004, 41, 647 CrossRef CAS.
- R. M. Abdel-Motaleb, A.-M. A. Makhloof, H. M. Ibrahim and M. H. Elnagdi, J. Heterocycl. Chem., 2006, 43, 931 CrossRef CAS.
- B. Al-Saleh, M. A. El-Apasery and M. H. Elnagdi, J. Chem. Res., 2004, 578 CrossRef CAS.
- M. M. Abdel-Khalik, S. M. Agamy and M. H. Elnagdi, Z. Naturforsch., 2000, 55, 1211 CAS.
- S. M. Agamy, J. Chem. Res., 2001, 349, 901 Search PubMed.
- R. Pichon, J. Le Saint and P. Courtot, Bull. Soc. Chim. Fr., 1980, 449 CAS.
- K. M. Al-Zaydi, R. M. Borik and M. H. Elnagdi, Molecules, 2003, 8, 910 CrossRef CAS.
- O. M. E. El-Dusouqui, M. M. AbdelKhalik, N. A. Al-Awadi, H. H. Dib, B. J. George and M. H. Elnagdi, J. Chem. Res., 2006, 295 CrossRef CAS.
- S. Al-Mousawi, M. M. Abdel-Khalik, S. El-Sherbiny, E. John and M. H. Elnagdi, J. Heterocycl. Chem., 2001, 38, 949 CrossRef CAS.
- F. Al-Omran and A. A. El-khair, J. Chem. Res., 2006, 6 CrossRef CAS.
- S. Makhseed, H. M. E. Hassaneen and M. H. Elnagdi, Z. Naturforsch., 2007, 62, 529 CAS.
- I. M. Kenawi and M. H. Elnagdi, Spectrochim. Acta, Part A, 2006, 65, 805 CrossRef PubMed.
- R. M. Abdel-Motaleb, A.-M. A.-S. Makhloof, H. M. Ibrahim and M. H. Elnagdi, J. Heterocycl. Chem., 2007, 44, 109 CrossRef CAS.
- B. Al-Saleh, M. A. El-Apasery, R. S. Abdel-Aziz and M. H. Elnagdi, J. Heterocycl. Chem., 2005, 42, 563 CrossRef CAS.
- A. Z. A. Hassanien, S. A. S. Ghozlan and M. H. Elnagdi, J. Heterocycl. Chem., 2003, 40, 225 CrossRef CAS.
- B. Al-Saleh, N. Al-Awadi, H. Al-Kandar, M. M. Abdel-Khalik and M. H. Elnagdi, J. Chem. Res., 2000, 16, 201 Search PubMed.
- B. Al-Saleh, H. Behbehani, M. A. El-Apasery and M. H. Elnagdi, J. Chem. Res., 2004, 575 CrossRef CAS.
- P. Schenone, L. Mosti and G. Menozzi, J. Heterocycl. Chem., 1982, 19, 1355 CrossRef CAS.
- K. M. Al-Zaydi, A. Al-Shamary and M. H. Elnagdi, J. Chem. Res., 2006, 408 CAS.
- T. A. Abdallah, A. M. Salaheldin and N. F. Radwan, Z. Naturforsch., 2007, 62, 261 CAS.
- S. Almazroa, M. H. Elnagdi and A. M. S. El-Din, J. Heterocycl. Chem., 2004, 41, 267 CrossRef CAS.
- T. A. Abdallah, J. Heterocycl. Chem., 2007, 44, 961 CrossRef CAS.
- S. M. Gomha, T. M. A. Eldebss, M. M. Abdulla and A. S. Mayhoub, Eur. J. Med. Chem., 2014, 82, 472 CrossRef CAS PubMed.
- T. M. A. Eldebss, S. M. Gomha, M. M. Abdulla and R. K. Arafa, MedChemComm, 2015, 6(5), 977 RSC.
- H. M. Gaber, Org. Prep. Proced. Int., 2008, 40, 365 CrossRef CAS.
- A. Al-Enezi, B. Al-Saleh and M. H. Elnagdi, J. Chem. Res., 1997, 4, 116 Search PubMed.
- A. Al-Etaibi, N. Al-Awadi, F. Al-Omran, M. M. Abdel-Khalik and M. H. Elnagdi, J. Chem. Res., 1999, 4, 151 Search PubMed.
- B. Al-Saleh and M. A. El-Apasery, J. Heterocycl. Chem., 2006, 43, 559 CrossRef CAS.
- S. M. Al-Mousawi, K. S. George and M. H. Elnagdi, Pharmazie, 1999, 54, 571 CAS.
- F. Al-Omran, N. Al-Awadi, M. M. Abdel-Khalik, K. Kaul, A. A. El-Khair and M. H. Elnagdi, J. Chem. Res., 1997, 84, 601 Search PubMed.
- F. Al-Omran, N. Al-Awadhi, A. A. Elassar and A. A. El-Khair, J. Chem. Res., 2000, 20, 237 Search PubMed.
- Z. E. Kandeel, K. M. Al-Zaydi, E. A. Hafez and M. H. Elnagdi, Phosphorus, Sulfur Silicon Relat. Elem., 2002, 117, 105 CrossRef.
- K. M. Al-Zaydi, M. A.-A. Al-Shiekh and E. A.-Z. Hafez, J. Chem. Res., 2000, 13, 173 Search PubMed.
- R. B. Toche, M. N. Jachak, T. S. Dalvi, R. W. Sabnis, H. Junek and T. Kappe, Org. Prep. Proced. Int., 1998, 30, 367 CrossRef CAS.
- N. A. Al-Awadi, M. M. Abdelkhalik, I. A. Abdelhamid and M. H. Elnagdi, Synlett, 2007, 2979 CrossRef CAS.
- J. B. Rasmussen, R. Shabana and S. O. Lawesson, Tetrahedron, 1982, 38, 1705 CrossRef CAS.
- S. I. El-Desoky, S. B. Bondock, H. A. Etman, A. A. Fadda and M. A. Metwally, Sulfur Lett., 2003, 26, 127 CrossRef CAS.
- B. Al-Saleh, M. M. Abdel-Khalik, A. M. Eltoukhy and M. H. Elnagdi, J. Heterocycl. Chem., 2002, 39, 1035 CrossRef CAS.
- M. M. Abdel-Khalik and M. H. Elnagdi, Synth. Commun., 2002, 32, 159 CrossRef CAS.
- M. A. Al-Shiekh, A. M. S. El-Din, E. A. Hafez and M. H. Elnagdi, J. Chem. Res., 2004, 174 CrossRef CAS.
- F. C. Pigge and F. Ghasedi, Tetrahedron Lett., 2000, 41, 6545 CrossRef CAS.
- K. Verma and P. Banerjee, Adv. Synth. Catal., 2016, 358, 2053 CrossRef CAS.
- S. Zhou, J. Wang, L. Wang, C. Song, K. Chen and J. Zhu, Angew. Chem., Int. Ed., 2016, 55, 1 CrossRef.
- A. R. Katritzky, Y. Fang, A. Donkor and J. Xu, Synthesis, 2000, 2029 CrossRef CAS.
- W. S. Murphy and M. J. Bertrand, J. Chem. Soc., Perkin Trans. 1, 1998, 4115 RSC.
- S. Kirschbaum and H. Waldmann, J. Org. Chem., 1998, 63, 4936 CrossRef CAS.
- A. W. Trautwein and G. Jung, Tetrahedron Lett., 1998, 39, 8263 CrossRef CAS.
- N. Nishimura, Y. Koyano, M. Sugiura and I. Maeba, Heterocycles, 1999, 51, 803 CrossRef CAS.
- I. Macba, Y. Nishiyama, S. Kanazawa and A. Sato, Heterocycles, 1995, 41, 507 CrossRef.
- Y. Nishiyama, N. Nishimura, N. Kuroyanagi and I. Maeba, Carbohydr. Res., 1997, 300, 283 CrossRef CAS.
- I. Macba, Y. Ito, M. Wakimura and C. Ito, Heterocycles, 1993, 36, 1617 CrossRef.
- F. M. A. A. El-Taweel and M. H. Elnagdi, J. Heterocycl. Chem., 2001, 38, 981 CrossRef CAS.
- S. Kantevari, R. S. Patpi, D. Addla, S. R. Putapatri, B. Sridhar, P. Yogeeswariand and D. Sriram, ACS Comb. Sci., 2011, 13, 427 CrossRef CAS PubMed.
- J. P. Wan and Y. J. Pan, Chem. Commun., 2009, 19, 2768 RSC.
- Y. Jiang, V. Z. Y. Khong, E. Lourdusamy and C. M. Park, Chem. Commun., 2012, 48, 3133 RSC.
- J. P. Wan, Y. Zhou, Y. Liu and S. Sheng, Green Chem., 2016, 18, 402 RSC.
- H. Behbehani, H. M. Ibrahim, S. Makhseed and H. Mahmoud, Eur. J. Med. Chem., 2011, 46, 1813 CrossRef CAS PubMed.
- S. Hernández, I. Moreno, R. SanMartin, G. Gómez, M. T. Herrero and E. Domínguez, J. Org. Chem., 2010, 75, 434 CrossRef PubMed.
- M. A. Abdallah, T. A. Farghali, M. M. Abdalla and Z. A. Mohammad, World J. Chem., 2011, 6, 96 CAS.
- J. P. Wan, S. Cao and Y. Liu, Org. Lett., 2016, 18, 6034 CrossRef CAS PubMed.
- A. Salomone, F. M. Perna, F. C. Sassone, A. Falcicchio, J. Bezenšek, J. Svete, B. Stanovnik, S. Florio and V. J. Capriati, Org. Chem., 2013, 78, 11059 CrossRef CAS PubMed.
- A. A. Hassanien, S. A. S. Ghozlan and M. H. Elnagdi, J. Heterocycl. Chem., 2003, 40, 225 CrossRef CAS.
- S. Al-Mousawi, E. John and N. Al-Kandery, J. Heterocycl. Chem., 2004, 41, 381 CrossRef CAS.
- M. Abdel-Megid, M. H. Elnagdi and A. M. Negm, J. Heterocycl. Chem., 2002, 39, 105 CrossRef CAS.
- T. V. Hughes, S. D. Hammond and M. P. Cava, J. Org. Chem., 1998, 63, 401 CrossRef CAS.
- F. Al-Omran, N. Al-Awadi, A. A. El-Khair and M. H. Elnagdi, Org. Prep. Proced. Int., 1997, 29, 285 CrossRef CAS.
- F. Al-Omran and A. A. El-Khair, J. Heterocycl. Chem., 2005, 42, 307 CrossRef CAS.
- E. Domínguez, E. Ibeas, E. Martínez de Marigorta, J. K. Palacios and R. San Martín, J. Org. Chem., 1996, 61, 5435 CrossRef.
- Y. W. Ho, J. Chin. Chem. Soc., 2007, 54, 1075 CrossRef CAS.
- H. Adams, S. R. Batten, G. M. Davies, M. B. Duriska, J. C. Jeffery, P. Jensen, J. Lu, G. R. Motson, S. J. Coles, M. B. Hursthouse and M. D. Ward, Dalton Trans., 2005, 1910 RSC.
- K. M. Al-Zaydi, Molecules, 2003, 8, 541 CrossRef CAS.
- B. Al-Saleh, M. M. Abdel-Khalik, E. Darwich, O. A.-M. Salah and M. H. Elnagdi, Heteroat. Chem., 2002, 13, 141 CrossRef CAS.
- F. Al-Omran, J. Heterocycl. Chem., 2000, 37, 1219 CrossRef CAS.
- A. R. Katritzky, C. W. Rees and E. F. Scriven, In Compr. Heterocycl. Chem. II, Pergamon Press, New York, 1996, vol. 3, p. 227 Search PubMed.
- Aldrich Handbook of Fine Chemicals and Laboratory Equipment, Germany, 2000–2001, p. 1368 Search PubMed.
- H. M. Ibrahim, S. Makhseed, R. M. Abdel-Motaleb, A.-M. A.-S. Makhloof and M. H. Elnagdi, Heterocycles, 2007, 71, 1951 CrossRef CAS.
- C. Reidlinger, R. Dworczak and H. Junek, Monatsh. Chem., 1998, 129, 1207 CAS.
- K. M. Dawood, A. M. Farag and Z. E. Kandeel, J. Chem. Res., 1999, 88, 537 Search PubMed.
- K. M. Al-Zaydi and A. Z. Hafez, J. Chem. Res., 1999, 360, 1621 Search PubMed.
- A. O. Abdelhamid and A. A. Al-Atoom, Synth. Commun., 2006, 36, 97 CrossRef CAS.
- M. A. M. Gad-Elkareem and A. O. Abdelhamid, Afinidad, 2004, 61, 427 CAS.
- A. O. Abdelhamid and M. A. M. Alkhodshi, J. Heterocycl. Chem., 2005, 42, 527 CrossRef CAS.
- T. S. Saleh, K. Narasimharao, N. S. Ahmed, S. N. Basahel, S. A. Al-Thabaiti and M. Mokhtar, J. Mol. Catal. A: Chem., 2013, 367, 122 CrossRef.
- H. M. Gaber, K. M. ElSawy and S. M. Sherif, Afinidad, 2008, 65, 61 CAS.
- M. M. Abdel-Khalik, M. H. Elnagdi and S. M. Agamy, Synthesis, 2000, 1166 CrossRef CAS.
- A. Raghunadh, S. B. Meruva, R. Mekala, K. R. Rao, T. Krishna, R. G. Chary, L. V. Rao and U. K. S. Kumar, Tetrahedron Lett., 2014, 55, 2986 CrossRef CAS.
- S. M. Kim, S. W. Lee and Y. H. Song, Bull. Korean Chem. Soc., 2014, 35, 2859 CrossRef CAS.
- S. Al-Mousawi, M. S. Moustafa and M. H. Elnagdi, ARKIVOC, 2008, x, 17 Search PubMed.
- R. W. Sabnis, D. W. Rangnekar and N. D. Sonawane, J. Heterocycl. Chem., 1999, 36, 333 CrossRef CAS.
- T. I. Mukhanova, E. K. Panisheva, V. M. Lyubchanskaya, L. M. Alekseeva, Y. N. Sheinker and V. G. Granik, Tetrahedron, 1997, 53, 177 CrossRef CAS.
- M. A. P. Martins, G. C. Paveglio, L. V. Rodrigues, C. P. Frizzo, N. Zanatta and H. G. Bonacorso, New J. Chem., 2016, 40, 5989 RSC.
- S. Mousawi, M. S. Moustafa and M. H. Elnagdi, Arkivoc, 2008, x, 17 Search PubMed.
- S. M. Agamy, M. M. Abdel-Khalik, M. H. Mohamed and M. H. Elnagdi, Z. Naturforsch. B, 2001, 56, 1074 CrossRef.
- L. X. Zhao, J. Sherchan, J. K. Park, Y. Jahng, B. S. Jeong, T. C. Jeong, C. S. Lee and E. S. Lee, Arch. Pharmacal Res., 2006, 29, 1091 CrossRef CAS.
- F. Al-Omran, A. A. El-Khair and M. H. Elnagdi, J. Chem. Res., 1998, 798 RSC.
- N. Y. Wang, W. Q. Zuo, Y. Xu, C. Gao, X. X. Zeng, L. D. Zhang, X. Y. You, C. T. Peng, Y. She, S. Y. Yang, Y. Q. Wei and L. T. Yu, Bioorg. Med. Chem. Lett., 2014, 24, 1581 CrossRef CAS PubMed.
- J. Witherington, V. Bordas, A. Gaiba, N. S. Garton, A. Naylor, A. D. Rawlings, B. P. Slingsby, D. G. Smith, A. K. Takle and R. W. Ward, Bioorg. Med. Chem. Lett., 2003, 13, 3055 CrossRef CAS PubMed.
- Y. W. Ho and W. H. Yao, J. Chin. Chem. Soc., 2005, 52, 313 CrossRef CAS.
- M. M. AbdelKhalik, A. M. Eltoukhy, S. M. Agamy and M. H. Elnagdi, J. Heterocycl. Chem., 2004, 41, 431 CrossRef CAS.
- F. A. Abu-Shanab, A. M. Hessen and S. A. S. Mousa, J. Heterocycl. Chem., 2007, 44, 787 CrossRef CAS.
- J. Tois, M. Vahermo and A. Koskinen, Tetrahedron Lett., 2005, 46, 735 CrossRef CAS.
- K. Shankaraiah, G. Chandrasekhar, K. S. N. Reddy and G. Sabitha, Tetrahedron Lett., 2015, 56, 842 CrossRef CAS.
- J. P. Wan, S. F. Gan, G. L. Sun and Y. J. Pan, J. Org. Chem., 2009, 74, 2862 CrossRef CAS PubMed.
- S. Cao, L. Xin, Y. Liu, J. P. Wan and C. Wen, RSC Adv., 2015, 5, 27372 RSC.
- I. Elghamry and Y. Al-Faiyz, Tetrahedron Lett., 2016, 57, 110 CrossRef CAS.
- P. W. Manley, W. Breitenstein, J. Brűggen, S. W. Cowan-Jacob, P. Furet, J. Mestan and T. Meyer, Bioorg. Med. Chem. Lett., 2004, 14, 5793 CrossRef CAS PubMed.
- H. Alinezhad, M. Tajbakhsh, M. Zare and M. Mousavi, Heteroat. Chem., 2016, 1–5 Search PubMed.
- H. Alinezhad, M. Tajbakhsh, M. Zare and M. Mousavi, Chem. Pap., 2016, 70, 1126 CAS.
- R. J. Ingham, E. Riva, N. Nikbin, I. R. Baxendale and S. V. Ley, Org. Lett., 2012, 14, 3921 CrossRef PubMed.
- Y. F. Liu, C. L. Wang, Y. J. Bai, N. Han, J. P. Jiao and X. L. Qi, Org. Process Res. Dev., 2008, 12, 490 CrossRef CAS.
- V. Kepe, M. Kočevar and S. Polanc, J. Heterocycl. Chem., 1996, 33, 1707 CrossRef CAS.
- V. Kepe, M. Kocevar, S. Polanc, B. Vercek and M. Tisler, Tetrahedron, 1990, 46, 2081 CrossRef CAS.
- K. M. Al-Zaydi, R. M. Borik and M. H. Elnagdi, J. Heterocycl. Chem., 2007, 44, 1187 CrossRef CAS.
- A.-K. Pleier, H. Glas, M. Grosche, P. Sirsch and W. R. Thiel, Synthesis, 2001, 55 CrossRef CAS.
- H. Xiang and C. Yang, Org. Lett., 2014, 16, 5686 CrossRef CAS PubMed.
- Y. Igarashi, H. Kumazawa, T. Ohshima, H. Satomi, S. Terabayashi, S. Takeda, M. Aburada and K. Miyamoto, Chem. Pharm. Bull., 2005, 53, 1088 CrossRef CAS PubMed.
- S. A. S. Ghozlan and A. Z. A. Hassanien, Tetrahedron, 2002, 58, 9423 CrossRef CAS.
- S. M. Al-Mousawi, K. Kaul, M. A. Mohammad and M. H. Elnagdi, J. Chem. Res., 1997, 318, 2026 Search PubMed.
- S. M. Al-Mousawi, M. A. Mohammad and M. H. Elnagdi, J. Heterocycl. Chem., 2001, 38, 989 CrossRef CAS.
- B. Al-Saleh, M. M. Abdel-Khalik, A. Al-Enzy and M. H. Elnagdi, J. Chem. Res., 1999, 654, 2848 Search PubMed.
- Y.-W. Ho and C.-T. Yao, J. Chin. Chem. Soc., 2003, 50, 283 CrossRef CAS.
- M. A. M. Gad-Elkareem, A. M. Abdel-Fattah and M. A. A. Elneairy, Can. J. Chem., 2007, 85, 592 CrossRef CAS.
- S. M. Gomha, T. M. A. Eldebss, M. G. Badrey, M. M. Abdulla and A. S. Mayhoub, Chem. Biol. Drug Des., 2015, 86, 1292 CAS.
- E. M. H. Abbas, S. M. Gomha and T. A. Farghaly, Arabian J. Chem., 2014, 7, 623 CrossRef CAS.
- M. Mokhtar, T. S. Saleh and S. N. Basahela, J. Mol. Catal. A: Chem., 2012, 353, 122 CrossRef.
- J. Wan, S. Zhong and Y. Liu, Synthesis, 2015, 47, 3611 CrossRef CAS.
- M. S. Moustafa, S. M. Al-Mousawi, N. M. Hilmy, Y. A. Ibrahim, J. C. Liermann, H. Meier and M. H. Elnagdi, Molecules, 2013, 18, 276 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |