
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel nanoparticles supported on nitrogen-doped honeycomb-like carbon frameworks for effective methanol oxidation†
Juan Wanga,
Qi Zhaoa,
Hongshuai Houb,
Yifei Wua,
Weizhen Yua,
Xiaobo Ji *b and
Lidong Shao*a
*b and
Lidong Shao*a
aShanghai Key Laboratory of Materials Protection and Advanced Materials in Electric Power, Shanghai University of Electric Power, Shanghai 200090, PR China. E-mail: lidong.shao@shiep.edu.cn
bCollege of Chemistry and Chemical Engineering, Central South University, Changsha, 410083, PR China. E-mail: xji@csu.edu.cn
First published on 2nd March 2017
Abstract
In this work, nitrogen-doped honeycomb-like carbon framework (CNFs) is synthesized by a low-cost and scalable method. Ni nanoparticles are then supported on CNFs (CNFs–Ni) as a catalyst for methanol oxidation. In addition to the enhanced mass activity, both the onset and oxidation peak potentials of CNFs–Ni for methanol oxidation shift negatively in comparison to its non-doped counterpart, CFs–Ni. We propose that the small particle size, uniform dispersion, and electronic effect arising from the electron interactions between CNFs and Ni contribute to the improved electrocatalytic performance.
Introduction
Over the past several decades, significant progress has been made in developing carbon materials for catalytic and electrocatalytic applications.1,2 Carbon black has been widely used supporting material in electrocatalytic reactions. However, its agglomeration property reduces its active oxidant utilization and increases the transportation resistance in catalysis process.3 One or two dimensional graphitized carbon materials, such as carbon nanotubes, graphene have been used as supporting materials with unique electronic and chemical properties.4,5 For example, Gong et al. used oxygen functionalized carbon nanotubes supported NiO/Ni heterostructures for hydrogen generation from water electrolysis. And due to the interaction between the oxygen functionalized CNTs and nickel precursors, the reduction of nickel species into larger aggregated nanoparticles and less reactive pure phase nickel were impeded during the thermal treatment process6 which contributed to the excellent catalytic performance.Recently, porous carbon networks with high specific surface area, good conductivity, and additional ion diffusion channels have been extensively investigated as catalysts or supports. Zhang et al. used a porous carbon framework with a dense network for interdispersing MnO nanoparticles, with the obtained composite displaying an enhanced capability and cycling properties.7 Active Pd were supported on hierarchical porous carbon which derived from a zeolitic imidazolate framework (ZIF-8), and employed in methanol oxidation, excellent activity and stability were obtained.8
Nitrogen-doped carbon materials have been the subject of much investigation for catalytic reactions, mainly due to the abundance and accessibility of the required raw materials. In addition, the incorporation of nitrogen atoms into the carbon nanostructures resulted the tailoring of the surface chemical and electronic properties of carbon materials. After nitrogen doping, the carbon nanotubes displayed basic properties are beneficial for solid base catalysis.9 Nitrogen doping can facilitate the adsorption of oxygen and decomposition of hydroperoxide in oxygen reduction reactions.10 Sheng et al. reported an excellent oxygen reduction activity of nitrogen doped graphene, and the electronic effect arising from the interactions between the doped nitrogen atoms and the neighbored carbon atoms contributed to the catalytic activity.11
The lone electron pairs of nitrogen atoms can delocalise in the conjugated system of the sp2-hybridised carbon frameworks, and the N-doped carbon structure has already been employed to enhance the catalytic activities in electrochemical reactions.12 Pt nanoparticles modified nitrogen doped porous carbon nanocubes were prepared for methanol oxidation, and the good electrocatalytic performances were arising from the uniform particle dispersion, high surface area, and the interaction between the support and Pt nanoparticles.13 High nitrogen content doped mesoporous carbon supported Pd nanoparticles were used for biomass refining, achieving excellent catalytic conversion and selectivity.14 Nowadays, the development of low-cost, scalable, economically attractive and sustainable carbon materials based on renewable and highly abundant resources is extremely important.15 As low-cost and non-noble metal materials, nickel based catalysts with good surface catalytic oxidation activities in alkaline electrolyte have been the subject of much investigation.16,17 Kannan et al. prepared NiMoO4 nanorods with graphene by a facile hydrothermal process for methanol oxidation, and arising from the unique one dimensional structure and well distributed carbon, enhanced catalytic activity and high stability were obtained.18 Co3O4/NiO nanowire arrays with hierarchically porous core and branch nanoflakes shell was prepared for methanol oxidation and the electrode showed lower over-potential, high reactivity and stability in the electrocatalytic tests which was attributed to the synergistic effect and core–shell structure.19
Herein, a feasible, simplified and productive synthesis procedure to produce nitrogen-doped carbon frameworks (CNFs) with porous structure and morphology is proposed. And the nickel nanoparticles with small particle size and narrow size distribution and were supported on the CNFs. Methanol was chosen as the molecular target to investigate the electrocatalytic properties.
Experimental
Materials and methods
Potassium hydroxide (≥99.0%), nickel(II)nitrate hexahydrate (≥98.0%), sodium hydroxide (≥97.0%), acetone (≥99.5%), HCl (35–37 wt%), Nafion solution (5 wt%), and methanol (≥95%) were bought from Aldrich Chemical Co. (USA). A JEOL JSM-7400F instrument with an acceleration voltage of 3 kV was used for scanning electron microscopy (SEM) characterization.A Bruker DAVINCI D8 ADVANCE diffractometer with Cu Kα radiation (λ = 0.15406 nm) was used for X-ray diffraction (XRD) characterization. X-ray photoelectron spectroscopy (XPS) characterization was conducted on a Thermos ESCALAB 250 spectrometer with an Al Kα radiation source (passing energy: 30 eV). All binding energies were calibrated using the C 1s hydrocarbon peak at 284.60 eV. Inductively coupled plasma optical emission spectrometry (ICP-OES, PerkinElmer Optima 8000) was used for the Ni content measurement of samples. Transmission electron microscopy (TEM) and scanning transmission electron microscopy (STEM) characterizations was performed on a JEOL JEM-2100 High Resolution Transmission Electron Microscopy (HRTEM) instrument with 200 kV operation voltage. A potentiostat (SP-300, BioLogic Science Instruments, France) was used for investigating the electrochemical properties of catalysts. All electrochemical measurements were conducted in a three-electrode cell, with Pt wire as counter electrode, a glass carbon (GC) disk (3 mm diameter) as a working electrode and Hg/HgO as a reference electrode. The working electrode was prepared by dispersing 2 mg of the as-prepared catalyst in a mixture of 200 μL deionized water, 800 μL ethanol, and 120 μL of 5 wt% Nafion solution. And then, sonicated the mixture for about 20 min to form homogeneous ink, and 5.6 μL of the ink was placed on a GC disk electrode, which was then dried gently in air and ready for use.
Synthetic procedures
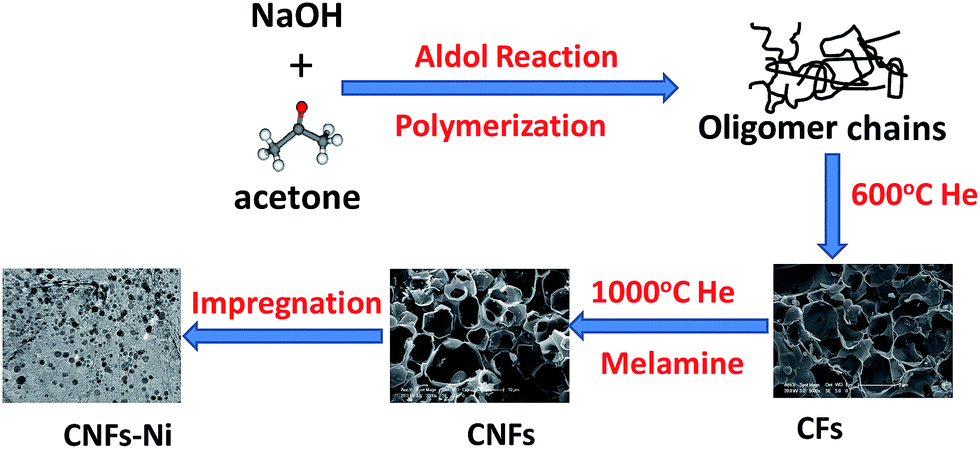 | ||
| Fig. 1 Schematic design and preparation process of the CNFs–Ni catalyst. | ||
Results and discussion
Structural characteristics
The morphology and microstructure of CNFs were characterised by SEM. Fig. 2 shows honeycomb-like porous frameworks composed of interconnected and well-arranged nanosheets, at low and high magnifications. The SEM and TEM images show porous carbon frameworks with different pore diameters composed of micropores and mesopores. It is generally considered that the large pore sizes and multi-charge transfer channels provided by the carbon frameworks facilitate diffusion of reactants to the electrocatalytic active sites. More specifically, microporous carbons with large specific surface areas contributed to the electrocatalytic activities;21–23 a porous structure with large mesopores instead of micropores is beneficial for contact with reactants, and reduce the mass-transport limitations.24–26 | ||
| Fig. 2 SEM images of CNFs at (a) low and (b) high magnification. | ||
TEM images reveal that graphene-like thin-layer CNFs and CFs with porous structures are uniformly covered with nanoparticles. The catalyst particle sizes were estimated from the TEM images, with the size distribution histograms shown in Fig. 3a–c. HR-TEM imaging results for CNFs–Ni are shown in Fig. 3d, and the Ni lattice spacing was 0.2 nm which corresponds to the 111 crystal plane. The particle size range of CNFs–Ni was 2–12 nm, with an average of 6.45 nm, while CF–Ni particles were 4–14 nm in size (average of 8.01 nm).
 | ||
| Fig. 3 TEM images and size distribution histograms of (a and b) CNFs–Ni, (c) CFs–Ni, and (d) HR-TEM image of CNFs–Ni. | ||
The smaller particle size and narrow size distributions of CNFs–Ni might originate from the nitrogen doping. After nitrogen doping, smaller particle size and narrow size distributions were obtained. Nitrogen species on the CNFs provides induced defects in the framework, in addition, nucleation is enhanced after nitrogen doping, thus, more nucleation sites generate for particle growth. The surface defects and electronic effects arising from the nitrogen doping, induced the interactions between nanoparticles and CNFs, thus, promotes the anchoring of nanoparticles.27 Better hydrophilicity also obtained by nitrogen doping into the carbon materials.28 During the impregnation process, facilitate the solvated and charged Ni2+ ions to the CNFs surface, thus better dispersion can obtained and avoid the Ni nanoparticles aggregates into larger ones.27
STEM imaging and energy dispersive X-ray spectroscopy (EDX) mapping results for CNFs–Ni are shown in Fig. 4, indicating a homogeneous distribution of C, N, O, and Ni in the catalyst. Fig. S1 (ESI†) shows the XRD patterns of CFs–Ni and CNFs–Ni composites. The broad strong diffraction peak around 23.8° is likely due to the presence of graphite carbon, while peaks at 2θ = 44.5, 51.8, and 76.3° correspond to Ni (111), (200), and (220) planes, respectively. No diffraction peaks corresponding to nickel oxide are observed which might due to the relative low content of the oxide layer at the catalysts surface which can be identified by XPS characterization. And the average crystallite sizes for CNFs–Ni and CFs–Ni catalysts were calculated using the Scherrer formula29 as 6.7 and 8.2 nm, respectively, based on the (111) diffraction peak. The nanoparticles highly dispersed on supports surfaces and no aggregates formed during the pyrolysis process indicating the strong anchoring effect of the supporting material. The above particle sizes were similar to those obtained by TEM characterisation.
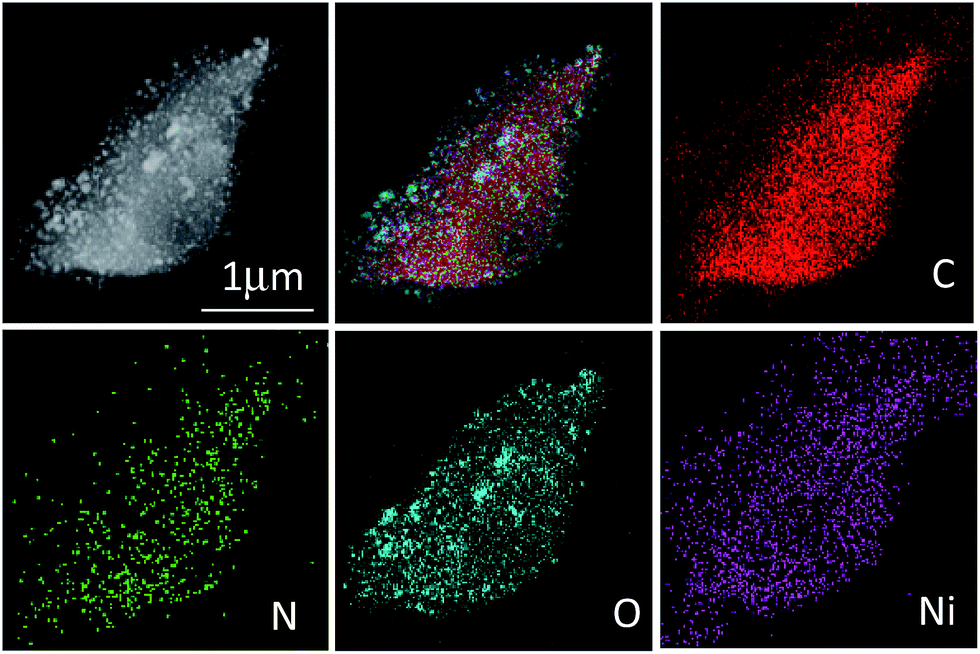 | ||
| Fig. 4 STEM image and EDX mappings of CNFs–Ni. | ||
In order to study the surface electronic properties, the surface chemical states of the catalysts were investigated by XPS. The Ni 2p spectra of the composites are shown in Fig. 5a and b. Arising from the multi-electron excitation, broad satellite peaks along with the main peaks were observed in Ni 2p core-level spectra.19 In addition to the shake-up satellite peaks, the Ni 2p peaks around 852.7 (2p3/2) and 869.8 eV (2p1/2) originate from metallic Ni. In addition, the Ni 2p3/2 peak around 855.8 eV and the Ni 2p1/2 peak around 873.5 eV indicate the presence of Ni2+ on the catalysts surfaces.30,31 Compared to those of CFs–Ni, the Ni 2p peaks of CNFs–Ni (Fig. 5b) are significantly shifted (about 0.3 eV) to higher binding energies which might be attributed to the interaction between Ni and CNFs, resulting in decreased electron density for Ni. The C 1s XPS spectrum of CNFs–Ni in Fig. 5c can be deconvoluted into four peaks: C1 (284.6 eV, C–C), C2 (285.3 eV, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), C3 (286.6 eV, C–N), and C4 (289.6 eV, O–CO).31,32 Fig. 5d shows that the N 1s XPS spectrum of CNFs–Ni can also be deconvoluted into four peaks: N1 (398.1 eV, pyridinic N), N2 (399.5 eV, pyrrolic N), N3 (400.8 eV, graphitic N), and N4 (402.0 eV, nitrogen oxides).33,34 The N content is 1.89 at% by XPS characterization.
N), C3 (286.6 eV, C–N), and C4 (289.6 eV, O–CO).31,32 Fig. 5d shows that the N 1s XPS spectrum of CNFs–Ni can also be deconvoluted into four peaks: N1 (398.1 eV, pyridinic N), N2 (399.5 eV, pyrrolic N), N3 (400.8 eV, graphitic N), and N4 (402.0 eV, nitrogen oxides).33,34 The N content is 1.89 at% by XPS characterization.
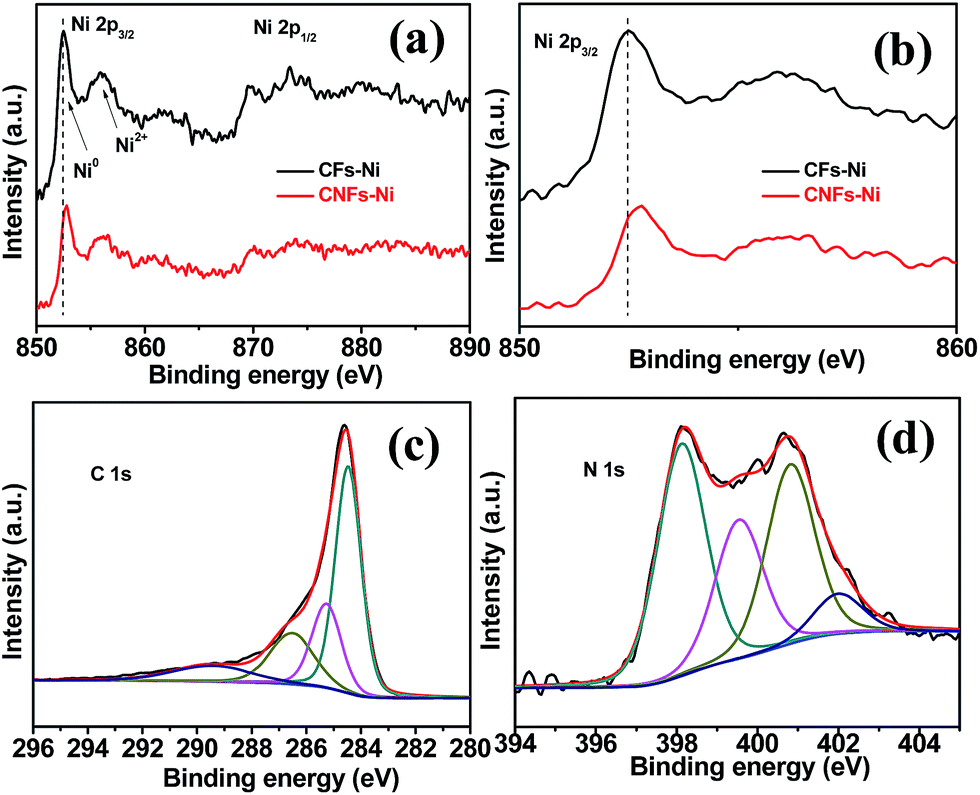 | ||
| Fig. 5 (a and b) Ni 2p3/2 XPS spectra and the deconvolution of (c) C 1s and (d) N 1s spectra for CNFs–Ni. | ||
Activity of catalysts in methanol oxidation
Cyclic voltammetry (CV) was used to investigate the electrocatalytic activity of the catalysts. As shown in Fig. 6a and b, a pair of redox peaks corresponding to Ni(II)/Ni(III) redox reaction were found. Furthermore, the cyclic voltammograms demonstrated the peak current increased with the scan rate, and the peak current present a linear relationship with the square root of the scan rate, in addition, the anodic peak potentials and cathodic peak potentials shift to more positive and negative potentials, respectively, thus, the transformation of Ni(OH)2 to NiOOH of the catalyst is a diffusion controlled process. The insets in Fig. 6a and b present the relative diffusion velocity of both catalysts. As shown in the insets, the slopes for CNFs–Ni and CFs–Ni are 0.0075 and 0.0044, respectively. The larger slope related to better diffusion properties, so that more electroactive NiOOH species formed on CNFs–Ni catalyst.35 The electrocatalytic oxidation of methanol by NiOOH is enabled by the unpaired d-electrons or empty d-orbitals of NiOOH, which are available for bond formation with absorbed species or redox intermediates.36,37 The mechanism of the electro-oxidation of methanol over Ni is shown below:38| Ni + 2OH− − 2e− → Ni(OH)2 | (1) |
| Ni(OH)2 + OH− − e− → NiOOH + H2O | (2) |
| NiOOH + CH3OH → Ni(OH)2 + products (HCOO− or CO32−) | (3) |
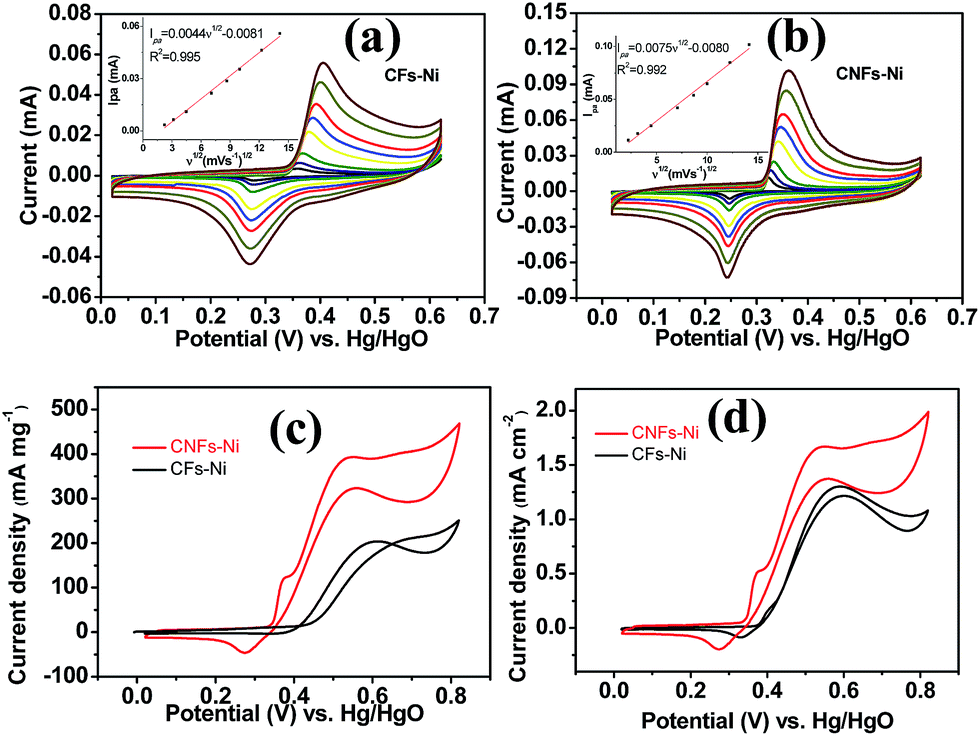 | ||
| Fig. 6 Linear relationship between the CV anodic peak current in 1.0 M KOH (Ipa) and the square root of the scan rate (ν1/2) for (a) CFs–Ni and (b) CNFs–Ni. (c) Mass activities and (d) specific activities of CFs–Ni and CNFs–Ni in 1.0 M KOH/0.5 M CH3OH (scan rate 50 mV s−1). | ||
It is worth mentioning that prior to the electrocatalytic activity tests, all catalysts were conditioned by potential cycling over 200 times at a scan rate of 50 mV s−1 in 1 M KOH, which was necessary to enrich the catalyst surface with hydroxides and oxyhydroxides, resulting in the thickening of electrocatalytic layers. Fig. 6c shows the mass activity of the samples, normalised to the Ni mass on the GC electrode. The catalyst mass loadings determined by ICP tests equal 3.27 and 3.32% for CNFs–Ni and CFs–Ni, respectively. The mass activity of CNFs–Ni at the peak potential can reach 400 mA mg−1, exceeding that of CFs–Ni (298 mA mg−1) 1.3-fold. More importantly, the onset potential of CNFs–Ni is negatively shifted by about 40 mV vs. Hg/HgO compared to that of CFs–Ni. The main oxidation peak of CNFs–Ni is located around 526 mV vs. Hg/HgO, while that of CFs–Ni was around 582 mV. The evident negative shift of peak and onset potentials demonstrates that nitrogen doping makes the catalyst more kinetically effective for methanol oxidation. For both catalysts, a small peak could be found before the main methanol oxidation peak, indicating that the electrocatalytic oxidation reaction of Ni(OH)2 occurs before methanol oxidation.
The good catalytic performance of CNFs–Ni catalyst arises from the nitrogen doped carbon frameworks which provide anchoring sites for active nickel nanoparticles, active sites and charge transfer channel for catalytic reactions. And the electronic effect caused by the incorporation of nitrogen atoms also contributes to the improved activity. Fig. 6d also shows the specific activity curves of the samples, normalised to the geometric surface area of the GC electrode. In this study, the methanol oxidation current barely increased at methanol concentrations above 0.9 M (Fig. 7a), which is caused by the reason that when the methanol concentration is up to relatively high, the methanol oxidation reaction is not controlled by the diffusion of methanol molecules to the catalyst/electrolyte interface, but the direct reaction of methanol and catalyst occurred. This is also reported for another Ni-based catalyst.39 When the methanol concentration is relatively lower, as shown in Fig. 7b, the methanol oxidation current changed with increasing scan rate, and the inset in Fig. 7b demonstrates the plot of linear relation between peak current and square root of the scan rate demonstrating that the oxidation reaction is a methanol diffusion-controlled reaction. Fig. S2† shows the Nyquist plots of CFs–Ni and CNFs–Ni measured at an open circuit potential (vs. Hg/HgO) in the frequency range from 10 mHz to 100 kHz by electrochemical impedance spectroscopy (EIS) in 0.5 M CH3OH/1.0 M KOH solution. Nyquist plots were fitted by the software EC-lab, and the inset is the equivalent electrical circuit used to fit the EIS data (Cdl, RS, RSEI and Rct are the double layer capacitance, electrolyte resistance, electrode/electrolyte interface resistance and charge-transfer resistance respectively). The impedance parameters of CFs–Ni and CNFs–Ni catalysts are listed in Table S1.† It can be observed that, both the charge transfer resistance and especially, the interface resistance decreased after nitrogen doping. The depressed arc radius in the high-frequency region of the Nyquist plot corresponds to the electron transfer limited process.40 After nitrogen doping, the arc radius was reduced, and revealing decreased interface impedance and accelerated electron transfer. This is related to the fact that after nitrogen doping, the Fermi level of carbon shift closer to the conduction band due to the extra electron of nitrogen, thus, the carbon matrix becoming more conductive.41,42 Chronoamperometric tests were conducted to assess the electrocatalytic stabilities of the catalysts. Fig. 8a displays the corresponding curves recorded in 0.5 M CH3OH/1.0 M KOH at 526 mV vs. Hg/HgO. At 3600 s, the mass activity of CNFs–Ni (395.5 mA mg−1) is still higher than that of CFs–Ni, with no evident activity loss observed with time. The long-term stability was also tested by recording CV curves in 0.5 M CH3OH/1.0 M KOH for up to 300 consecutive cycles (Fig. 8b and c). For CNFs–Ni, the current remained almost stable from the 50th to the 300th cycle; however, the current density for CFs–Ni clearly decreased compared to the initial value (Fig. 8d). Thus, the cycling durability test revealed that the stability of the CFs–Ni catalyst can be greatly enhanced by nitrogen doping. Due to the similar atom size of nitrogen and carbon, the strong delocalized bond formed due to the incorporation of nitrogen atoms which improves the stability.43 And the enhanced stability might also be confirmed by the strong binding of metal atoms and nitrogen doping supports.44,45
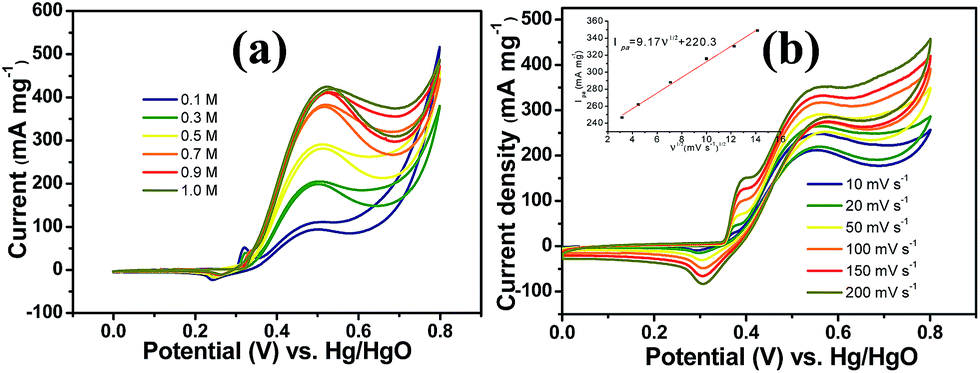 | ||
| Fig. 7 (a) Methanol oxidation current response of CNFs–Ni with increasing methanol concentration (scan rate 20 mV s−1) and (b) methanol oxidation current response of CNFs–Ni with increasing scan rate in 1.0 M KOH/0.5 M CH3OH. | ||
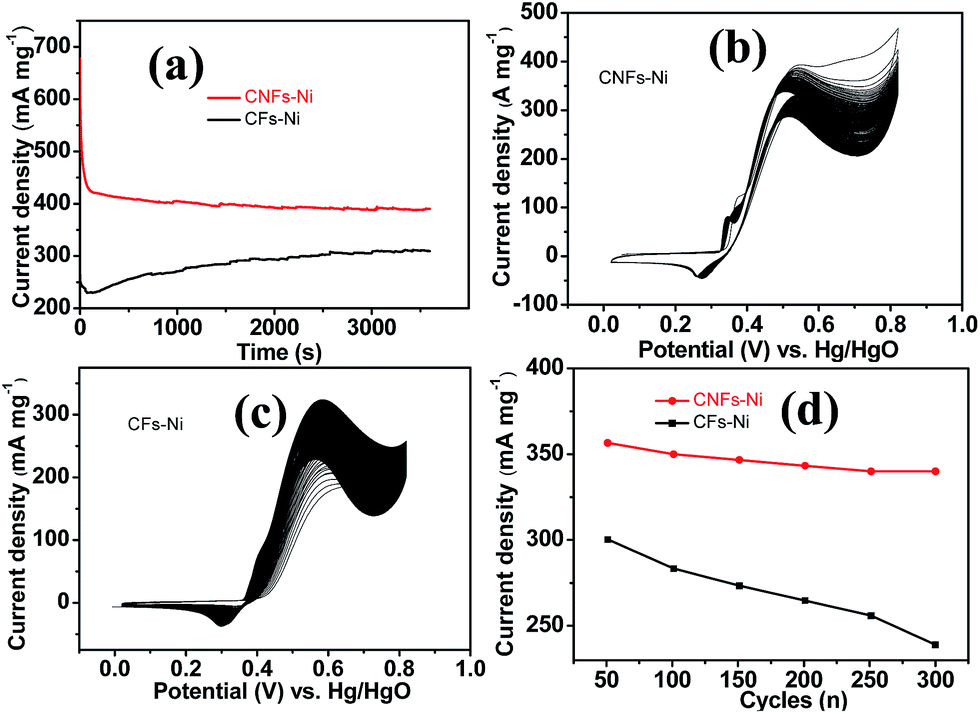 | ||
| Fig. 8 (a) Chronoamperometric study of CFs–Ni and CNFs–Ni at a constant potential of 5.26 V vs. Hg/HgO. (b and c) CV curves of catalysts in 1.0 M KOH/0.5 M CH3OH for 300 cycles (scan rate 50 mV s−1) and (d) the corresponding change in peak current density. | ||
The three dimensional carbon frameworks not only provide anchoring sites for active nickel particles but also the effective channel for ions and charges transfer which lead to the acceleration of electrocatalytic reaction. However, both the CNFs–Ni and CFs–Ni catalysts have the same three dimensional structures due to the similar preparation processes, the good electrocatalytic activity cannot be attributed to the structure of the support alone. The nitrogen atoms doping into the carbon layers facilitates the reactivity of the neighbourly located carbon atoms might also contributed to the enhanced electrocatalytic activity.46 It is reported that pyridinic and pyrrolic nitrogen atoms resulted in the disordered structure. It is generally considered that support–precursor interactions inevitably affect the morphology, particle size and distribution, and the defects located at the edge of graphene layers, thus, more nucleation sites for nanoparticle attach generated.47 In our case, from the XPS results, the pyridinic and pyrrolic nitrogen accounts for more than 60% of the all nitrogen containing species, and this might contribute to the enhanced activity of catalysts. In this study, the N-containing surface functionalities of CNFs were identified by XPS, and when nickel nanoparticles interact with CNFs, nickel would donate electrons to CNFs resulting in the decreased electrons density of nickel nanoparticles in CNFs–Ni. The lower electrons density of nickel resulted in weaker adsorption of reaction intermediates on catalysts surface, so the surface coverage of intermediates reduced which might also affect the activity and stability. In addition, the incorporation of nitrogen atoms not only influenced the electronic structure of carbon materials, but also affected the charge transfer properties at the electrolyte/catalyst interface. EIS measurements reveal that nitrogen doping decreased the interface impedance and accelerated the electron transfer, enhancing the catalytic properties.
Hydrolysis was performed after the coordination of metal ions to crystallise the precursors that nucleate and form nanocrystals.48 During thermal treatment, the reduction of nickel-containing species into larger particles or aggregates via Ostwald ripening was impeded due to their interaction with CNFs. From the TEM characterization, after nitrogen doping, the average particle size of CNFs–Ni (6.45 nm) is smaller than that of CFs–Ni (8.01 nm), and better dispersion can also obtained. Smaller particle size and better dispersion of nickel nanoparticles also contributed to the improved activity.
Conclusions
In summary, an effective CNFs–Ni catalyst with enhanced catalytic activity and stability was developed. We used a simple, productive method to synthesis nitrogen-doped carbon frameworks with honeycomb-like porous structure. This catalyst showed excellent stability compared to its non-doped counterpart. The excellent electrochemical performance of this composite is attributed to its continuous honeycomb-like network with ultrathin walls doped with nitrogen, which facilitates the uniform distribution and anchoring of Ni nanoparticles. We proposed that the decreased interface and charge transfer impedance, uniform dispersion and smaller size of the active nanoparticles, and interactions between the supporting CNFs and Ni nanoparticles which leaded to lower electrons density of nickel and weaker adsorption of reaction intermediates contribute to the improved electrocatalytic performance.Acknowledgements
This work was supported by the Science and Technology Commission of Shanghai Municipality (No. 14DZ2261000).References
- Y. Zheng, Y. Jiao, L. H. Li, T. Xing, Y. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2014, 8, 5290–5296 CrossRef CAS PubMed.
- W. Qi and D. S. Su, ACS Catal., 2014, 4, 3212–3218 CrossRef CAS.
- M. S. Risbud, S. Baxter and M. Skyllas-Kazacos, Open Fuels Energy Sci. J., 2012, 5, 9–20 CrossRef CAS.
- Y. S. Wang, S. Y. Yang, S. M. Li, H. W. Tien, S. T. Hsiao, W. H. Liao, C. H. Liu, K. H. Chang, C. C. M. Ma and C. C. Hu, Electrochim. Acta, 2013, 87, 261–269 CrossRef CAS.
- N. Jha, R. I. Jafri, N. Rajalakshmi and S. Ramaprabhu, Int. J. Hydrogen Energy, 2011, 36, 7284 CrossRef CAS.
- M. Gong, W. Zhou, M. C. Tsai, J. G. Zhou, M. Y. Guan, M. C. Lin, B. Zhang, Y. F. Hu, D. Y. Wang, J. Yang, S. J. Pennycook, B. J. Hwang and H. J. Dai, Nature, 2016, 5, 4695 Search PubMed.
- S. B. Wang, Y. A. Xing, H. Z. Xu and S. C. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 12713–12718 CAS.
- J. Li, Q. L. Zhu and Q. Xu, Chem. Commun., 2015, 51, 10827–10830 RSC.
- S. Dommele and K. P. áde Jong, Chem. Commun., 2016, 46, 4859–4861 Search PubMed.
- S. Maldonado and K. J. Stevenson, J. Phys. Chem. B, 2005, 109, 4707–4716 CrossRef CAS PubMed.
- Z. H. Sheng, L. Shao, J. J. Chen and W. J. Bao, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed.
- K. N. Wood, R. O. Hayre and S. Pylypenko, Energy Environ. Sci., 2014, 7, 1212–1249 CAS.
- Y. Liang, J. Wei, X. Y. Zhang, J. Zhang, S. P. Jiang and H. T. Wang, ChemCatChem, 2016, 8, 1901–1904 CrossRef CAS.
- X. Xu, Y. Li, Y. T. Gong, P. F. Zhang, H. R. Li and Y. Wang, J. Am. Chem. Soc., 2012, 134, 16987–16990 CrossRef CAS PubMed.
- L. D. Shao, X. Huang and D. Teschner, ACS Catal., 2014, 4, 2369–2373 CrossRef CAS.
- H. He, P. Xiao, M. Zhou, Y. H. Zhang, Q. Lou and X. Dong, Int. J. Hydrogen Energy, 2012, 37, 4967–4973 CrossRef CAS.
- L. R. Zhang, J. Zhao, M. Li, H. T. Ni, J. L. Zhang, X. M. Feng, Y. W. Ma, Q. L. Fan, X. Z. Wang, Z. Hu and W. Huang, New J. Chem., 2012, 36, 1108–1113 RSC.
- P. R. Jothi, S. Kannan and G. Velayutham, J. Power Sources, 2015, 277, 350–359 CrossRef CAS.
- J. B. Wu, Z. G. Li, X. H. Huang and Y. Lin, J. Power Sources, 2013, 224, 1–5 CrossRef CAS.
- H. S. Hou, G. E. Banks, M. J. Jing, Y. Zhang and X. B. Ji, Adv. Mater., 2015, 16, 7861–7866 CrossRef PubMed.
- X. Zhao, H. Zhao, T. Zhang, X. Yan, Y. Yuan, H. H. Zhao, D. Zhang, G. Zhu and X. Yao, J. Mater. Chem. A, 2014, 2, 11666–11671 CAS.
- J. Y. Choi, R. S. Hsu and Z. Chen, J. Phys. Chem. C, 2010, 114, 8048–8053 CAS.
- Y. He, X. Han, Y. Du, B. Song, P. Xu and B. Zhang, ACS Appl. Mater. Interfaces, 2015, 8, 3601–3608 Search PubMed.
- N. Daems, X. Sheng, I. F. J. Vankelecom and P. P. Pescarmona, J. Mater. Chem. A, 2014, 2, 4085–4110 CAS.
- N. Gavrilov, I. A. Pašti, M. Mitrić, J. Travas-Sejdić, G. Ćirić-Marjanović and S. V. Mentus, J. Power Sources, 2012, 220, 306–316 CrossRef CAS.
- Y. L. Liu, C. X. Shi, X. Y. Xu, P. C. Sun and T. H. Chen, J. Power Sources, 2015, 283, 389–396 CrossRef CAS.
- Y. G. Chen, J. J. Wang, H. Liu, M. N. Banis, R. Y. Li, X. L. Sun, T. K. Sham, S. Y. Ye and S. N. Knights, J. Phys. Chem. C, 2011, 115, 3769–3776 CAS.
- Y. Zhou, R. Pasquarelli, T. Holme, J. Berry, D. Ginleyc and R. O'Hayre, J. Mater. Chem., 2009, 19, 7830–7838 RSC.
- S. N. Anitha and I. Jayakumari, J. Nanosci. Nanotechnol., 2015, 1, 26–31 Search PubMed.
- L. L. Wang, D. F. Zhang and L. Guo, Nanoscale, 2014, 6, 4635–4641 RSC.
- Y. S. Li and Y. L. He, RSC Adv., 2014, 4, 16879–16884 RSC.
- Y. L. Huang, H. W. Tien, C. C. M. Ma, S. Y. Yang, S. Y. Wu, H. Y. Liu and Y. W. Mai, J. Mater. Chem., 2011, 21, 18236–18241 RSC.
- A. P. Dementjev, A. D. Graaf, M. C. M. V. D. Sanden, K. I. Maslakov, A. V. Naumkin and A. A. Serov, Diamond Relat. Mater., 2000, 9, 1904–1907 CrossRef CAS.
- Z. H. Sheng, L. Shao, J. J. Chen, W. J. Bao, F. B. Wang and X. H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed.
- M. S. Kim, T. S. Hwang and K. B. Kim, J. Electrochem. Soc., 1997, 144, 1537–1543 CrossRef CAS.
- M. Vidotti, C. D. Cerri, R. F. Carvalhal, J. C. Dias, R. K. Mendes, S. I. Córdoba de Torresi and L. T. Kubota, J. Electroanal. Chem., 2009, 636, 18–23 CrossRef CAS.
- L. A. Hutton, M. Vidotti, A. N. Patel, M. E. Newton, P. R. Uniwin and J. V. Macpherson, J. Phys. Chem. C, 2011, 115, 1649–1658 CAS.
- W. Huang, Z. L. Li, Y. D. Peng, S. Chen, J. F. Zheng and Z. J. Liu, J. Solid State Electrochem., 2005, 9, 284–289 CrossRef CAS.
- S. Xie, X. L. Tong, G. Q. Jin, Y. Qin and X. Y. Guo, J. Mater. Chem. A, 2013, 1, 2104–2109 CAS.
- W. Sugimoto, H. Iwata, K. Yokoshima, Y. Murakami and Y. Takasu, J. Phys. Chem. B, 2005, 109, 7330–7338 CrossRef CAS PubMed.
- J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Catal., 2015, 5, 5207–5234 CrossRef CAS.
- S. Maldonado, S. Morin and K. J. Stevenson, Carbon, 2006, 44, 1429–1437 CrossRef CAS.
- H. Kim, K. Lee, S. I. Woo and Y. Jung, Phys. Chem. Chem. Phys., 2011, 13, 17505–17510 RSC.
- Y. H. Li, T. H. Hung and C. W. Chen, Carbon, 2009, 47, 850 CrossRef CAS.
- W. An and C. H. Turner, J. Phys. Chem. C, 2009, 113, 7069–7078 CAS.
- C. H. Wang, H. Y. Du, Y. T. Tsai, C. P. Chen, C. J. Huang, L. C. Chen, K. H. Chen and H. C. Shih, J. Power Sources, 2007, 171, 55–62 CrossRef CAS.
- R. L. Jia, C. Y. Wang and S. M. Wang, J. Mater. Sci., 2006, 41, 6881–6888 CrossRef CAS.
- Z. L. Xin, J. Wang, X. Huang, Y. Y. Yao and L. D. Shao, J. Electrochem. Soc., 2015, 162, H898–H902 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: XRD, Nyquist plots. See DOI: 10.1039/c7ra00590c |
| This journal is © The Royal Society of Chemistry 2017 |