
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Understanding the domino reaction between 1-diazopropan-2-one and 1,1-dinitroethylene. A molecular electron density theory study of the [3 + 2] cycloaddition reactions of diazoalkanes with electron-deficient ethylenes†
Luis R. Domingo *a,
Mar Ríos-Gutiérreza and
Saeedreza Emamianb
*a,
Mar Ríos-Gutiérreza and
Saeedreza Emamianb
aDepartment of Organic Chemistry, University of Valencia, Dr. Moliner 50, E-46100 Burjassot, Valencia, Spain. E-mail: domingo@utopia.uv.es; Web: http://www.luisrdomingo.com
bChemistry Department, Shahrood Branch, Islamic Azad University, Shahrood, Iran
First published on 9th March 2017
Abstract
The reaction between 1-diazopropan-2-one and 1,1-dinitroethylene has been studied using the Molecular Electron Density Theory (MEDT) at the B3LYP/6-31G(d,p) computational level. This reaction comprises two domino processes initialised by a common [3 + 2] cycloaddition (32CA) reaction yielding a 1-pyrazoline, which participates in two competitive reaction channels. Along channel I, 1-pyrazoline firstly tautomerises to a 2-pyrazoline, which by a proton abstraction and spontaneous loss of nitrite anion yields the final pyrazole, while along channel II, the thermal extrusion of the nitrogen molecule in 1-pyrazoline gives a very reactive diradical intermediate which quickly yields the final gem-dinitrocyclopropane. Analysis of the conceptual DFT reactivity indices permits an explanation of the participation of 1-diazopropan-2-one in polar 32CA reactions. A Bonding Evolution Theory (BET) study along the more favourable regioisomeric reaction path associated to the 32CA reaction allows an explanation of its molecular mechanism. The present MEDT study sheds light on these complex domino reactions as well as on the participation of diazoalkanes in polar 32CA reactions towards strong electrophilic ethylenes via a two-stage one-step mechanism.
1. Introduction
Heterocyclic compounds are easily synthesised in a highly regio- and stereoselective fashion by a [3 + 2] cycloaddition (32CA) reaction between a three atom-component (TAC) and an ethylene derivative (see Scheme 1).1 Substitution of a, b and c in the TAC, and d and e in the ethylene by C, N, O, P or S atoms has proven to be a powerful synthetic tool in the construction of a great diversity of five-membered heterocyclic compounds.1 TACs can be structurally classified into two categories: allylic type (A-TAC) and propargylic type (P-TAC) structures.2 While A-TACs such as nitrone I are bent, P-TACs such as nitrile oxide II have a linear structure (see Scheme 1).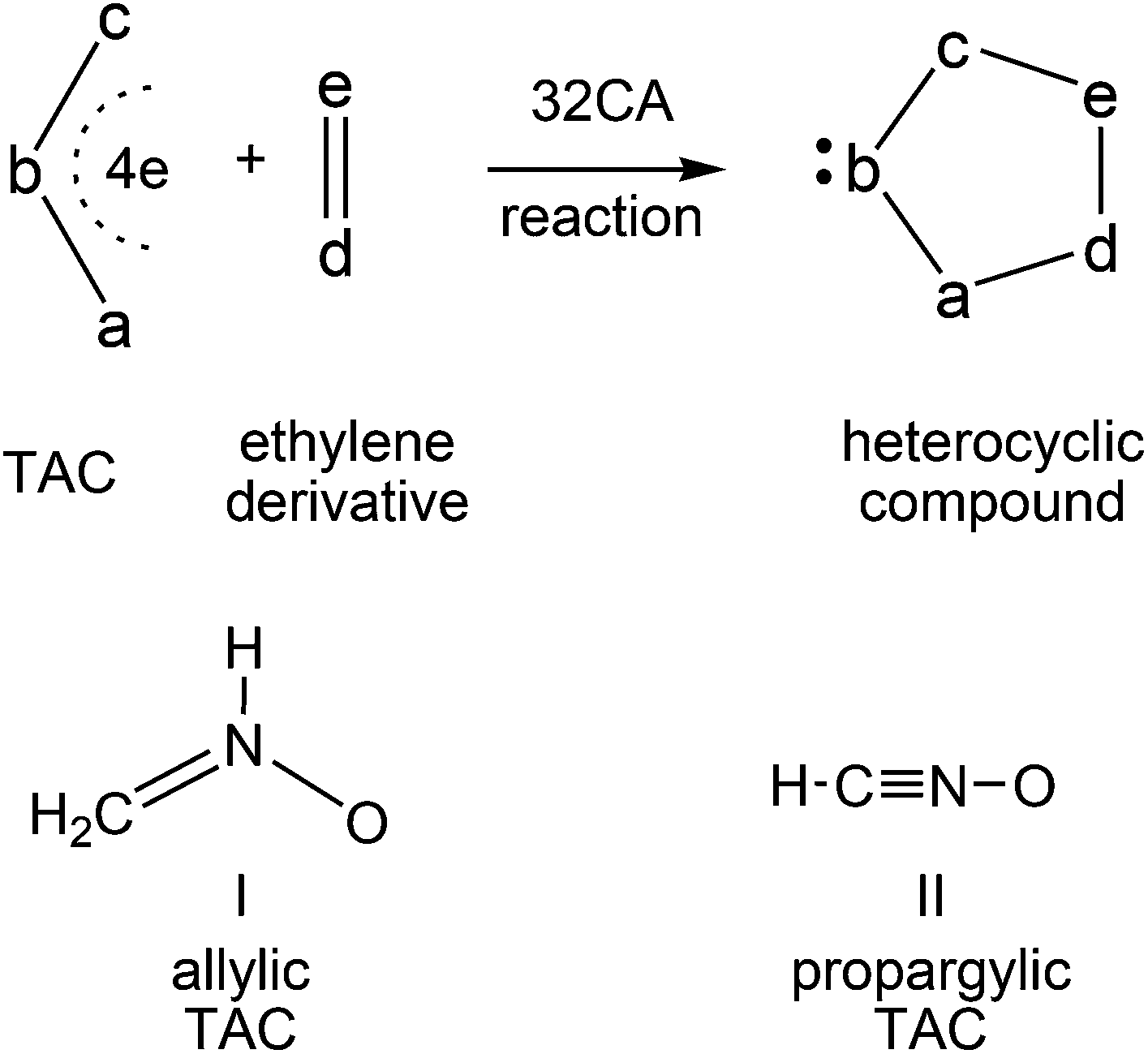 | ||
| Scheme 1 Construction of five-membered heterocyclic compounds by a 32CA reaction and structural classification of TACs. | ||
Diazoalkanes (DAAs) are P-TACs in which four electrons are distributed in a linear C–N–N structure. After reporting the cycloadditions of diazoacetate and diazomethane toward C–C multiple bonds by Buchner and von Pechmann in the 1890s,3 many 32CA reactions of DAAs have been reported. In contrast to many TACs which are generated as transient species in the reaction medium, mono- and di-substituted DAAs have been extensively prepared and isolated in pure form.1c The participation of DAA 1 in a 32CA reaction toward non-symmetrically substituted ethylene 2 yields two regioisomeric 1-pyrazolines, 3 and 4, depending on the approach mode of the reagents (see Scheme 2).
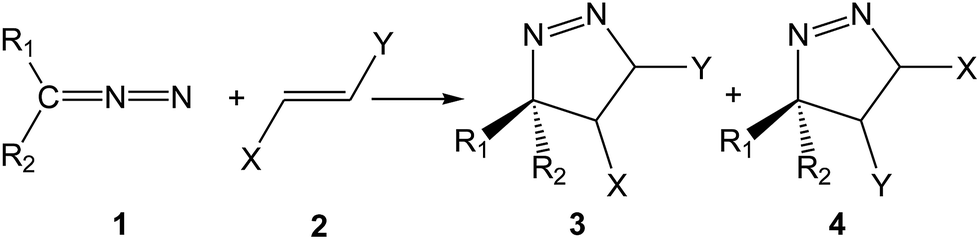 | ||
| Scheme 2 Participation of DAAs in 32CA reactions toward non-symmetrically substituted ethylenes to generate two regioisomeric 1-pyrazolines. | ||
Pyrazolines play a key role in medicinal and agricultural chemistry due to their potent biological activities. Antimicrobial, anticancer, anti-tubercular, anti-inflammatory, antiviral, antitumor and antiangiogenic activities turn the synthesis of pyrazolines into an attractive and valued research area.4 Due to the strong repulsion caused by the lone pairs of two adjacent sp2 hybridised nitrogen atoms, 1-pyrazolines potentially tend to lose a nitrogen molecule.5 In addition, 1-pyrazolines resulting from a 32CA reaction between DAAs 1 and electron-deficient (ED) ethylenes 5 can experience different subsequent transformations, depending on the electronic nature of the substituents present in the DAA and the ethylene. As shown in Scheme 3, cyclopropanes 7, 2-pyrazolines 8 or pyrazoles (PYZ) 9 can be obtained, respectively, via the extrusion of the nitrogen molecule, tautomerisation or HY elimination at the resulting 1-pyrazolines 6.
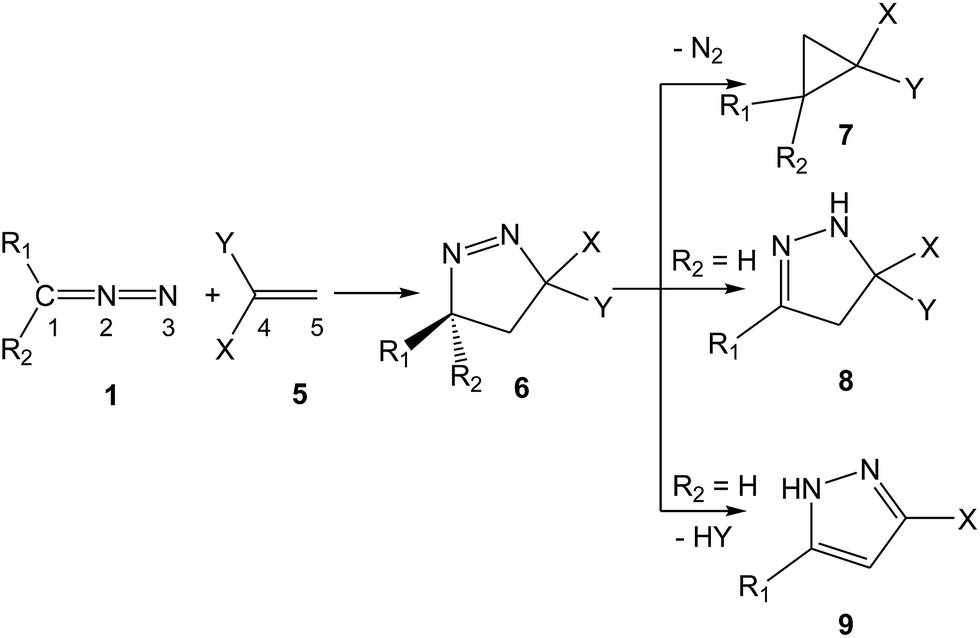 | ||
| Scheme 3 Transformation of 1-pyrazolines 6 into cyclopropanes 7, 2-pyrazolines 8 or PYZs 9 when appropriate substitutions are present in DAAs 1 and ethylenes 5. | ||
Establishing a relationship between the molecular electronic structure and reactivity is the main goal of theoretical organic chemistry. In this sense, very recently, Domingo proposed a new reactivity model in organic chemistry named Molecular Electron Density Theory (MEDT),6 in which changes in the electron density along an organic reaction, and not molecular orbital interactions, are responsible for its feasibility. Various MEDT studies devoted to the understanding of the relationship between the electronic structure of TACs and their reactivity in 32CA reactions have allowed establishing a useful classification of these cycloaddition reactions into pseudodiradical-type7 (pr-type), carbenoid-type8 (cb-type) and zwitterionic-type7 (zw-type) reactions (see Scheme 4). TACs with a pseudodiradical character participate in pr-type 32CA reactions taking place easily through earlier transition state structures (TSs) with very low activation energies and even non-polar character,7 while TACs with a carbenoid or zwitterionic character participate in cb- or zw-type 32CA reactions whose feasibility depends on the polar character of the reaction, i.e. the nucleophilic character of the TAC and the electrophilic character of the ethylene derivative, or vice versa.7,8
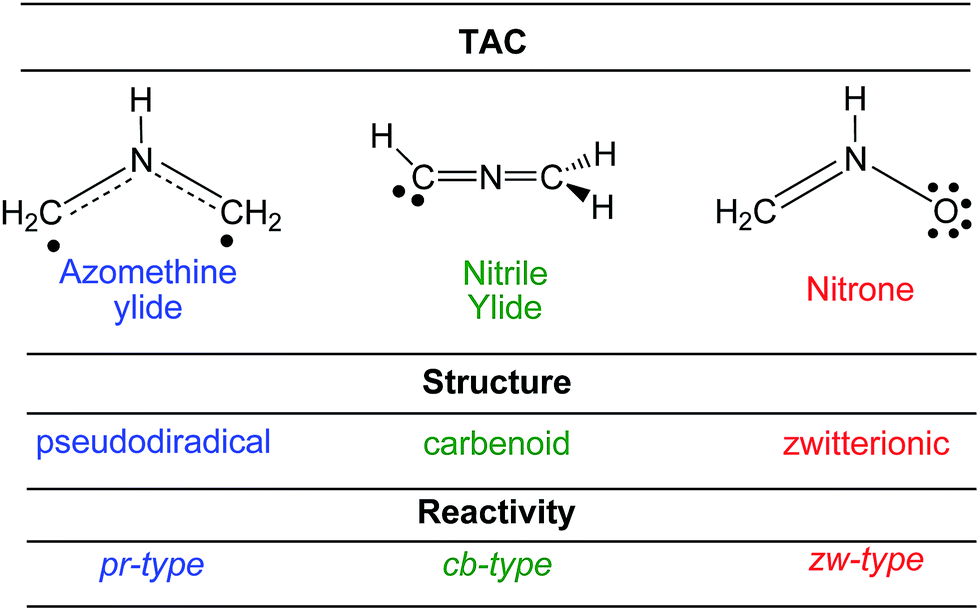 | ||
| Scheme 4 Electronic structure of TACs and the proposed reactivity types in 32CA reactions. | ||
Recently, Ivanova et al.9 experimentally studied some 32CA reactions of various DAAs containing an α-hydrogen atom with 1,1-dinitroethylene (DNE) 11, an ED ethylene, in the presence as well as in the absence of hexacarbonylmolybdenum, Mo(CO)6. Thus, when DAA 10, 1-diazopropan-2-one, was treated with DNE 11 in benzene at 60 °C for 6 h, a mixture of PYZ 12 and gem-dinitrocyclopropane (DNCP) 13 was obtained in 38% and 42% yields, respectively (see Scheme 5). Similar results were obtained in the presence of the Mo(CO)6 complex.9
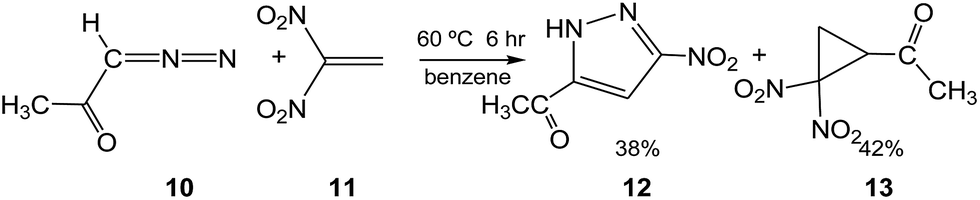 | ||
| Scheme 5 Generation of PYZ 12 and DNCP 13 via the 32CA reaction of DAA 10 with DNE 11 experimentally studied by Ivanova et al.9 | ||
Herein, an MEDT study of the reaction of DAA 10 with DNE 11 yielding PYZ 12 and DNCP 13, experimentally reported by Ivanova et al.,9 is carried out using quantum chemical procedures at the B3LYP/6-31G(d,p) computational level in order to understand this complex process. A Bonding Evolution Theory10 (BET) study of the more favourable regioisomeric channel associated with the 32CA reaction between DAA 10 and DNE 11 is performed in order to characterise the bonding changes along the studied 32CA reaction, and thus to establish the molecular mechanism of the reaction.
2. Computational details
All stationary points were optimised using the B3LYP functional11 together with the 6-31G(d,p) basis set.12 The Berny analytical gradient optimisation method13 was employed in geometry optimisation steps. The stationary points were characterised by frequency calculations in order to verify that TSs have one and only one imaginary frequency. Intrinsic Reaction Coordinate (IRC) curves14 were traced in order to check the energy profiles connecting each TS to the two associated minima of the proposed mechanism using the second order González–Schlegel integration method.15 Solvent effects of benzene were taken into account through single point energy computations at the gas phase optimised stationary points using the Polarisable Continuum Model (PCM) developed by Tomasi's group16 in the framework of the Self-Consistent Reaction Field (SCRF).17 The electronic structures of the stationary points were analysed by a Natural Population Analysis (NPA) within the Natural Bond Orbital (NBO) method.18 The global electron density transfer19 (GEDT) is computed by the sum of the natural atomic charges (q) of the atoms belonging to each framework (f) at the TSs; GEDT = ∑qf. The sign indicates the direction of the electron density flux in such a manner that positive values mean a flux from the considered framework to the other one. All computations were carried out with the Gaussian 09 suite of programs.20The global electrophilicity ω index21 is given by the following expression, ω = μ2/2η, based on the electronic chemical potential, μ, and the chemical hardness, η. Both quantities may be approached in terms of the one-electron energies of the frontier molecular orbitals HOMO and LUMO, εH and εL, such as μ ≈ (εH + εL)/2 and η ≈ (εL − εH).22 The global nucleophilicity N index23 based on the HOMO energies obtained within the Kohn–Sham scheme24 is defined as N = εHOMO(Nu) − εHOMO(TCE), in which Nu denotes the given nucleophile. This relative nucleophilicity N index is referenced to tetracyanoethylene (TCE). The pr index, which has recently been introduced in order to characterise the participation of pseudodiradical TACs in a pr-type 32CA reaction,7 comprises the chemical hardness η and the nucleophilicity N index of the TAC, as pr = N/η. Electrophilic Pk+ and nucleophilic Pk− Parr functions25 were obtained through the analysis of the Mulliken atomic spin densities (ASD) of the radical anion of DNE 11 and the radical cation of DAA 10. DFT reactivity indices were computed at the B3LYP/6-31G(d) level.
The topological analysis of the Electron Localisation Function (ELF), η(r),26 was performed with the TopMod program27 using the corresponding B3LYP/6-31G(d,p) monodeterminantal wavefunctions. For the BET study,10 the reaction paths were followed using the IRC procedure in mass-weighted internals. Steps of 0.1 [amu1/2 bohr] along the IRCs were assumed. A total of 300 points along each side of the IRC was analysed.
3. Results and discussion
The present theoretical study is divided into four parts: (i) first, an analysis of the electronic structure of experimental DAA 10 and the simplest DAA 14 is performed; (ii) then, an analysis of Conceptual DFT (CDFT) reactivity indices at the ground state of the reagents involved in the 32CA reaction between DAA 10 and DNE 11 is performed in order to predict the reactivity and regioselectivity in this 32CA reaction; (iii) next, the reaction paths involved in these domino processes initialised by the 32CA reaction between DAA 10 and DNE 11 yielding PYZ 12 and DNCP 13 are studied; and (iv) finally, a BET study along the more favourable regioisomeric channel associated with the 32CA reaction of DAA 10 with DNE 11 is carried out in order to characterise the molecular mechanism of this cycloaddition.3.1. Analysis of the electronic structures of DAAs 10 and 14
As commented in the Introduction, the reactivity of TACs can be correlated with their electronic structure. Consequently, first, an ELF topological analysis of the experimental DAA 10 and the simplest DAA 14 was performed. The representation of ELF attractors, natural atomic charges, obtained through an NPA, ELF valence basins and the proposed Lewis structures for DAAs 10 and 14 are shown in Fig. 1.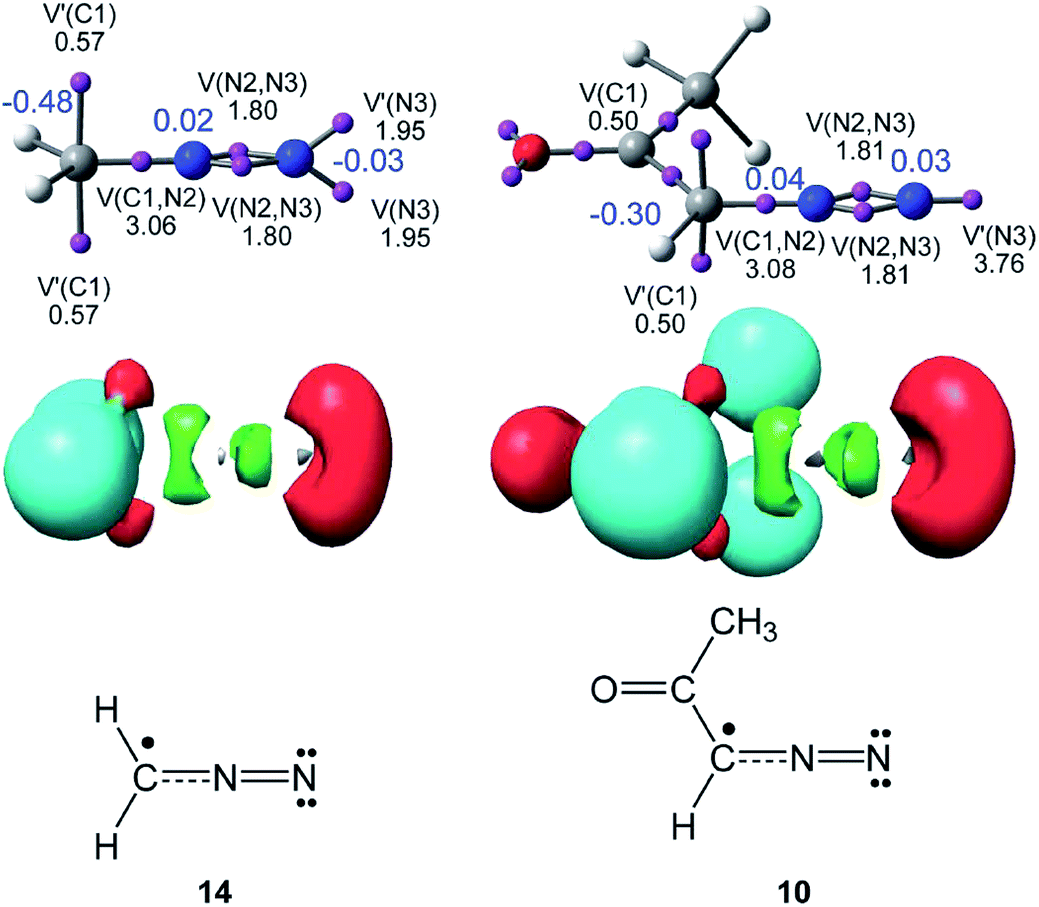 | ||
| Fig. 1 Representation of ELF attractors, valence basin populations, in e, natural atomic charges, in e and in blue, ELF valence basins and proposed pseudoradical Lewis structures for DAAs 10 and 14. | ||
As can be seen in Fig. 1, ELF topology of the simplest DAA 14 shows the presence of two monosynaptic basins, V(C1) and V′(C1), integrating a total of 1.04 e, at the sp2 hybridised C1 carbon. The C1–N2 bonding region appears characterised by the presence of one V(C1,N2) disynaptic basin integrating 3.06 e, while the presence of two disynaptic basins, V(N2,N3) and V′(N2,N3), integrating 1.80 e each one, suggests the existence of a N2–N3 double bond according to the Lewis bonding model. Finally, ELF topology of DAA 14 shows the presence of two monosynaptic basins, V(N3) and V′(N3), with a total electron density of 3.90 e, associated with two lone pairs at the N3 nitrogen also according to the Lewis bonding model.
ELF topology of the experimental DAA 10 shows a very similar electronic structure to that found in the simplest DAA 14 (see Fig. 1). In this TAC, the two monosynaptic basins, V(C1) and V′(C1), present a population of 0.50 e each one. On the other hand, the C1–N2–N3 bonding region shows a similar bonding pattern than that in DAA 14. The only topological difference between the two DAAs is that at DAA 10 the non-bonding electron density associated to the N3 nitrogen atom is represented by one single V(N3) monosynaptic basin integrating 3.76 e.
According to the Lewis structures, V(C) monosynaptic basins integrating ca. 1 e are associated to pseudoradical centers,28,29 while those integrating ca. 2 e in neutral molecules are associated to carbenoid centers.30 TACs presenting two pseudoradical centers have been classified as pseudodiradical TACs,7 while those presenting a carbenoid center have been classified as carbenoid TACs.8 Finally, TACs that neither present pseudoradical nor carbenoid centers have been classified as zwitterionic TACs.7 Consequently, ELF topology of the experimental DAA 10 and simplest DAA 14 indicates that these TACs neither presents a pseudodiradical,7 nor a carbenoid8 nor a zwitterionic7 electronic structure that would enable them to participate in pr-, cb- or zw-type 32CA reactions (see Scheme 4),7,8 but they have a pseudoradical structure.
Although the ELF topological analysis of DAAs 10 and 14 allows establishing a similar bonding pattern in these TACs, NPA does not allow characterising any zwitterionic structure for these two DAAs. It is noteworthy that the C1 carbon has a high negative charge, −0.48 e (14) and −0.30 e (10), while the N2 and N3 nitrogen atoms have a negligible charge. These findings disagree with the commonly accepted Lewis structure of DAAs represented by a 1,2-zwitterionic structure.
3.2. Analysis of the global and local CDFT reactivity indices at the ground state of the reagents involved in the 32CA reaction
Global reactivity indices defined within CDFT31 are powerful tools to explain the reactivity in cycloaddition reactions. Since the global electrophilicity and nucleophilicity scales are given at the B3LYP/6-31G(d) level, the present analysis has been performed at this computational level. The global indices, namely, electronic chemical potential (μ), chemical hardness (η), global electrophilicity (ω) and global nucleophilicity (N) for DAAs 10 and 14 and DNE 11, and the pr index of DAAs 10 and 14, are presented in Table 1.| μ | η | ω | N | pr | |
|---|---|---|---|---|---|
| DNE 11 | −5.98 | 5.03 | 3.56 | 0.62 | |
| DAA 14 | −3.64 | 4.73 | 1.40 | 3.11 | 0.66 |
| DAA 10 | −4.40 | 4.66 | 2.07 | 2.39 | 0.51 |
As shown in Table 1, the electronic chemical potential of DAA 10, −4.40 eV, is higher than that of DNE 11, −5.98 eV. Consequently, along the polar 32CA reaction of DAA 10 with DNE 11, the GEDT19 will take place from DAA 10 toward ED DNE 11.
Along a polar reaction, there is an electron density transfer from the nucleophilic to the electrophilic species, which is measured by the GEDT19 value computed at the TS of the reaction; the larger the GEDT at the TS, the more polar the reaction. Note that the GEDT concept comes from the observation that the electron density transfer taking place from the nucleophile to the electrophile along a polar reaction is not a local process, but a global one involving the two interacting frameworks and depending on the electrophilic/nucleophilic interactions taking place between them.19
The simplest DAA 14 has an electrophilicity ω index of 1.40 eV and a nucleophilicity N index of 3.11 eV, being classified on the borderline of strong electrophiles32 and as a strong nucleophile.33 Inclusion of a carbonyl group at the C1 carbon atom of the simplest DAA 14 increases the electrophilicity ω index of the experimental DAA 10 to 2.07 eV and decreases the nucleophilicity N index to 2.39 eV, being classified as a strong electrophile and as a moderate nucleophile. Note that most TACs are strong nucleophilic species.34 Consequently, DAA 10 will participate as a strong electrophile or as a moderate nucleophile in polar 32CA reactions.
On the other hand, DNE 11 has a global electrophilicity ω index of 3.56 eV and a nucleophilicity N index of 0.62 eV, being classified as a strong electrophile and as a marginal nucleophile.
In order to characterise the participation of pseudodiradical TACs in a pr-type 32CA reaction, the pr index has recently been introduced.7 A-TACs with pr values larger than 0.90 can be related to species having a very soft character, i.e. with low hardness η values, and low stabilised frontier electrons, i.e. with low ionisation potential, participating in pr-type 32CA reactions, while P-TACs with low pr values should participate in zw-type 32CA reactions. P-TACs 10 and 14 have pr values of 0.51 and 0.66, indicating that in spite of their pseudoradical structure they will have a low pr-type reactivity.7 This analysis is in clear agreement with the high activation energy found in the 32CA reaction between the simplest DAA 14 and ethylene 17, 16.6 kcal mol−1.34 Consequently, DAA 10 will not be able to participate in pr-type 32CA reactions, and will thus participate as a strong electrophile or as a moderate nucleophile in polar 32CA reactions.
By approaching a non-symmetric electrophilic/nucleophilic pair along a polar or ionic process, the most favourable reactive channel is that associated with the initial two-center interaction between the most electrophilic center of the electrophile and the most nucleophilic center of the nucleophile. Recently, Domingo proposed the nucleophilic Pk− and electrophilic Pk+ Parr functions,25 derived from the changes of spin electron-density reached via the GEDT process from the nucleophile to the electrophile, as a powerful tool in the study of the local reactivity in polar or ionic processes.
Accordingly, the nucleophilic Pk− Parr functions of DAA 10 as well as the electrophilic Pk+ Parr functions of DNE 11 were analysed in order to characterise the most nucleophilic and electrophilic centers of the species involved in this 32CA reaction (see Fig. 2).
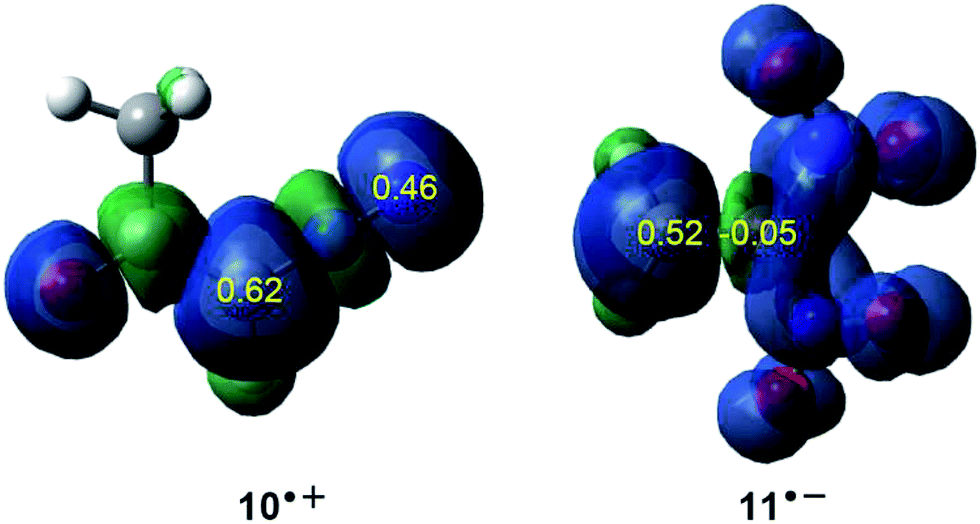 | ||
| Fig. 2 3D representations of the ASD of the radical cation 10˙+ and the radical anion 11˙−, together with the nucleophilic Pk− Parr functions of DAA 10 and the electrophilic Pk+ Parr functions of DNE 11. | ||
Analysis of the nucleophilic Pk− Parr functions at the reactive sites of DAA 10 indicates that the C1 carbon atom, with a maximum Pk− value of 0.62, is the most nucleophilic center. On the other hand, the electrophilic Pk+ Parr functions at the reactive sites of DNE 11 indicate that the most electrophilic center is the C5 carbon atom, possessing the maximum value of Pk+ = 0.52. Note that the C4 carbon atom is electrophilically deactivated, possessing a negative Pk+ value of −0.05. Therefore, it is predictable that the most favourable electrophile–nucleophile interaction along the nucleophilic attack of DAA 10 on DNE 11 in a polar process will take place between the most nucleophilic center of DAA 10, the C1 carbon atom, and the most electrophilic center of DNE 11, the C5 carbon atom. This prediction is in complete agreement with the experimental outcomes9 favouring the formation of 1-pyrazoline 15 which, in turn, participates in the subsequent reactions to generate PYZ 12 and DNCP 13 (see Scheme 6).
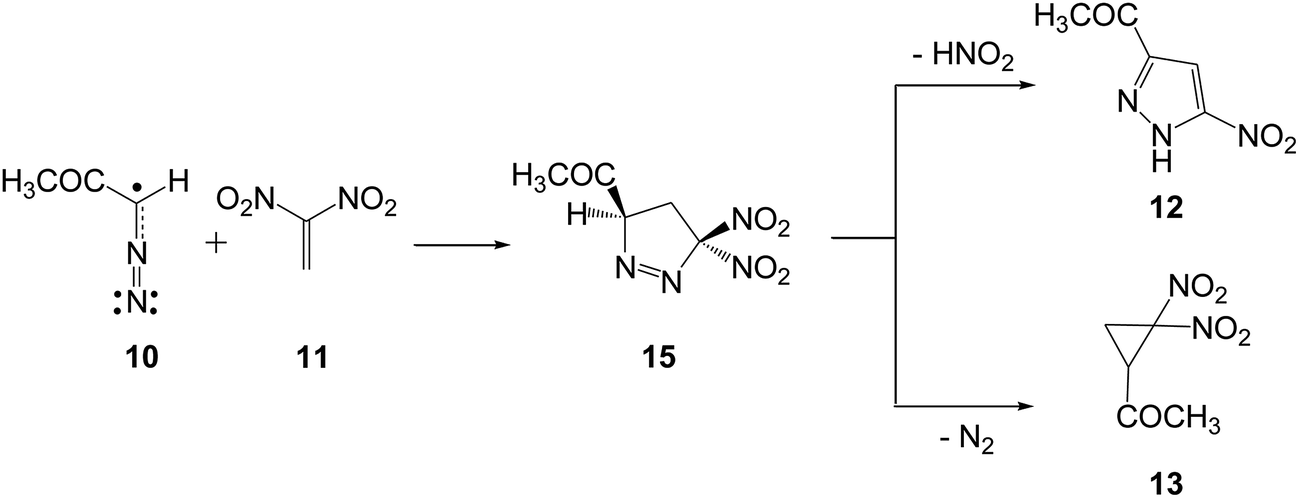 | ||
| Scheme 6 Domino reactions between DAA 10 and DNE 11 yielding PYZ 12 and DNCP 13. | ||
3.3. Study of the domino reaction between DAA 10 and DNE 11 giving PYZ 12 and DNCP 13
The reaction between DAA 10 and DNE 11 giving PYZ 12 and DNCP 13 is a domino process that comprises several consecutive reactions (see Scheme 6). The first one is a 32CA reaction between DAA 10 and DNE 11 yielding 1-pyrazoline 15. Then, 15 may experience two competitive reactions: (i) a tautomerisation and the subsequent loss of nitrous acid to yield PYZ 12; or (ii) the extrusion the nitrogen molecule and a rapid ring closure resulting in DNCP 13.The first reaction of these domino processes is a 32CA reaction between DAA 10 and DNE 11 yielding 1-pyrazolines 15 and/or 16. Formation of these pyrazolines depends on the regioisomeric approach mode of the reagents, i.e. the initial formation of the C1–C5 or C1–C4 single bonds. An analysis of the stationary points involved in the two regioisomeric paths indicates that this 32CA reaction takes place through a one-step mechanism. Consequently, two reactive molecular complexes (MCs), MC1 and MC2, two regioisomeric TSs, TS1 and TS2, and the two corresponding 1-pyrazolines 15 and 16, were located and characterised (see Scheme 7). B3LYP/6-31G(d,p) relative energies in gas phase and in benzene for the 32CA reaction between DAA 10 and DNE 11 are given in Scheme 7, while total energies are given in the ESI.†
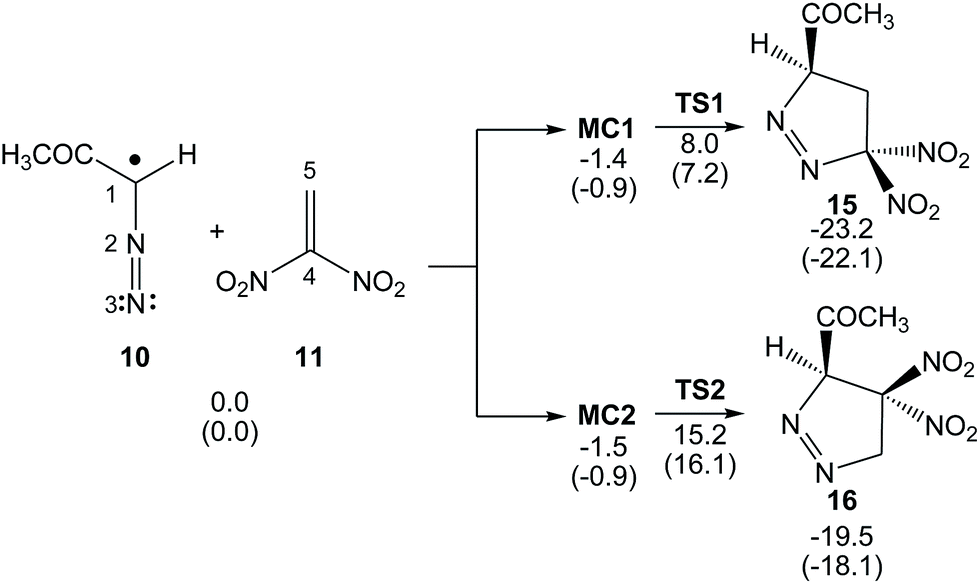 | ||
| Scheme 7 32CA reaction between DAA 10 and DNE 11. Relative energies are given in kcal mol−1. Energies in benzene are given in parentheses. | ||
When DAA 10 and DNE 11 gradually approach each other, the energy is reduced until the formation of two MCs located 1.4 (MC1) and 1.5 (MC2) kcal mol−1 below the separated reagents takes place. Further approach of the two DAA and DNE frameworks leads to the formation of the TSs, which are located 9.4 (TS1) and 16.7 (TS2) kcal mol−1 above the corresponding MCs. Moreover, formation of 1-pyrazolines 15 and 16 becomes exothermic by 23.2 and 19.5 kcal mol−1, respectively. Some appealing conclusions can be drawn from these energy results: (i) the activation energy associated with TS1 is 7.4 kcal mol−1 lower in energy than that associated with the 32CA reaction of the simplest DAA 14 with ethylene 17; 16.6 kcal mol−1.34 It is interesting to note that in spite of the pseudoradical structure of DAAs 10 and 14 (see Fig. 1), the low pr index of these TACs (see Table 1) together with the very high activation energy of the 32CA reaction of DAA 14 with ethylene 17 indicates that they do not participate in pr-type 32CA reactions; (ii) this 32CA reaction is completely regioselective, TS2 being 7.9 kcal mol−1 above TS1, in complete agreement with the experimentally observed regioselectivity; and (iii) the strong exothermic character of this reaction makes the formation of 1-pyrazolines 15 and 16 irreversible. Consequently, 1-pyrazoline 15 is obtained through the kinetic control of the reaction.
Inclusion of the solvent effects of benzene does not significantly modify the relative gas phase energies (see Scheme 7). In benzene, the activation energy of the reaction slightly decreases to 8.1 kcal mol−1, while the 32CA reaction remains completely regioselective.
After the formation of 1-pyrazoline 15, this species can participate in the competitive reaction channels I and II (see Scheme 8). Along channel I, 1-pyrazoline 15 first tautomerises to 2-pyrazoline 18, which by a loss of nitrous acid yields PYZ 12, while along channel II, the thermal extrusion of the nitrogen molecule in 1-pyrazoline 15 gives the diradical intermediate IN2, which quickly yields the final DNCP 13. (U)B3LYP/6-31G(d,p) relative energies in gas phase and in benzene for the competitive reactions of 1-pyrazoline 15 are given in Scheme 8, while total energies are given in the ESI.†
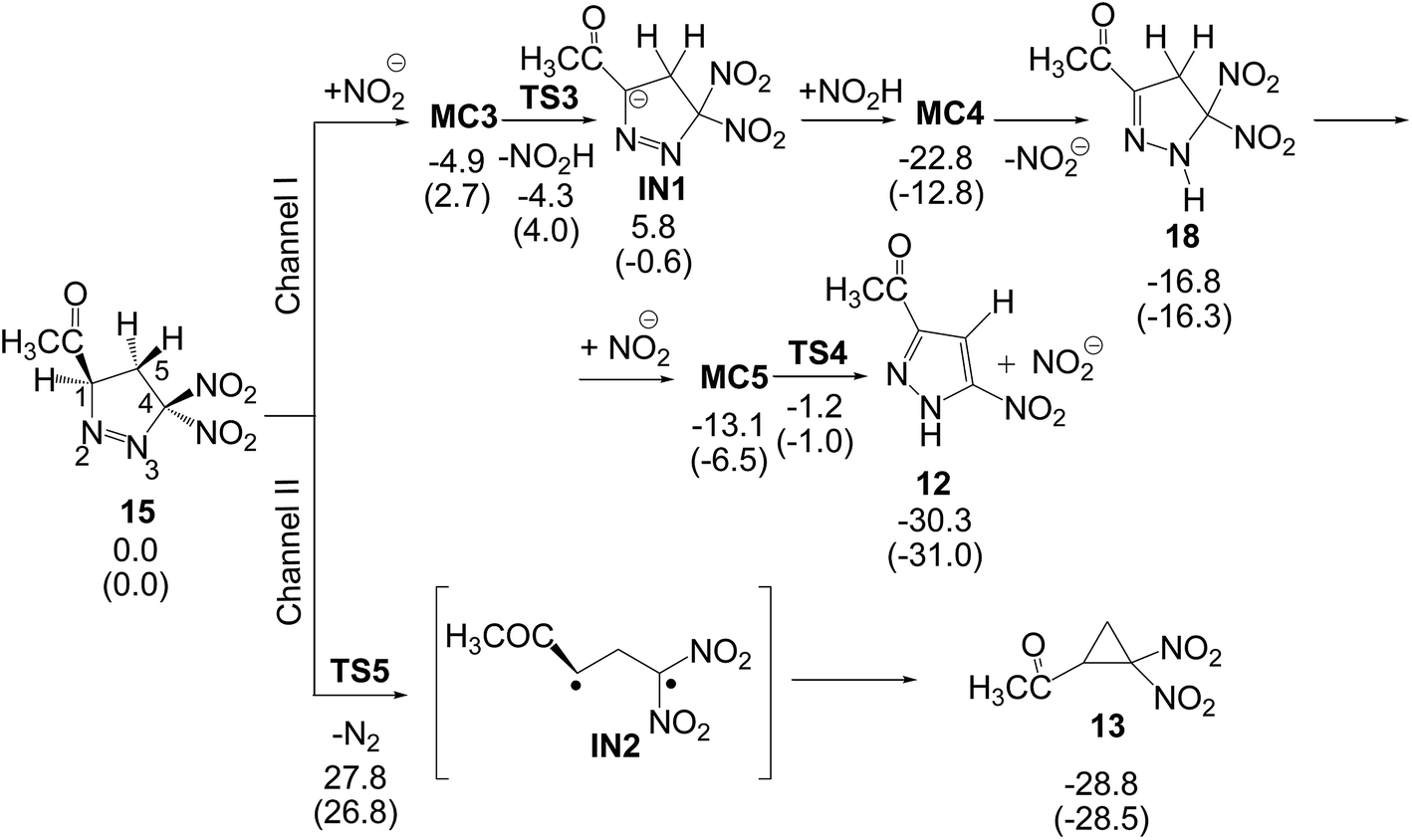 | ||
| Scheme 8 Proposed reaction channels for the conversion of 1-pyrazoline 15 into PYZ 12 and DNCP 13. Relative energies are given in kcal mol−1. Energies in benzene are given in parentheses. | ||
The two consecutive reactions involved in channel I can be promoted by any acid/basic species present in the reaction medium. In this study, the HNO2/NO2− pair was selected as the acid/basic species since nitrous acid is a reaction product. Conversion of 1-pyrazoline 15 into 2-pyrazoline 18 is a tautomerisation process associated to a 1,3-hydrogen shift. Several studies have shown that the direct 1,3-hydrogen shift is energetically very unfavourable due to the formation of a strained four-membered cyclic TS.35 Consequently, the tautomerisation should take place trough a stepwise mechanism. The first step consists in the H1 proton abstraction by the nitrite anion acting as a base yielding anionic intermediate IN1. From MC3, in which the nitrite anion forms a hydrogen-bond with the H1 hydrogen atom, this step has a very low activation energy, 1.3 kcal mol−1 (TS3); formation of intermediate IN1 is slightly exothermic by 0.6 kcal mol−1. The subsequent proton transfer from nitrous acid to the N3 nitrogen atom has no activation barrier, formation of 2-pyrazoline 18 being exothermic by 16.3 kcal mol−1; note that this step is a thermodynamically controlled acid/base process (since the nitrite anion is involved in some steps of channel I, relative energies in benzene are discussed). Conversion of 1-pyrazoline 15 into 2-pyrazoline 18 is thermodynamically very favourable as it is exothermic by 16.3 kcal mol−1. This finding is in agreement with the experimental observation that in 32CA reactions involving DAAs containing an α-hydrogen atom, 2-pyrazolines are obtained as a reaction product (see 2-pyrazolines 8 in Scheme 3).
Due to the relative acid character of the H5 hydrogen atoms of 2-pyrazoline 18, the corresponding proton abstraction process from C5 by the nitrite anion presents a low activation energy of 7.5 kcal mol−1. Interestingly, after the H5 proton abstraction, the subsequent loss of the nitrite anion has not activation energy; this last step being exothermic by 24.5 kcal mol−1. Along channel I, formation of final PYZ 12 plus nitrous acid from 1-pyrazoline 15 is exothermic by 31.0 kcal mol−1. Consequently, conversion of 1-pyrazoline 15 into final PYZ 12 through the studied reaction channel I can be considered irreversible.
Along channel II, the thermal extrusion of the nitrogen molecule at 1-pyrazoline 15 gives a diradical intermediate IN2, which is quickly converted into the final DNCP 13 by a ring closure through a C-to-C coupling of the C1 and C4 radical centers present in intermediate IN2. Due to the diradical structure of intermediate IN2, channel II was studied using unrestricted UB3LYP calculations. The activation energy associated with the extrusion of the nitrogen molecule via TS5 is 26.8 kcal mol−1. In spite of this high activation energy, this unimolecular process is not entropically unfavourable. The IRC from TS5 to products discontinues at species 19, which is 17.0 kcal mol−1 higher in energy than 1-pyrazoline 15. However, full optimisation of this species yields the final DNCP 13 in a straightforward manner, and the extrusion of the nitrogen molecule and subsequent ring closure being exothermic by 28.5 kcal mol−1. Consequently, the strong exothermic character of the formation of DNCP 13 as well as PYZ 12 makes these domino reactions irreversible.
In 1978, Engel studied the extrusion of the nitrogen molecule from cyclic and bicyclic azo compounds.36 For the extrusion of the nitrogen molecule from cyclic azo compound 20 yielding cyclopropane 21, an activation enthalpy of 39.0 kcal mol−1 was experimentally estimated (see Scheme 9). Interestingly, a relatively similar gas phase activation energy of 42.1 kcal mol−1 has been obtained at the UB3LYP/6-31G(d,p) level, asserting our computational level to study this reaction channel. In spite of this high activation enthalpy, the favourable activation entropy experimentally estimated for this extrusion reaction, 8.8 cal mol−1 K, favours this thermal reaction.
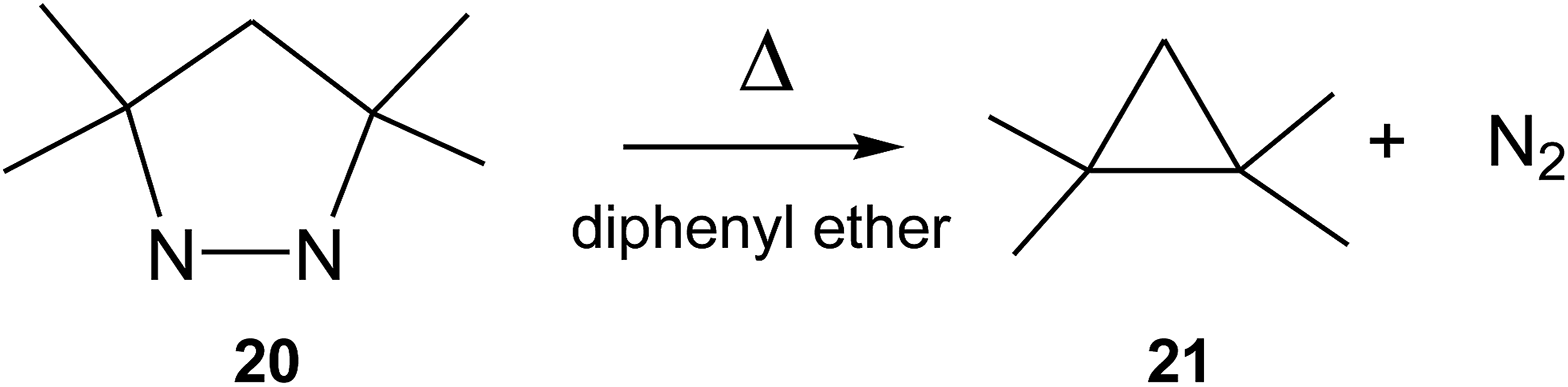 | ||
| Scheme 9 Thermal extrusion of the nitrogen molecule from cyclic azo compound 20. | ||
Gas-phase optimised TSs involved in the domino reactions between DAA 10 and DNE 11 (see Schemes 7 and 8), including some selected distances, are given in Fig. 3. At the TSs associated with the 32CA reaction between DAA 10 and DNE 11, the distances between the atoms involved in the formation of the C–C and C–N single bonds are: 2.025 Å (C1–C5) and 2.653 Å (N3–C4) at TS1, and 1.941 Å (N3–C5) and 2.369 Å (C1–C4) at TS2. Some appealing conclusions can be drawn from these geometrical parameters; (i) the more favourable TS1 is associated with a highly asynchronous bond-formation process; (ii) this 32CA reaction takes place via a two-stage one-step mechanism37 (see below). Thus, TS1 is associated with the nucleophilic attack of the C1 carbon of DAA 10 on the β-conjugated position of DNE 11, in clear agreement with the analysis of the Parr functions; and (iii) at the two regioisomeric TSs, the single-bond formation involving the most electrophilic center of DNE 11, the C5 carbon, is more advanced than that involving the C4 carbon.
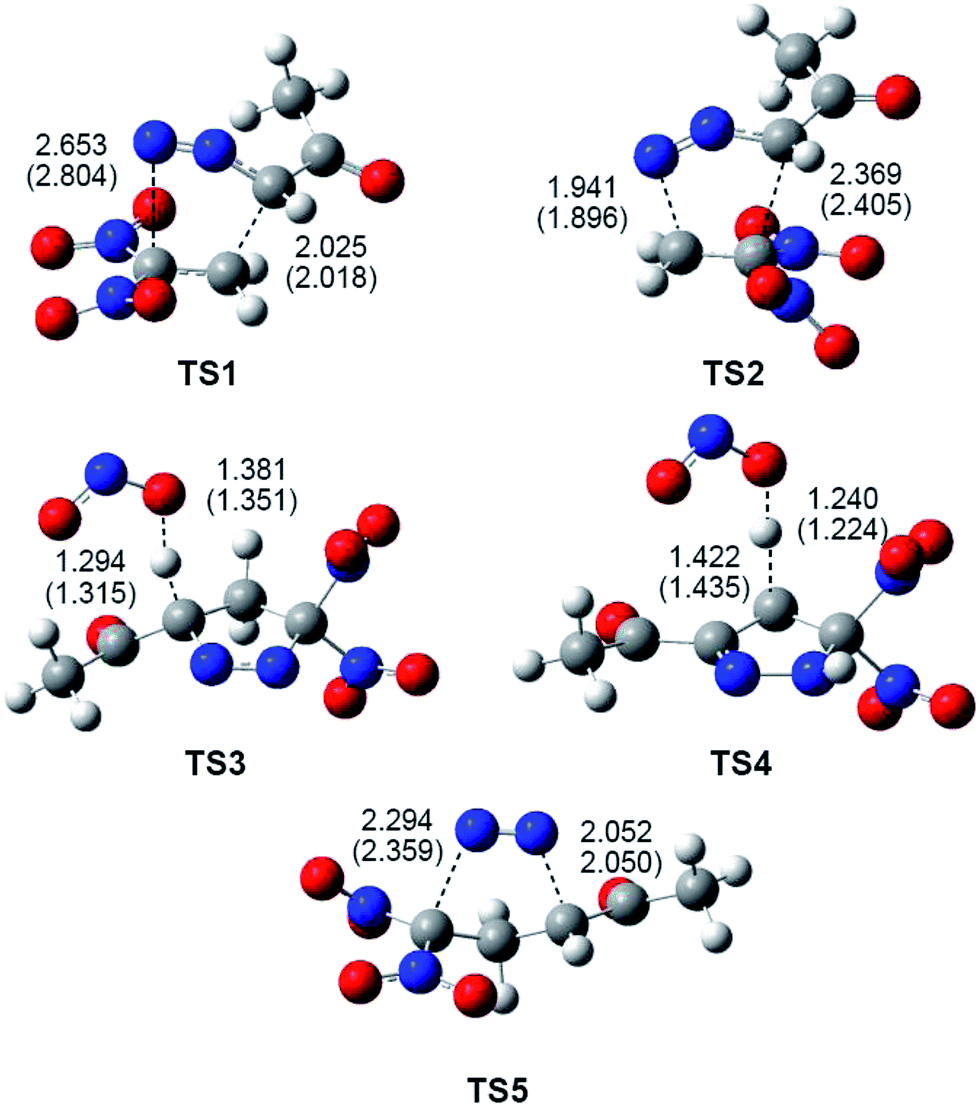 | ||
| Fig. 3 B3LYP/6-31G(d,p) optimised geometries of the TSs involved in the domino reaction between DAA 10 and DNE 11, see Schemes 7 and 8. Distances are given in Angstroms. Distances in benzene are given in parentheses. | ||
At TS3, associated with the proton abstraction at 1-pyrazole 15, the length of the C1–H1 breaking bond is 1.294 Å, while the length of the H1–O forming bond is 1.381 Å. At TS4, associated with the proton abstraction at 2-pyrazoline 18, the length of the C5–H5 breaking bond is 1.422 Å, while the length of the H5–O forming bond is 1.240 Å. Finally, at TS5, associated with the extrusion of the nitrogen molecule in 1-pyrazole 15, the lengths of the C1–N2 and N2–C3 breaking bonds are 2.052 Å and 2.294 Å, respectively. These lengths are relatively similar to those obtained at TS6, associated to the extrusion of the nitrogen molecule from cyclic azo compound 20, namely 2.23 Å (see TS6 in the ESI†).
The inclusion of the solvent effects of benzene does not substantially modify the TS geometries. At the most favourable regioisomeric TS1 associated with the 32CA reaction, benzene slightly increases the asynchronicity as the C3–N4 distance is increased, but this change has no chemical significance.
Both, the single imaginary frequency associated to TS4, −1005.6 cm−1, and the atomic movements associated to this imaginary frequency, indicate that this TS is mainly associated with the H5 proton abstraction. However, the IRC from TS4 towards the products shows that the extrusion of nitrite anion takes place in the same elementary step after the complete proton abstraction. On the other hand, as commented before, the IRC from TS5 towards the products discontinues at species 19 (see Fig. 4). After removing the nitrogen molecule, full UB3LYP/6-31G(d,p) optimisation of a feasible diradical intermediate yields DNCP 13 in a straightforward manner.
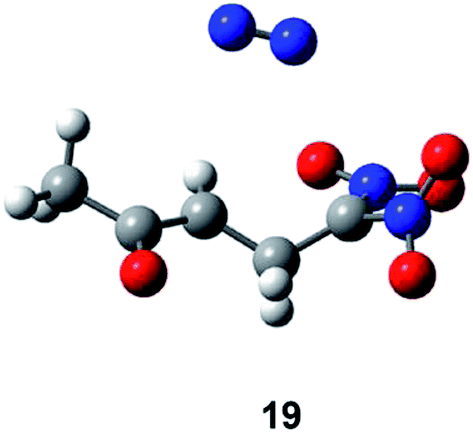 | ||
| Fig. 4 Diradical species 19 resulting from the IRC from TS5 towards the products. | ||
The polar nature of the 32CA reaction between DAA 10 and DNE 11 was analysed by computing the GEDT19 at the corresponding TSs. In order to calculate the GEDT, the natural atomic charges, obtained through an NPA, at TS1 and TS2, involved in the studied 32CA reaction, were computed at the ethylene as well as the DAA frameworks. In gas phase, the GEDT that fluxes from the DAA moiety toward the ED ethylene one is 0.34 e at TS1 and 0.24 e at TS2. These values indicate that this 32CA reaction has a strong polar character. The higher GEDT value found at the more favourable regioisomeric TS1 is in clear agreement with the relative low activation energy associated with this 32CA reaction (see Scheme 7).38 This high polar character arises from the strong electrophilic character of DNE 11, in spite of the moderate nucleophilic character of DAA 10 (see Section 3.2).
3.4. BET study of the 32CA reaction between DAA 10 and DNE 11
In order to understand the molecular mechanism of the 32CA reaction between DAA 10 and DNE 11, a BET study of the more favourable C1–C5 regioisomeric channel was performed. The complete BET study is given in the ESI.†Some appealing conclusions can be drawn from this BET study: (i) ten differentiated phases associated with the creation or disappearance of valence basins are distinguished along the C1–C5 regioisomeric reaction channel (see Fig. 5); (ii) TS1 is found in the very short phase VI presenting a bonding pattern similar to that found at P5 (see ELF valence basins of P5 in Fig. 6). TS1 is characterised by the presence of three monosynaptic basins, V(C1), V(C4) and V(C5). While the V(C1) monosynaptic basin was already present at DAA 10, the V(C4) and V(C5) monosynaptic basins present at the DNE 11 framework are formed along the reaction path on going from MC1 to TS1. A significant amount of the electron density of these monosynaptic basins comes from the high GEDT that takes place at TS1, 0.35 e.38 (iii) formation of the C1–C5 single bond begins at phase VII at a C1–C5 distance of 2.01 Å, following the recently proposed pattern:19 (a) depopulation of the C1–N2 and C4–C5 bonding regions, (b) formation of two non-bonding V(C1) and V(C5) monosynaptic basins (see P5 in Fig. 6), and (c) formation of a new V(C1,C5) disynaptic basin through the merger of the electron density of the aforementioned monosynaptic basins (see the V(C1,C5) disynaptic basin at P6 in Fig. 6); (iv) a different behaviour is found for the formation of the N3–C4 single bond. Formation of the N3–C4 single bond begins with the creation of a new V(N3,C4) disynaptic basin at the last phase X at the very short N3–C4 distance of 1.69 Å (see V(N3,C5) disynaptic basin at P9 in Fig. 6). The electron population of this new disynaptic basin proceeds from the electron density of one V(C4) monosynaptic basin created at the α position of the two nitro groups and one V′(N3) monosynaptic basin associated with a N3 pseudoradical center (see P8 in Fig. 6); (v) at P9, the V(C1,C5) disynaptic basin has reached 97% of its population in 1-pyrazoline 15. This behaviour indicates that this 32CA reaction takes place through a two-stage one-step mechanism;37 and (vi) along the more favourable regioisomeric channel, formation of the first C1–C5 single bond takes place through a two-center interaction involving the most nucleophilic center of DAA 10, the C1 carbon, and the most electrophilic center of DNE 11, the C5 carbon, a behaviour anticipated by the analysis of the electrophilic and nucleophilic Parr functions.25
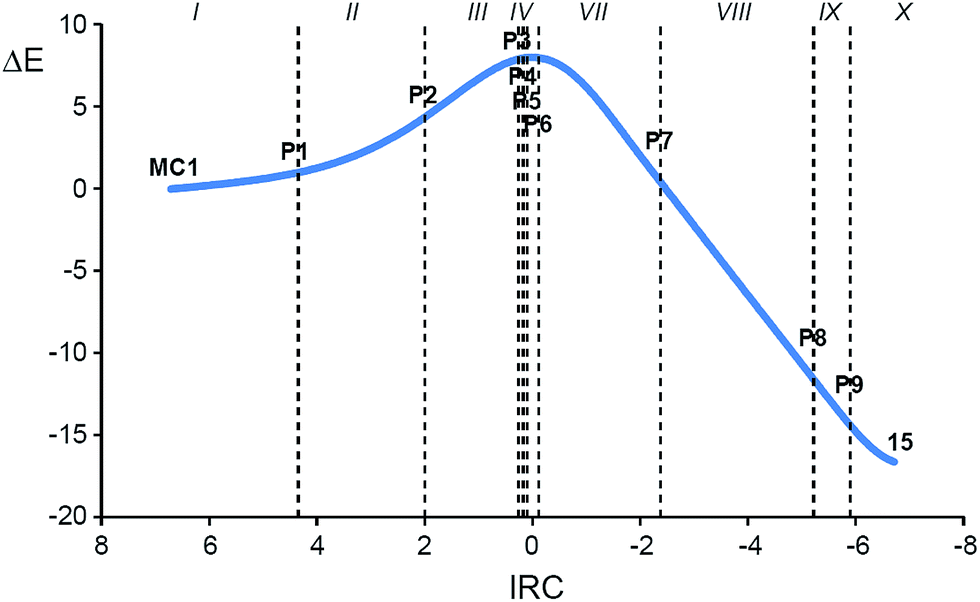 | ||
| Fig. 5 Relative energy (ΔE, in kcal mol−1) variations along the IRC (amu1/2 bohr) associated with the 32CA reaction between DAA 10 with DNE 11 showing the relative positions of the selected points separating the ten topological phases along the reaction path. | ||
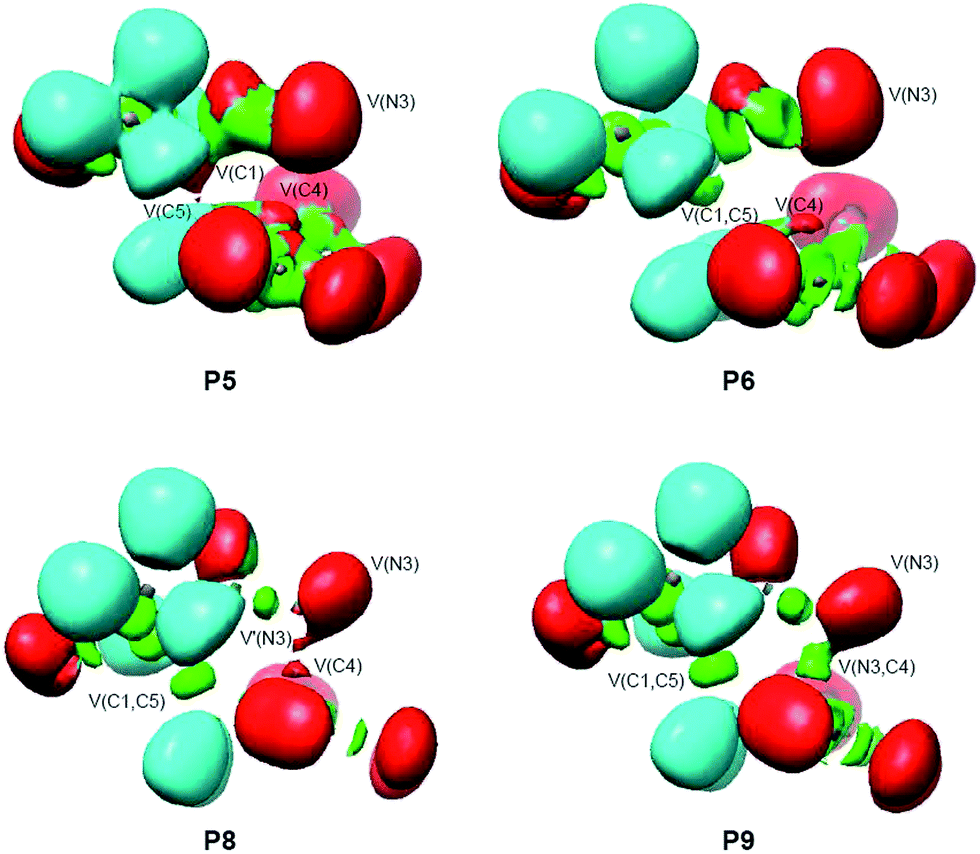 | ||
| Fig. 6 ELF valence basin representations of selected points involved in the C1–C5 and N3–C5 single bond formation processes along the IRC path of the 32CA reaction between DAA 10 with DNE 11. | ||
4. Conclusions
The reaction between DAA 10 and DNE 11 yielding PYZ 12 and DNCP 13 has been theoretically studied using the MEDT at the B3LYP/6-31G(d,p) computational level. Formation of PYZ 12 and DNCP 13 takes place through two domino processes initialised by a polar 32CA reaction between DAA 10 and DNE 11 yielding 1-pyrazoline 15, which experiences two successive competitive reactions. Along channel I, 1-pyrazoline 15 first tautomerises to 2-pyrazoline 18, which by a proton abstraction and spontaneous loss of nitrite anion yields PYZ 12, while along channel II, the thermal extrusion of the nitrogen molecule in 1-pyrazoline 15 gives a diradical intermediate IN2, which quickly closes yielding the final DNCP 13. Although the activation energies of the two competitive channels are not comparable, due to the need to model the corresponding acid/basic species demanded for the tautomerisation of 1-pyrazolines into 2-pyrazolines, and the further conversion of those into PYZs (see Scheme 8), the present MEDT study makes it possible to understand the chemical conversion of 1-pyrazolines obtained from a 32CA reaction of DAAs with ED ethylenes into the different reaction products experimentally observed.ELF analysis of the electronic structures of DAAs 10 and 14 shows that they have a pseudoradical structure. Analysis of the CDFT reactivity indices indicates that although DAA 10 is not a strong nucleophile, the strong electrophilic character of DNE 11 favours the 32CA reaction to take place through a polar mechanism with high polar character and with relatively low activation energy, 9.4 kcal mol−1. Note that the low pr index of these TACs together with the high activation energy associated with the non-polar 32CA reaction of the simplest DAA 14 with ethylene 17, 15.4 kcal mol−1,34 indicate that these pseudoradical TACs do not participate in pr-type 32CA reactions.
A BET study of the bonding changes along the more favourable regioisomeric channel associated with the 32CA reaction between DAA 10 and DNE 11 allows concluding that: (i) formation of the first C1–C5 single bond takes place at a C1–C5 distance of 2.01 Å by a C-to-C coupling of two pseudoradical centers, one already present in DAA 10 and another generated at the most electrophilic center of DNE 11 as a consequence of the high GEDT taking place in this polar process;38 (ii) formation of the second N3–C4 single bond begins at the end of the reaction path at the very short N3–C4 distance of 1.69 Å through sharing the electron density of two N3 and C4 pseudoradical centers. This model for the formation of carbon–heteroatom single bonds is similar to that found for the C–O single bond formation in the second stage of the cycloaddition reactions of carbenoid intermediates with CO2;39 and (iii) the high asynchronicity found in the formation of the C–C and C–N single bonds indicates that this polar 32CA reaction takes place through a two-stage one-step mechanism.37
The present MEDT study sheds light on these complex domino reactions as well as on the participation of DAAs in polar 32CA reactions. The electronic structures of the studied DAAs as well as their reactivity towards ethylene do not permit the classification of these TACs into one of the three groups in which TACs are currently classified in Scheme 4. Currently, further studies devoted to the reactivity of pseudoradical TACs are being carried out by our group.
Acknowledgements
This research was supported by the Ministry of Economy and Competitiveness (MINECO) of the Spanish Government, project CTQ2016-78669-P. M. R.-G. also thanks MINECO for a pre-doctoral contract co-financed by the European Social Fund (BES-2014-068258).References
- (a) W. Carruthers, Some Modern Methods of Organic Synthesis, Cambridge University Press, Cambridge, 2nd edn, 1978 Search PubMed; (b) W. Carruthers, Cycloaddition Reactions in Organic Synthesis, ed. J. E. Baldwin and P. D. Magnus, Pergamon, Oxford, 1990 Search PubMed; (c) Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products, ed. A. Padwa and W. H. Pearson, John Wiley & Sons, Inc, 2002, vol. 59 Search PubMed.
- K. V. Gothelf and K. A. Jorgensen, Chem. Rev., 1998, 98, 863 CrossRef CAS PubMed.
- (a) E. Buchner, Ber. Dtsch. Chem. Ges., 1888, 21, 2637 CrossRef; (b) H. von Pechmann, Ber. Dtsch. Chem. Ges., 1898, 31, 2950 CrossRef CAS.
- (a) J. V. Mehta, S. B. Gajera, P. Thakor, V. R. Thakkar and M. N. Patel, RSC Adv., 2015, 5, 85350 RSC; (b) K. A. Kumar and M. Govindaraju, Int. J. ChemTech Res., 2015, 8, 313 CAS.
- P. S. Engel, Chem. Rev., 1980, 80, 99 CrossRef CAS.
- L. R. Domingo, Molecules, 2016, 21, 1319 CrossRef PubMed.
- L. R. Domingo and S. R. Emamian, Tetrahedron, 2014, 70, 1267 CrossRef CAS.
- L. R. Domingo, M. Ríos-Gutiérrez and P. Pérez, Tetrahedron, 2016, 72, 1524 CrossRef CAS.
- O. Ivanova, E. M. Budynina, E. B. Averina, T. S. Kuznetsova, Y. K. Grishin and N. S. Zefirov, Synthesis, 2007, 13, 2009 Search PubMed.
- X. Krokidis, S. Noury and B. Silvi, J. Phys. Chem. A, 1997, 101, 7277 CrossRef CAS.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- X. Li and M. J. Frisch, J. Chem. Theory Comput., 2006, 2, 835 CrossRef CAS PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161 CrossRef CAS.
- (a) H. B. Schlegel, J. Comput. Chem., 1982, 2, 214 CrossRef; (b) H. B. Schlegel, in Modern Electronic Structure Theory, ed. D. R. Yarkony, World Scientific Publishing, Singapore, 1994 Search PubMed.
- (a) J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027 CrossRef CAS; (b) B. Y. Simkin and I. Sheikhet, Quantum Chemical and Statistical Theory of Solutions – Computational Approach, Ellis Horwood, London, 1995 Search PubMed.
- (a) E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef CAS; (b) M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327 CrossRef CAS; (c) V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404 CrossRef CAS.
- (a) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- L. R. Domingo, RSC Adv., 2014, 4, 32415 RSC.
- M. J. Frisch, et al., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- R. G. Parr, L. von Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922 CrossRef CAS.
- (a) R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512 CrossRef CAS; (b) R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- (a) L. R. Domingo, E. Chamorro and P. Pérez, J. Org. Chem., 2008, 73, 4615 CrossRef CAS PubMed; (b) L. R. Domingo and P. Pérez, Org. Biomol. Chem., 2011, 9, 7168 RSC.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, 1133 CrossRef.
- L. R. Domingo, P. Pérez and J. A. Sáez, RSC Adv., 2013, 3, 1486 RSC.
- A. D. Becke and K. E. Edgecombe, J. Chem. Phys., 1990, 92, 5397 CrossRef CAS.
- S. Noury, K. Krokidis, F. Fuster and B. Silvi, Comput. Chem., 1999, 23, 597 CrossRef CAS.
- L. R. Domingo, E. Chamorro and P. Pérez, Lett. Org. Chem., 2010, 7, 432 CrossRef CAS.
- L. R. Domingo and J. A. Sáez, J. Org. Chem., 2011, 76, 373 CrossRef CAS PubMed.
- M. Ríos-Gutiérrez, L. R. Domingo and P. Pérez, RSC Adv., 2015, 5, 84797 RSC.
- (a) P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793 CrossRef CAS PubMed; (b) L. R. Domingo, M. Ríos-Gutérrez and P. Pérez, Molecules, 2016, 21, 748 CrossRef PubMed.
- L. R. Domingo, M. J. Aurell, P. Pérez and R. Contreras, Tetrahedron, 2002, 58, 4417 CrossRef CAS.
- P. Jaramillo, L. R. Domingo, E. Chamorro and P. Pérez, J. Mol. Struct.: THEOCHEM, 2008, 865, 68 CrossRef CAS.
- L. R. Domingo, M. J. Aurell and P. Pérez, Tetrahedron, 2014, 70, 4519 CrossRef CAS.
- S. R. Emamian, L. R. Domingo and S. F. Tayyari, J. Mol. Graphics Modell., 2014, 49, 47 CrossRef CAS PubMed.
- P. S. Engel, R. A. Hayes, L. Keifer, S. Szilagyi and J. W. Timberlake, J. Am. Chem. Soc., 1978, 100, 1876 CrossRef CAS.
- L. R. Domingo, J. A. Saéz, R. J. Zaragozá and M. Arnó, J. Org. Chem., 2008, 73, 8791 CrossRef CAS PubMed.
- L. R. Domingo, M. Ríos-Gutérrez and P. Pérez, Tetrahedron, 2017 DOI:10.1016/j.tet.2017.02.012.
- L. R. Domingo, M. Ríos-Gutiérrez, E. Chamorro and P. Pérez, Theor. Chem. Acc., 2017, 136, 1 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Theoretical background of the topological analysis of ELF and the BET. BET study of the 32CA reaction between DAA 10 and DNE 11. Geometry of TS6. B3LYP/6-31G(d,p) total electronic energies of the stationary points involved in the domino process between DAA 10 and DNE 11, in gas phase and in the presence of benzene. See DOI: 10.1039/c7ra00544j |
| This journal is © The Royal Society of Chemistry 2017 |