
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of magnetic mesoporous carbon from polystyrene-grafted magnetic nanoparticles for rapid extraction of chlorophenols from water samples†
Shan Xue,
Chaozhan Wang * and
Yinmao Wei
* and
Yinmao Wei
Synthetic and Natural Functional Molecule Chemistry of Ministry of Education Key Laboratory, Key Laboratory of Modern Separation Science in Shaanxi Province, College of Chemistry and Materials Science, Northwest University, 1 Xue Fu Avenue, Changan District, Xi'an 710127, Shannxi, China. E-mail: czwang@nwu.edu.cn; Fax: +86-29-81535026; Tel: +86-29-81535026
First published on 20th February 2017
Abstract
A magnetic mesoporous carbon material (Fe3O4@C) was fabricated by carbonizing polystyrene grafted polydopamine-coated magnetic nanoparticles. The chemical composition, morphology and magnetism of the mesoporous carbon materials were characterized. The as-prepared magnetic mesoporous carbon was employed for enrichment of chlorophenols from water samples coupled with high-performance liquid chromatography with UV detection (HPLC-UV). Various parameters affecting the enrichment were investigated. A good linearity was obtained in the range of 10–200 ng mL−1 for 2-CP, 4-CP and 2,4-DCP, and 5–200 ng mL−1 for 2,4,6-TCP. The recoveries of CPs were in the range of 84.2–120% with intra- and inter-relative standard deviations (RSD) lower than 11.0%. The proposed method is fast, convenient and environmentally friendly. The results revealed the suitability of Fe3O4@C nanoparticles as adsorbents for enrichment of CPs from environmental water samples.
1. Introduction
Chlorophenols (CPs) as important raw materials are widely used in the production of petrochemicals, resins, pharmaceuticals, paints, herbicides and insecticides.1 During the production and application procedures, chlorophenols are introduced into the environment, inevitably. Owing to its toxic, biorefractory, and bioaccumulative characteristics, the United States Environmental Protection Agency has listed most CPs as priority pollutants.2,3 Therefore, it is very necessary to develop convenient and effective analytical methods for determination of these compounds in environment samples.Because of the low concentrations of CPs in environmental samples, extraction and preconcentration processes are usually required. Several pre-treatment methods for enrichment of CPs have been reported, including solid-phase-extraction (SPE),4–6 solid phase microextraction (SPME),7–9 liquid–liquid–liquid-microextraction (LLLME),10,11 and stir bar sorptive extraction (SBSE).12 Among them, the sorbent-based techniques, mainly SPE, SPME and magnetic solid-phase-extraction (MSPE), have become widely used for the enrichment of chlorophenols from environmental samples owing to the variety of the adsorbent materials.13,14 Compared with traditional SPE, MSPE can greatly simplify the phase separation procedure and increase the contact areas between analytes and adsorbents, which increase the mass transfer rate and extraction equilibrium. In the MSPE procedure, the magnetic sorbent is essential, which determines the extraction efficiencies of analytes. So, the exploration of new types of the MSPE adsorbents to improve the enrichment capability for different kinds of analytes has become an active field in analytical chemistry.15
With the properties of high surface area, tunable surface chemistry, and high chemical and physical stability,16 nanoporous carbon materials are currently finding its wide application in many fields including adsorbents, drug deliver, electrode materials, and catalyst supports.17–20 To prepare the nanoporous carbon materials, the most common and effective routes are template methods, including soft- and hard-template.21 There are some drawbacks with both methods. The hard-template method often involves silica as hard-template, which requires harsh chemical treatment, such as HF etch, to remove the sacrificial silica after carbonization.22 By contrast, the soft-template method which use of block copolymers as precursor, remove the template by thermal decomposition prior to carbonization. But the interactions between soft templates and carbon sources are usually too weak to prepare well-defined mesostructures.23 And both methods are complicated, expensive.24
Dopamine molecules (DA) can self-polymerize to form polydopamine (PDA) under alkaline conditions.25 Owing to the adhesion of PDA, PDA can be easily coated on the surface of substrates. Such properties can allow PDA to serve as functional layer, which contains catechol and amine function groups. This provides a route to modify the surface chemistry of materials. In addition, PDA has been proved as promising carbon precursors, owing to the nontoxic and good carbon yield.26 Surface-initiated atom transfer radical polymerization (SI-ATRP) has been explored as one of the most successful controlled/living radical polymerization techniques due to its mild reaction conditions, excellent tolerance to functional groups and impurities, and precise control on molecular weight.27 It also has been proved to be an excellent tool to introduce organic (co) polymers with precise molar mass, composition, and functionality onto nanoparticles.28,29 Recently, organic polymer chains grafted from particles by using SI-ATRP have been used as carbon precursors to prepare mesocarbon materials.22,23
Herein, we fabricated carbon precursors by using ATRP technology to graft polystyrene from polydopamine-encapsulated magnetic nanoparticles. After carbonization, the magnetic mesoporous carbon materials were obtained. The prepared magnetic carbon material was applied as adsorbent for the enrichment and analysis of chlorophenols from environmental aqueous samples.
2. Materials and methods
2.1 Chemical and materials
Anhydrous ferric chloride, anhydrous sodium acetate, trisodium citrate dihydrate, 2-chlorophenol (2-CP), 4-chlorophenol (4-CP) were obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Dopamine hydrochloride (98%) was purchased from Macklin Biochemical Co., Ltd. (Shanghai, China). 2-Bromoisobutyryl bromide (2-BiBB), 2,4-dichlorophenol (2,4-DCP), 2,4,6-trichlorophenol (2,4,6-TCP), copper(I) bromide (>98%) and 2,2-bipyridyl (Bpy) (>99.5%) were all supplied from Aladdin Chemistry Co., Ltd (Shanghai, China). Styrene (St) was obtained from Tianli Chemical Reagent Co., Ltd. (Tianjin, China), and was passed over a column of basic alumina prior to use. Acetonitrile (HPLC grade) was purchased from Fisher (USA). HPLC grade methanol and acetic acid were obtained from Kermel Chemical Reagent Co. (Tianjin, China). Other reagents were of analytical grade.Stock solutions of CPs were prepared in methanol, with concentration levels of 1 mg mL−1 for each compound, and were stored in a freezer at 4 °C. Working solutions were freshly diluted with 20 mM NaAC-HAC solution at given concentrations. The tap water was collected from our laboratory, the river water from Ju River in Chang'an District, Xi'an. The water samples were stored at 4 °C and filtered using 0.45 μm Nylon membranes before use.
2.2 Instrumentation
The Fourier-transform infrared spectrometer (FT-IR, TENSOR27, Bruker, Germany) was used to determine the functional group of nanoparticles surface. The X-ray photoelectron spectroscopy (XPS) analysis (K-Alpha Thermo Fisher Scientific) was used to determine the contents of the elements and chemical state in the nanospheres. The morphology and size were observed by scanning electron microscopy (SEM, Quanta 600FEG, America) and transmission electron microscopy (TEM, H-600, 75 kV, Hitachi, Japan). The magnetic properties were determined via a SQUID magnetometer (MPMS-XL-7, Quantum Design, USA). Powder X-ray diffraction (XRD) measurements were made with CuKα radiation on a D8 ADVANC (Bruker, Germany). Brunauer–Emmett–Teller (BET) specific surface measurements were performed using ASAP 2460 (Micromeritics, USA).2.3 Synthesis of Fe3O4@PDA
Magnetic Fe3O4 nanoparticles were prepared via solvothermal method following the reference with minor modification.30 Briefly, 1.35 g of FeCl3 and 0.45 g of trisodium citrate dehydrate were added in 30 mL of ethylene glycol. The mixture was heated to dissolve the solid. After cooling to room temperature, 2.40 g of sodium acetate was added with magnetic stirring for 30 min. The mixture was sealed in a Teflon-lined stainless-steel autoclave. After reaction for 12 h at 200 °C, the autoclave was cooled down to room temperature. The resulting Fe3O4 nanoparticles (NPs) were washed with ethanol and distilled water several times respectively, and then dried under vacuum at 45 °C.The Fe3O4@PDA core–shell NPs were fabricated according to the literature with minor modification.31 200 mg Fe3O4 was dispersed in 100 mL of Tris–HCl solution (10 mM, pH 8.5) under ultrasonication for 30 min, followed by addition of dopamine hydrochloride (200 mg). After mechanical stirring at room temperature for 24 h, Fe3O4@PDA NPs were collected by magnetic separation and washed with distilled water and ethanol respectively. The product was dried under vacuum.
2.4 Immobilization of initiator on Fe3O4@PDA NPs
1.0 g of Fe3O4@PDA NPs were dispersed in 20 mL of anhydrous tetrahydrofuran (THF) and sonicated for 5 min. After stirred the mixture for 30 min in an ice bath, 2 mL of triethylamine (TEA) and 2 mL of 2-BiBB were drop-wise added into the solution and maintained in an ice bath for 3 h under vigorous stirring. The reaction was left at 35 °C for another 12 h. Afterwards, the obtained initiator-functionalized nanospheres, denoted as Fe3O4@PDA–Br, were extensively rinsed with THF, methanol and water in sequence.2.5 Synthesis of Fe3O4@PDA@St by ATRP
0.5 g of Fe3O4@PDA–Br nanospheres were dispersed in a mixture of 2.4 mL of styrene (21 mmol), 66.5 mg of 2,2-bipyridyl (0.42 mmol) and 10 mL anisole in a 50 mL three-neck flask. After the mixture was deoxygenated via two freeze–pump–thaw cycles, CuBr (30.5 mg, 0.21 mmol) was quickly added to the flask under an nitrogen atmosphere. After two freeze–pump–thaw cycles, the polymerization was allowed to carry out at 90 °C for 16 h with continuous stirring. The resulting nanospheres were extensively washed with anisole, methanol and water in sequence. To remove the residual catalyst completely, the obtained nanoparticles were re-dispersed into a mixture of methanol and 0.25 M EDTANa2 solution (1/1, v/v), and the mixture was stirred at 40 °C for 4 h. After magnetic separation, the nanoparticles was washed with water, methanol and dried at 45 °C under vacuum.2.6 Synthesis of Fe3O4@C
Fe3O4@C was obtained by carbonizing the as-synthesized Fe3O4@PDA@St in an N2 atmosphere. The Fe3O4@PDA@St NPs were heated from ambient temperature to 600 °C with a heating rate of 3 °C min−1, and was maintained at this temperature for 1 h. The whole preparation process is shown in Fig. 1.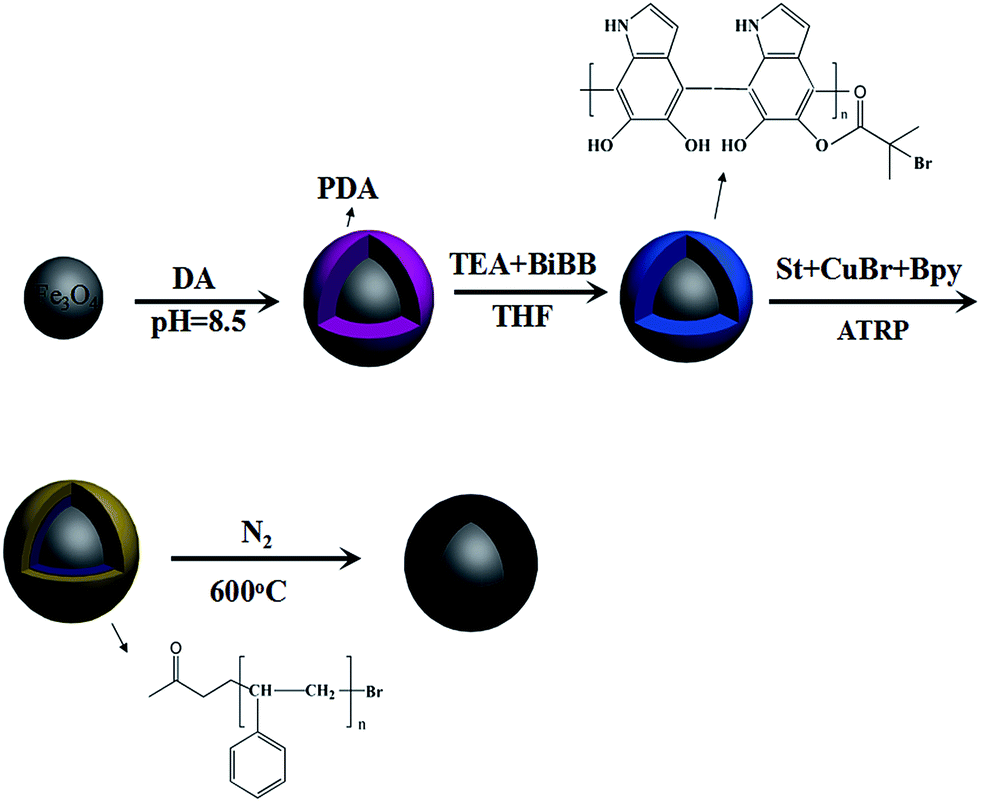 | ||
| Fig. 1 Schematic illustration of the preparation of Fe3O4@C. | ||
2.7 MSPE procedures
20 mg of Fe3O4@C was added to a 20 mL sample solution. The mixture was shaken for 1 min. Subsequently, the adsorbent was separated from the sample solution using a magnet, and the supernatant was discarded. Next, 0.6 mL of alkaline methanol was added into the centrifuge tube to elute the chlorophenols by shaking for 1 min. The desorption solution was also separated under a magnet, then the elute solution was adjusted to neutral by HCl.2.8 Analytical conditions
The HPLC analyses were conducted on a LC-20A system from Shimadzu (Tokyo, Japan), equipped with two LC-20AD pumps, a SPD-20A ultraviolet detector, an injector with a 20 μL sample loop and a CTO-20AC column oven. Chromatographic separations were performed on an Agilent TC-C18 (150 mm × 4.6 mm i.d., 5 μm) column. The mobile phase consisted of acetonitrile/water containing 1% acetic acid (20/80, v/v, solvent A) and acetonitrile containing 1% acetic acid (solvent B). The gradient elution program was as follows: started at 20% B and kept for 10 min, then increased B to 70% in 10 min and kept for 5 min. Finally the mobile phase was returned to initial condition. The flow rate was set at 1 mL min−1. The UV monitoring wavelength was chosen at 285 nm and the column oven temperature was maintained at 25 °C. The injection volume was 20 μL.3. Results and discussion
3.1 Synthesis and characterizations of material
In this study, polystyrene chains were grafted from magnetic nanoparticles via SI-ATRP, and the polymer chains were then taken as carbon resource to fabricate the Fe3O4@C at a carbonization temperature of 600 °C.The morphology of the adsorbent was observed by SEM and TEM. As can be seen from the SEM (Fig. 2A) and TEM (Fig. 2C) images, the Fe3O4 NPs are spherical with an average diameter of 220 nm and have a coarse surface. The SEM (Fig. 2B) and TEM (Fig. 2D) images of Fe3O4@C show that the resulting microspheres are clearly core–shell structure, with a thickness of carbon coating ∼40 nm.
 | ||
| Fig. 2 SEM images of Fe3O4 (A), Fe3O4@C (B); TEM images of Fe3O4 (C), Fe3O4@C (D). | ||
The functional group of Fe3O4, Fe3O4@PDA, Fe3O4@PAD@St, and Fe3O4@C were investigated with FT-IR. As shown in Fig. 3A, the peak at 590 cm−1 (Fig. 3A(a–d)) is assigned to the vibration of Fe–O. Compared with Fe3O4 (Fig. 3A(a)), the IR spectrum of Fe3O4@PDA (Fig. 3A(b)) shows the characteristic adsorption of the stretching of C–O bond at 1291 cm−1, the stretching vibration of the aromatic rings at 1620 cm−1 and the O–H stretching vibration and N–H stretching vibration at 3380 cm−1 from PDA. After grafting St (curve c in Fig. 3A), new bands appeared at 1442 cm−1 and 1110 cm−1, which are ascribed to the stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and in-plane bending vibrations of C–H from aromatic rings. After carbonization (Fig. 3A(d)), most of the characteristic peaks of organic groups are greatly weakened, which confirm that the polymer is successfully carbonized.
C and in-plane bending vibrations of C–H from aromatic rings. After carbonization (Fig. 3A(d)), most of the characteristic peaks of organic groups are greatly weakened, which confirm that the polymer is successfully carbonized.
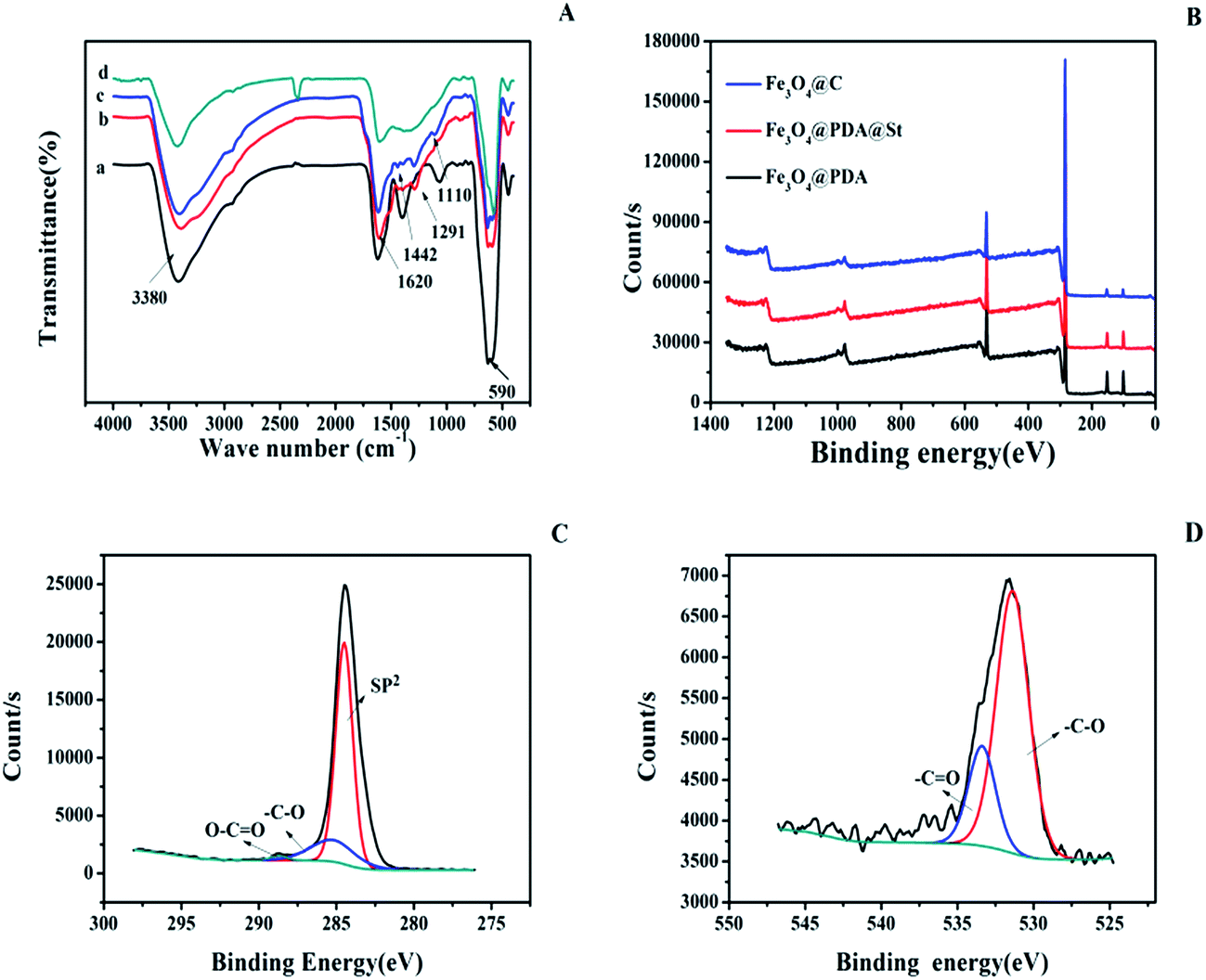 | ||
| Fig. 3 FT-IR spectrum (A) of Fe3O4 (a), Fe3O4@PDA (b), Fe3O4@PDA@St (c) and Fe3O4@C (d); XPS survey spectrum (B) of Fe3O4@PDA, Fe3O4@PDA@St, and Fe3O4@C; high-resolution XPS spectrum of C 1s(C), and O 1s (D) of Fe3O4@C. | ||
Surface chemical compositions of the Fe3O4@PDA, Fe3O4@PDA@St, and Fe3O4@C are further investigated by XPS. The survey spectrum (Fig. 3B) shows the two main peaks for C 1s and O 1s, and the content of the each element is showed in the Table 1. From Table 1, the content of C element increases after carbonization, while the content of O element decreases. For high-resolution C 1s spectra of Fe3O4@C in Fig. 3C, there are three peaks at around 284.4 eV, 285.8 eV, 289.1 eV, which are ascribed to –CC, –C–O and –CO, respectively.32 From the high-resolution XPS spectra of O 1s for Fe3O4@C (Fig. 3D), the binding energy peak at 531.6 eV belongs to –C–O and the peak at 533.7 eV belongs to –CO can be found.33
| C 1s (at%) | N 1s (at%) | O 1s (at%) | |
|---|---|---|---|
| Fe3O4@PDA | 75.18 | 1.32 | 14.40 |
| Fe3O4@PDA@St | 80.82 | 1.22 | 11.60 |
| Fe3O4@C | 87.24 | 1.49 | 7.73 |
The magnetic properties of Fe3O4 and Fe3O4@C were measured via SQUID magnetometer at 300 K (Fig. S1A†). The saturated magnetization values of Fe3O4, Fe3O4@PDA, Fe3O4@PDA@St and Fe3O4@C are 26.7, 21.2, 18.5 and 26.5 emu g−1, respectively. It indicated that after polydopamine coating and polystyrene grafting, the magnetism of the nanoparticles decreased gradually, and then increased after carbonization. The fact that the Fe3O4@C has stronger magnetism than Fe3O4@PDA@St maybe due to the Fe3O4@C nanoparticles prepared at 600 °C exhibit ferromagnetic properties (Fig. S1A,† insert).34 Fig. S1B† shows the XRD pattern of Fe3O4 and Fe3O4@C. All of the diffraction peaks of Fe3O4@C are the same as Fe3O4, indicting that no new phase formed during the subsequent modification process.
The nitrogen adsorption/desorption isotherm (77 K) and pore size distribution curve (Fig. S2†) indicate that the BET surface area of Fe3O4@C is 8.78 m2 g−1; the pore volume is 0.0171 cm3 g−1 and the pore size is 7.73 nm. The adsorption/desorption isotherms are type IV35 with H4 hysteresis loops, which indicate the carbon materials is mesoporous.
3.2 Optimization of MSPE conditions
The prepared magnetic carbon materials were then employed as an adsorbent for enrichment of four chlorophenols (2-CP, 4-CP, 2,4-DCP and 2,4,6-TCP). Principally, hydrophobic interactions and π–π interactions should be involved during adsorption. To achieve the maximal extraction efficiency, several important parameters, such as solution pH, type and volume of elution solvent, amounts of adsorbent, salt concentration, adsorption time and elution time were optimized.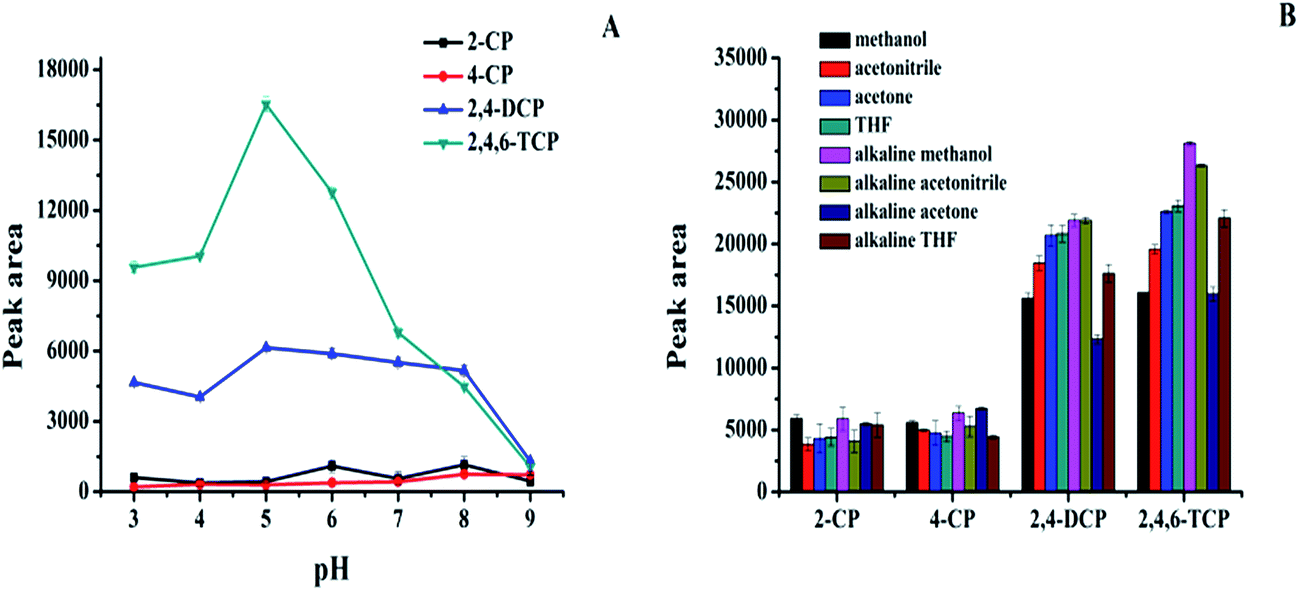 | ||
| Fig. 4 Effect of several parameters for adsorption of chlorophenols. Sample solution pH (A); desorption solvents (B); CPs concentrations are all at 1.0 μg mL−1. Sample volume, 10 mL; amount of adsorbent, 5 mg. | ||
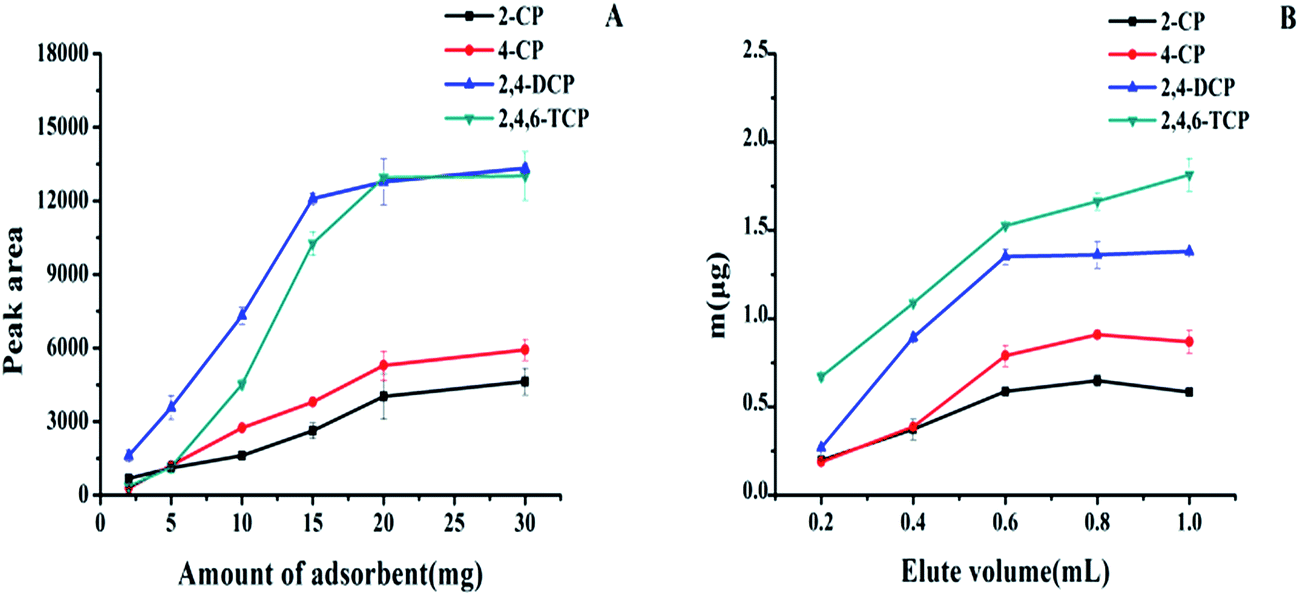 | ||
| Fig. 5 Effect of the amount of adsorbent (A) and volume of elution solution (B) on the extraction of CPs. CPs concentrations are all at 200 ng mL−1. Salt concentration, 10% (m/v); pH values, 5; extraction and desorption time, 1 min; sample volume, 20 mL; elution solution, alkaline methanol. | ||
3.3 Validations of the method
Under the optimal experimental conditions, the linearity, limit of detection (LOD) and limit of quotation (LOQ) were evaluated. All the parallel experiments were repeated three times and the results were listed in Table 2. A good linearity was obtained in the range of 10–200 ng mL−1 for 2-CP, 4-CP, and 2,4-DCP, and 5–200 ng mL−1 for 2,4,6-TCP, with correlation coefficient (r) ranging from 0.9913 to 0.9987. The LODs were ranging from 0.88–2.50 ng mL−1 (LOD = 3Sb/S, Sb is the SD of three replicated runs of spiked samples at the lowest concentration, S is the slope of calibration curve). The LOQs were ranging from 2.95–8.34 ng mL−1 (LOQ = 10 Sb/S).| Analytes | Calibration curve | r | Linear range (ng mL−1) | LOD (ng mL−1) | LOQ (ng mL−1) |
|---|---|---|---|---|---|
| 2-CP | y = 15.951x + 223.39 | 0.9935 | 10–200 | 1.41 | 4.70 |
| 4-CP | y = 21.836x + 107.7 | 0.9913 | 10–200 | 0.88 | 2.95 |
| 2,4-DCP | y = 64.155x + 775.43 | 0.9971 | 10–200 | 2.50 | 8.34 |
| 2,4,6-TCP | y = 66.827x + 11.061 | 0.9987 | 5–200 | 1.32 | 4.40 |
3.4 Real sample analysis
The method was applied for extraction chlorophenols from tap water and river water under optimized conditions. The chromatograms of river water sample are shown in Fig. 6. The analytical figure merits are shown in Table 3, none of four CPs was found in tap and river water. The recoveries of chlorophenols spiked at different concentration were ranging from 84.2–120% with RSDs less than 11.0%, which indicated that the method is applicable for real sample analyses.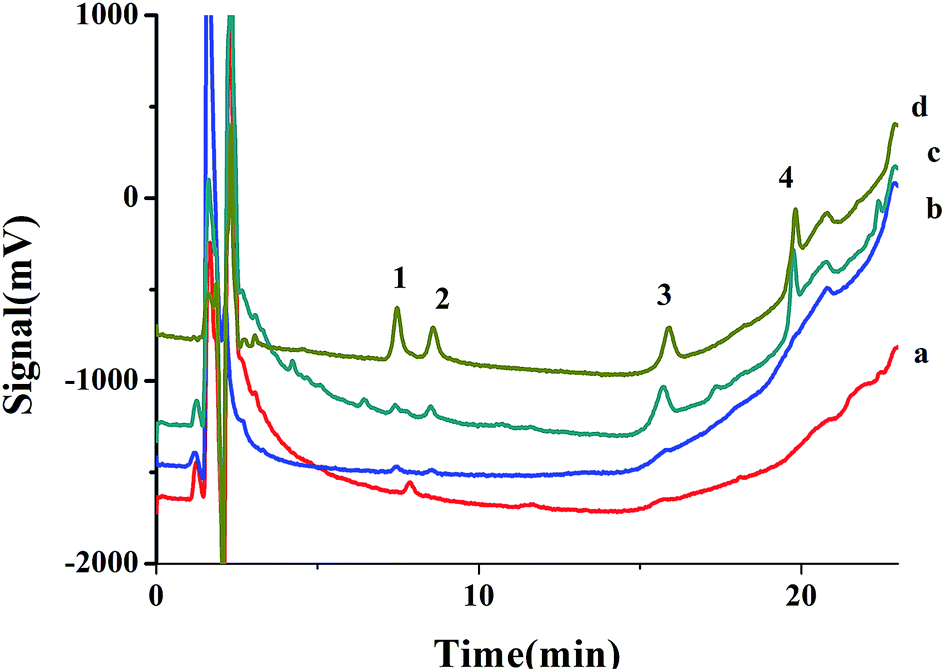 | ||
| Fig. 6 Chromatograms of (a) blank river water after extraction, (b) river water spike with CPs (50 ng mL−1 for each CPs), (c) river water spike with CPs at 50 ng mL−1 after extraction, and (d) standard solution of CPs at 500 ng mL−1. Peaks: (1) 2-CP, (2) 4-CP, (3) 2,4-DCP, (4) 2,4,6-TCP. | ||
| Compound | Added (ng mL−1) | Recovery (%) | Precision (RSD, %) | ||||
|---|---|---|---|---|---|---|---|
| Tap water | River water | Tap water | River water | ||||
| (n = 4) | (n = 4) | Intra-day (n = 4) | Inter-day (n = 3) | Intra-day (n = 4) | Inter-day (n = 3) | ||
| a nd: not detect. | |||||||
| 2-CP | 0 | nd | nd | ||||
| 10 | 107 | 84.2 | 4.85 | 4.48 | 7.52 | 9.84 | |
| 20 | 107 | 117 | 6.79 | 3.94 | 8.28 | 0.39 | |
| 200 | 117 | 95.5 | 4.14 | 1.52 | 2.82 | 6.32 | |
| 4-CP | 0 | nd | nd | ||||
| 10 | 113 | 107 | 5.22 | 2.88 | 6.00 | 1.20 | |
| 20 | 110 | 117 | 5.75 | 0.67 | 5.94 | 2.77 | |
| 200 | 111 | 95.7 | 6.35 | 2.92 | 6.39 | 2.28 | |
| 2,4-DCP | 0 | nd | nd | ||||
| 10 | 84.2 | 86.1 | 7.82 | 6.61 | 10.1 | 6.10 | |
| 20 | 96.3 | 101 | 10.9 | 4.47 | 9.28 | 6.27 | |
| 200 | 120 | 99.2 | 2.80 | 1.62 | 6.93 | 4.49 | |
| 2,4,6-TCP | 0 | nd | nd | ||||
| 10 | 113 | 112 | 3.34 | 1.13 | 4.51 | 3.81 | |
| 20 | 115 | 112 | 3.08 | 2.51 | 6.64 | 1.68 | |
| 200 | 114 | 103 | 0.84 | 2.13 | 3.32 | 5.70 | |
3.5 Compare with other methods
The comparison of different methods for the determination of CPs is displayed in Table 4. Compared with other methods, the recovery of current method is comparable, while the sample preparation time (only 2 min) of the proposed method are greatly shorter than the previous method,6,38–41 which indicates that the extraction procedure is greatly fast. In addition, the amount of the sorbent (20 mg for 20 mL sample) is much lower than reported methods (40 mg for 10 mL sample,38 150 mg for 80 mL sample,39 100 mg for 10 mL,40 100 mg for 70 mL sample41), meaning that the adsorbent has a higher extraction capability for the CPs. Compared with other carbon materials,6,41 the as-prepared sorbent shows short sample preparation time, less adsorbent dosage, and good recovery; in addition, the as-prepared carbon possess magnetism, which makes the extraction process greatly simple, avoid the need of centrifugation or filtration. These results suggested that the proposed method is very rapid, sensitive, and convenient method to enrichment of CPs in environmental water samples.| Method | Adsorbent | Amount of sorbent (mg) | Sample preparation time (min) | LOD (ng mL−1) | Recovery | Ref. |
|---|---|---|---|---|---|---|
| a SPME: solid phase microextraction. SPE: solid phase extraction. GC-MS: gas chromatography-tandem mass spectrometry. MWCNT: multiwalled carbon nanotube. | ||||||
| MSPE-GC-MS | Fe3O4@C@PANI | 40 | 25 | 3.22–5.55 | 87.7–103.1 | 38 |
| MSPE-HPLC-UV | OMMT-Fe3O4@PSF | 150 | 35 | 0.17–0.22 | 90.9–115 | 39 |
| SPE-HPLC-UV | Si–Ti@CN/IL | 100 | 10 | 0.83–0.95 | 73.9–105.54 | 40 |
| SPE-HPLC-UV | Cyano-functionalized MWCNT | 100 | >67 | 0.45–3 | 82.53–102.13 | 41 |
| SPE-HPLC-UV | Graphene | 20 | >35 | 0.1–0.4 | 77.2–116.6 | 6 |
| MSPE-HPLC-UV | Fe3O4@C | 20 | 2 | 0.88–2.50 | 84.2–120 | This work |
4. Conclusion
In this study, magnetic mesoporous carbon materials were fabricated by carbonization of polystyrene-grafted magnetic nanoparticles. The Fe3O4@C NPs were used as adsorbents to determination of chlorophenols from environmental water. The as-prepared adsorbent showed a good recovery for extraction chlorophenols from water samples because of hydrophobic interactions and π–π interactions. In addition, this method not only proposed a greatly fast, convenient and simple extraction process, but was environmental friendly because of using less organic solvent. These advantages reveal that the proposed method is applicable for real water sample analysis.Acknowledgements
This work was supported by the National Natural Science Foundation in China (No. 21575114 and 21475104) and the Scientific Research Program Funded by Shaanxi Provincial Education Department (No. 16JS114).References
- J. P. Wang, J. Chen, Y. F. Sun, J. T. Dai and Y. Y. Wei, Asian J. Chem., 2013, 25, 6209–6212 CAS.
- X. Cui, W. Zuo, M. Tian, Z. Dong and J. Ma, J. Mol. Catal. A: Chem., 2016, 423, 386–392 CrossRef CAS.
- Z. Dong, X. Le, C. Dong, W. Zhang, X. Li and J. Ma, Appl. Catal., B, 2015, 162, 372–380 CrossRef CAS.
- L. Elci, N. Kolbe, S. G. Elci and J. T. Anderson, Talanta, 2011, 85, 551–555 CrossRef CAS PubMed.
- A. H. El-Sheikh, M. K. Al-Jafari and J. A. Sweileh, Int. J. Environ. Anal. Chem., 2012, 92, 190–209 CrossRef CAS.
- Q. Liu, J. Shi, L. Zeng, T. Wang, Y. Cai and G. Jiang, J. Chromatogr. A, 2011, 1218, 197–204 CrossRef CAS PubMed.
- R. Morales, L. A. Sarabia, M. Sagrario Sánchez and M. Cruz Ortiz, J. Chromatogr. A, 2013, 1296, 179–195 CrossRef CAS PubMed.
- M. Anbia, A. Haghi and S. Shariati, Anal. Methods, 2012, 4, 2555–2561 RSC.
- X. J. Huang, Y. Zhang, M. Mei and D. X. Yuan, J. Sep. Sci., 2014, 37, 1185–1193 CrossRef CAS PubMed.
- Y. Chao, Y. Tu, Z. Jian, H. Wang and Y. Huang, J. Chromatogr. A, 2013, 1271, 41–49 CrossRef CAS PubMed.
- D. Ge and H. K. Lee, Talanta, 2015, 132, 132–136 CrossRef CAS PubMed.
- M. Ghani, M. Saraji, F. Maya and V. Cerdà, J. Chromatogr. A, 2016, 1445, 10–18 CrossRef CAS PubMed.
- G. Li, J. Lan and G. Li, RSC Adv., 2015, 5, 1705–1711 RSC.
- L. Hao, X. L. Liu, J.-T. Wang, C. Wang, Q. H. Wu and Z. Wang, Chin. Chem. Lett., 2016, 27, 783–788 CrossRef CAS.
- S. Huo and X. Yan, Analyst, 2012, 137, 3445–3451 RSC.
- W. Xin and Y. Song, RSC Adv., 2015, 5, 83239–83285 RSC.
- Y. Zhao, L. Zhao, Ke X. Yao, Y. Yang, Q. Zhang and Y. Han, J. Mater. Chem., 2012, 22, 19726–19731 RSC.
- L. Guo, H. Shi, H. Wu, Y. Zhang, X. Wang, D. Wu, L. An and S. Yang, Carbon, 2016, 107, 87–99 CrossRef CAS.
- H. Xing, F. Zhang, Y. Lu, B. Zhai, S. Zhai, Q. An and C. Yu, RSC Adv., 2016, 6, 79366–79371 RSC.
- K. Ai, Y. Liu, C. Ruan, L. Lu and G. Lu, Adv. Mater., 2013, 25, 998–1003 CrossRef CAS PubMed.
- D. Wu, H. Dong, J. Pietrasik, E. K. Kim, C. M. Hui, M. Zhong, M. Jarnoiec, T. Kowalewsik and K. Matyjaszewski, Chem. Mater., 2011, 23, 2024–2026 CrossRef CAS.
- C. Tang, L. Bombalski, M. Kruk, M. Jaroniec, K. Matyjaszewski and T. Kowalewski, Adv. Mater., 2008, 20, 1516–1522 CrossRef CAS.
- D. Wu, Z. Li, M. Zhong, T. Kowalewski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2014, 53, 3957–3960 CrossRef CAS PubMed.
- J. Zhou, Z. Zhang, Z. Li, T. Zhu and S. Zhuo, RSC Adv., 2015, 5, 46947–46954 RSC.
- Y. Liu, K. Ai and L. Lu, Chem. Rev., 2014, 114, 5057–5115 CrossRef CAS PubMed.
- R. Liu, S. M. Mahurin, C. Li, R. R. Unocic, J. C. Idrobo, H. Gao, S. J. Pennycook and S. Dai, Angew. Chem., Int. Ed., 2011, 50, 6799–6802 CrossRef CAS PubMed.
- Y. Zhu, F. Jiang, P. Zhang and H. Tang, Chin. Chem. Lett., 2016, 27, 910–914 CrossRef CAS.
- J. Pyun, S. Jia, T. Kowalewski, G. D. Patterson and K. Matyjaszewski, Macromolecules, 2003, 36, 5094–5104 CrossRef CAS.
- J. Pyun, K. Matyjaszewski, T. Kowalewski, D. Savin, G. Patterson, G. Kickelbick and N. Huesing, J. Am. Chem. Soc., 2001, 123, 9445–9446 CrossRef CAS PubMed.
- J. Liu, Z. Sun, Y. Deng, Y. Zou, C. Li, X. Guo, L. Xiong, Y. Gao, F. Li and D. Zhao, Angew. Chem., Int. Ed., 2009, 48, 5875–5879 CrossRef CAS PubMed.
- R. Liu, Y. Guo, G. Odusote, F. Qu and R. D. Priestley, ACS Appl. Mater. Interfaces, 2013, 5, 9167–9171 CAS.
- S. Xian, F. Xu, Z. Zhao, Y. Li, Z. Li, Q. Xia, J. Xiao and H. Wang, AIChE J., 2016, 62, 3730–3738 CrossRef CAS.
- X. Wang, Y. Wu, X. Zhou, J. Xiao, Q. Xia, H. Wang and Z. Li, Chem. Eng. Sci., 2016, 155, 338–347 CrossRef CAS.
- L. Ding, M. Zhang, Y. Zhang, J. Yang, J. Zheng and J. Xu, Green Chem., 2016, 18, 6282–6290 RSC.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquérol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- B. Yang, Y. Liu, Z. Li, L. Lei, X. Zhang and J. Zhou, Environ. Sci. Pollut. Res., 2016, 23, 1482–1491 CrossRef CAS PubMed.
- X. Li, A. Xue, H. Chen and S. Li, J. Chromatogr. A, 2013, 1280, 9–15 CrossRef CAS PubMed.
- J. Meng, C. Shi, B. Wei, W. Yu, C. Deng and X. Zhang, J. Chromatogr. A, 2011, 1218, 2841–2847 CrossRef CAS PubMed.
- X. Liu, J. Yin, L. Zhu, G. Zhao and H. Zhang, Talanta, 2011, 85, 2451–2457 CrossRef CAS PubMed.
- S. Bakhshaei, M. A. Kamboh, S. Mohamad, S. Md Zain and A. Ma'amor, RSC Adv., 2016, 6, 49358–49369 RSC.
- W. Gao, X. Sun, T. Chen, Y. Lin, Y. Chen, F. Lu and Z. Chen, J. Sep. Sci., 2012, 35, 1967–1976 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00523g |
| This journal is © The Royal Society of Chemistry 2017 |