
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polypeptide-participating complex nanoparticles with improved salt-tolerance as excellent candidates for intelligent insulin delivery†
Yuqiang Lia,
Yunyan Zhanga,
Junjiao Yangb and
Jing Yang *a
*a
aState Key Laboratory of Chemical Resource Engineering, Beijing Key Laboratory of Bioprocess, College of Life Science and Technology, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: yangj@mail.buct.edu.cn
bCollege of Science, Beijing University of Chemical Technology, Beijing 100029, China
First published on 2nd March 2017
Abstract
A facile strategy toward improving the salt-tolerance of a glucose-responsive delivery system to realize self-regulated release of insulin in a physiological environment (0.15 M phosphate buffer solution, pH 7.4 and 37 °C) is reported in this study. A type of glucose-responsive complex nanoparticle (CNP) is fabricated by co-assembly of two types of amphiphilic copolymers, poly(ethylene glycol)-block-poly(2-phenylboronic ester-1,3-dioxane-5-ethyl)methylacrylate (MPEG-b-PPBDEMA) and poly(ethylene glycol)-block-poly(γ-benzyl-L-gluamate) (MPEG-b-PBLG). With the help of circular dichroism (CD) and FTIR, it is found that the highly ordered structures (α-helix and β-sheet) affected the stability of CNPs in 0.15 M salt concentration. The CNPs with 75% weight fraction of MPEG-b-PBLG exhibit self-regulated release of insulin in response to changes in the glucose concentration under physiological conditions, enabling the repeated on–off release of insulin regulated by normoglycemic and hyperglycemic levels. The probable mechanism of the controllable responsiveness is reasonably deduced based on an investigation of the change of the secondary structure of CNPs during the process of insulin release.
Introduction
Diabetes mellitus has become one of the most serious threats to human health worldwide.1,2 Although daily injection of insulin is by far the most effective way to treat insulin-dependent diabetes mellitus, it brings long-suffering pain and inconvenience to diabetic patients. Recently, great efforts have been devoted to exploring glucose-responsive materials for controlled closed-loop insulin delivery systems like mimicking pancreatic function, which can automatically “secrete” insulin in response to the fluctuation of blood glucose concentration.3–5 Amongst these self-regulated insulin delivery systems, synthetic phenylboronic acid (PBA)-based polymeric materials with high stability have shown more promising applications than glucose oxidase (GOD) and lectin (Con A) based systems.6–8 Until now, a variety of PBA-based glucose-responsive materials particularly in the form of nanoparticles including nanogels (microgels),9–11 micelles,12–15 vesicles,16–20 and mesoporous silica nanoparticles21,22 have been broadly explored and shown to exhibit effective glucose-responsiveness at physiological pH and reasonable glucose-sensitivity.Besides the controlled glucose-responsiveness at physiological pH which is the main concern in the current research, another important issue in designing drug carriers for biomedical applications is their stability in physiological environment, particularly salt-tolerance of the drug carriers prior to their glucose-responsiveness. Although a series of PBA-based nanoparticles exhibited controlled glucose-responsive release of insulin under physiological pH,23–40 these nanocarriers with relatively low salt stability would undergo the direct disassembly at physiological salt concentration [0.15 M phosphate buffer solution (PBS)] and release the encapsulated cargo in the absence of sugar molecules.37–42 Such requirement of low salt concentration would significantly prevent further investigation of these nanocarriers as potential candidates for smart release of insulin under physiological salt environment.
Synthetic polypeptides have drawn increasing attention as building blocks for construction of nanomaterials,43 and been widely studied for various biomedical applications including drug delivery, biosensors and diagnostics,44–49 due to their excellent biocompatibility and biodegradability. For example, copolymers containing a polypeptide backbone and PBA as pendant groups have been prepared to study glucose-dependent insulin release behavior at physiological pH. Chen and his coworkers reported the preparation of a glucose-responsive polymeric micelles using monomethoxy poly(ethylene glycol)-b-poly(L-glutamic acid-co-N-3-L-glutamyl amidophenyl boronic acid).50 Shi's group developed a glucose-responsive core–shell–corona micelles through co-assembly of two amphiphilic copolymers containing poly(aspartic acid-co-aspartamido phenylboronic acid) block, and reported the insulin release behavior at 0.05 M PBS with varying glucose concentrations.51 Lately, Shi and his coworkers further studied the glucose-sensitivity of nanoparticles formed from poly(N-isopropylacrylamide)-block-poly(aspartic acid-co-aspart amido phenylboronic acid)/poly(aspartic acid-co-aspartamido glucose)-block-poly(ethylene glycol).52 However, in those cases, it is mainly focused on the impact of the modifiable carboxyl side group of the synthetic polypeptides on the glucose-responsiveness and biocompatibility of the overall materials.
Compared to traditional amphiphilic copolymers, the synthetic polypeptides usually have more complex and specific molecular interactions in addition to nonspecific hydrophobic interaction.53,54 In particular, the molecular hydrogen-bonding interactions would construct the unique higher-ordered structures of polypeptides, which significantly enhanced the stability of polypeptide under the physiological conditions. In fact, synthetic polypeptides have been utilized as tools to mimic hierarchical ordered structures of proteins, providing beneficial information for rational design of materials with protein-mimicking properties.55 Understanding the importance of rigidly ordered structures of synthetic polypeptides in physiological environment would intrigue us to design the biomaterials for better satisfying actual need. To the best of our knowledge, few were reported on the secondary structures of synthetic polypeptides impacting on the salt-tolerance of PBA-based nanocarriers under the physiological conditions.
The co-assembly of amphiphilic copolymers is one highly efficient and convenient method to obtain the nanoparticles with the special properties from the individual copolymer. The self-assembled complex nanoparticles (CNP) with hierarchical structures have boosted further achievements in understanding natural self-assembly as well as biomedical applications over the past decade.56,57 In this study, we design a kind of CNP by co-assembly of two amphiphilic copolymers, poly(ethylene glycol)-block-poly(γ-benzyl-L-gluamate) (MPEG-b-PBLG) and poly(ethylene glycol)-block-poly(2-phenylboronate ester-1,3-dioxane-5-ethyl)methylacrylate (MPEG-b-PPBDEMA) (Scheme 1). The hierarchical PBLG blocks and hydrophobic PPBDEMA blocks from these two copolymers would associate as the core with inherent secondary structure of polypeptide and glucose-responsiveness under physiological pH. On the basis of hydrophobic interactions of PBLG and PPBDEMA blocks, the hydrophobic core was sturdily strengthened by higher-ordered structure of polypeptide blocks due to molecular hydrogen bonding, which significantly enhanced salt-tolerance of these CNPs. By adjusting the weight ratio between PBLG and PPBDEMA (WPBLG/WPPBDEMA) in the core, such CNP exhibited exciting stability under normal physiological conditions (0.15 M PBS and pH 7.4), and had great disparity in glucose-responsive behavior between normoglycemic and hyperglycemic condition. This strategy of introducing synthetic polypeptides with hierarchical ordered structures into glucose-responsive materials may make exploring insulin intelligent administration better match actual requirement.
 | ||
| Scheme 1 Synthesis routes of the amphiphilic copolymers. | ||
Experimental section
Materials
α-Methoxy-ω-amino poly(ethylene glycol) (MPEG-NH2) (Mn = 5000, degree of polymerization (DP) = 114 and polydispersity index (PDI) = 1.05) was purchased from Fluka. Monomers (2-phenylboronic esters-1,3-dioxane-5-methyl)methylacrylate (PBDEMA),38 γ-benzyl-L-gluamate N-carboxyanhydride (BLG-NCA)58 and macroinitiator monomethoxy poly(ethylene glycol)-Br (MPEG-Br, Mn = 5100)37 were synthesized according to the reported procedure. Insulin (27 UI mg−1) purchased from Genview was labeled by fluorescein isothiocyanate (FITC) based on the previous report.59 Anisole was dried with sodium using benzophenone as an indicator. Anhydrous N,N-dimethylformamide (DMF) purchased from Aldrich was used without any treatment. All other reagents with analytical grade were used without further purification unless otherwise stated. NIH 3T3 mouse fibroblast cells were provided by Beijing Tumor Resource Institute. DMEM (Dulbecco's Modified Eagle Medium), fetal bovine serum, penicillin and streptomycin purchased from Aldrich were used without treatment.Characterization
The chemical structures of the monomeres and copolymers were characterized by 1H NMR carrying out on a 400 MHz NMR instrument (Bruker Corporation, Germany) at room temperature using CDCl3 as solvent. The chemical shifts were measured against the solvent signal of CDCl3 as internal standard. GPC measurements of the block polymers were carried out by Waters 515-2410 instrument equipped with a differential refractive-index detector. The measurements were performed using THF as eluent (flow rate of 1.0 mL min−1 at 30 °C), and polystyrene standards were employed for calibration. A Hitachi F-4600 Fluorescence instrument (Hitachi High-Technologies Corporation, Tokyo Japan) was used to measure the steady-state fluorescence emission. Laser scattering measurements were performed at 660 nm by using a laser light scattering spectrometer (ZEN3600, Malvern) with Zetasizer software. TEM was performed on a Hitachi H800 electron microscopy (TEM) operated with 100 kV. FTIR measurements were performed on Nicolet 10 with EverGlo light and KBr plate, and the scanning wavelength was ranged between 4000 and 400 cm−1. Circular dichroism (CD) measurements were performed on a Jasco J-810 CD spectropolarimeter at 25 °C with a cell length of 0.1 cm. The scanning speed was 50 nm min−1, and wavelength range was between 260 and 190 nm.Synthesis of MPEG-b-PPBDEMA (P-1)
The preparation procedure was similar to our previously reported method.37 Briefly, a Schlenk flask charged with CuBr (5.45 mg, 0.038 mmol), MPEG-Br (196.7 mg, 0.038 mmol) and CuBr2 (0.43 mg, 0.002 mmol) was degassed using three vacuum–nitrogen cycles, followed by adding PMDETA (7.8 mg, 0.045 mmol), monomer PBDEMA (2.1 g, 7.6 mmol) and anisole (1.0 mL) by syringes under nitrogen atmosphere. The reaction system further degassed using three freeze–pump–thaw cycles was immersed in an oil bath at 90 °C under thermostat control for 40 h. The cooled reaction solution was diluted with CHCl3 and passed through a neutral alumina column to remove the catalyst. The concentrated reaction solution was dropwise added into the mixed solvent of hexane/diethyl ether (v/v = 4/1), and 1.1 g product was afforded in yield of 48.3%. 1H NMR (400 MHz, CDCl3) δ (ppm): for PPBDEMA block: 7.75 (d, J = 8 Hz, 2H, o-C6H5), 7.37 (m, 1H, p-C6H5), 7.29 (m, 2H, m-C6H5), 3.76–3.99 (m, 6H, CH2OOC, CH2OBOCH2 and CH2OBOCH2), 2.29 (br, 1H, CHCH2), 1.60–1.91 (br, 2H, CHCH2), 1.35 (m, 2H, CH2CH3), 0.79 (m, 3H, CH2CH3); for MPEG block: 3.64 (s, 4H, CH2CH2O), 3.38 (s, 3H, CH3O).Synthesis of MPEG-b-PBLG (P-2)
MPEG-NH2 (0.5 g, 0.1 mmol), BLG-NCA (0.79 g, 3.0 mmol) and 2.0 mL anhydrous DMF were added into 25 mL Schlenk flask in the glovebox. The reaction mixture was stirred at 40 °C for 3 d, and the resulting solution was precipitated into 10-fold excess of cold diethyl ether. The crude solid was washed twice with diethyl ether to obtain 1.08 g of white solid in yield of 83.5%. 1H NMR (400 MHz, CDCl3), δ (ppm): 7.89 (1H, –NH–), 7.41–7.16 (5H, C6H5–), 5.13–5.09 (2H, –CH2–C6H5), 4.62 (1H, –CO–CH–NH–), 3.95–3.68 (–CH2CH2O–), 2.46 (2H, BnO–CO–CH2–), 2.13–1.93 (2H, –CH2–CH–).Preparation and characterization of CNP
10 mg mixture of two block polymers with six different P-2 weight fraction including 0, 10%, 30%, 50%, 75%, 100% were dissolved in 1.0 mL THF, respectively. CNPs were carried out by slowly adding the mixed polymer solutions into 10 mL ultrapure water, followed by the evaporation of THF with stirring. The nanoparticles solution was dialyzed against ultrapure water in a dialysis bag with a molecular weight cutoff of 3500 for 48 h, during which dialysis water was refreshed every 6 h. The concentrations of the resulting CNP were all fixed at 1.0 mg mL−1.The secondary structure of CNP aqueous solution was determined by CD measurement. The CNP solutions with different P-2 content at 0.2 mg mL−1 were scanned from 190 to 260 nm, and accumulated 5 times at a resolution of 1.0 nm and scanning speed of 700 nm min−1. The secondary structure population was calculated using software CD Pro. After CNP solution was lyophilized, the freshly-ground mixture of 2.0 mg CNPs and 110 mg anhydrous KBr was used for FTIR measurements.
FITC-insulin loading and entrapment capacity evaluation of CNPs
Firstly, a stock of FITC-insulin (3.0 mg) dissolved in 10 mL HCl (0.01 M) was adjusted to pH 6.0 using NaOH (0.1 M). Subsequently, 10 mg mL−1 mixture of two block polymers with the given weight ratio of P-2 in THF was dropwise added into the above mentioned FITC-insulin aqueous solution with stirring in ice bath. After THF evaporation, the purification of CNPs encapsulating FITC-insulin was performed by dialysis method (MWCO, 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Da). The encapsulated insulin mass was calibrated according to the measured standard curve of fluorescence intensity against insulin concentration. The insulin entrapment efficiency (EE) and loading capacity (LC) were calculated using the following equations:
000 Da). The encapsulated insulin mass was calibrated according to the measured standard curve of fluorescence intensity against insulin concentration. The insulin entrapment efficiency (EE) and loading capacity (LC) were calculated using the following equations: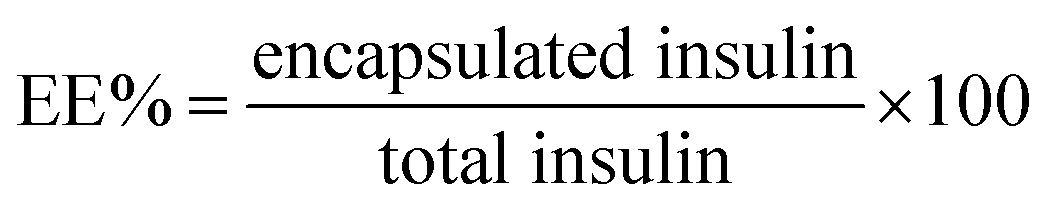
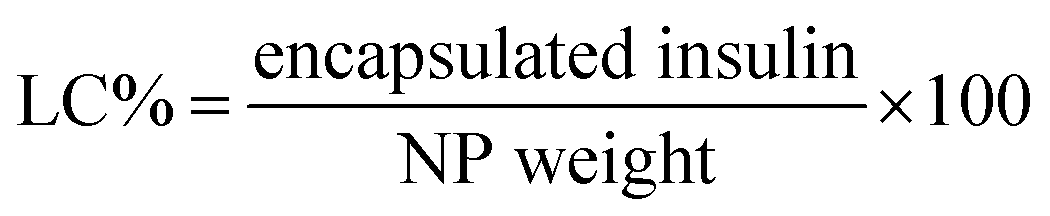
Salt-tolerance of CNPs
The release of FITC-insulin from CNPs under the stimuli of different salt concentrations (0.01, 0.03, 0.05, 0.10 and 0.15 M PBS) was used to evaluate their salt stability. The release of insulin was carried out by dialysis. The dialysis bag with a molecular weight cutoff 14 kD filled with 1.0 mg mL−1 of purified insulin-encapsulated CNPs were immersed in 40 mL of the given PBS concentrations (pH = 7.4) at 37 °C. The cumulative released FTIC-insulin outside of the dialysis bag was assayed by fluorescence spectrometry at an emission wavelength of 525 nm upon excitation at 494 nm.Glucose-responsive release
Based on the results of salt stability, CNPs with 50% and 75% weight fractions of P-2 were selected to investigate insulin release behavior with varying glucose concentrations of 0, 1.0, 2.0 and 3.0 mg mL−1 at 0.15 M PBS, pH 7.4 and 37 °C. Other operations were similar to the salt stability.The insulin release by pulsed triggering of glucose concentration was performed by immersing drug-loaded CNPs alternatively in 1.0 and 5.0 mg mL−1 glucose for 9 h under physiological condition (0.15 M PBS, pH 7.4 and 37 °C), and the release percentage of insulin was monitored by fluorescence spectrometry every 3 h.
In vitro cytotoxicity of the nanoparticles
The in vitro cytotoxicity was evaluated by MTS assay (Cell Titer 96 Aqueous One Solution Cell Proliferation assay). NIH3T3 mouse fibroblast cells were cultured in monolayer in DMEM (Dulbecco's Modified Eagle Medium) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin in 5% CO2/95% air at 37 °C. The cells were seeded into a 96-well plate at a density of 1.0 × 104 cells per well. The prepared CNP solutions were diluted to give a range of final concentration from 25 to 500 mg L−1 using DMEM solutions. The plates were maintained in the incubator for 24, 48 and 72 h, respectively. After incubation, 20 μL of MTS solution was added to each well. After the predetermined incubation time, 20 μL of MTS was added into each well and incubated for a further 2 h. The optical density of the solution was measured at 490 nm using a microplate reader (Labsystem, Multiskan, Ascent, Finland) in triplicate. The relative cell proliferation was calculated as the percentage of cell viability as compared with that of an untreated control.Results and discussion
Synthesis and characterization of two amphiphilic block copolymers
The amphiphilic copolymers MPEG-b-PPBDEMA (P-1) and MPEG-b-PBLG (P-2) were respectively synthesized by atom transfer radical polymerization (ATRP)37 and ring-opening polymerization (ROP) as reported52 (Scheme 1). The chemical structures of the block copolymers P-1 and P-2 were confirmed by 1H NMR and GPC measurements. From 1H NMR results shown in Fig. S1 and S2,† the degree of polymerization (DP) of PPBDEMA and PBLG blocks in these copolymers, comparing the peak intensities between the phenyl protons of PPBDEMA (δ = 7.78 ppm) or methylene of PBLG block (δ = 5.07 ppm) and the methyl protons of MPEG block (δ = 3.43 ppm), were calculated to be 75 and 20, respectively (Table 1). GPC measurements exhibited unimodal and relatively narrow distribution without macroinitiator MPEG-Br or MPEG-NH2 (Fig. S3 and S4†), further indicating the successful preparation of these copolymers. Using fluorescence technique with pyrene as probe, critical micellar concentrations (CMC) of P-1 and P-2 were measured to be 9.8 and 2.4 mg L−1 (Fig. S5†). The low and close CMCs suggested that these two copolymers had similar and strong hydrophobic aggregation force, which provided great opportunity to form CNPs instead of two single nanoparticles self-assembled by individual copolymer in aqueous solution.| Sample No. | Amphiphilic polymera | Mnb (1H NMR) | GPCc | CMCd (mg L−1) | |
|---|---|---|---|---|---|
| Mn | PDI | ||||
| a The numbers at the footnote showing the average repeating unit number of each moiety were determined by 1H NMR.b The number-average molecular weights of the block copolymers were calculated from 1H NMR results.c Determined by GPC measurements.d Measured by fluorescence technique using pyrene as probe. | |||||
| P-1 | MPEG110-b-PPBDEMA75 | 25500 |
28200 |
1.29 | 9.8 |
| P-2 | MPEG110-b-PBGL20 | 9380 | 5800 | 1.16 | 2.4 |
Fabrication of complex nanoparticles (CNP)
To obtain glucose-responsive CNPs, four different CNPs with varying weight ratios of P-2 were fabricated through simple co-assembly of P-1 and P-2, in which the contents of P-2 in CNPs were respectively 10%, 30%, 50% and 75%. TEM technology was employed to study the nanostructures of the CNPs as related to the P-2 composition ratio. TEM image of the CNPs with 30% content of P-2 showed closely spherical micelles with the average diameter of 40 nm (Fig. S6A†). With increasing P-2 weight fraction in CNPs to 50% (Fig. S6B†) and 75% (Fig. 1A), the polymeric vesicles with outer diameter of 30 nm and wall thickness of 5 nm were interestingly observed. DLS measurement was performed to investigate the size and distribution of the nanoparticles via co-assembly of the two copolymers in aqueous solution. As shown in Fig. 1B, the sizes of CNPs with P-2 content ranged from 10–75% were below 50 nm at pH 7.4 and 25 °C. Compared to the single nanoparticles formed from P-1, the average diameters of CNPs were slightly decreased with an increase of P-2 weight ratio in the whole nanoparticles, which is possibly associated with the shorter chain length of hydrophobic PBLG block. The relatively narrow PDI of CNPs suggested the successful formation of complex nanoparticles by co-assembly.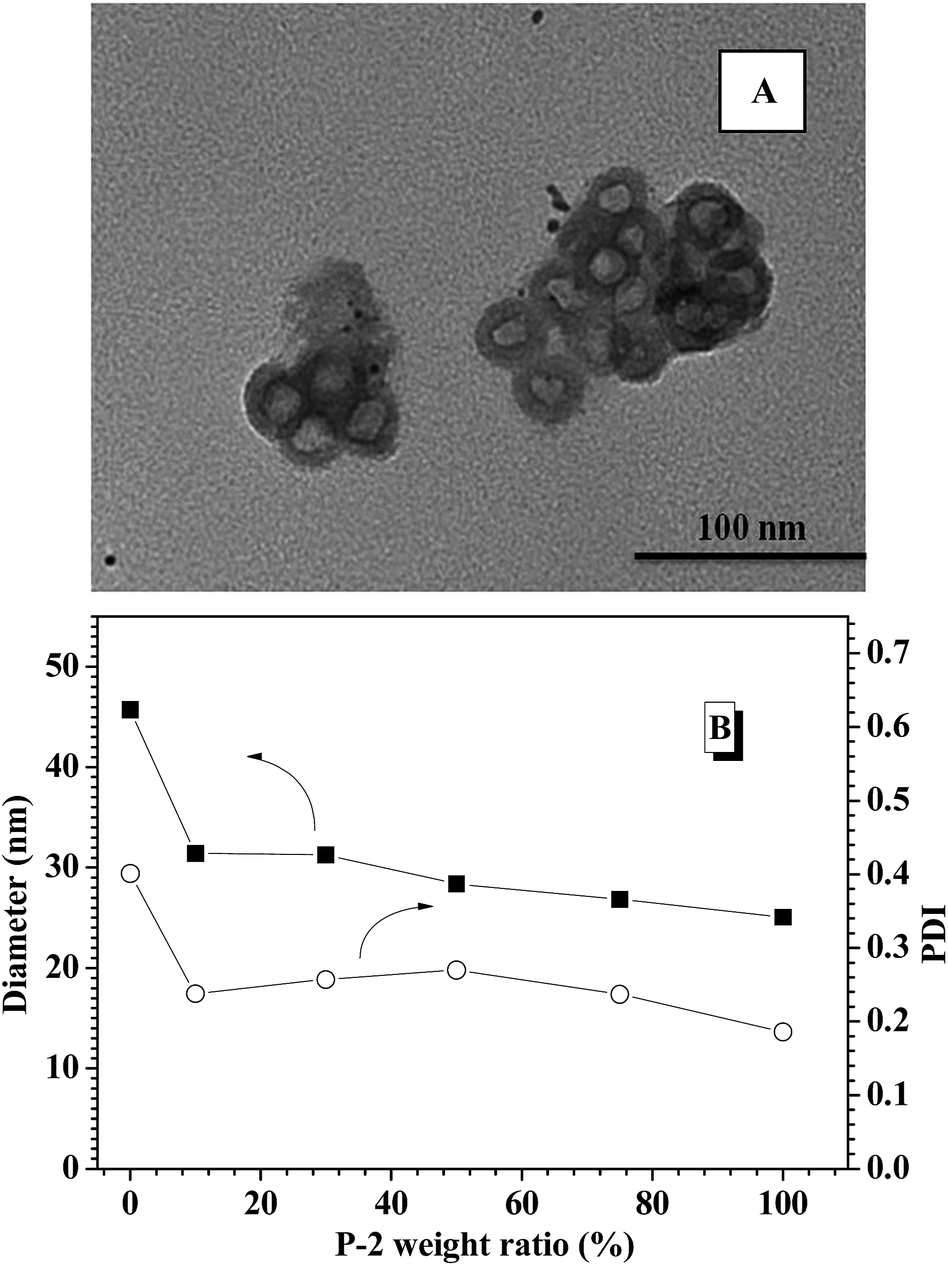 | ||
| Fig. 1 (A) TEM images of CNPs with 75% content of P-2; (B) DLS results of polymeric nanoparticles with different P-2 weight ratio. | ||
Secondary structure analysis
CD spectroscopy is one of the most convenient ways to estimate the secondary structures of proteins. Therefore, the secondary structures of CNPs with varying P-2 weight ratio at 0.2 mg mL−1 in water were first examined with CD spectroscopy. It is well known that α-helical structure can be characterized by two negative bands at 208 and 222 nm and a strong positive band about 192 nm, and the β-sheet structure was characterized by a negative minimum band near 218 nm and a positive maximum between 185 and 200 nm.48 For the CNPs by co-assembly of P-1 and P-2, Fig. 2A shows the gradual transformation of the α-helical conformation was evidenced by the slight change of the ellipticity ratio ([θ]222/[θ]208), upon increasing P-2 content from 10% to 100%. The quantitative analyses of secondary structures were further conducted by employing Spectra software CD Pro. For each CNP, the secondary structure was composed of α-helix, β-sheet and other secondary structures (Fig. 2B), regardless of the difference in the chemical compositions. As the weight fraction of P-2 in CNPs increased from 10% to 75%, the β-sheet content decreased systematically from 41.8% to 15.6% of total components, and the α-helical chains raised gradually from 4.2% to 36.9%. The α-helix contribution to secondary structure became dominant over the β-sheet contribution with P-2 weight fraction greater than 60% in CNPs. Other secondary structure percentage was slightly reduced for all formulations.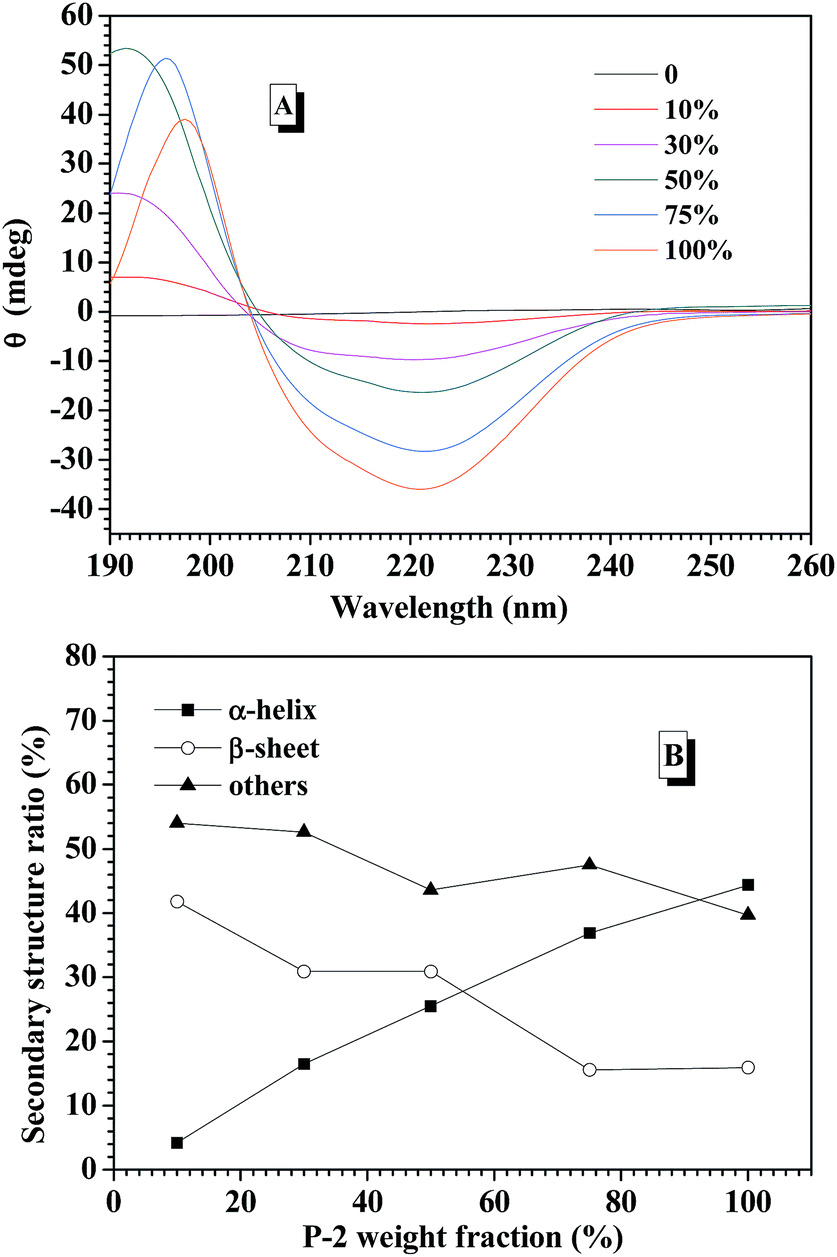 | ||
| Fig. 2 (A) Circular dichroism spectra of CNPs varying P-2 content in aqueous solution at the concentration of 0.2 mg mL−1. (B) The calculated secondary structure populations of CNPs versus the weight fraction of P-2 in deionized water. | ||
Freeze-dried complex nanoparticles were further studied by FTIR spectroscopy, and the characteristic absorbance of the secondary structures was clearly observed in Fig. S7.† The absorbance at 1653 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration, amide I) and at 1547 cm−1 (N–H bending vibration, amide II)60 were attributed to α-helical conformations. Meanwhile, the absorption bands of amide I appeared at 1626 cm−1, indicating the presence of the β-sheet conformation.61 The peak at 1733 cm−1 corresponded to the CO stretch of the benzyl ester group in the side chain of BLG.62 Relative to the benzyl ester group at 1733 cm−1, the absorption intensities assigned to the peaks of α-helical conformation increased with higher weight fraction of P-2 in the CNPs, which clearly indicates that the secondary structure of the complex nanoparticles went through a change toward more compact α-helix conformation.
O stretching vibration, amide I) and at 1547 cm−1 (N–H bending vibration, amide II)60 were attributed to α-helical conformations. Meanwhile, the absorption bands of amide I appeared at 1626 cm−1, indicating the presence of the β-sheet conformation.61 The peak at 1733 cm−1 corresponded to the CO stretch of the benzyl ester group in the side chain of BLG.62 Relative to the benzyl ester group at 1733 cm−1, the absorption intensities assigned to the peaks of α-helical conformation increased with higher weight fraction of P-2 in the CNPs, which clearly indicates that the secondary structure of the complex nanoparticles went through a change toward more compact α-helix conformation.
Effect of P-2 content on salt-tolerance of CNPs
Prior to intelligent releasing its cargos under special stimuli, the stability of drug delivery systems in normal physiological environment is one important factor to measure their safety for the medical application. Amongst others, the salt tolerance of nanocarriers at physiological concentration (0.15 M PBS) is a major concern. To evaluate the salt-tolerance of the CNPs, in this study, FITC-labeled insulin was encapsulated into the complex nanoparticles by co-assembly of P-1 and P-2 as reported,36 and the release behavior of the loaded insulin from the CNPs was tested upon varying salt concentration. As shown in Fig. 3A, although they exhibited nearly zero release of FITC-insulin at the salt concentration of 0.01 M and 37 °C for 75 h incubation, one distinct cumulative release of insulin was investigated when the salt concentration was slightly increased to 0.03 M. Moreover, the release efficiency of insulin was accelerated with increasing salt concentration. Above 0.10 M salt concentration, the cumulative release of insulin rapidly reached 80% during the first 40 h. These results demonstrated that the nanocarriers from P-1 had low salt-stability and salt-dependent responsive release behavior. However, when the nanocarriers were formed via the co-assembly of P-1 and P-2, their salt stability was significantly improved upon an increase of P-2 weight fraction in CNPs. For example, the CNPs with only 10 percentage of P-2 content exhibited better salt-tolerance than that from P-1 alone. The quite low cumulative release of insulin was monitored at the salt concentration of 0.03 M, and the release efficiency was obviously slowed down at 0.05 M (Fig. 3B). When P-2 weight fraction in CNPs was increased to 30%, the low release of insulin from the CNP was detected at salt concentration of 0.05 M, and the insulin release was clearly depressed even at 0.15 M (Fig. 3C). With further increasing P-2 weight ratio to 50% in CNPs, it was excitingly found that the CNPs showed perfect stability in the measured range of salt concentration except the slight release behavior at 0.15 M during 75 h incubation (Fig. 3D). As P-2 content was enhanced to 75%, the CNPs exhibited high salt stability at 0.15 M (Fig. 3E). The cumulative release of insulin from the CNPs with different P-2 weight fraction at the salt concentration of 0.15 M was further summarized in Fig. 3F. It is clearly seen that with P-2 content increasing systematically from 0 to 75%, the insulin release trend of the complex nanoparticles was gradually decreased, and the cumulative release amount of insulin was finally reduced to less 10%. These results indicate that the introduction of P-2 into the self-assembly system of P-1 can efficiently enhance the stability of the overall nanocarriers in the physiological salt environment, and the salt-tolerance of the resulting CNPs would be adjusted by a change of P-2 weight fraction.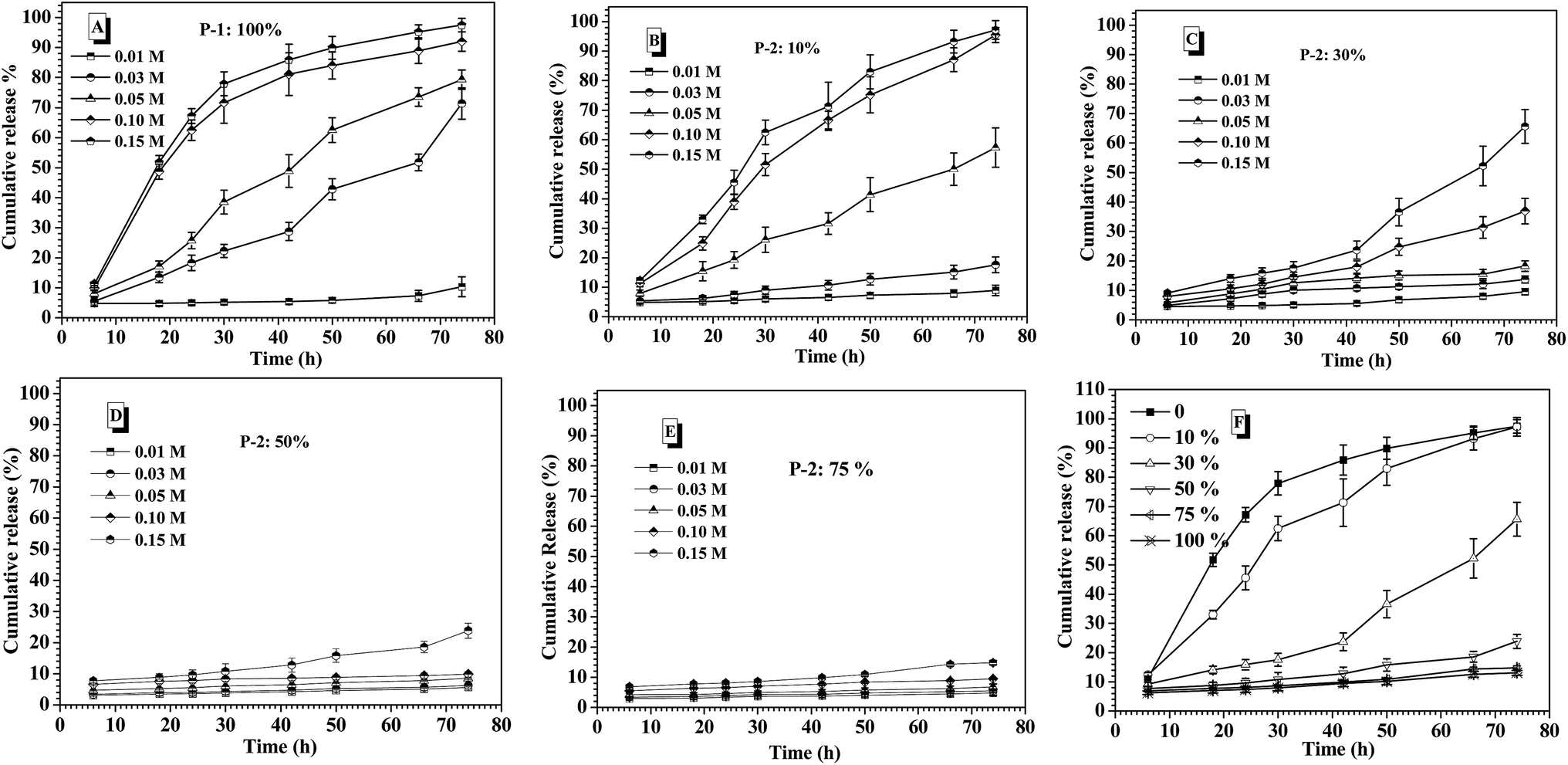 | ||
| Fig. 3 Salt concentration-dependent release behavior of insulin from the nanoparticles of single P-1 (A); the CNPs with 10% P-2 content (B); with 30% P-2 content (C); with 50% P-2 content (D); with 75% P-2 content (E) at pH 7.4 and 37 °C. (F) The cumulative release of insulin from CNPs with varying P-2 weight fraction at 0.15 M salt concentration and 37 °C. | ||
To ascertain the significance of P-2 enhancing the salt stability of CNPs, as control, polyethylene glycol-block-poly(lactic acid) [MPEG5000-b-PLA, Mn (1H NMR) = 25600, PDI measured by GPC is 1.35] with biodegradable polyester as hydrophobic block, and polyethylene glycol-block-polystyrene [MPEG5000-b-PS, Mn (1H NMR) = 9000, PDI measured by GPC is 1.31] with strong hydrophobic block were respectively prepared, as displayed in Scheme S1.† The new complex nanoparticles were formed via the co-assembly of P-1 and MPEG-b-PLA or MPEG-b-PS with different weight ratio, respectively. The effect of the different chemical structure and component ratios on the stability of the complex nanoparticles was comparatively investigated at 0.15 M salt concentration. As shown in Fig. S9A,† as an increase of MPEG-b-PLA weight fraction in CNPs, the release of insulin was slowed down in some degree. However, the CNPs with 75% weight fraction of MPEG-b-PLA still exhibited 50% cumulative release amount of insulin for 75 h incubation. The CNPs formed via the co-assembly of P-1 and MPEG-b-PS displayed high cumulative release of insulin at 0.15 M salt concentration, and even CNPs with 75% weight fraction of MPEG-b-PS showed rapid and high release behavior of insulin in the first 30 h (Fig. S9B†). Moreover, there was no great disparity in the enhancement of salt stability among different MPEG-b-PS contents. Although PLA and PS blocks had strong hydrophobic aggregation, these CNPs maintained by only hydrophobic interaction didn't exhibit satisfying the stability at 0.15 M PBS.
To explore the contribution of P-2 to stabilize the CNPs in the physiological salt environment, the secondary structure change of the CNPs with different P-2 content at 0.15 M salt concentration was carefully investigated by CD measurement. After the CNPs containing the given P-2 content were incubated in 0.15 M salt concentration for 10 min, their CD curves were subsequently collected (Fig. S10A†). The calculated secondary structure populations of CNPs with different P-2 contents were displayed in Fig. 4A. The change trend of secondary structure in 0.15 M salt concentration was similar to that in deionized water (Fig. 2B). With an increase of P-2 weight fraction in CNPs from 10% to 75%, the content of β-sheet conformation decreased from 40.1% to 14.7%, corresponding to an obvious increase in α-helical chains from 4% to 34.2%. The content of other secondary structure (β-turn and random coil) was gradually decreased with P-2 content increasing, indicating the enhancement of the ordered structure in the overall conformation. The more highly ordered structure of CNPs benefits to their stability at physiological salt concentration.
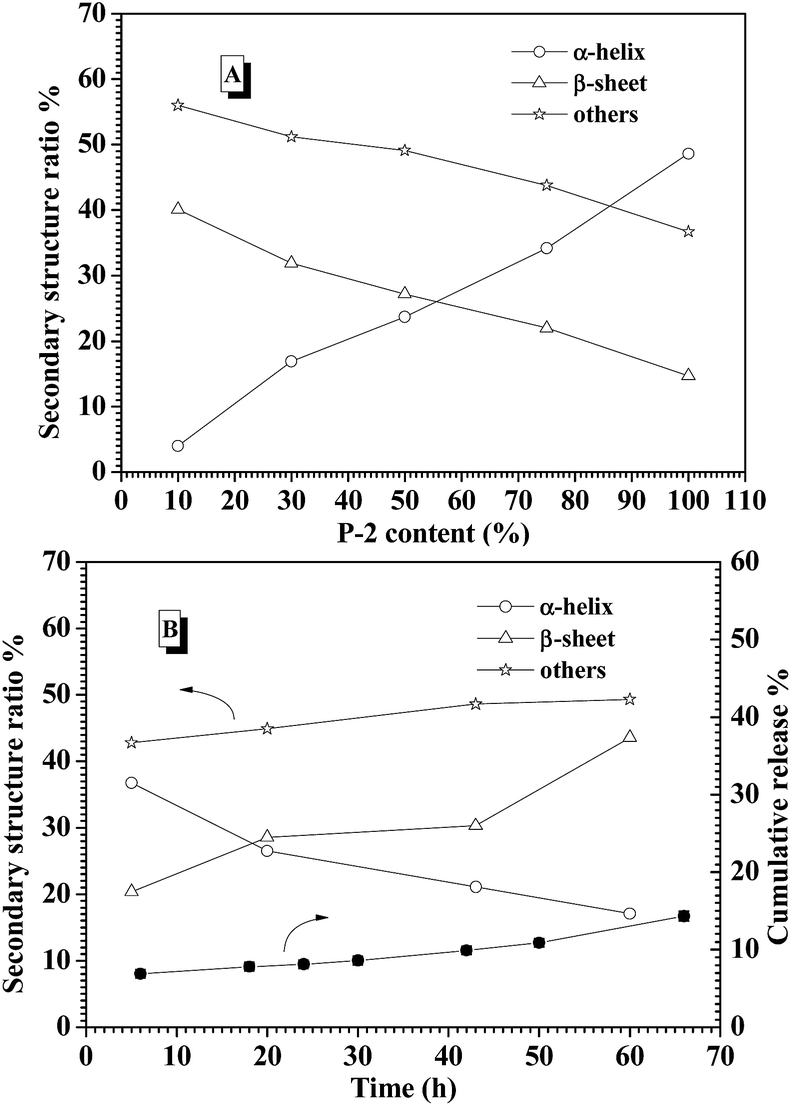 | ||
| Fig. 4 (A) The calculated secondary structure populations of CNP versus the weight fraction of P-2 at 0.15 M salt concentration. (B) The secondary structure populations change of the CNPs with 75% content of P-2 at 0.15 M salt concentration and 37 °C for 60 h. | ||
The secondary structure of CNPs with 75% content of P-2 was further monitored by CD measurements at 0.15 M salt concentration and 37 °C for 60 h incubation. As shown in Fig. 4B, the decrease of α-helix content was accompanied by an increase of β-sheet structure. The sum of α-helix and β-sheet was kept above 50% of the whole secondary structures during 60 h incubation. Meanwhile, it is monitored that the cumulative release of insulin from the CNPs with 75% of P-2 content was well controlled to be about 10% in the measured time. This low release of insulin is very likely attributed to the constant content of the highly ordered structure in CNPs.
Insulin-loading of the CNPs
From the above results, the CNPs with 75% of P-2 content were selected to investigate the entrapment efficiency (EE) and loading capacity (LC) of FITC-insulin in the CNP's depending on the concentrations of the nanoparticle and FITC-insulin, as shown in Fig. 5. For example, when FITC-insulin concentration was controlled at 0.1 mg mL−1, EE of insulin was dramatically enhanced from 30% to 70% with a change in CNPs' concentration from 0.5 to 1.0 mg mL−1 (Fig. 5A). However, LC leveled off at the CNPs' concentration of 1.0 mg mL−1 and slightly decreased when CNPs' concentration was increased to 1.5 mg mL−1 (Fig. 5B). Although the high nanoparticles' concentration can strengthen the interaction with the loaded molecules, leading to high EE of insulin, the too high nanocarriers' mass would result in LC lowering. When the given insulin concentration was increased from 0.1 to 0.2 mg mL−1, both EE and LC of insulin was simultaneously enhanced, particularly, LC was obviously increased to 12%. However, when the given insulin concentration was further enhanced to 0.3 mg mL−1, EE started to drop off at the CNPs' concentration of 1.0 and 1.5 mg mL−1, and LC was only slightly increased. In contrast to the nanoparticles formed from only P-1, the CNPs exhibited higher EE and LC of insulin in the experimental range. As discussed above, the CNPs' concentration of 1.0 mg mL−1 and the given insulin concentration of 0.2 mg mL−1 was selected as the encapsulation condition of insulin for the following glucose-responsiveness.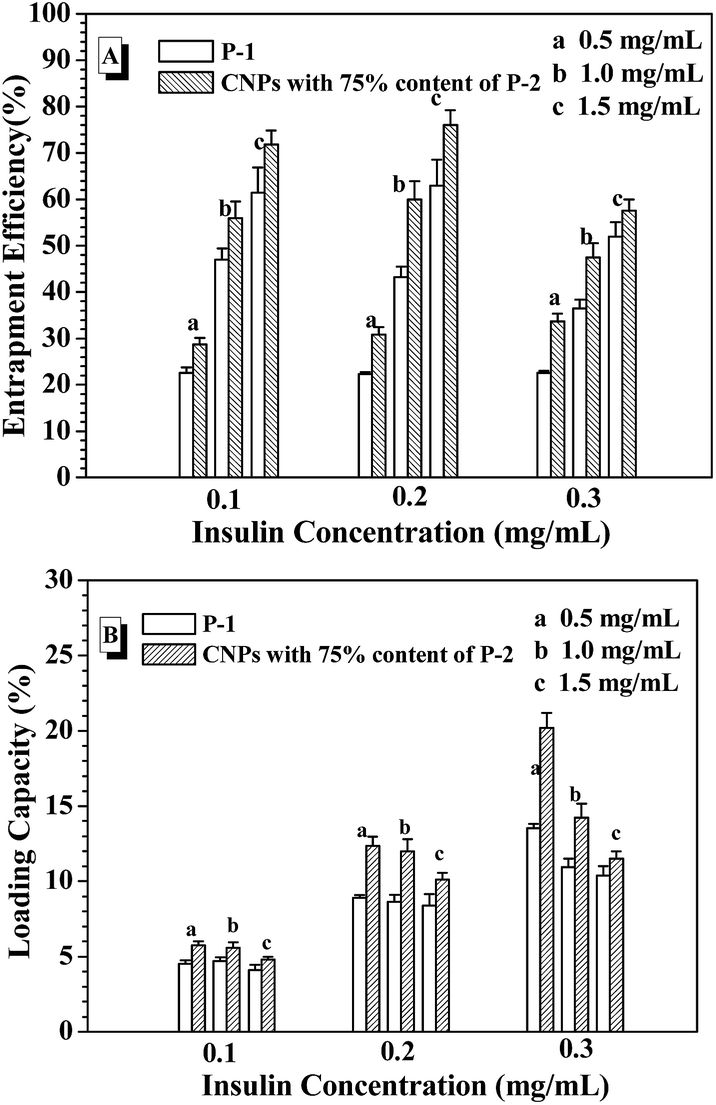 | ||
| Fig. 5 EE (A) and LC (B) with varying the nanoparticle concentration and the given insulin concentration. | ||
Glucose-responsive release of FITC-insulin from the CNPs
In the human body, the normal blood glucose level fluctuates in the range of 0.6 and 1.2 mg mL−1. Over 2.0 mg mL−1 glucose concentration is generally considered as hyperglycemia, which needs to be treated. Besides, hypoglycemia effect induced by excess insulin release from the drug carriers under normoglycemia should be strictly avoided, because hypoglycemia happens rapidly and easily threatens the life. In the consideration of the self-regulated insulin release demand upon the change of blood glucose concentration, the glucose-responsiveness of these CNPs by co-assembly of P-1 and P-2 was evaluated under the simulated physiological environment (pH 7.4, 0.15 M PBS and 37 °C) in this study. The cumulative release of insulin from these nanoparticles was calculated by monitoring the changes in the fluorescence intensity of the external fluid. Fig. 6A shows the insulin release profiles from the CNPs with 50% P-2 weight fraction upon varying glucose concentration at physiological environment. In the absence of glucose, less 20% insulin was released from the CNPs for 90 h measurement, exhibiting good stability of the complex nanoparticles. When the FITC-insulin loaded CNPs was incubated at the glucose concentration of 1.0 mg mL−1 (normoglycemic condition), low release of insulin was observed in the first 60 h, however, the subsequently rapid release led to over 50% of the insulin escaping from the CNPs after 90 h incubation. Although the CNPs with 50% of P-2 content exhibited excellent performance of glucose-responsive behavior at glucose concentration of 2.0 and 3.0 mg mL−1, excessive release of insulin at normal physiological condition makes the CNPs with 50% content of P-2 deviate from the basic demand of insulin delivery systems.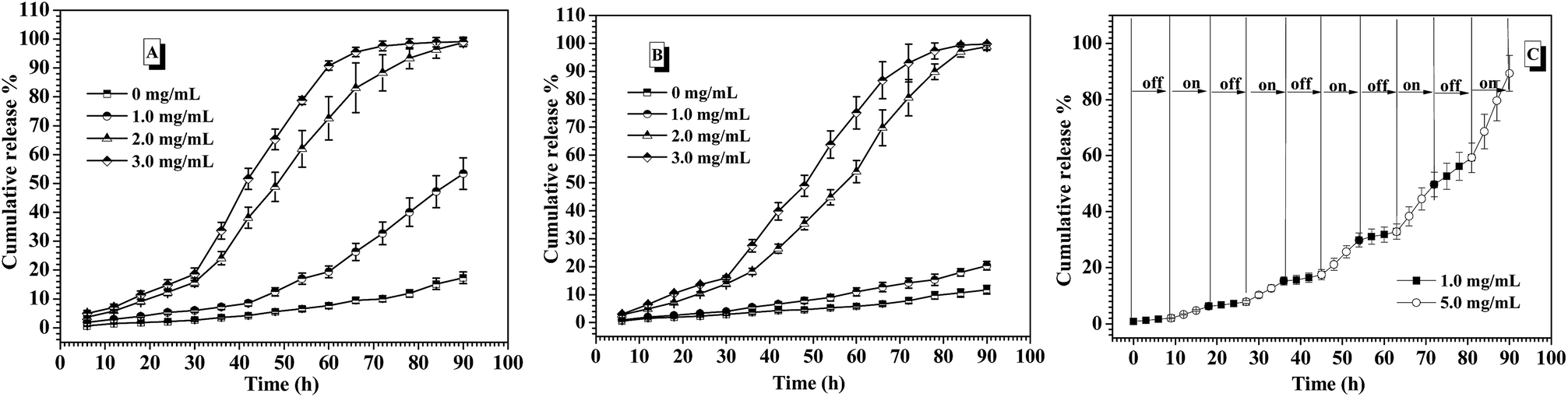 | ||
| Fig. 6 Release profiles of FITC-insulin from the CNPs under varying concentrations of glucose at 0.15 M PBS, pH 7.4 and 37 °C. (A) 50% P-2; (B) 75% P-2. (C) The glucose-responsive insulin release from CNPs with 75% content of P-2 with alternant glucose concentrations of 1.0 and 5.0 mg mL−1 at 0.15 M PBS, pH 7.4 and 37 °C. | ||
Concurrently, the insulin release from CNPs with 75% of P-2 content was investigated under the simulated physiological environment, and the exciting results upon varying glucose concentration were shown in Fig. 6B. Due to the increase of P-2 content in CNPs, the remarkable and sustained insulin release was not only observed at a relatively stable release rate under hyperglycemic condition, more importantly, the release of insulin at glucose concentration of 1.0 mg mL−1 was efficiently controlled. During 90 h measurement, less than 20% release of insulin demonstrated the better stability of the CNPs with 75% of P-2 content under normal physiological condition, decreasing the risk of the hypoglycemia occurrence. Besides, the CNPs with 75% of P-2 content also displayed the faster release of insulin at 3.0 mg mL−1 glucose than 2.0 mg mL−1, indicating that the CNPs had the ability of self-regulated insulin release upon the glucose concentration fluctuation.
Furthermore, the on–off insulin release ability of the CNPs with 75% weight fraction of P-2 upon the physiological demand was evaluated through the consecutive switch of glucose concentration between 1.0 and 5.0 mg mL−1 every 9 h at 0.15 M PBS, pH 7.4 and 37 °C (Fig. 6C). The insulin-loaded CNPs were first placed at physiological solution with 1.0 mg mL−1 glucose, and almost zero release of insulin was observed in the first 9 h. After that, the culture medium was changed to the phosphate buffer with 5.0 mg mL−1 glucose, then, relatively obvious insulin release, ca. 4.8% release amount, was seen for the subsequent 9 h. When the release media was switched back to the physiological solution with 1.0 mg mL−1 glucose, insulin release was immediately paused and release plateau was obtained in the following 9 h. Once the release medium was alternated to 5.0 mg mL−1 glucose, the release behaviour of insulin was triggered again, which was demonstrated by release of 5.5% insulin in the fourth 9 h. Such on–off insulin release was more clearly observed in the next several cycles of alternate glucose concentration between 1.0 and 5.0 mg mL−1, and the cumulative release amount was close to 100% after 90 h measurements. Notably, no burst release of insulin occurred each time when the CNPs were placed back to 5.0 mg mL−1 glucose medium. The multiple and smoothly pulsed release behaviour indicates that the CNPs had potential to realize close-loop and consecutive insulin administration in response to the fluctuation of blood glucose concentration. These CNPs with 75% of P-2 content that can undergo the self-regulated glucose-switchable release under physiological environment (0.15 M PBS, pH 7.4 and 37 °C) show promising application for potential clinical used glucose-responsive insulin delivery system.
In the previous work, it was found that pH value of environmental solution was rapidly dropped from 7.4 to 6.0 or lower with triggering the glucose-responsiveness of the nanocarriers formed from PBA-based polymers series at low salt environment (such as 0.01 M and 0.03 M) or deionized water. In order to maintain the efficient glucose-responsiveness of those nanocarriers, the continuous adjusting pH value back to 7.4 by dropwise adding base into the environment solution was necessary. In this study, when the release of insulin from the CNPs with 75% of P-2 content was performed at the simulated physiological environment (3.0 mg mL−1 glucose, 0.15 M PBS, pH 7.4 and 37 °C), the corresponding pH value change was concurrently monitored (Fig. 7A). It was interestingly found that the pH value of the environment solution was almost kept stable at pH 7.4 for 90 h measurements, which guarantees the successful performance of glucose-responsiveness of the CNPs without the aid of additional base. The buffer ability of such salt concentration endowed the whole glucose-responsive process with stable pH environment. This improved glucose-responsive insulin delivery system would better satisfy the physiological demand.
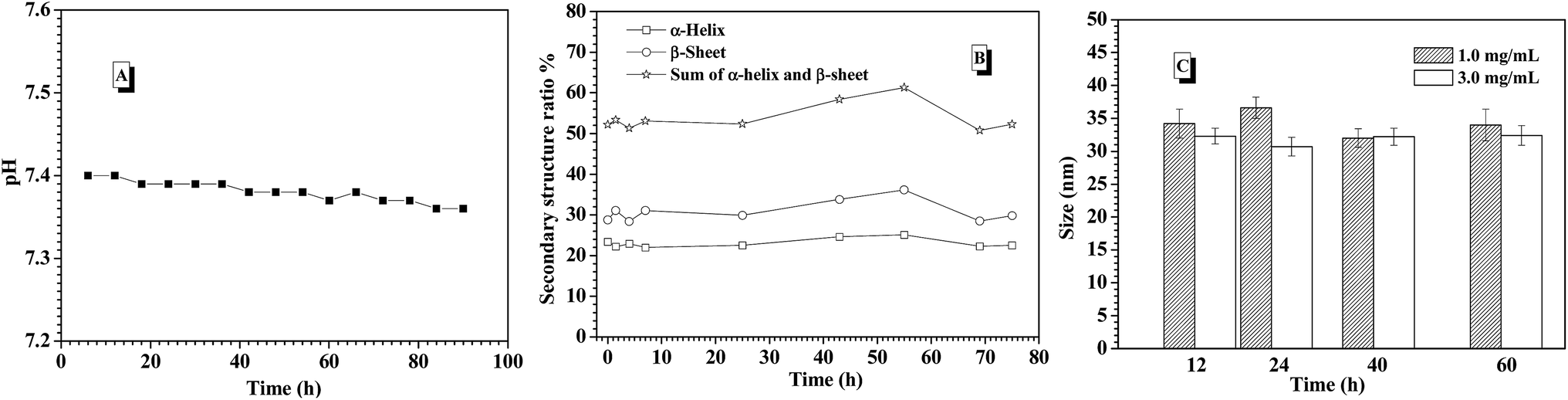 | ||
| Fig. 7 (A) pH change of the environmental solution; (B) the secondary structure populations change of the CNPs with 75% content of P-2; (C) size change of the CNPs with 75% content of P-2 during the process of insulin release under the stimuli of 1.0 and 3.0 mg mL−1 glucose at 0.15 M PBS, pH 7.4 and 37 °C for 90 h incubation. | ||
To further understand the probable reason of the self-regulated glucose-responsiveness of the CNPs, the secondary structure change of the CNPs with 75% weight fraction of P-2 during the intelligent response process at 3.0 mg mL−1 glucose, 0.15 M PBS, pH 7.4 and 37 °C was monitored by CD technology. As shown in Fig. 7B, the contents of α-helix and β-sheet in the whole secondary structure conformation did not have great change for 70 h measurements, and the sum of α-helix and β-sheet content was kept above 50%. Meanwhile, the sizes of CNPs were monitored by DLS under the stimuli of 1.0 and 3.0 mg mL−1 glucose at 0.15 M PBS, pH 7.4 and 37 °C for 90 h incubation. As a result, only slight size fluctuation was observed in the range of 35 and 40 nm (Fig. 7C). This indicates that the dominant framework constructed by P-2 in the CNPs was well maintained, and the chemical structure change of P-1 under the stimuli of glucose didn't bring great impact on the whole skeleton of the CNPs in the glucose response process.
Combined with the above experimental results, the salt-tolerance of CNPs with 75% content of P-2 and their self-regulated glucose-responsiveness were tried to deduce as shown in Fig. 8. Besides the hydrophobic interaction of the amphiphilic copolymers P-1 and P-2, the hydrogen bonding, which maintained the secondary structures of the hydrophobic polypeptide block, further supported the whole CNPs. The combination of hydrophobic interaction with hydrogen bonding significantly enhanced the stability of the complex nanoparticles in salt solution. The higher content of P-2 in CNPs, the more contribution from hydrogen bonding to maintain the nanoparticles, and the more stable the CNPs in 0.15 PBS. The polypeptides formed one solid skeleton in the CNPs to sustain the whole nanoparticles and guard glucose-responsive segments of P-1.
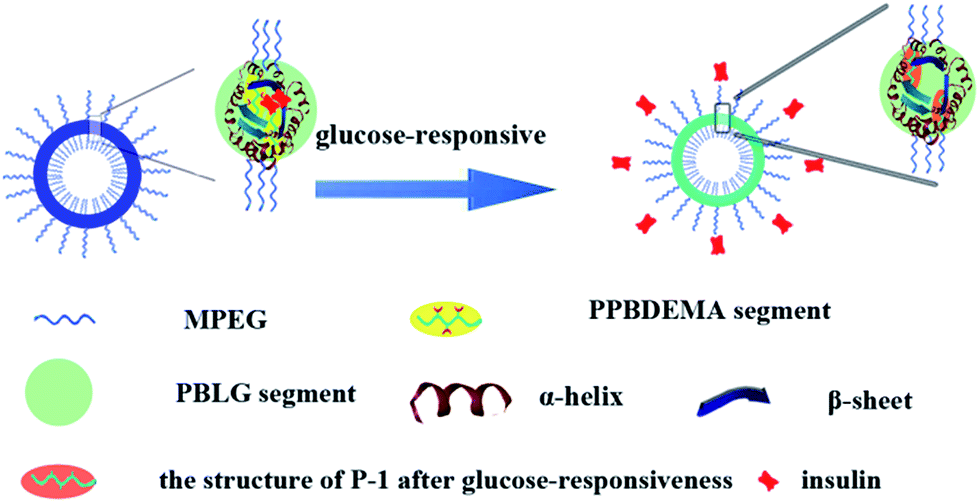 | ||
| Fig. 8 Illustration of CNPs' structure and their glucose responsiveness. | ||
This special hydrophobic structure improved the glucose-sensitive capacity of CNPs to distinguish normoglycemia and hyperglycemia, trying to avoid the release behavior of insulin at normal or low glucose concentration. Although the insulin release was induced by glucose molecules reacting with phenylboronate ester units of hydrophobic blocks, similarly to the previous work,37,38 the hydrophobic frameworks of the CNPs were completely retained by the dominant polypeptides in the whole process of glucose response. This was greatly different from the reported nanoparticles with only phenylboronate ester as hydrophobic core, in which the hydrophobic core would be rapidly destroyed with response to glucose molecules. This firmly hydrophobic skeleton allows the self-regulated release of insulin with glucose concentration fluctuating.
Cell toxicity assays of the CNPs
In vitro cytotoxicity assays of the CNPs with 75% content of P-2 were performed using familiar NIH 3T3 mouse fibroblast cells and analyzed by MTS assay (Fig. 9). For all of the culture, the relative cell proliferation rates were close to 100% for 24 h, irrespective of the CNPs' concentration. The NIH3T3 cell viability was still maintained more than 80% after 48 and 72 h cultivation as the nanoparticle concentration increasing from 25 to 500 mg L−1, demonstrating that the CNPs did not negatively impact cell proliferation. Compared to the nanoparticles formed from only P-1 (Fig. S13†), the CNPs exhibited higher cell viability in the experimental range, suggesting that the introduction of polypeptide segment successfully enhanced biocompatibility of the nanocarriers.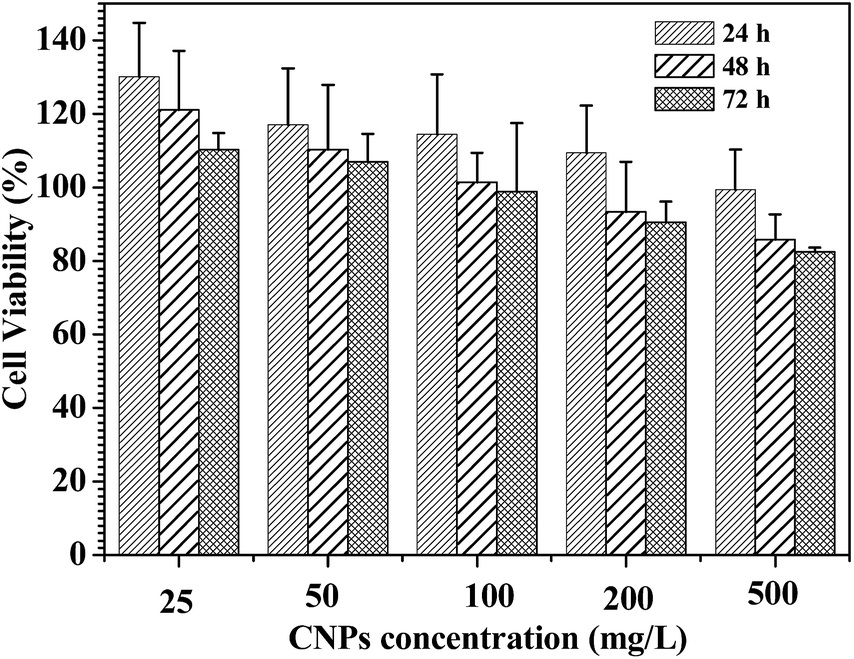 | ||
| Fig. 9 Cell viability assay in NIH3T3 mouse fibroblast cell. The cells were treated with the CNPs with 75% content of P-2 at various concentrations at 37 °C for 24, 48 and 72 h, respectively. | ||
Conclusions
In summary, glucose-responsive CNPs were prepared via co-assembly of the amphiphilic polymers MPEG-b-PPBDEMA and MPEG-b-PBLG. Due to the introduction of hierarchical PBLG blocks, the stability of the whole CNPs was significantly strengthened by hydrogen bonding of polypeptide block on the basis of hydrophobic aggregation of the hydrophobic blocks. As a result, the salt tolerance of the CNPs was concomitantly enhanced in 0.15 M PBS with increasing the weight fraction of MPEG-b-PBLG in CNPs. When MPEG-b-PBLG content reached 75%, the solid skeleton formed by the polypeptides in the CNPs sustained the whole nanoparticles in the process of glucose response, and greatly improved the glucose-sensitive capacity of CNPs to distinguish normoglycemia and hyperglycemia under physiological condition (0.15 M PBS, pH 7.4 and 37 °C), efficiently avoiding the release behavior of insulin at normal or low glucose concentration. This kind of intelligent nanoparticles may be a promising glucose-responsive nanocarrier and find applications in insulin delivery. A further study on animal models is underway in our laboratory.Acknowledgements
This work was supported by National Natural Science Foundation of China (NSFC, Grant No. 21174013; 21374005), the New Century of Ministry of Education (NCET-12-0754), Beijing National Laboratory for Molecular Sciences (BNLMS20150127).Notes and references
- American Diabetes Association, Diabetes Care, 2012, 35, S11 CrossRef PubMed.
- D. R. Owens, B. Zinman and G. B. Bolli, Lancet, 2001, 358, 739 CrossRef CAS.
- R. Ma and L. Shi, Polym. Chem., 2014, 5, 1503 RSC.
- W. Wu and S. Zhou, Macromol. Biosci., 2013, 11, 1464 CrossRef PubMed.
- R. Mo, T. Jiang, J. Di, W. Tai and Z. Gu, Chem. Soc. Rev., 2014, 43, 3595 RSC.
- I. Otsuka, T. Hongo, H. Nakade, A. Narumi, R. Sakai, T. Satoh, H. Kaga and T. Kakuchi, Macromolecules, 2007, 40, 8930 CrossRef CAS.
- W. Qi, X. Yan, J. Fei, A. Wang, Y. Cui and J. Li, Biomaterials, 2009, 30, 2799 CrossRef CAS PubMed.
- T. Ye, X. Bai, X. Jiang, Q. Wu, S. Chen, A. Qu, J. Huang, J. Shen and W. Wu, Polym. Chem., 2016, 7, 2847 RSC.
- A. Matsumoto, T. Ishii, J. Nishida, H. Matsumoto, K. Kataoka and Y. Miyahara, Angew. Chem., Int. Ed., 2012, 124, 2166 CrossRef.
- D. Mandal, S. K. Mandal, M. Ghosh and P. K. Das, Chem.–Eur. J., 2015, 21, 12042 CrossRef CAS PubMed.
- X. Zou, X. Zhao and L. Ye, RSC Adv., 2015, 5, 96230 RSC.
- W. Yuan, L. Li and H. Zou, RSC Adv., 2015, 5, 80264 RSC.
- Q. Guo, Z. Wu, X. Zhang, L. Sun and C. Li, Soft Matter, 2014, 10, 911 RSC.
- H. Guo, H. Li, L. Ling, B. Wang, Q. Guo, Y. Gu and C. Li, Polym. Chem., 2016, 7, 3189 RSC.
- D. Shi, M. Ran, L. Zhang, H. Huang, X. Li, M. Chen and M. Akashi, ACS Appl. Mater. Interfaces, 2016, 8, 13688 CAS.
- H. Kim, Y. J. Kang, E. S. Jeong, S. Kang and K. T. Kim, ACS Macro Lett., 2012, 1, 1194 CrossRef CAS.
- I. Dasgupta, E. A. Tanifum, M. Srivastava, S. S. Phatak, C. N. Cavasotto, M. Analoui and A. Annapragada, PLoS One, 2012, 7(1), e29585 CAS.
- M. Zhang, W. Wang, R. Xie, X. J. Ju, L. Liu, Y. Y. Gu and L. Y. Chu, Soft Matter, 2013, 9, 4150 RSC.
- H. Yang, C. Zhang, C. Li, Y. Liu, Y. An, R. Ma and L. Shi, Biomacromolecules, 2015, 16, 1372 CrossRef CAS PubMed.
- Q. Guo, T. Zhang, J. An, Z. Wu, Y. Zhao, X. Dai and X. Zhang, Biomacromolecules, 2015, 16, 3345 CrossRef CAS PubMed.
- J. Liang, Y. Ma, S. Sims and L. Wu, J. Mater. Chem. B., 2015, 3, 1281 RSC.
- Z. Zou, D. He, L. Cai, X. He, K. Wang, X. Yang, L. Li, S. Li and X. Su, ACS Appl. Mater. Interfaces, 2016, 8, 8358 CAS.
- A. Matsumoto, S. Ikeda, A. Karada and K. Kataoka, Biomacromolecules, 2003, 4, 1410 CrossRef CAS PubMed.
- A. Matsumoto, R. Yoshida and K. Kataoka, Biomacromolecules, 2004, 5, 1038 CrossRef CAS PubMed.
- A. Matsumoto, K. Yamamoto, R. Yoshida, K. Kataoka, T. Aoyagi and Y. Miyahara, Chem. Commun., 2010, 46, 2203 RSC.
- A. Matsumoto, T. Ishii, J. Nishida, H. Matsumoto, K. Kataoka and Y. Miyahara, Angew. Chem., Int. Ed., 2012, 51, 2124 CrossRef CAS PubMed.
- C. Cheng, X. G. Zhang, J. X. Xiang, Y. X. Wang, C. Zheng, Z. T. Lu and C. X. Li, Soft Matter, 2012, 8, 765 RSC.
- C. Cheng, X. Zhang, Y. Wang, L. Sun and C. Li, New J. Chem., 2012, 36, 1413 RSC.
- Y. Kotsuchibashi, R. V. C. Agustin, J. Y. Lu, D. G. Hall and R. Narain, ACS Macro Lett., 2013, 2, 260 CrossRef CAS.
- T. Hoare and R. Pelton, Macromolecules, 2007, 40, 670 CrossRef CAS.
- B. L. Wang, R. J. Ma, G. Liu, Y. Li, X. J. Liu, Y. L. An and L. Q. Shi, Langmuir, 2009, 25, 12522 CrossRef CAS PubMed.
- B. L. Wang, R. J. Ma, G. Liu, Y. Li, X. J. Liu, Y. H. Gao, J. Y. Shen, Y. L. An and L. Q. Shi, Macromol. Rapid Commun., 2010, 31, 1628 CrossRef CAS PubMed.
- R. J. Ma, H. Yang, Z. Li, G. Liu, X. C. Sun, X. J. Liu, Y. L. An and L. Q. Shi, Biomacromolecules, 2012, 13, 3409 CrossRef CAS PubMed.
- H. Yang, X. Sun, G. Liu, R. Ma, Z. Li, Y. An and L. Shi, Soft Matter, 2013, 9, 8589 RSC.
- L. Sun, X. Zhang, C. Zheng, Z. Wu, X. Xia and C. Li, RSC Adv., 2012, 2, 9904 RSC.
- A. Matsumoto, K. Yamamoto, R. Yoshida, K. Kataoka, T. Aoyagi and Y. Miyahara, Chem. Commun., 2010, 46, 2203 RSC.
- Y. Yao, X. M. Wang, T. W. Tan and J. Yang, Soft Matter, 2011, 7, 7948 RSC.
- Y. Yao, L. Y. Zhao, J. J. Yang and J. Yang, Biomacromolecules, 2012, 13, 1837 CrossRef CAS PubMed.
- Y. Yao, H. Shen, G. Zhang, J. Yang and X. Jin, J. Colloid Interface Sci., 2014, 431, 216 CrossRef CAS PubMed.
- L. Zhao, X. Zhang, Y. Yao, C. Yu and J. Yang, Macromol. Chem. Phys., 2014, 215, 1609 CrossRef CAS.
- G. Zhang, X. Zhang, H. Shen, J. J. Yang and J. Yang, RSC Adv., 2014, 4, 49964 RSC.
- X. Zhang, L. Zhao, J. J. Yang and J. Yang, RSC Adv., 2016, 6, 21486 RSC.
- C. Deng, J. Wu, R. Cheng, F. Meng, H. A. Klok and Z. Zhong, Prog. Polym. Sci., 2014, 39, 330 CrossRef CAS.
- M. A. Quadir, M. Martin and P. T. Hammond, Chem. Mater., 2014, 26, 461 CrossRef CAS.
- J. Fan, J. Zou, X. He, F. Zhang, S. Zhang, J. E. Raymond and K. L. Wooley, Chem. Sci., 2014, 5, 141 RSC.
- J. Ding, C. Li, Y. Zhang, W. Xu, J. Wang and X. Chen, Acta Biomater., 2015, 11, 346 CrossRef CAS PubMed.
- Y. Shen, X. Fu, W. Fu and Z. Li, Chem. Soc. Rev., 2015, 44, 612 RSC.
- L. Y. Lin, P. C. Huang, D. J. Yang, J. Y. Gao and J. L. Hong, Polym. Chem., 2016, 7, 153 RSC.
- Y. Liu, P. Chen and Z. Li, Macromol. Rapid Commun., 2012, 33, 287 CrossRef CAS PubMed.
- L. Zhao, J. Ding, C. Xiao, P. He, Z. Tang, X. Pang, X. Zhuang and X. Chen, J. Mater. Chem., 2012, 22, 12319 RSC.
- G. Liu, R. Ma, J. Ren, Z. Li, H. Zhang, Z. Zhang, Y. An and L. Shi, Soft Matter, 2013, 9, 1636 RSC.
- H. Yang, R. Ma, J. Yue, C. Li, Y. Liu, Y. An and L. Shi, Polym. Chem., 2015, 6, 3837 RSC.
- M. Danial and H. A. Klok, Chem. Soc. Rev., 2010, 39, 3541 RSC.
- D. W. P. M. Löwik, E. H. P. Leunissen, M. van den Heuvel, M. B. Hansen and J. C. M. van Hest, Chem. Soc. Rev., 2010, 39, 3394 RSC.
- N. T. Ross, W. P. Katt and A. D. Hamilton, Philos. Trans. R. Soc., A, 2010, 368, 989 CrossRef CAS PubMed.
- Z. B. Li, E. Kesselman, Y. Talmon, M. A. Hillmyer and T. P. Lodge, Science, 2004, 306, 98 CrossRef CAS PubMed.
- I. K. Voets, P. M. Moll, A. Aqil, C. Jerome, C. Detrembleur, P. de Waard, A. de Keizer and M. Stuart, J. Phys. Chem. B, 2008, 112, 10833 CrossRef CAS PubMed.
- W. H. Daly and D. Poché, Tetrahedron Lett., 1988, 29(46), 5859 CrossRef CAS.
- M. G. Li, W. Lu, L. J. Wang, C. X. Zhang, X. Q. Wang, A. P. Zheng and Q. Zhang, Int. J. Pharm., 2007, 329, 182 CrossRef CAS PubMed.
- S. W. Kuo, H. F. Lee, W. J. Huang, K. U. Jeong and F. C. Chang, Macromolecules, 2009, 42, 1619 CrossRef CAS.
- A. Aggeli, M. Bell, N. Boden, J. N. Kenn, P. F. Knowles, T. C. B. Mcleish, M. Pitkeathly and S. E. Radford, Nature, 1997, 386, 259 CrossRef CAS PubMed.
- V. K. Kotharangannagari, A. Sanchez-Ferrer, J. Ruokolainen and R. Mezzenga, Macromolecules, 2012, 45, 1982 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00418d |
| This journal is © The Royal Society of Chemistry 2017 |