
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Experimental and theoretical studies on solvation in aqueous solutions of ionic liquids carrying different side chains: the n-butyl-group versus the methoxyethyl group†
Jéssica C. de Jesusa,
Paulo A. R. Piresa,
Rizwana Mustafab,
Naheed Riazb and
Omar A. El Seoud *a
*a
aInstitute of Chemistry, The University of São Paulo, 748 Professor Lineu Prestes Av., 05508-000 São Paulo, SP, Brazil. E-mail: elseoud@usp.br
bDepartment of Chemistry, The University of Bahawalpur, Bahawalpur 63100, Pakistan
First published on 10th March 2017
Abstract
We used solvatochromic compounds to probe solvation in mixtures of water, W, and four ionic liquids (ILs), 1-R-3-methylimidazoliumX, where R = n-butyl or methoxyethyl and X = acetate and chloride; these are denoted as (C4MeImAc), (C3OMeImAc), (C4MeImCl), and (C3OMeImCl). Our aim was to investigate the effects on solvation when an ether linkage is substituted for a –CH2– group in the IL side chain. We used the solvatochromic probes 2,6-dichloro-4-(2,4,6-triphenylpyridinium-1-yl)phenolate (WB) and 5-nitroindoline, 1-methyl-5-nitroindoline to determine the solvent polarity, ET(WB), and Lewis basicity, SB, respectively. From UV-Vis spectral data, we calculated ET(WB) as a function of the water mole fraction (χW) at different temperatures; from 15 to 60 °C for WB in IL–acetate–W; 25 °C for SB and WB in IL–chloride–W. For all IL–W mixtures, the dependence of ET(WB) on χW is non-linear and, surprisingly, shows negligible dependence on the nature of the side chain. Values of the SB of IL–W were higher for C4MeImX–W than for C3OMeImX. A rationale for these results is the deactivation of the ether oxygen due to the formation of intramolecular hydrogen bonds with the hydrogens of the imidazolium ring. Our hypothesis is confirmed by quantum chemistry and molecular dynamics calculations (energy of the conformers and radial distribution functions), density, and 1H NMR data (chemical shifts, line widths). We attributed the non-linear dependence of the solvatochromic parameters on χW to preferential solvation of the dyes. We treated ET(WB) data with a model that includes the formation of the complex solvent IL–W. Equilibrium constants for solvent exchange in the solvation layer of WB were calculated; their values showed that IL–W is the most efficient solvent species present.
1 Introduction
Ionic liquids (ILs) are composed only of ions and have, by operational definition, melting points <100 °C. The most extensively studied ILs are derivatives of imidazole that are substituted at both nitrogen atoms. Their structural versatility can be demonstrated by the fact that it is possible to synthesize 144 ILs based on 1,3-dialkylimidazolium bromides, chlorides, and iodides, with C1 to C4 as n-alkyl groups. This structural versatility results in different physicochemical properties including, inter alia, viscosity, polarity, and miscibility with water and other molecular solvents.1–4ILs with an ether linkage in the side chain have attracted considerable attention. This is because the introduction of this functional group leads to clear modifications of the physicochemical properties of the IL relative to that with an alkyl group, e.g., four carbon atoms of the n-butyl group versus three carbons plus oxygen of the 2-methoxyethyl group. For example, introduction of the ether functional group in the IL leads to a reduction in viscosity relative to the corresponding IL with an alkyl side chain with the same anion.5–10 Due to their low viscosities, high conductivities, and electrochemical stability, ILs with ether functional groups are used as electrolytes in lithium batteries, electric double layer capacitors, dye-sensitized photoelectrochemical solar cells,11–14 absorption of acidic combustion gases,15,16 and the dissolution of a variety of carbohydrates.17,18 In the latter application, it was shown that polar ILs with a large difference between their solvent Lewis basicity, SB, and Lewis acidity, SA, can dissolve cellulose.19 Note that functionalized ILs are task-specific; i.e., they may perform an additional role besides acting as solvents.20–22
Mixtures of ionic liquids, water (W), and other molecular solvents are used for practical reasons. These include “tuning” of the properties of a binary mixture by changing its composition, increasing the reactant diffusion coefficients due to the concomitant decrease in viscosity (as indicated by the Stokes–Einstein diffusion equation), and cost reduction. The effect of decreasing viscosity on reactivity was nicely demonstrated by the observed increase in the rates of Diels–Alder reactions as a result of decreasing the viscosity of the medium, both for pure ILs and their aqueous solutions.23,24 The use of aqueous ILs for pulp fractionation, fiber spinning,25 and as additives for improving dying of natural and synthetic fibers26 are interesting examples of the potential applications of green chemistry to improve these important industrial processes.
The rational use of mixtures of ILs with W and other molecular solvents, however, is deceptively simple. The reason is that the dependence of mixture properties on composition is usually non-ideal.4 This non-ideality covers both macroscopic properties, e.g., viscosity, surface tension, and conductivity,27–29 and microscopic ones, e.g., solvent empirical polarity, SA, and SB.30,31 The latter properties are determined by the use of solvatochromic dyes (hereafter designated as “probes”).32–34 These are compounds whose UV-Vis spectra, absorption or emission, are particularly sensitive to a specific solvent property. An example is solvent empirical polarity, ET(probe), which is calculated from the dependence of λmax of the intramolecular charge transfer band on the nature of the solvent or the composition of the solvent mixture.3,22,35
ET(probe)/(kcal mol−1) = 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 590.5/λmax (nm) 590.5/λmax (nm)
| (1) |
In the present work, we used the following solvatochromic probes to study solvation in several IL–W binary mixtures (probe name and acronym, binary mixture property): 2,6-dichloro-4-(2,4,6-triphenylpyridinium-1-yl)phenolate (WB), empirical solvent polarity ET(WB); 5-nitroindoline (NI) and 1-methyl-5-nitroindoline (MeNI), SB. The ionic liquids were of the form 1-R-3-methylimidazoliumX, where R = n-butyl and methoxyethyl and X = acetate or chloride, and are denoted as (C4MeImAc), (C3OMeImAc), (C4MeImCl), and (C3OMeImCl); see Fig. 1. We examined ET(WB) as a function of χW over the entire composition range (χW from 0.1 to 0.95) at four different temperatures, 15, 25, 40, and 60 °C (IL–acetates). Measurements of the ET(WB) of IL–chlorides and the SB of all IL–W mixtures were carried out at 25 °C.
 | ||
| Fig. 1 Molecular structures of substances of interest in this work. These are: 2,6-dichloro-4-(2,4,6-triphenylpyridinium-1-yl)phenolate, WB, 1; 5-nitroindoline, NI, 2; 1-methyl-5-nitroindoline, MeNI, 3; 1-(n-butyl)-3-methylimidazolium, C4MeImAc, 4; and 1-(methoxyethyl)-3-methylimidazolium acetate, C3OMeImAc, 5. IL–chlorides are not depicted. | ||
All plots of ET(WB) and SB versus χW were non-linear at all temperatures. Surprisingly, pure C3OMeImAc is only slightly more polar than pure C4MeImAc, although C3OMeImAc–W is slightly less polar than C4MeImAc–W in the χW range 0.1–0.67, becoming more polar at higher χW. Similarly, at 25 °C, C3OMeImCl–W is more polar than C4MeImCl–W in the χW range 0.4–0.95. Mixtures of C4MeImX–W are more basic than the corresponding C3OMeImX–W, where X = acetate and chloride, within the χW ranges studied.
These results are analyzed in terms of “deactivation” of the ether functionality of C3OMeImX due to its hydrogen bonding with the relatively acidic hydrogens of the imidazolium ring, in particular C2–H. This interpretation is corroborated by theoretical calculations (quantum chemistry and molecular dynamics simulations, MD), density, and 1H NMR spectral data (chemical shifts and line widths).
We analyzed the ET(WB) data using a solvation model where the medium consists of the two pure solvents plus a “complex” one, IL–W, formed by interactions (dipolar and hydrogen bonding) between its two components. We calculated the concentrations of these complex solvents from accurate density data; see Fig. ESI-1† (Fig. 1 of the ESI†). The effective concentrations of all binary mixture components were then employed to calculate the local compositions in the solvation layer of WB. Our calculations indicated that IL–W are the most efficient solvent species; i.e., they displace IL and W from the solvation layer of WB. Recently, a paper was published on the solvatochromism of Reichardt betaine (RB; 2,6-diphenyl-4-(2,4,6-triphenylpyridinium-1-yl)phenolate) in the same solvent mixture at 25 °C. In addition to studying at a single temperature, the emphasis in this publication was to fit the Redlich–Kister equation to the data without extracting information about the local composition of the solvation layer of RB.36
2 Experimental
2.1 Material
We purchased the solvents and reactants from Merck or Synth (São Paulo) and purified them as recommended elsewhere.37 The probes WB, NI, and MeNI were available from previous studies;4,38 deionized water was used throughout.2.2 Synthesis of RMeImX (R = n-butyl and methoxyethyl; X = acetate and chloride)
We synthesized these ILs according to Scheme 1: | ||
| Scheme 1 Synthesis of the ILs; R represents 1-chlorobutane (C4MeImX) or 1-chloro-2-methoxyethane (C3OMeImX); X = acetate and chloride. | ||
We transformed the IL–Cl into IL–OH by Cl−/OH− anion exchange and neutralized the latter IL with acetic acid. Briefly, C4MeImCl, was prepared by reacting 19.5 mL of 1-methylimidazole (0.25 mol) with 24.0 mL of 1-chlorobutane (0.23 mol) in 75 mL of acetonitrile in a PTFE-coated stainless-steel reactor (10 bar, N2 atmosphere, 110 °C, 6 h). After cooling the product and removing acetonitrile, the resultant C4MeImCl was washed with cold ethyl acetate (thrice; each 75 mL; vigorous agitation) and dried at 40 °C under reduced pressure over P4O10 for 72 h. The product (94.4% yield) is a white solid, mp 40–41 °C, which gave the expected 1H NMR spectrum (Bruker DRX-500 spectrometer, 500 MHz for 1H, solution in CDCl3, see Table ESI-1, Table 1 of ESI†).39
We converted C4MeImCl into C4MeImOH by ion exchange on a macroporous resin (Purolite SGA-550-OH; hydroxide form). In a typical experiment, we dissolved 15 g of C4MeImCl (0.1 mol) in 500 mL of methanol and slowly passed the solution through a column containing 180 mL of the above-mentioned resin (0.144 mol OH−; equilibrated with methanol). We tested the completeness of Cl−/OH− exchange using AgNO3/HNO3 solution. The produced C4MeImOH was promptly neutralized with a cold solution of acetic acid in methanol (expanded-scale pH paper) before removing the alcohol under reduced pressure and drying, which yielded a clear, slightly yellowish liquid that gave the expected 1H NMR spectrum; see Table ESI-1.†
A similar procedure was employed for 1-(methoxyethyl)-3-methylimidazolium chloride, C3OMeImCl, by reacting 1-methylimidazole and 1-chloro-2-methoxyethane in acetonitrile under pressure, except that the reactant volumes were different. The product (93.2% yield) is a white solid, mp 71–72 °C, that gave the expected 1H NMR spectrum (Bruker DRX-500 spectrometer, solution in CDCl3, see Table ESI-1†). We converted C3OMeImCl into C3OMeImAc by the same procedure used for C4MeImAc; the final yield was 88.5%.
2.3 Sample preparation for solvatochromic measurements
Binary mixtures of IL–W (7 for IL–Cl and 16 for IL–Ac) were prepared by weight at 25 °C to cover a χW range of 0.10 to 0.95. WB solutions in acetone were pipetted into 1 mL volumetric tubes and dried under reduced pressure over P4O10. We added pure IL, pure W, and binary IL–W mixtures to the solid WB samples; the probe was dissolved with the aid of a tube rotator (Labquake, Lab Industries; 30 min). The same procedure was employed for the solvatochromic homomorphic pair NI and MeNI.2.4 Spectrophotometric determination of ET(WB) and SB
The final probe concentration was 2–5 × 10−4 mol L−1 for all probes. UV-Vis spectra of the probe solutions showed no changes in lmax or the shape of the charge-transfer band as a function of probe concentration in the range 1 × 10−4 to 5 × 10−4 mol L−1. This shows that there is no detectable probe aggregation under our experimental conditions. We used a Shimadzu UV-2550 UV-Vis spectrophotometer provided with a digital thermometer (model 4000A, Yellow Springs Instrument) that measured the temperature (±0.05 °C) inside the cell-holder. We recorded each spectrum 3 times at a resolution of 0.2 nm and determined the values of λmax from the first derivative of the absorption spectrum. The uncertainty in ET(WB) is ≤0.04 kcal mol−1 (spectral range = 400–500 nm).2.5 Density measurements
We measured the densities of the above-mentioned liquids (pure solvents and binary mixtures), at 15, 25, 40, and 60 °C, using a DMA 4500M resonating-tube density meter (Anton Paar).2.6 1H NMR study of IL solutions in CDCl3
The ILs were weighed in 2 mL volumetric tubes, dried under reduced pressure for 6 h in the presence of P4O10, and weighed again; the volume was made up to 2 mL with dry CDCl3. The solutions were transferred to a Wilmad 535 NMR-tube and the spectra were recorded at 25 °C using a Bruker DRX-500 spectrometer (64 scans; 2 s acquisition time). We calculated chemical shifts (δ) and line widths at half heights (Δν) from the spectra using the MestRe-C program package version 4.5.9.1 (Mestrelab Research, Santiago de Compostela).2.7 Theoretical calculations
The geometries of these structures were optimized in the gas phase using the hybrid density functional B3LYP in conjunction with the 6-311++G(d,p) basis set. Subsequently, we scanned the dihedral angles formed by atoms C2–N1–C7–C8 of conformers A and C in Fig. 6, (see atom numbering in Fig. 1) from 0 to 360° in 15° steps, and calculated the corresponding electronic energy after each step. We repeated the same calculation steps for the IL-cation in water using the implicit solvation model (PCM) as implemented in the Gaussian program package. We present the results of all these calculations in Fig. 5 below.
Simulation strategy. We optimized the (gas phase) geometry of WB and IL using density functional theory (DFT) calculations, employing the “good-opt” parameter implemented in the Orca 2.9 program.43 Partial charges on the atoms were calculated using the RESP (Restrained ElectroStatic Potential fit) approach44,45 as calculated with the RED on-line server (RESP ESP charge Derive).46 The topology files for GAFF (General Amber Force Field) were generated using Acpype47 and Antechamber 12 programs.48 The GAFF-optimized geometry and topology of SPC/E water were obtained with the Gromacs package. The simulation boxes were generated by using the Packmol program.49
Considering that all binary solvent mixtures are viscous and have an ionic component, we proceeded as follows in order to guarantee the robustness of the calculation results: in the first or equilibration phase, we equilibrated each system using a canonical ensemble (NVT) for 100 ps followed by an isothermal–isobaric ensemble (NpT) for another 100 ps. We carried a subsequent production phase for 40 ns and then examined the system potential energy and density as a function of simulation time. Normally, both properties reached essentially constant values after ca. 10 ns from the start of the production phase. The system was then “annealed” as follows: the simulation boxes were heated from 298 to 370 K over 2 ns under constant volume, kept at 370 K for 6 ns, cooled to 298 K over 2 ns with an additional pressure equilibration to 1 bar for 0.25 ns (under NpT ensemble), and subjected to a production phase for 40 ns. The results of this “annealing” were usually in excellent agreement with those of the first run. Therefore, each system reached equilibrium during the first run; we report here such results (first run).
We used the radial distribution function (RDF) of pairs of atoms and minimum distances of residues to calculate the extension of the solvation layer of WB in nm and the number of solvent species in this layer; vide infra.
Validation and analysis of simulation data. We used densities to validate the simulation results. After the first production phase, the calculated system densities were ρ = 1.06030 (A1), 1.08662 (B1), 1.11129 (C1), 1.17290 (A2), 1.13555 (B2), and 1.18104 (C2) g mL−1; see Table ESI-2† for the compositions of mixtures A1, B1, etc. The respective experimental densities for solutions with the same composition but without WB, at 25 °C, were (density in g mL−1; % difference between calculated and experimental values) 1.06153 (−0.1%); 1.06314 (+2.2%), 1.07669 (+3.1%), 1.12865 (+3.8%), 1.12083 (+0.6%), and 1.15273 (+2.4%) g mL−1. The agreement between the theoretically calculated and experimental densities is therefore excellent.
3 Results and discussion
Note: we discuss the calculation of all the parameters required to treat the solvatochromic data in the Calculations section of the ESI.†3.1 Dependence of ET(WB) and SB on binary mixture composition
Fig. 2 shows the dependence of ET(WB) on χW for C4MeImAc and C3OMeImAc at 15, 25, 40, and 60 °C. In this figure, we exemplify the ideal solvation behavior by straight lines that connect the data for pure IL and W. Fig. 3 and 4 show the (non-linear) dependence of ET(WB) and SB on χW at 25 °C; the maximum deviations from linearity occur at χW values of 0.77 (ET(WB)) and 0.71 (SB) for C4MeImAc and C3OMeImAc. For IL–Cl at the same temperature (25 °C), the maximum deviations from linearity occur at χW = 0.83 (for ET(WB) and (SB) and for both C4MeImCl and C3OMeImCl).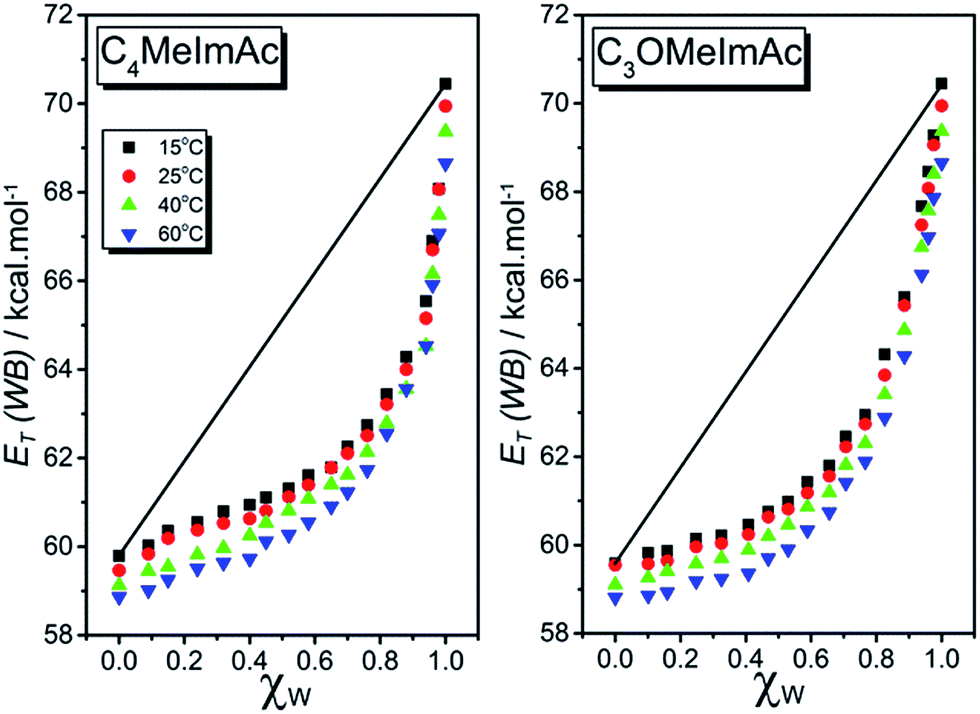 | ||
| Fig. 2 Dependence of the calculated empirical polarity, ET(WB) on the composition of binary solvent mixtures at 15, 25, 40, and 60 °C for C4MeImAc–W (left) and C3OMeImAc–W (right). The ideal solvation behavior of WB is depicted by the straight lines drawn between ET(WB) of the pure solvents, shown for the data at 15 °C. | ||
 | ||
| Fig. 3 Dependence of solvent polarity, ET(WB), on the mole fraction of water in the IL–W binary mixtures, χW, at 25 °C for IL–acetates (left) and IL–chlorides (right). Similar to Fig. 2, the ideal solvation behavior of WB is depicted by the straight line drawn between ET(WB) of the pure solvents. As both IL–chlorides are solids, ET(WB) for pure ILs at 25 °C cannot be determined. The limited solubility of IL–chlorides in water precluded examination of the entire χW range. | ||
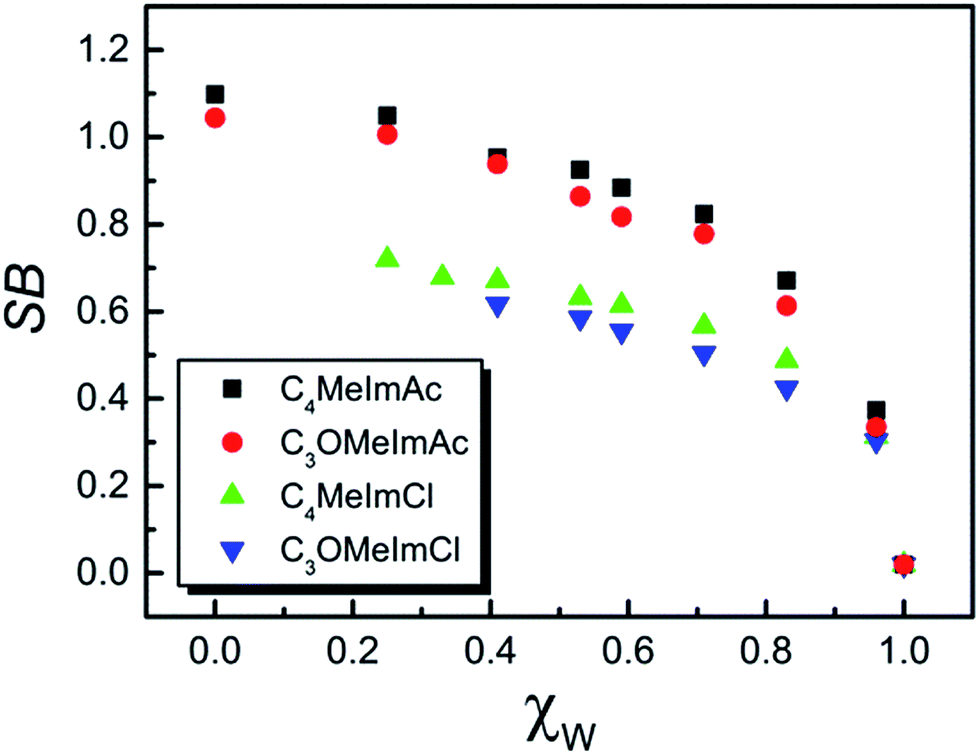 | ||
| Fig. 4 Dependence of solvent Lewis basicity (SB) on the mole fraction of water, χW, at 25 °C in the IL–W binary mixtures. | ||
We calculated the (polynomial) dependence of ET(WB) on the analytical mole fraction of W; the data is presented in Table ESI-3.† The quality of the fit is indicated by the values of the regression coefficient, r2, and ΣQ2, the sum of the squares of the residuals. The degree of polynomial employed is that which gave the best data fit as indicated by these statistical criteria.
3.2 Effect of introducing an ether linkage in the IL side chain on the solvatochromic parameters
The literature shows that the empirical polarity and Lewis basicity of a solvent carrying ether oxygen are higher than those of a hydrocarbon with a related structure except for the absence of oxygen(s). This is shown by comparing ET(RB) for diethyl ether and n-pentane (34.5, 31.0), tetrahydrofuran and cyclohexane (37.4, 30.9), and 1,2-dimethoxyethane and n-heptane (38.2, 31.1) kcal mol−1. Likewise, solvents with ether linkages are more basic than hydrocarbons with related structures as shown by the SB values of diethyl ether and n-pentane (0.562, 0.07) and of tetrahydrofuran and n-heptane (0.591, 0.08).3,50Based on the preceding paragraph, the values of ET(WB) and SB of pure C3OMeImAc and C3OMeImAc–W mixtures are expected to be higher than the corresponding values for pure C4MeImAc and C4MeImAc–W mixtures. Surprisingly, the values of both solvatochromic parameters for the pure ILs are practically the same; i.e., they are insensitive to the presence of ether linkages at all temperatures. Fig. 3 and 4 show that C4MeImAc–W is more polar than C3OMeImAc–W at χW values from 0.1 to 0.67. On the other hand, the former binary mixture is more basic than the latter one over the entire range of χW. A similar negligible effect of the side chain structure on ET(RB) was reported for C4MeImX–acetonitrile and C3OMeImX–acetonitrile, where X = Cl−, BF4−, and PF6−.51
Regarding the possible reasons for this insensitivity, we consider below the basicity of the anion and interactions of the ether oxygen with the hydrogens of the imidazolium cation:
(i) A consequence of the fact that the pyridinium nitrogens of zwitterionic solvatochromic probes, e.g., RB and WB, are sterically hindered is that their solvatochromic response is governed by hydrogen bonding to the phenolate oxygen, as deduced from NMR data52 and theoretical calculations on the solvation of RB.53 The species that participate in this bonding include water (most certainly hydrogen bonded to the IL-anion) and the relatively acidic hydrogens of the imidazolium heterocycle, in particular C2–H.54–57 Fig. ESI-1† showing the solvation of WB is consistent with this conclusion: the RDF curves for the interactions of the probe phenolate oxygen with water and with C2–H of the IL cation are sharp, and their first maxima are located at distances that allow for efficient hydrogen-bonding (parts C and D; Fig. ESI-2†). This contrasts with corresponding RDF plots for the probe quaternary nitrogen (parts A and B; Fig. ESI-2†), which are much less structured with maxima located at distances that do not allow for efficient interactions (see below for a detailed discussion on the results of MD calculations).
(ii) We assess the effect of the anion by decreasing its hydrogen bonding ability with water. The chloride ions of C4MeImCl and C3OMeImCl are less basic than the acetate ion. As a first approximation, the order of ET(WB) and SB should be C3OMeImCl–W > C4MeImCl–W because of the attenuated effect of the anion. Fig. 3 shows that this is the case for ET(WB) in the χW range investigated. We were unable to investigate the entire χW range due to the insolubility of these ILs at low χW. Therefore, the order of polarity in the water-poor IL–Cl–W mixtures cannot be assessed. Fig. 4 shows that C4MeImCl–W mixtures are more basic than C3OMeImCl–W. Therefore, it is unlikely that the solvatochromic response of the probes to the structure of the IL side chain is dominated by the nature of these particular anions; we therefore focused on the cation.
(iii) An obvious possibility is the formation of intramolecular hydrogen bonds in the C3OMeIm+ cation between the ether oxygen and the acidic hydrogens of the imidazolium ring, in particular with (more acidic) C2–H. This bonding, if it occurs, decreases the Lewis basicity of the ether oxygen and the Lewis acidity of the imidazolium ring hydrogens with concomitant attenuation of the difference between solvation by the two ILs. Theoretical chemistry results (DFT calculations and MD simulations) indicated the formation of such intramolecular hydrogen bonds, in particular with C2–H in 1-(2-methoxyethyl)-3-methylimidazoliumX (X = Br−, Cl−, I−)58 and the structurally related 1-(2-hydroxyethyl)-3-methyl imidazoliumX (Br−, Cl−, NO3−, BF4−).59
We carried out two types of theoretical calculations: computations for C3OMeIm+ (gas phase; Fig. 5) to determine the dependence of the electronic energy of conformers on their structures and MD simulations on the solvation of WB in IL–W mixtures (Fig. 6); see Fig. 1 for the IL atom numbering.
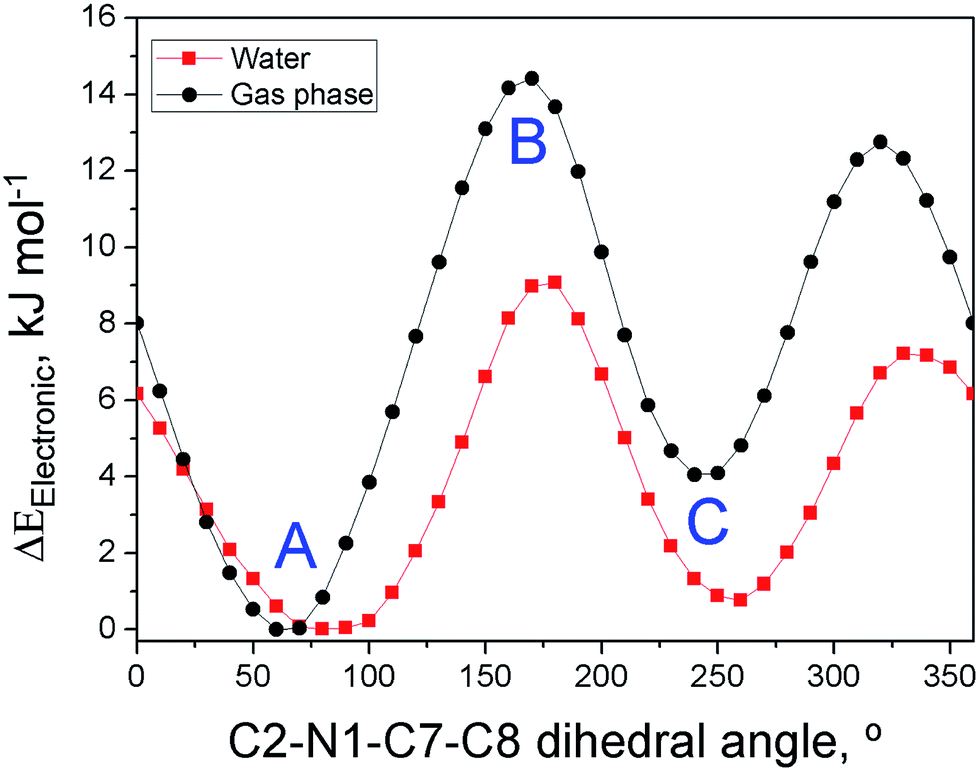 | ||
| Fig. 5 Dependence of the electronic energy of C3OMeIm+ on the C2–N1–C7–C8 dihedral angle calculated for the IL-cation in the gas phase (black) and solvated in a water continuum (red). The conformations of the points marked as (A), (B), and (C) are similar to those depicted in Fig. 6. | ||
 | ||
| Fig. 6 MD simulation-based snapshots of some limiting conformations of the cation of C3OMeImAc in WB in IL–W at χW = 0.67. The colors used to denote the atoms are red, cyan, white, and navy blue for oxygen, carbon, hydrogen, and nitrogen, respectively. The distances between the atoms are (in nm) O⋯H–C2 = 0.239 (A); O⋯H–C2 = 0.468 nm (B; open structure); O⋯H–C5– = 0.243 (C). | ||
Fig. 5 shows the variation of the electronic energy of C3OMeIm+ as a function of the C2–N1–C7–C8 dihedral angle, which is calculated for the ion in the gas phase (black points) and for the ion solvated in a water continuum (red points). Solvation by water lowers the electronic energy and changes the dihedral angles of the conformers to correspond to energy minima and maxima to some extent. As the two curves are qualitatively similar, we dwell on the curve in the gas phase. The two energy minima (left to right) correspond to A and C in Fig. 6 and refer to the conformations with dihedral angles of 60° and 260°, respectively. In both conformations, there is a possibility of intramolecular hydrogen bonding between (–O–) and C2–H (distance O⋯H–C2 = 0.239 nm) or with C5–H (distance O⋯H–C5 = 0.243 nm). As C2–H is more acidic than C5–H,54 the former hydrogen-bond is associated with the smallest electronic energy in the curve. In Fig. 5, the two energy maxima (left to right) correspond to conformations with dihedral angles of 170° and 320°, respectively; the geometry of the latter is similar to that of (B) in Fig. 6. Therefore, the more stable conformations of the IL-cation are those where the ether oxygen is hydrogen-bonded to the relatively acidic hydrogens of the imidazolium ring, which is in agreement with the literature.58 This bonding attenuates the Lewis acidity of the imidazolium ring hydrogens and the Lewis basicity of the ether oxygen and leads to the above-mentioned insensitivity of probe solvation to the nature of the IL side chain.
A short comment on MD simulations is in order. These provide the radial distribution function, g(r), that describes the probability of finding an atom at a distance (r) from another one, which is chosen as a reference point. For the present case, we extract information about the solvation of WB from the first g(r) peak (first probe solvation layer); the distances between pairs of atoms, where short distances indicate strong interactions; and the number of interacting species, calculated from the (normalized) areas under the g(r) curve. Note that the MD data calculated were based on the solvation of WB by pure solvents; i.e., there is no provision for the presence of the IL–W complex solvent. This limitation, however, is acceptable because we compare pairs of ILs (with or without oxygen; same anion) to draw conclusions about the effects of the IL side chain on solvation.
The Gromacs program allows calculation of the RDF curves for the intra- and intermolecular interactions between any two atoms, as shown in Fig. ESI-4.† In the latter, the black curve refers to both types of interactions, whereas the red and blue curves represent inter- and intramolecular interactions, respectively. The ratio of integration of the areas of both peaks (after normalization) gives the fraction of molecules with the conformations that produce these peaks. We extended the same treatment to the pair C5–H and (–O–); all the results are listed in Table 1.
| RDF between C2–H and (–O–) | |||||
|---|---|---|---|---|---|
| First peak | Second peak | ||||
| Peak maximum, nm | Area, arbitrary units | Peak maximum, nm | Area, arbitrary units | Molar fractiona | |
| a The system simulated is WB in IL–W. Molar fraction = area of the first peak/(total area of peaks). | |||||
| C3OMeImAc χW = 0.33 | 0.264 | 0.109 | 0.429 | 0.207 | 0.35 |
| C3OMeImAc χW = 0.67 | 0.259 | 0.147 | 0.445 | 0.226 | 0.39 |
| C3OMeImCl χW = 0.67 | 0.261 | 0.161 | 0.431 | 0.192 | 0.46 |
| RDF between C5–H and (–O–) | |||||
|---|---|---|---|---|---|
| First peak | Second peak | ||||
| Maximum, nm | Area, arbitrary units | Maximum, nm | Area, arbitrary units | Molar fractiona | |
| C3OMeImAc χW = 0.33 | 0.261 | 0.058 | 0.431 | 0.258 | 0.18 |
| C3OMeImAc χW = 0.67 | 0.259 | 0.082 | 0.428 | 0.291 | 0.22 |
| C3OMeImCl χW = 0.67 | 0.260 | 0.067 | 0.435 | 0.287 | 0.19 |
The following is relevant from the results of Table 1:
(iv) Independently of the value of χW, 35 ± 5% and 20 ± 2% of the cations of C3OMeImAc have intermolecular hydrogen bonding between (–O–) and C2–H and C5–H, respectively. This agrees with the energy minima in Fig. 5 and with the fact that C2–H is more acidic than C5–H.
(v) For all RDF curves, the maxima of the second peaks lie at ca. 0.43 nm, which is clearly well above the range for efficient hydrogen-bonding.60 The reported O⋯H–C2 distances are not far from that of conformation (B) in Fig. 6 (0.468 nm).
In summary, the results in Fig. ESI-4† and Table 1 show that a considerable fraction of the cations of C3OMeImAc is involved in intramolecular hydrogen bonding between (–O–) and the acidic hydrogens of the heterocyclic ring. In contrast, a small fraction is involved in efficient intermolecular hydrogen bonding, which is indicated by the shoulder (at ca. 0.25 nm) of the red curve in Fig. ESI-4.† We corroborate these conclusions with two pieces of evidence: the density of the ILs and the 1H NMR data of their solutions in CDCl3.
The (experimental) density of pure C3OMeImAc is larger than that of C4MeImAc (1.13048 and 1.05780 g mL−1, respectively at 25 °C), which is in agreement with published data on other ILs with ether side chains.56 These densities correspond to molar volumes of 177.12 and 187.43 cm3 mol−1, indicating that the preferential conformations of pure C3OMeImAc are more compact (i.e., cyclic) than the corresponding ones of (acyclic) C4MeImAc. Admittedly, the IL molar volumes should be different because of the volume difference between (–O–) and a methylene group, but the ratio of molar volumes of C3OMeImAc/C4MeImAc is larger (0.945) than those of compounds where intramolecular hydrogen bonding does not occur, e.g., diethyl ether/n-pentane (0.903), tetrahydrofuran/cyclopentane (0.869), and tetrahydropyran/cyclohexane (0.906).
Further evidence for the formation of intramolecular hydrogen bonding can be deduced from 1H NMR chemical shifts and line widths of the hydrogens of the heterocyclic ring and side chain. We depict the data for solutions of ILs in CDCl3 in Table 2.
| Data | Unit | C4MeImCl | C3OMeImCl | C4MeImAc | C3OMeImAc |
|---|---|---|---|---|---|
| a Δν = peak width at half-height.b Calculated using Bothner-By equation: 3JHH = 7 − cosθ + 5cos2θ, where θ is the dihedral angle between the hydrogens.61 |
|||||
| δC2H | (ppm) | 10.53 | 10.40 | 11.25 | 10.89 |
| ΔνC2Ha | (Hz) | 3.8 | 4.4 | 3.9 | 5.8 |
| δC7H | (ppm) | 4.34 | 4.60 | 4.30 | 4.54 |
| ΔνC7Ha | (Hz) | 2.4 | 3.0 | 1.6 | 2.8 |
| δC8H | (ppm) | 1.91 | 3.78 | 1.87 | 3.74 |
| 3JC7H | (Hz) | 7.52 | 4.77 | 7.33 | 4.76 |
| Dihedral angle θ; C7H–C7–C8–C8Hb | (°) | 148.9 | 60.7 | 131.0 | 55.3 |
Regarding these NMR data, the following is relevant:
(vi) The orders of δC2–H are C4MeImCl > C3OMeImCl and C4MeImAc > C3OMeImAc. Assuming equal diamagnetic shielding/deshielding of this hydrogen in each pair of ILs, these orders of δ may be attributed to the attenuated positive charge on C2–H due to hydrogen-bonding with electron-rich species, e.g., the acetate ion and the ether oxygen.62 The order of the chemical shifts of C7–H and C8–H is the opposite and merely reflects the electron-withdrawing inductive effect of the oxygen atom (relative to –CH2–).
(vii) The indicated values of Δν are larger for ILs with an (–O–) side chain. From the relationship between Δν and the transverse relaxation time T2 (Δν = 1/πT2), C2–H of C3OMeImCl and C3OMeImAc are less mobile (relax faster) than the corresponding hydrogens of C4MeImCl and C4MeImAc, which is in agreement with their participation in intramolecular hydrogen-bonding with (–O–). The same argument applies to the values of ΔνC7H, which are larger for ILs with an (–O–) side chain, indicating the participation of the attached (–O–) in hydrogen bonding. We were unable to analyze Δν for C8–H comparatively because of the different multiplicities of the ILs (triplet for C3OMeImX and quintet for C4MeImX).
(viii) The dihedral angles calculated from 3JC7H show that the cations of ILs without (–O–), have the side chain, on average, fully extended. Conversely, IL cations with (–O–) have folded side chains with oxygen approaching C2–H and C5–H of the imidazolium ring.
3.3 Analysis of the solvatochromic data in IL–W mixtures
The observed non-ideal behavior shown in Fig. 2 and 4 may originate from so-called “dielectric enrichment”, i.e., enrichment of the probe solvation layer in the solvent of higher relative permittivity, ε.63 The value of ε is 78.5 (25 °C), whereas it is between 7 and 17.2 (25 °C) for, respectively, W and ILs with different cations (including C4MeIm+) and anions (BF4−, PF6−, Cl−, and methyl- to n-octyl sulfate).64–66 If this solvation mechanism were operative, then the ET(WB) versus χW plots would have shown positive deviation; i.e., they would have been above the straight line connecting the ET(WB) values of IL and W; this is not the case. Therefore, we seek another solvation mechanism to explain the non-ideal behavior.Previously, we introduced a model for describing the solvation of a probe in a binary mixture of two solvents, e.g., IL and W. According to this model, the solvatochromic data are analyzed in terms of the effective (not analytical) concentrations of IL, W, and a “complex” solvent (IL–W). As detailed in the ESI,† the latter is formed by the interaction of the two solvents, e.g., via hydrogen bonding, dipolar, and hydrophobic interactions.30,35,38,54–57 Based on this treatment, three equilibrium constants (or “fractionation factors”, φ) are calculated for the solvent exchange equilibria in the solvation layer of WB. They describe the composition of the probe solvation layer (in W, IL, and IL–W) relative to that of the bulk solvent. We deduce information regarding preferential solvation from the value of φ. For example, φW/IL > 1 indicates preferential solvation by water. In other words, water displaces the IL from the probe solvation layer; i.e., the latter is richer in W than the bulk solvent. The converse is true for φW/IL < 1; i.e., the probe is preferentially solvated by IL. Finally, a solvent fractionation factor of unity indicates ideal solvation; i.e., the solvation layer and bulk solvent have the same composition. The same line of reasoning applies to φIL–W/IL (complex solvent displaces IL in the solvation layer) and φIL–W/W (complex solvent displaces W). These solvent-exchange equilibria are given by eqn ESI-(5) to ESI-(7).† We carried out these calculations for ET(WB) in IL–acetate–W because the model fit to the data is reliable due to the large number of binary mixtures examined (18 at each temperature). Table 3 shows the results of these calculations.
| T, °C | m | φ(W/IL) | φ(IL–W/IL) | φ(IL–W/W) | ET(WB)IL | ET(WB)W | ET(WB)IL–W | r2′ | χ2 |
|---|---|---|---|---|---|---|---|---|---|
| a Analysis according to eqn ESI-(5) to ESI-(7). The symbols (m), φ(W/IL), φ(IL–W/IL), and φ(IL–W/W) refer to the number of solvent molecules that perturb the intramolecular charge transfer within the probe, and the equilibrium constants of solvent exchange within the probe solvation layer, where W/IL, IL–W/IL, and IL–W/W refer to water displacing the IL and the complex solvent displacing the IL and W, respectively. See the Calculations section of the ESI.b For pure solvents, the values within parentheses refer to the difference: (experimental ET(WB) − calculated ET(WB)).c The values reported for the mixed solvent IL–S were calculated by regression analysis. | |||||||||
| WB in C4MeImAc–water | |||||||||
| 15 °C | 1.34 | 0.056 | 13.327 | 237.982 | 59.8 (+0.3) | 70.45 (+0.01) | 62.9 | 0.99727 | 0.01932 |
| 25 °C | 1.15 | 0.068 | 7.932 | 116.647 | 59.5 (+0.1) | 69.8 (−0.1) | 63.3 | 0.99785 | 0.01344 |
| 40 °C | 1.09 | 0.072 | 6.824 | 94.778 | 59.18 (−0.02) | 69.37 (−0.01) | 63.0 | 0.99872 | 0.00853 |
| 60 °C | 1.00 | 0.115 | 4.111 | 35.748 | 58.6 (+0.1) | 68.66 (−0.04) | 62.1 | 0.99758 | 0.01616 |
|
|||||||||
| WB in C3OMeImAc–water | |||||||||
| 15 °C | 1.37 | 0.066 | 1.983 | 29.960 | 59.67 (−0.07) | 70.39 (+0.06) | 62.9 | 0.99781 | 0.02858 |
| 25 °C | 1.20 | 0.105 | 1.256 | 11.994 | 59.54 (+0.003) | 69.92 (+0.02) | 61.5 | 0.99899 | 0.01256 |
| 40 °C | 1.12 | 0.096 | 0.864 | 9.007 | 59.18 (−0.08) | 69.36 (+0.01) | 62.2 | 0.99936 | 0.00759 |
| 60 °C | 1.08 | 0.116 | 0.739 | 6.387 | 58.76 (+0.05) | 68.67 (−0.02) | 60.7 | 0.99927 | 0.00825 |
Regarding the data in Table 3, the following is relevant:
(ix) The quality of fit of the above-discussed solvation model to our data is shown by values of (r2) and ΣQ2 and by the excellent agreement between experimental and calculated ET(WB) in pure solvents at different temperatures.
(x) The second column of Table 3 shows that the values of (m) are close to unity. That is, a small number of solvent molecules perturbs the intramolecular charge transfer between the phenolate oxygen and the quaternary nitrogen of WB, leading to the observed nonlinear dependence of ET(WB) on χW; see Fig. 2.
(xi) The values of all φ(W/IL) are much smaller than unity; i.e., the IL is a better solvent for WB than W, probably because of solvation of the probe (phenolate oxygen) by the relatively acidic hydrogens of the imidazolium ring, which is in agreement with Fig. ESI-2.†
(xii) At any temperature, all values of IL–W/IL and IL–W/W are much larger than unity; i.e., the complex solvent is more efficient than its precursors, which is in agreement with our previous data on solvation of different probes (WB and merocyanines) in aqueous ILs.31,67 We discuss this relative efficiency by considering the interactions involved in the solvation of the phenolate oxygen of WB. IL–W is more efficient than IL and W because it offers more possibilities for hydrogen bonding, e.g., (C5–H⋯−O–WB) and (C2–H⋯O(H)H⋯−O–WB) simultaneously.
(xiii) Table 3 shows that all fractionation factors are larger for C4MeImAc–W than for C3OMeImAc–W; i.e., the former is a more efficient binary solvent. We discuss this result by considering the last column of Table 1 and Fig. 7. Based on the former, we conclude that a sizeable fraction of the C3OMeImAc is hardly involved in hydrogen bonding to (−O–WB) because some of the atoms concerned (–O–, C2–H and C5–H) are involved in intramolecular hydrogen bonding. The compositions of the first solvation layers of WB are shown in Fig. 7; the solvent species are given on the mole fraction scale. At each χW, there are more IL cations than anions. While the interactions of the IL-cation affect ET(WB) directly, e.g., via (C5–H⋯−O–WB), and indirectly, e.g., via (C2–H⋯O(H)H⋯−O–WB), the anion affects solvation indirectly, e.g., via (CH3CO2−⋯H–O–H⋯−O–WB), as shown in Fig. 8. Accordingly, interactions with the IL-cation are probably more important than those with the corresponding anion (see also point ii). As discussed above, intramolecular hydrogen bonding deactivates solvation by the cation (with –O–), leading to the observed insensitivity of solvation to the presence of the ether linkage. It is interesting that replacement of (more basic) acetate by chloride results in a reduction in χcation and an increase in χW in the solvation layer, as shown in Fig. 7.
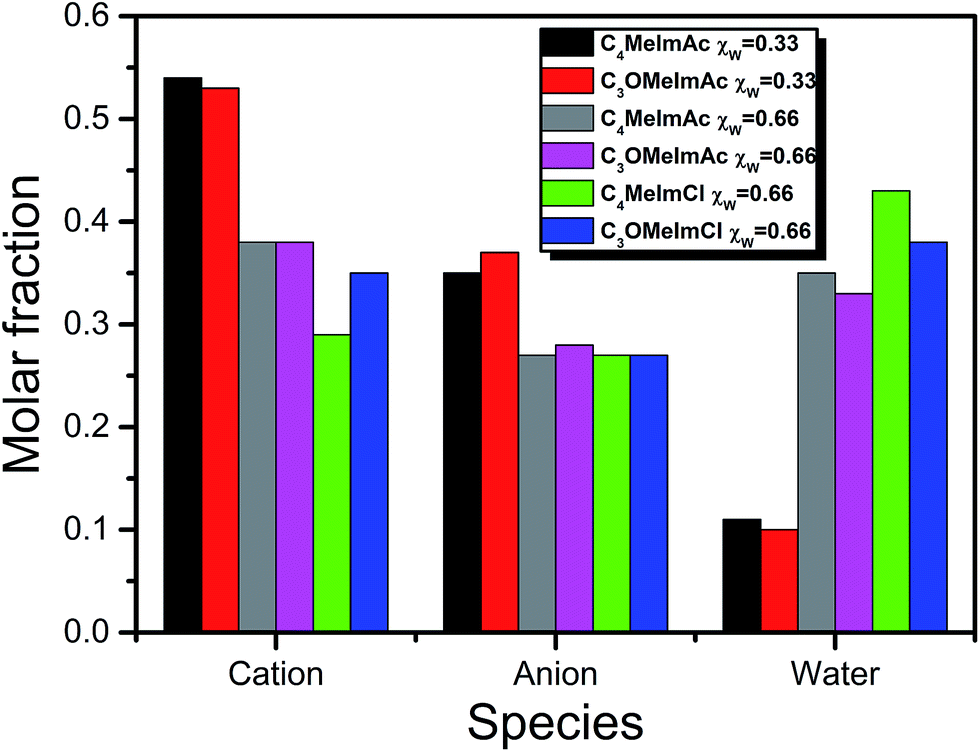 | ||
| Fig. 7 Composition of the first solvation layer of WB in IL–X (X = acetate and chloride) at different water mole fractions, χW. | ||
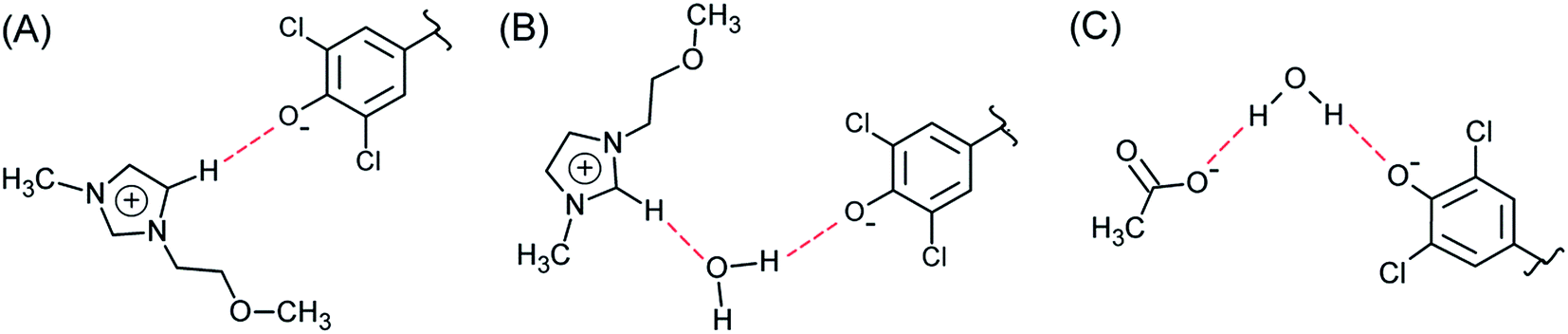 | ||
| Fig. 8 Schematic of hydrogen bonding between (−O–WB) and the binary mixture components. (A) = (C5–H⋯−O–WB), (B) = (C2–H⋯O(H)H⋯−O–WB), and (C) = (CH3CO2−⋯H–O–H⋯−O–WB); see Fig. 1 for the complete structure of WB. | ||
4 Conclusions
Depending on the application, the use of mixtures of ILs with molecular solvents rather than pure ILs is advantageous. Rational use of these mixtures requires understanding of the interactions between the components of the medium and between these and the species of interest (solvatochromic probe, biopolymer, and reactant and transition state). We investigated these interactions using solvatochromic probes, an appropriate solvation model, and theoretical calculations.In order to delineate the effects of the IL cation side chain on solvation (due to replacement of –CH2– by –O–), we studied the solvatochromism of WB in mixtures of water with C4MeImX and C3OMeImX (X = acetate and chloride) at different compositions and temperatures. All values of ET(WB) and SB showed non-linear dependence on χW, with negative deviation from the ideal behavior. We attributed this to preferential solvation of WB by one of the medium components. Because the ether oxygen is more polar and basic than the methylene group, we expected that the ET(WB) and SB of pure C3OMeImAc and C3OMeImAc–W should be larger than the corresponding values for pure C4MeImAc and C4MeImAc–W. This was not the case for ET(WB) in the χW range 0.1–0.67 for IL–acetate and for SB over the entire range of χW. We attributed these unexpected results to attenuation of the effect of (–O–) and the hydrogens of the imidazolium ring on solvation resulting from the formation of intramolecular hydrogen bonding. We corroborated this explanation with: quantum chemical calculations that indicated that conformations of C3OMeImAc with intramolecular hydrogen-bonding are low energy (Fig. 5 and 6); densities of the pure ILs; and 1H NMR chemical shifts and line widths that indicated the relative immobility of C2–H, C5–H, and C7–H due to this hydrogen bonding (Fig. 1 and Table 2). Regarding the solvatochromic data, we assessed the effect of the IL-anion by decreasing its basicity (chloride instead of acetate). Fig. 4 shows that pure C4MeImCl and its aqueous solutions are more basic than pure C3OMeImCl, and C3OMeImCl–W. Therefore, it is unlikely that the solvatochromic responses of the probes to the structure of the IL side chain are dominated by the nature of these particular anions. A solvation model based on the formation of the complex solvent IL–W fitted the data nicely. Values of φ (equilibrium constants for solvent exchange in the probe solvation layer) indicated that IL–W displaces IL and W efficiently. IL–W offers more possibilities than IL and W for hydrogen bonding with the phenolate oxygen of WB, e.g., via (C5–H⋯−O–WB) and indirectly, e.g., via (C2–H⋯O(H)H⋯−O–WB). In summary, C4MeImX–W is more polar (at least within a certain χW range) and more basic than the corresponding C3OMeImX–W (X = acetate and chloride) because participation of (–O–) and the imidazolium hydrogens in solvating WB is suppressed due to the above-mentioned intramolecular hydrogen bonding. Hydrogen NMR, solvatochromic data, and the results of MD simulations confirmed this conclusion.
Acknowledgements
J. C. de Jesus and O. A. El Seoud thank FAPESP (grants 2016/13932-7, 2014/22136-4) for the research fellowship and financial support of this work, respectively; O. A. El Seoud thanks CNPq for a research productivity fellowship (grant 307022/2014-5); R. Mustafa thanks CNPq and TWAS for a research fellowship. We thank Dr P. Farahani and Prof. K. Coutinho for helpful comments.References
- C. Chiappe and D. Pieraccini, Ionic liquids: solvent properties and organic reactivity, J. Phys. Org. Chem., 2005, 18, 275–297 CrossRef CAS.
- G. G. Fortunato, P. M. Mancini, M. V. Bravo and C. G. Adam, New Solvents Designed on the Basis of the Molecular-Microscopic Properties of Binary Mixtures of the Type (Protic Molecular Solvent + 1-Butyl-3-methylimidazolium-Based Ionic Liquid), J. Phys. Chem. B, 2010, 114, 11804–11819 CrossRef CAS PubMed.
- C. Reichardt and T. Welton, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, New York, 3rd edn, 2003, p. 534 Search PubMed.
- C. G. Adam, M. V. Bravo, P. M. E. Mancini and G. G. Fortunato, Solvatochromic dipolarity micro-sensor behaviour in binary solvent systems of the (water + ionic liquid) type: application of preferential solvation model and linear solvation energy relationships, J. Phys. Org. Chem., 2014, 27, 841–849 CrossRef CAS.
- Z. Z. Bin, H. Matsumoto and K. Tatsumi, Low-melting, low-viscous, hydrophobic ionic liquids: aliphatic quaternary ammonium salts with perfluoroalkyltrifluoroborates, Chem.–Eur. J., 2005, 11, 752–766 CrossRef PubMed.
- K. Tsunashima and M. Sugiya, Physical and electrochemical properties of low-viscosity phosphonium ionic liquids as potential electrolytes, Electrochem. Commun., 2007, 9, 2353–2358 CrossRef CAS.
- S. V. Dzyuba and R. A. Bartsch, Expanding the polarity range of ionic liquids, Tetrahedron Lett., 2002, 43, 4657–4659 CrossRef CAS.
- J. D. Holbrey, M. B. Turner, W. M. Reichert and R. D. Rogers, New ionic liquids containing an appended hydroxyl functionality from the atom-efficient, one-pot reaction of 1-methylimidazole and acid with propylene oxide, Green Chem., 2003, 5, 731–736 RSC.
- P. Petiot, C. Charnay, J. Martinez, L. Puttergill, F. Galindo, F. Lamaty and E. Colacino, Synthesis of a new hydrophilic poly(ethylene glycol)–ionic liquid and its application in peptide synthesis, Chem. Commun., 2010, 46, 8842–8844 RSC.
- Y. Deng, P. Besse-Hoggan, M. Sancelme, A. M. Delort, P. Husson and M. F. Costa Gomes, Influence of oxygen functionalities on the environmental impact of imidazolium based ionic liquids, J. Hazard. Mater., 2011, 198, 165–174 CrossRef CAS PubMed.
- S. Fang, L. Yang, J. Wang, M. Li, K. Tachibana and K. Kamijima, Ionic liquids based on functionalized guanidinium cations and TFSI anion as potential electrolytes, Electrochim. Acta, 2009, 54, 4269–4273 CrossRef CAS.
- S. Fang, Z. Zhang, Y. Jin, L. Yang, S. I. Hirano, K. Tachibana and J. Kamijima, New functionalized ionic liquids based on pyrrolidinium and piperidinium cations with two ether groups as electrolytes for lithium battery, J. Power Sources, 2011, 196, 5637–5644 CrossRef CAS.
- A. J. R. Rennie, N. Sanchez-Ramirez, R. M. Torresi and P. J. Hall, Ether-bond-containing ionic liquids as supercapacitor electrolytes, J. Phys. Chem. Lett., 2013, 4, 2970–2974 CrossRef CAS PubMed.
- M. Díaz, A. Ortiz and I. Ortiz, Progress in the use of ionic liquids as electrolyte membranes in fuel cells, J. Membr. Sci., 2014, 469, 379–396 CrossRef.
- G. Cui, C. Wang, J. Zheng, Y. Guo, X. Luo and H. Li, Highly efficient SO2 capture by dual functionalized ionic liquids through a combination of chemical and physical absorption, Chem. Commun., 2012, 48, 2633–2635 RSC.
- G. B. Damas, A. B. A. Dias and L. T. Costa, A quantum chemistry study for ionic liquids applied to gas capture and separation, J. Phys. Chem. B, 2014, 118, 9046–9064 CrossRef CAS PubMed.
- H. Zhao, C. L. Jones and J. V. Cowins, Lipase dissolution and stabilization in ether-functionalized ionic liquids, Green Chem., 2009, 11, 1128–1138 RSC.
- H. Zhao, G. A. Baker and J. V. Cowins, Fast enzymatic saccharification of switchgrass after pretreatment with ionic liquids, Biotechnol. Prog., 2010, 26, 127–133 CAS.
- A. Parviainen, A. W. T. King, I. Mutikainen, M. Hummel, C. Selg, L. K. J. Hauru, H. Sixta and I. Kilpeläinen, Predicting cellulose solvating capabilities of acid–base conjugate ionic liquids, ChemSusChem, 2013, 6, 2161–2169 CrossRef CAS PubMed.
- S. Tang, G. A. Baker and H. Zhao, Ether- and alcohol-functionalized task-specific ionic liquids: attractive properties and applications, Chem. Soc. Rev., 2012, 41, 4030–4066 RSC.
- C. Chiappe and C. S. Pomelli, Point-functionalization of ionic liquids: an overview of synthesis and applications, Eur. J. Org. Chem., 2014, 28, 6120–6139 CrossRef.
- V. G. Machado, R. I. Stock and C. Reichardt, Pyridinium N-Phenolate Betaine Dyes, Chem. Rev., 2014, 114, 10429–10475 CrossRef CAS PubMed.
- S. Tiwari, N. Khupse and A. Kumar, Intramolecular Diels–Alder reaction in ionic liquids: effect of ion-specific solvent friction, J. Org. Chem., 2008, 73, 9075–9083 CrossRef CAS PubMed.
- A. Manna and A. Kumar, Invoking Pairwise Interactions in Water-Promoted Diels–Alder Reactions by using Ionic Liquids as Cosolvents, ChemPhysChem, 2014, 15, 3067–3077 CrossRef CAS PubMed.
- A. M. Stepan, A. Michud, S. Hellstén, M. Hummel and H. Sixta, Pulp Fractionation and Fiber Spinning with Ionic Liquids, Ind. Eng. Chem. Res., 2016, 55, 8225–8233 CrossRef CAS.
- R. Bianchini, G. Cevasco, C. Chiappe, C. S. Pomelli and M. J. Rodríguez Douton, Ionic Liquids Can Significantly Improve Textile Dyeing: An Innovative Application Assuring Economic and Environmental Benefits, ACS Sustainable Chem. Eng., 2015, 3, 2303–2308 CrossRef CAS.
- A. Stoppa, J. Hunger and R. Buchner, Conductivities of Binary Mixtures of Ionic Liquids with Polar Solvents, J. Chem. Eng. Data, 2009, 54, 472–479 CrossRef CAS.
- J. N. C. Lopes, M. F. C. Gomes, P. Husson, A. H. P. Agílio, L. P. N. Rebelo, S. Sarraute and M. Tariq, Polarity, Viscosity, and Ionic Conductivity of Liquid Mixtures Containing [C4C1Im][Ntf2] and a Molecular Component, J. Phys. Chem. B, 2011, 115, 6088–6099 CrossRef PubMed.
- V. Govinda, P. Attri, P. Venkatesu and P. Venkateswarlu, Thermophysical properties of dimethylsulfoxide with ionic liquids at various temperatures, Fluid Phase Equilib., 2011, 304, 35–43 CrossRef CAS.
- B. M. Sato, C. G. G. de Oliveira, C. T. Martins and O. A. El Seoud, Thermo-solvatochromism in binary mixtures of water and ionic liquids: on the relative importance of solvophobic interactions, Phys. Chem. Chem. Phys., 2010, 12, 1764–1771 RSC.
- B. M. Sato, C. T. Martins and O. A. El Seoud, Solvation in aqueous binary mixtures: consequences of the hydrophobic character of the ionic liquids and the solvatochromic probes, New J. Chem., 2012, 36, 2353–2360 RSC.
- L. K. J. Hauru, M. Hummel, A. W. T. King, I. Kilpeläinen and H. Sixta, Role of solvent parameters in the regeneration of cellulose from ionic liquid solutions, Biomacromolecules, 2012, 13, 2896–2905 CrossRef CAS PubMed.
- K. A. Le, R. Sescousse and T. Budtova, Influence of water on cellulose–EMIMAc solution properties: a viscometric study, Cellulose, 2012, 19, 45–54 CrossRef CAS.
- T. A. Bioni, E. P. G. Areas, L. G. Couto, G. Favarin and O. A. El Seoud, Dissolution of cellulose in mixtures of ionic liquid and molecular solvents: relevance of solvent–solvent and cellulose–solvent interactions, Nord. Pulp Pap. Res. J., 2015, 30, 105–111 CAS.
- O. A. El Seoud, Understanding solvation, Pure Appl. Chem., 2009, 81, 697–707 CrossRef CAS.
- A. V. Ladesov, D. S. Kosyakov, K. G. Bogolitsyn and N. S. Gorbova, Solvatochromic Polarity Parameters for Binary Mixtures of 1-Butyl-3-Methylimidazolium Acetate with Water, Methanol, and Dimethylsulfoxide, Russ. J. Phys. Chem. A, 2015, 89, 1814–1820 CrossRef CAS.
- W. L. F. Armarego and C. L. L. Chai, Purification of Laboratory Chemicals, Elsevier, New York, 5th edn, 2003 Search PubMed.
- C. T. Martins, B. M. Sato and O. A. El Seoud, First study on the thermo-solvatochromism in aqueous 1-(1-butyl)-3-methylimidazolium tetrafluoroborate: a comparison between the solvation by an ionic liquid and by aqueous alcohols, J. Phys. Chem. B, 2008, 112, 8330–8339 CrossRef CAS PubMed.
- S. A. Dharaskar, M. N. Varma, D. Z. Shende, C. K. Yoo and K. L. Wasewar, Synthesis, characterization and application of 1-butyl-3 methylimidazolium chloride as green material for extractive desulfurization of liquid fuel, Sci. World J., 2013, 1–9 CrossRef PubMed.
- Gaussian-09, revision D-01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, GROMACS: Fast, flexible, and free, J. Comput. Chem., 2005, 26, 1701–1718 CrossRef CAS PubMed.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, The Missing Term in Effective Pair Potentials, J. Phys. Chem., 1987, 91, 6269–6271 CrossRef CAS.
- F. Neese, U. Becker, D. Ganiouchine, S. Koßmann, T. Petrenko, C. Riplinger and F. Wennmohs, Orca – an ab initio, DFT and semiempirical SCF-MO package, University of Bonn, Germany, 2011 Search PubMed.
- C. C. I. Bayly, P. Cieplak, W. D. Cornell and P. A. Kollman, A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model, J. Phys., 1993, 97, 10269–10280 CAS.
- M. Kohagen, M. Brehm, Y. Lingscheid, R. Giernoth, J. Sangoro, F. Kremer, S. Naumov, C. Jacob, J. Kärger, R. Valiulins and B. Kirchner, How Hydrogen Bonds Influence the Mobility of Imidazolium-Based Ionic Liquids. A Combined Theoretical and Experimental Study of 1-n-Butyl-3-methylimidazolium Bromide, J. Phys. Chem. B, 2011, 115, 15280–15288 CrossRef CAS PubMed.
- E. Vanquelef, S. Simon, G. Marquant, E. Garcia, G. Klimerak and J. C. Delepine, R. E. D. server: a web service for deriving RESP and ESP charges and building force field libraries for new molecules and molecular fragments, Nucleic Acids Res., 2011, W511–W517 CrossRef CAS PubMed.
- A. W. S. Silva and W. F. Vranken, ACPYPE – AnteChamber PYthon Parser interfacE, BMC Res. Notes, 2012, 5, 367 CrossRef PubMed.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, Automatic atom type and bond type perception in molecular mechanical calculations, J. Mol. Graphics Modell., 2006, 25, 247–260 CrossRef CAS PubMed.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, Packmol: A package for building initial configurations for molecular dynamics simulations, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- J. Catalán, Toward a generalized treatment of the solvent effect based on four empirical scales: dipolarity (SdP, a new scale), polarizability (SP), acidity (SA), and basicity (SB) of the medium, J. Phys. Chem. B, 2009, 113, 5951–5960 CrossRef PubMed.
- R. M. Couto, C. Lourenço, J. C. Lima, P. C. Simões and L. C. Branco, Studies of the influence in acetonitrile polarity using imidazolium ionic liquids as additives, J. Chem. Eng. Data, 2013, 58, 1449–1453 CrossRef CAS.
- J. G. Dawber and R. A. Williams, A Nuclear Magnetic Resonance Study of the Solvatochromism of a Pyridinium Betaine, J. Chem. Soc., Faraday Trans. 1, 1986, 82, 3097–3112 RSC.
- C. Chiappe and C. S. Pomelli, The first solvation shell of Reichardt's dye in ionic liquids: a semiempirical study, Theor. Chem. Acc., 2012, 131, 1195–2001 CrossRef.
- S. Sowmiah, V. Srinivasadesikan, M. C. Tseng and Y. H. Chu, On the chemical stabilities of ionic liquids, Molecules, 2009, 14, 3780–3813 CrossRef CAS PubMed.
- A. Samanta, Solvation dynamics in ionic liquids: What we have learned from the dynamic fluorescence Stokes shift studies, J. Phys. Chem. Lett., 2010, 1, 1557–1562 CrossRef CAS.
- Z. J. Chen, T. Xue and J.-M. Lee, What causes the low viscosity of ether-functionalized ionic liquids? Its dependence on the increase of free volume, RSC Adv., 2012, 2, 10564–10574 RSC.
- Z.-D. Ding, Z. Chi, W.-X. Gu, S.-M. Gu and H.-J. Wang, Theoretical and experimental investigation of the interactions between [emim]Ac and water molecules, J. Mol. Struct., 2012, 1015, 147–155 CrossRef CAS.
- Z. Fei, W. H. Ang, D. Zhao, R. Scopelliti, E. E. Zvereva, S. A. Katsyuba and P. J. Dyson, Revisiting Ether-Derivatized Imidazolium-Based Ionic Liquids, J. Phys. Chem. B, 2007, 111, 10095–10108 CrossRef CAS PubMed.
- M. Fakhraee, B. Zandkarimi, H. Salari and M. R. Gholami, Ionic Liquids: Thermodynamic and Structural Properties using Molecular Dynamics Simulations and ab Initio Calculations, J. Phys. Chem. B, 2014, 118, 14410–14428 CrossRef CAS PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Defining the hydrogen bond: An account (IUPAC Technical Report), Pure Appl. Chem., 2011, 83, 1619–1636 CAS.
- E. D. Becker, High Resolution NMR: Theory and Chemical Applications, Academic Press, San Diego, 2000, p. 119 Search PubMed.
- W. Beichel, N. Trapp, C. Hauf, O. Kohler, G. Eickerling, W. Scherer and I. Krossing, Charge-scaling effect in ionic liquids from the charge–density analysis of N,N′-dimethylimidazolium methylsulfate, Angew. Chem., Int. Ed., 2014, 53, 3143–3146 CrossRef CAS PubMed.
- P. Suppan and N. Ghoneim, in Solvatochromism, The Royal Society of Chemistry, Cambridge, 1997, pp. 21–67 Search PubMed.
- P. W. Khirade, A. Chaudhari, J. B. Shinde, S. N. Helambe and S. C. Mehrotra, Static dielectric constant and relaxation time measurements on binary mixtures of dimethyl sulfoxide with ethanol, 2-ethoxyethanol, and propan-1-ol at 293, 303, 313, and 323 K, J. Chem. Eng. Data, 1999, 44, 879–881 CrossRef CAS.
- M. Uematsu and E. U. Frank, Static Dielectric Constant of Water and Steam, J. Phys. Chem. Ref. Data, 1980, 9, 1291–1306 CrossRef CAS.
- T. Singh and A. Kumar, Static Dielectric Constant of Room Temperature Ionic Liquids: Internal Pressure and Cohesive Energy Density Approach, J. Phys. Chem. B, 2008, 112, 12968–12972 CrossRef CAS PubMed.
- P. L. Silva, M. S. Trassi, C. T. Martins and O. A. El Seoud, Solvatochromism in binary mixtures: first report on a solvation free energy relationship between solvent exchange equilibrium constants and the properties of the medium, J. Phys. Chem. B, 2009, 113, 9512–9519 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00273d |
| This journal is © The Royal Society of Chemistry 2017 |