DOI:
10.1039/C7RA00130D
(Paper)
RSC Adv., 2017,
7, 41573-41584
Conformational stability, spectroscopic signatures and biological interactions of proton pump inhibitor drug lansoprazole based on structural motifs†
Received
4th January 2017
, Accepted 18th August 2017
First published on 25th August 2017
Abstract
Structure based biological and chemical properties of Lansoprazole (LSP) have been studied by spectroscopic and quantum chemical methods. The geometrical parameters of the title compound obtained by DFT calculation are compared with single crystal XRD data. The conformational flexibility of the title compound has been discussed on the basis of the potential energy profile achieved from the rotation of various groups present in the molecule and the minimum energy conformer has been determined. The spectroscopic fingerprints are studied by variety of experiments (IR, Raman, UV, and NMR). Normal mode analysis is performed to assign the vibrational frequencies according to the potential energy distribution (PED). Simulation of infrared and Raman spectra has led to an excellent overall agreement with the observed spectral patterns by refinement of scale factors. TD-DFT approach is used to investigate the excited states of molecule and prediction of electronic absorption spectra. The 1H nuclear magnetic resonance (NMR) chemical shifts of the molecule are calculated by the gauge independent atomic orbital (GIAO) method and compared with experimental results. Prediction of Activity Spectra analysis (PASS) of the title compound has been done to explore several biological and toxic effects with high probability. The lipophilicity and aqueous solubility have been calculated to get insight into cell membrane penetration and drug absorption processes in biological systems. The molecular docking is performed to identify the binding energy of the ligand with the active site of protein. These studies show that several sites in the molecule are crucial for bonding and these results lead us to the conclusion that the compound might be metabolized by human protein. Electron density-based local reactivity descriptors such as Fukui functions have been calculated to describe the chemical reactivity sites within the molecule.
1. Introduction
Drug design is the inventive process of finding new medications based on the knowledge of a biological target. The ever increasing use of drugs has prompted pharmaceutical scientists to explore various approaches in finding and developing bioactive compounds suitable for the treatment of diseases and often for the maintenance of human health. During the pre-formulation stage of drug development, a great number of characterization methodologies can be employed and each has its associated utility and function. However when a large amount of information is to be gleaned from a small amount of sample, spectroscopic methods of analysis can be of great use. Our aim is to study the biological and chemical properties of the pharmaceutical sample lansoprazole using different spectroscopic techniques. Lansoprazole, a substituted benzimidazole belongs to a group of drugs called proton pump inhibitors. The benzimidazole scaffold is an accepted pharmacophore and represents an important synthetic precursor in new drug discovery.1–3 Lansoprazole suppresses gastric acid secretion by specific inhibition of the H+/K+ ATPase enzyme system at the secretary surface of the gastric parietal cell.4 An increased dose of lansoprazole is to be recommended for the extensive metabolizer (particularly homEM) patients who are refractory to the usual dose of a proton pump inhibitor.5 Literature survey reveals that no experimental and theoretical spectral analyses for LSP molecule have been performed yet. A study on spectral characterization, conformational distribution, chemical reactivity and biological interactions analysis will be useful in predicting several properties of compound. The spectroscopic techniques used here are FT-IR, Raman, UV-vis and NMR spectroscopy. Experimental spectroscopic features have been correlated with the theoretical results obtained from density functional theory (DFT) computations. The thermodynamically most stable configuration of the compound has been analyzed and the structure based properties are determined. The prospective biological activities and toxicity are evaluated by the predictions made by PASS software which predicts pharmacological effects on the basis of structural formula. As the biological activity of a drug compound is affected by the ability of the drug to be transported and absorbed, the lipophilicity and aqueous solubility were calculated. The study of drug–target interactions is important for the understanding of biological processes and for the efforts to develop clinical performance of drug molecules. Docking simulations have been done to determine the aspects of drug–target interactions. The global reactivity indices are calculated for interpreting and predicting diverse aspects of chemical bonding and reaction mechanism whereas local reactivity indices are employed to probe site selectivity of molecule.
2. Experimental details
The compound under investigation is purchased from Sigma-Aldrich chemicals, USA with spectroscopic grade and it is used as such without any further purification for recording the spectra.
2.1 Fourier transform infrared spectroscopy
The Infrared spectrum was recorded in the solid phase in the region 4000–400 cm−1 on Bruker FT-IR spectrometer, model ALPHA equipped with Diamond ATR module by averaging 24 scans with spectral resolution of 4 cm−1.
2.2 Fourier transform Raman spectroscopy
The Raman spectrum was recorded in the region 4000–100 cm−1 on a BRUKER RFS-100/S FT-Raman spectrometer. A Nd:YAG laser operating at 1064 nm with an output of 30 mW was used as the exciting source. The detector was a Ge diode cooled to liquid nitrogen temperature. 64 scans were accumulated at 4 cm−1 resolution.
2.3 UV-vis spectroscopy
The ultraviolet absorption spectrum of LSP was examined using a Shimadzu UV 1800 Spectrophotometer. The UV pattern is recorded from a 10−5 M solution prepared by dissolving LSP in ethanol solvent. The spectrum is shown in Fig. S1 of the ESI.†
2.4 NMR spectroscopy
The 1H NMR spectrum was recorded on a Bruker DRX-300 spectrometer operating at 300 MHz. The measurements were done in CDCl3 solution. The solution was prepared by dissolving about 2 mg of the sample in 0.6 mL of CDCl3. The spectrum has following experimental parameters: number of scans 32; spectral width 9615.385 Hz; acquisition time 3.40 s. Chemical shifts were reported in ppm relative to TMS. The spectrum is shown in Fig. S2 of the ESI.†
3. Computational details
The molecular geometry of the compound was optimized by DFT method with a hybrid functional B3LYP (Becke's three parameter hybrid functional using the LYP correlation functional) supplemented with standard 6-311++G(d,p) basis set. The way the functional is derived and the lack of empirical parameters fitted to specific properties make this functional widely applicable, both to quantum chemistry as well as to condensed matter physics. It has shown to predict a wide range of molecular properties successfully.6–8
The equilibrium geometry corresponding to the true minima on the Potential Energy Surface (PES) has been obtained by solving self consistent field equation effectively. The optimized structural parameters were used in the vibrational frequency calculations. The entire calculations were performed using Gaussian 09W9 program package, invoking gradient geometry optimization.10 Due to systematic errors caused by basis set incompleteness, neglect of electron correlation and vibrational anharmonicity effects, the calculated harmonic vibrational frequencies were scaled by the wavenumber linear scaling (WLS) procedure of Yoshida et al.11
| | |
νobs = (1.0087 − 0.0000163νcal)νcal cm−1
| (1) |
The WLS method employs essentially only one parameter to scale wavenumbers in the whole region and is applicable to a variety of compounds, except the ones for which the effect of dispersion forces is significant.11 It has excellently reproduced the experimental wavenumbers of pharmaceutical compounds studied by authors previously.12–14 Time dependent density functional theory (TD-DFT)15 is used to find the low-lying excited states and their nature within molecule. The solvent effects are taken care of by means of the polarizable continuum model (PCM)16 as there is no specific interaction, such as hydrogen bonds, between the solute and the solvent.17 The Gaussian software generated output file for NMR calculations using guage-independent atomic orbital (GIAO) approach18 provides the computation of absolute chemical shielding due to the electronic environment of the individual nuclei. In order to obtain the calculated results comparable with the experimental data, the absolute shielding returned by the program is transformed to chemical shift by subtracting it from the absolute shielding of tetramethylsilane (TMS).
| | |
δrel = δTMSabs − δabs
| (2) |
To predict biological activity of LSP, an online structure activity relationship (SAR) tool ‘PASS’ (Prediction of Activity Spectra)19 has been used. Lipophilicity and aqueous solubility of LSP has been calculated using ALGOPS 2.1 program.20 This program combines electronic and topological characters to predict the lipophilicity and aqueous solubility of the analyzed molecule. In order to get an insight into ligand–receptor interactions, molecular docking calculations were performed using Auto Dock Vina software21 and Discover studio 4.0 Software.22
4. Results and discussion
4.1 Structural analysis
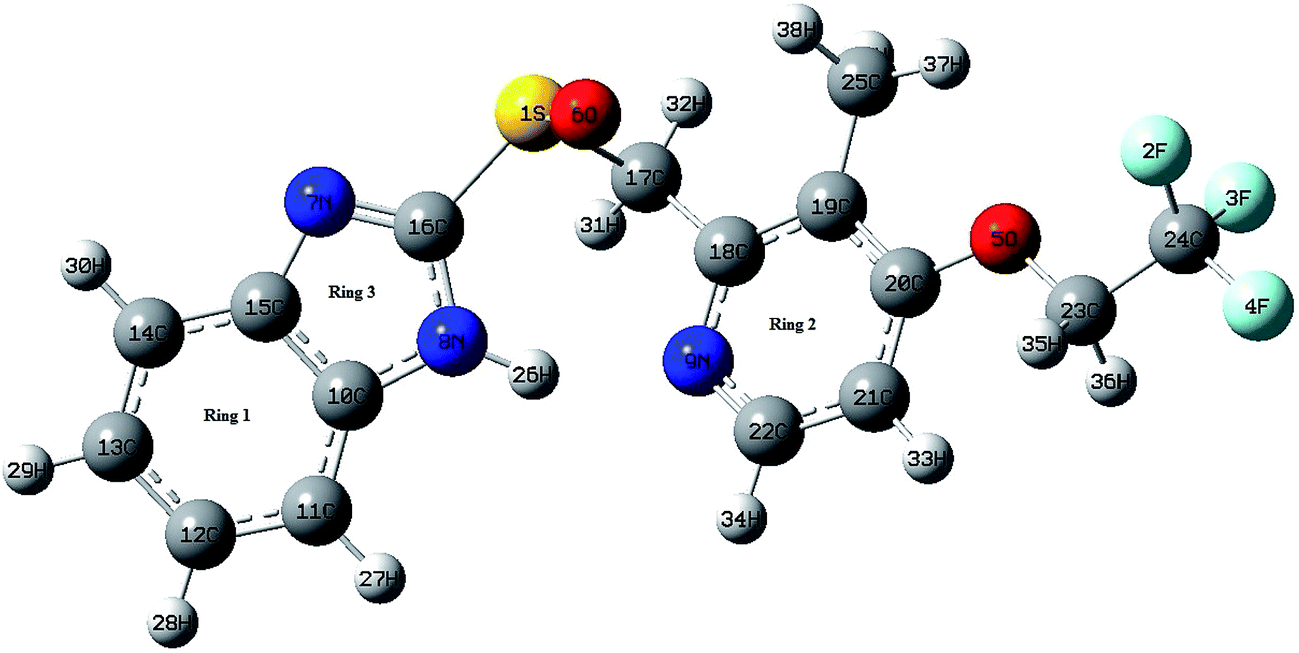
4.1.1 Structural description. Geometry optimization of LSP was done by DFT/B3LYP using the X-ray experimental atomic position crystallographic information file (CIF) as input. The optimized molecular geometry along with the atom numbering scheme of the molecule is shown in Fig. 1. The optimized parameters are listed in Table S1 of ESI† along with the available XRD data.23 The optimized and experimental structures of LSP are compared by superimposing them using a least squares algorithm that minimizes the distances of the corresponding non-hydrogen atoms and are shown in Fig. S3 of ESI.† The agreement between the optimized and experimental structural parameters is quite good which supports the accuracy of functional B3LYP. However some discrepancies may be attributed to the fact that the experimental results belong to solid phase and the theoretical calculations belong to gaseous phase. The pyridine ring is heterocyclic. The similarity of the optimized C–C bond lengths in the pyridine ring in the range 1.407–1.389 Å, to the C–C bond lengths in the benzene ring is indicative of the resonance effect in the pyridine ring. These values are also in good agreement with the experimentally reported values. The resonance in pyridine ring also affects the C–N bond lengths (1.348 Å, 1.329 Å) and these bond lengths appear between the single (C–N) and the double (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) bond lengths. Similar observations are also found for C–N bond lengths in the benzimidazole ring. The replacement of C by N in pyridine ring distorts the hexagonal symmetry, and the ring angle C18–N9–C22 becomes 118.389° rather than 120°. Also the ring carbon atom exerts a large attraction on valence electron cloud of the nitrogen atom resulting in an increase in C–N force constant and a decrease in the corresponding bond length. Trifluoroethoxy group is almost coplanar with pyridine ring with dihedral angles C19–C20–O5–C23, C20–O5–C23–C24 and O5–C23–C24–F4 having values 178.154°, 179.617°, and 179.743° respectively. The DFT calculations give shortening of angle C19–C20–O5 by 3.734° and an enhancement of angle C21–C20–O5 by 3.474° from 120°; this asymmetry of exocyclic angles reveals the attraction between the methyl group and O5 atom of trifluoroethoxy group. The shortening of C20–O5 bond length (1.363 Å) is mainly due to the conjugation interaction between the pyridine ring and the strong electron withdrawing trifluoroethoxy group. The bond length of S1–C17 (1.907 Å) is greater than the bond length S1–C16. Elongation of S1–C17 bond is due to the induction effect of attachment of electron donating methylene group. In benzimidazole ring, the bond angle N7–C16–N8 is the largest (114.415°). This may be due to the electron donating substitution at C16 atom. Closer examination of the bond lengths relative to the XRD values shows the larger deviation in the bond length of N8–H26 and S1–O6 because of intramolecular N–H⋯N hydrogen bonding between the benzimidazole and pyridine ring of same molecule and intermolecular N–H⋯O hydrogen bonding between benzimidazole ring and S–O group of adjacent molecules. These intra and intermolecular bondings are shown in Fig. S4 of ESI.†
N) bond lengths. Similar observations are also found for C–N bond lengths in the benzimidazole ring. The replacement of C by N in pyridine ring distorts the hexagonal symmetry, and the ring angle C18–N9–C22 becomes 118.389° rather than 120°. Also the ring carbon atom exerts a large attraction on valence electron cloud of the nitrogen atom resulting in an increase in C–N force constant and a decrease in the corresponding bond length. Trifluoroethoxy group is almost coplanar with pyridine ring with dihedral angles C19–C20–O5–C23, C20–O5–C23–C24 and O5–C23–C24–F4 having values 178.154°, 179.617°, and 179.743° respectively. The DFT calculations give shortening of angle C19–C20–O5 by 3.734° and an enhancement of angle C21–C20–O5 by 3.474° from 120°; this asymmetry of exocyclic angles reveals the attraction between the methyl group and O5 atom of trifluoroethoxy group. The shortening of C20–O5 bond length (1.363 Å) is mainly due to the conjugation interaction between the pyridine ring and the strong electron withdrawing trifluoroethoxy group. The bond length of S1–C17 (1.907 Å) is greater than the bond length S1–C16. Elongation of S1–C17 bond is due to the induction effect of attachment of electron donating methylene group. In benzimidazole ring, the bond angle N7–C16–N8 is the largest (114.415°). This may be due to the electron donating substitution at C16 atom. Closer examination of the bond lengths relative to the XRD values shows the larger deviation in the bond length of N8–H26 and S1–O6 because of intramolecular N–H⋯N hydrogen bonding between the benzimidazole and pyridine ring of same molecule and intermolecular N–H⋯O hydrogen bonding between benzimidazole ring and S–O group of adjacent molecules. These intra and intermolecular bondings are shown in Fig. S4 of ESI.†
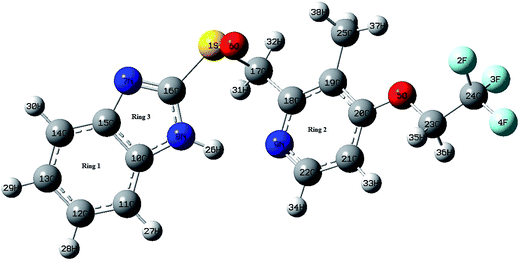 |
| | Fig. 1 Optimized structure of LSP at B3LYP/6-311++G(d,p). | |
In the benzene ring the bond lengths C10–C15 (1.417 Å), C12–C13 (1.411 Å), C14–C15 (1.400 Å) are appreciably greater than other C–C bonds (C10–C11/C11–C12/C13–C14 = 1.397/1.388/1.387 Å). The internal bond angles C10–C11–C12 = 116.756° and C13–C14–C15 = 117.847° are consistently smaller than the normal bond angle of the phenyl ring. These are due to the intramolecular charge transfer from N7 and N8 to the ring system.
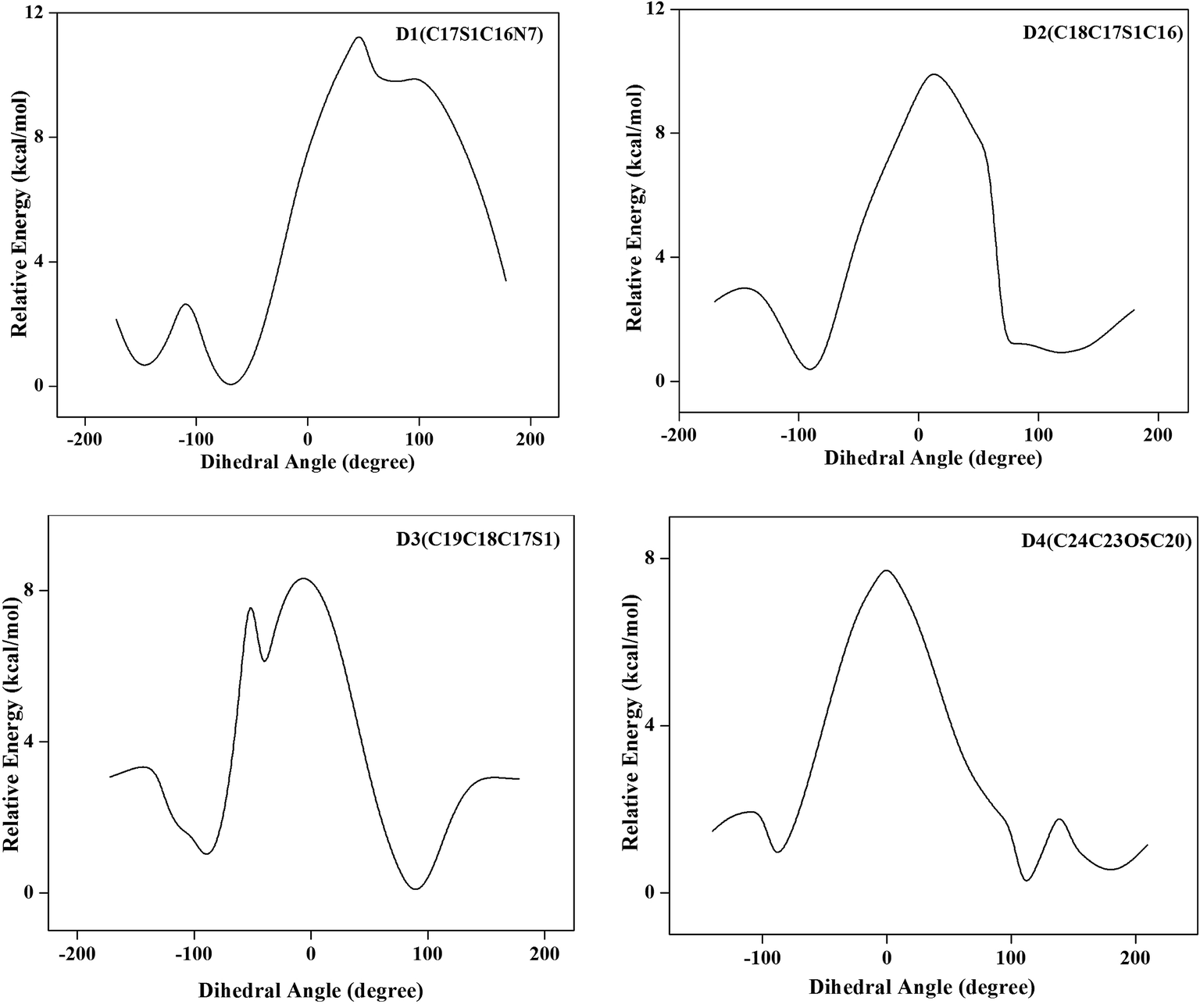
4.1.2 Conformational analysis. Conformational behavior of the molecule provides meaningful information relevant to drug action. In order to ascertain the most stable geometry, the conformational analysis of LSP was carried out through the potential surface scan with B3LYP method using 6-31G basis set. It is seen that the energy trends are same for both 6-31G and 6-311++G(d,p) basis sets. So smaller basis set has been used in order to reduce the computational cost. During the scan, all the geometrical parameters were simultaneously relaxed while the dihedral angles D1(C17S1C16N7), D2(C18C17S1C16), D3(C19C18C17S1) and D4(C24C23O5C20) were varied in steps of 10° ranging from 0° to 360°. The energy profiles as a function of angle of rotation with respect to the dihedral angles derived from potential energy surface scan are shown in Fig. 2. The minimum point structures located on the potential surface scan (PES) of the title compound were submitted to optimization using B3LYP/6-31G computational level of theoretical approximation. The potential energy scans about the dihedral angles predict two minima each for D1, D2 and three minima each for D3, D4. The values −152.02°, −90.25°, −92.06° of dihedrals D1, D2 and D3 respectively provide favorable condition for intramolecular hydrogen bonding N8–H26⋯N9 in the molecule and stabilize the system. The energy profile of the dihedral angle D4 shows that the minimum corresponds to the value 109.62° but it is seen that the steric repulsion between the pyridine ring and the trifluoroethoxy group is the least at the torsional angle 179.62° and hence this conformation is more stable. This value also corresponds to that in the experimental conformation. The rotation of different groups, results in fourteen conformational possibilities. Full geometry optimization of these structures was performed by B3LYP/6-31G method and the optimized are shown in Fig. S5 of the ESI.† On comparing the theoretical and experimental geometries of the title compound (Table S1 of ESI†), it is evident that the optimized geometry of conformer 1, reproduces the experimental one fairly well. So the discussion will be based on conformer 1. The energy difference between conformer 1 and 2 is lower than kT (at room temperature) so conformer 2 may also exist at room temperature. However there is no experimental evidence of existence of any conformer other than conformer 1.
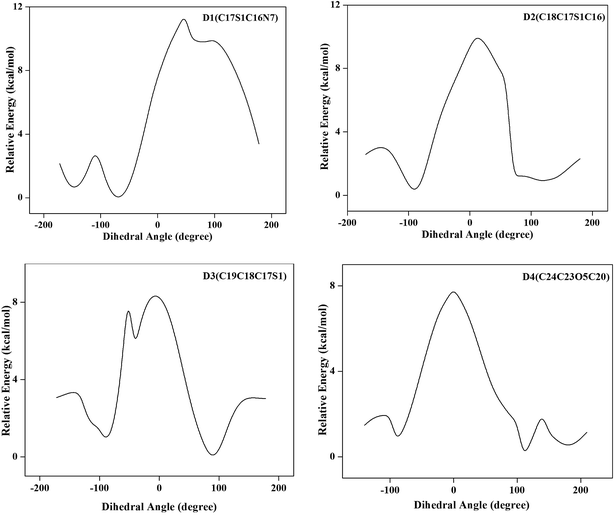 |
| | Fig. 2 One dimensional potential energy scans for dihedral angles D1(C17S1C16N7), D2(C18C17S1C16), D3(C19C18C17S1) and D4(C24C23O5C20). | |
4.2 Spectral analysis
4.2.1 Vibrational spectra. In order to study the vibrational properties of LSP, normal mode analysis was performed. The assignments of the normal modes were done on the basis of the PED calculations by using the GAR2PED program.24 PED was calculated for each of the internal coordinates using localized symmetry and is represented in Table S2 of ESI.† For this purpose a complete set of internal coordinates was defined using Pulay's recommendations.25,26 The experimental FT-IR and FT-Raman spectra and the simulated spectra are shown in Fig. 3 and 4. The detailed analyses of vibrational wavenumbers are discussed below.
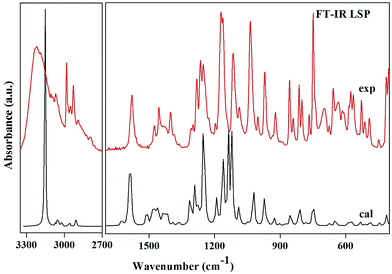 |
| | Fig. 3 Experimental and calculated FT-IR spectrum of LSP. | |
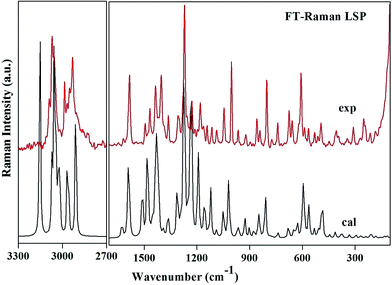 |
| | Fig. 4 Experimental and calculated FT-Raman spectrum of LSP. | |
The scaled wavenumbers obtained showed respectable agreement with the observed FT-IR and FT-Raman spectra and also with the literature values. The deviation of the scaled wavenumbers from the experimental ones was less than 10 cm−1, with exception in few modes where intra and intermolecular interactions come into play. This reveals that B3LYP functional is suitable for the study of pharmaceutical compounds.
4.2.2 Electronic spectra. In order to understand electronic transitions of compound, TD-DFT calculation were done in the gas phase as well as in solvent (ethanol). The computed values, such as absorption wavelength (λ), excitation energies (E), and oscillator strengths (f) are tabulated in Table S3 of ESI† along with the experimental absorption spectral data. Calculations predict intense electronic transition at 287.10 nm with a weak oscillator strength f = 0.0794, that shows good agreement with recorded spectrum showing intense band at 284 nm. This electronic transition corresponds to transition from HOMO-2 → LUMO. The plots of the molecular orbitals are shown in Fig. S6 of the ESI.† On the basis of calculated molecular orbital coefficients analysis and molecular orbital plots, the nature of this transition is assigned to be π → π*. The energy gap between HOMO and LUMO determines the kinetic stability, chemical reactivity, optical polarizability and chemical hardness–softness of a molecule. The hard molecules are less polarizable than the soft ones as they need big energy to excitation. HOMO–LUMO energy gap is calculated to be 4.7436 eV. The formation of organic radical ions by direct electron exchange between a donor molecule and an acceptor molecule is becoming increasingly important, particularly with regard to reaction mechanisms involving intermediate radical ions. All redox processes also involve electron exchange. In simple molecular orbital theory approach, the HOMO energy is related to the ionization potential (I = −EHOMO) and the LUMO energy is used to estimate the electron affinity (A = −ELUMO). The ionization potential for LSP is 6.5407 eV; this value suggests that the molecule is likely to be reactive. The electron affinity measures the energy to form a negative ion. Large value of A suggests formation of stable negative ion. Electron affinity for LSP is 1.7971 eV; this small value indicates that a negative ion with low stability is formed. Substitution by methyl decreases the electron affinity: 1,3,5-trinitrobenzene > 1,3,5-trinitrotoluene > 1,3,5-trinitro-rr-xylenae.39
4.2.3 Nuclear magnetic resonance spectra. Nuclear magnetic resonance spectroscopy has evolved into an important technique in support of structure-based drug design. It can provide crucial information that often resolves conflicting results obtained with other analyses, such as IR, X-ray powder diffraction, and thermal methods. Chemical shifts are recognized as an imperative part of the information contained in NMR spectra. The recorded and calculated chemical shifts of LSP are gathered in Table S4 of ESI.† The proton chemical shift (1H NMR) of organic molecules generally varies greatly with the electronic environment of the proton. The hydrogen attached to or having nearby electron withdrawing atom or group is accompanied by a decrease in shielding resulting in movement of the resonance of proton towards to a higher frequency. By contrast electron donating atom or group increases the shielding and moves the resonance towards to a lower frequency. In LSP the hydrogen atom H26 of the benzimidazole ring is the most downfield having the largest chemical shift value 11.882 ppm. This is due to deshielding by the electron withdrawing property of nitrogen atom N8, the formation of hydrogen bond N8H26⋯N9 also contributes to the downfield of the chemical shift. The chemical shifts of other protons of benzimidazole ring calculated in the range 7.372–7.960 ppm are in good agreement with the experimental values. In the pyridine ring, the presence of nitrogen atom causes the nearby protons to have high chemical shifts. The chemical shift value of hydrogen atom H34 (8.343 ppm) near to nitrogen N9 atom is greater than that of H33 (6.659 ppm). The methyl protons at C25 appear as a singlet with three protons integral at 2.196 ppm and show moderate agreement with computed shifts at 1.849, 2.372, 3.051 ppm. The quartet chemical shifts in ranges 4.315–4.393 ppm and 4.721–4.864 ppm designate presence of the methylene groups and coincide well with computed chemical shifts.
4.3 Biological activities
4.3.1 Biological activity spectra. Biological properties study help to control the delivery of drug either topically or systemically and to control the drug release rate. Estimation of different types of molecular biological activities and toxic/side effects was done for LSP on the basis of Prediction of Activity Spectra for Substances (PASS) prediction results and the analysis was done by Pharma Expert software. PASS provides simultaneous prediction of many types of biological activities based on the structure of compounds. Thus, PASS can be used to estimate the biological activity profiles for molecules, prior to their chemical synthesis and biological testing. A biological activity spectrum for a substance is a list of biological activity types for which the probability to be revealed (Pa) and the probability not to be revealed (Pi) are calculated. Pa and Pi values are independent and their values vary from 0 to 1 and in general Pa + Pi < 1, since these probabilities are calculated independently.40If for any particular activity Pa > 0.7 the substance is very likely to exhibit that activity in experiment. Activities for title molecule analysed by Pharma Expert are listed in Table 1 with Pa > 0.7. The highest probability for LSP is to show the Mucositis treatment effect with Pa value 0.784. We also predicted the toxic effects of the compound. The prediction for toxicity shows that the title compound has high probability to show toxic/side effect bronchoconstrictor with Pa value 0.772. It means that toxic effects of LSP should be scrutinized in further investigation.
Table 1 Some biological activities of LSP molecule predicted by PASS with Pa > 0.7
| Effect |
Pa |
Pi |
| Mucositis treatment |
0.784 |
0.020 |
| Ankylosing spondylitis treatment |
0.757 |
0.017 |
| Phobic disorders treatment |
0.736 |
0.070 |
| Alopecia treatment |
0.726 |
0.009 |
| Kidney function stimulant |
0.718 |
0.020 |
| Antimutagenic |
0.712 |
0.004 |
| Proliferative diseases treatment |
0.701 |
0.027 |
4.3.2 Solubility and diffusion. The lipophilicity and aqueous solubility of the compounds, expressed as log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P and logS respectively are the key determinants of pharmacokinetic properties used in the quantitative structure–activity relationship (QSAR) studies and drug design, since these properties are related to drug absorption, metabolism, bioavailability, and toxicity. Many of the processes of drug disposition depend on the capability to cross membranes and hence these have a high correlation with measures of lipophilicity. On the basis of review of the relevant literature41 high lipophilicity (>5) frequently leads to compounds with rapid metabolic turnover, low solubility, and poor absorption however if lipophilicity is too low, a drug will generally display poor ADMET (absorption, distribution, metabolism, elimination and toxicology) properties. The average lipophilicity value (between ∼1 and 3) ensures optimal physiochemical properties. The calculated value of logP for LSP is 2.84. This suggests that the drug LSP may penetrate through the apolar cell membranes so that it is distributed well within the body after adsorption and is rapidly metabolized and excreted. This value of logP is in the range of other benzimidazole derivatives.42,43 The high value of lipophilicity ensures the high plasma level that provides the good gastrointestinal absorption. Aqueous solubility limits the amount of compound that can be present in solution in the gut and in the circulation. Solubility can limit the bioavailability of a compound and lead to high patient-to-patient and fed-to-fasted variation. Solubility of a compound must be accurately determined to assess the concentrations that the drug will achieve in the target area, to establish the therapeutic level, and to prevent toxicity. The value of logS for LSP is −3.2, which is in the range of good solubility describing the balanced pharmacokinetic properties of LSP. Most of the marketed drugs are found in this category.44 High solubility of LSP leads to high hydrophilicity that supports absorption across the biological membrane and distribution to reach the target organ or tissue. Lower solubility can hinder the absorption and distribution of compound.
P and logS respectively are the key determinants of pharmacokinetic properties used in the quantitative structure–activity relationship (QSAR) studies and drug design, since these properties are related to drug absorption, metabolism, bioavailability, and toxicity. Many of the processes of drug disposition depend on the capability to cross membranes and hence these have a high correlation with measures of lipophilicity. On the basis of review of the relevant literature41 high lipophilicity (>5) frequently leads to compounds with rapid metabolic turnover, low solubility, and poor absorption however if lipophilicity is too low, a drug will generally display poor ADMET (absorption, distribution, metabolism, elimination and toxicology) properties. The average lipophilicity value (between ∼1 and 3) ensures optimal physiochemical properties. The calculated value of logP for LSP is 2.84. This suggests that the drug LSP may penetrate through the apolar cell membranes so that it is distributed well within the body after adsorption and is rapidly metabolized and excreted. This value of logP is in the range of other benzimidazole derivatives.42,43 The high value of lipophilicity ensures the high plasma level that provides the good gastrointestinal absorption. Aqueous solubility limits the amount of compound that can be present in solution in the gut and in the circulation. Solubility can limit the bioavailability of a compound and lead to high patient-to-patient and fed-to-fasted variation. Solubility of a compound must be accurately determined to assess the concentrations that the drug will achieve in the target area, to establish the therapeutic level, and to prevent toxicity. The value of logS for LSP is −3.2, which is in the range of good solubility describing the balanced pharmacokinetic properties of LSP. Most of the marketed drugs are found in this category.44 High solubility of LSP leads to high hydrophilicity that supports absorption across the biological membrane and distribution to reach the target organ or tissue. Lower solubility can hinder the absorption and distribution of compound.
4.3.3 Drug–protein interaction. In drug discovery process, drug metabolism plays a major role and determines the fate of the prospective drugs. Drug metabolism take place only after the drugs reach their specific target site and produce the desired effects. Human P450 protein CYPs are the major enzymes involved in drug metabolism, accounting for about 75% of the total metabolism.45 There are approximately 60 CYP genes in humans. To examine the interaction mechanisms between CYPs and LSP, we used molecular docking method taking cytochrome CYP3A4. The molecular docking studies provide the low-energy binding modes of a small molecule, or ligand, within the active site of a macromolecule, or receptor, of known structure. 3D crystal structure of CYP3A4 was obtained from Protein Data Bank (PDB ID: 2V0M). Docking studies of LSP provides best orientation of ligand which would form a complex with overall minimum energy and possible ligand–protein interaction. The ligand binds at the active site of the protein by non-covalent interactions most prominent of which are H-bonding, π–π stacking and alkyl–π interactions (Fig. 5). Pro23, Leu22 and lle28 holds the molecule by forming multiple alkyl–pi bonds with benzimidazole ring. Methyl group shows alkyl–pi and alkyl–alkyl interactions with Phe24, Val203 and Leu25 respectively. Binding pattern shows that fluorine atom is also crucial for binding by forming alkyl–alkyl interaction with Pro205. Thus there are several sites in the molecule that play important role in stabilizing the ligand–protein complex. Binding free energy (ΔG in kcal mol−1) of −7.8, as predicted by autodock vina (Table 2) suggests good binding affinity between the ligand and the target protein. These results draw us to the conclusion that the compound metabolize well by CYP3A4 enzyme.
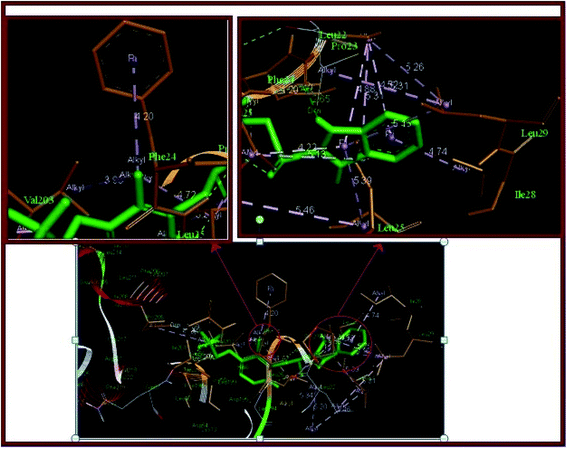 |
| | Fig. 5 Docked ligand conformation at the catalytic site of the enzyme. | |
Table 2 The binding affinity values of different poses of the ligand (LSP)
| S. No. |
Affinity (kcal mol−1) |
Distance from the best mode |
| RMSD (lower bound) |
RMSD (upper bound) |
| 1 |
−7.8 |
0 |
0 |
| 2 |
−7.2 |
2.512 |
1.726 |
| 3 |
−7.1 |
2.199 |
1.409 |
| 4 |
−7.1 |
8.391 |
4.393 |
| 5 |
−6.9 |
20.648 |
19.238 |
| 6 |
−6.9 |
18.425 |
17.183 |
| 7 |
−6.8 |
9.203 |
4.871 |
| 8 |
−6.8 |
14.061 |
12.976 |
| 9 |
−6.6 |
19.924 |
18.407 |
4.4 Reactivity predictions
4.4.2 Atomic reactive sites. The electron density-based local reactivity descriptors such as Fukui functions are proposed to explain the chemical selectivity or reactivity at a particular site of a chemical system. Local reactivity descriptors have been studied extensively in recent years for characterizing the selectivity of a specific site in a molecule and to characterize reactivity of a molecule in chemical reaction. Reactivity to specific agents, e.g. electrophilic and nucleophilic can be described using the above descriptors. The Fukui functions can be defined using finite differences of the electronic density| | |
f−k = ρ(N) − ρ(N − 1)
| (6) |
| | |
f+k = ρ(N + 1) − ρ(N)
| (7) |
where the densities of the cation (N − 1), the neutral (N), and the anion (N + 1) are used. This approach does not take into account the change in geometry from the neutral to the anion/cation species. It is often convenient to describe chemical reactivity on an atom-by-atom basis via a “condensed” Fukui function. The condensed Fukui function may be evaluated as| | |
f+k = qk(N + 1) − qk(N), for a nucleophilic attack
| (8) |
| | |
f−k = qk(N) − qk(N − 1), for an electrophilic attack
| (9) |
where qk is the atomic charge at the kth atomic site in the neutral (N), anionic (N + 1) or cationic (N − 1) chemical species.The atom with the largest f value is the most reactive during reaction: f+k for nucleophilic attack, f−k for electrophilic attack. From Table 3 it has been found that the most prone site for electrophilic attack is C17 having highest f−k value. The reactivity order for the electrophilic case is C17 > C16 > C15 > C19 > C21 > C23 > C10 > C12 > N9. The possible sites for nucleophilic attack are predicted by calculating f+k values and the reactivity order for nucleophilic case is C16, C19, C20, C23, C15, C13 and C24. It is seen that sites C16, C19, C23 and C15 are involved in both electrophilic and nucleophilic attack. To overcome this difficulty Morell et al.50 proposed a local reactivity descriptor called the dual descriptor f(2)(k) ≡ Δf(k) to reveal nucleophilic and electrophilic sites on a molecular system at the same time. The general working equation to obtain dual descriptor is given by the arithmetic difference between nucleophilic and electrophilic Fukui functions.
Table 3 Nucleophilic f+k and electrophilic f−k condensed Fukui functions, condensed dual descriptor f(2)(k) and condensed local hypersoftness s(2)(k) over the atoms of LSP
| kth atom |
f+k |
f−k |
f(2)(k) |
s(2)(k) |
| 1 S |
−0.2798 |
−0.0893 |
−0.1905 |
−0.0339 |
| 2 F |
−0.0117 |
−0.0059 |
−0.0058 |
−0.0010 |
| 3 F |
−0.0133 |
−0.0086 |
−0.0047 |
−0.0008 |
| 4 F |
−0.0185 |
−0.0125 |
−0.0059 |
−0.0011 |
| 5 O |
−0.0149 |
−0.0077 |
−0.0072 |
−0.0013 |
| 6 O |
−0.0035 |
−0.1609 |
0.1573 |
0.0279 |
| 7 N |
−0.0529 |
−0.0771 |
0.0241 |
0.0043 |
| 8 N |
−0.0173 |
−0.0764 |
0.0591 |
0.0105 |
| 9 N |
−0.0451 |
0.0037 |
−0.0488 |
−0.0087 |
| 10 C |
−0.0298 |
0.0129 |
−0.0426 |
−0.0076 |
| 11 C |
−0.0306 |
−0.0827 |
0.0521 |
0.0093 |
| 12 C |
−0.0085 |
0.0044 |
−0.013 |
−0.0023 |
| 13 C |
0.01318 |
−0.0268 |
0.0400 |
0.0071 |
| 14 C |
−0.0319 |
−0.0557 |
0.0238 |
0.0042 |
| 15 C |
0.01399 |
0.0165 |
−0.0025 |
−0.0005 |
| 16 C |
0.14010 |
0.0234 |
0.1167 |
0.0207 |
| 17 C |
−0.0343 |
0.0617 |
−0.0959 |
−0.0171 |
| 18 C |
−0.0332 |
−0.0437 |
0.0105 |
0.0019 |
| 19 C |
0.05399 |
0.0163 |
0.0377 |
0.0067 |
| 20 C |
0.02920 |
−0.0089 |
0.03822 |
0.0068 |
| 21 C |
−0.1097 |
0.0157 |
−0.1255 |
−0.0223 |
| 22 C |
−0.0343 |
−0.0215 |
−0.0128 |
−0.0023 |
| 23 C |
0.02368 |
0.0140 |
0.0096 |
0.0017 |
| 24 C |
0.00018 |
−0.0059 |
0.0062 |
0.0011 |
| 25 C |
−0.0046 |
−0.0259 |
0.0213 |
0.0038 |
When f(2)(k) > 0, the process is driven by a nucleophilic attack on atom k and then that atom acts as an electrophilic species; conversely, when f(2)(k) < 0, the process is driven by an electrophilic attack over atom k and therefore atom k acts as a nucleophilic species. However, dual descriptor is a sub intensive property; its condensed values become insignificant as the size of the molecule increases. To overcome the intrinsic behavior of this local reactivity descriptor, another local reactivity descriptor has been defined which permits one to measure local reactivity according to the molecular size. Such a descriptor is the local hyper softness which is expressed as follows:
where
S stands for the global softness.
The results for the calculation of the condensed electrophilic Fukui function f−(r), nucleophilic Fukui function f+(r), and dual descriptor f(2)(r) along with the local hyper softness s(2)(r) over the atoms (with the exception of hydrogen atoms) of the LSP molecule are presented in Table 3. On the basis of dual descriptor the order for the nucleophilic species is as follows O6 > C16 > N8 > C11 > C13 > C20 > C19 > N7 > C14 > C25 > C18 > C23 > C24, evidently O6 is more likely to undergo a nucleophilic attack, whereas electrophilic attacks will take place preferentially on atom S1 followed by atoms in order given as S1 > C21 > C17 > N9 > C10 > C12 > C22 > O5 > F4 > F2 > C15 > F3.
The dual descriptor is positive at S1 and negative at O6. This shows that LSP undergoes both electrophilic substitution (at S1) and nucleophilic substitution (at O6) at different sites. Therefore it is a dual molecule. Fukui functions are important in deciphering the initial stage of covalent bonding. As S1 and O6 are the most reactive atoms for receiving electrophilic and nucleophilic attacks, respectively and they are pretty close to each other in the middle of the molecule, so the most intense covalent interactions can be concentrated in this part of molecule. For isolated molecules, f(2)(k) and s(2)(k) should give similar reactivity descriptions. The observation of the trend of the reactive sites in LSP by s(2)(k) is found identical to f(2)(k).
4.4.3 Molecular reactive sites. To predict reactive molecular sites, the molecular electrostatic potential (MEP) surface was plotted and is shown in Fig. 6. The electrostatic potential is well suited for analyzing processes based on the ‘‘recognition’’ of one molecule by another, as in drug–receptor, and enzyme–substrate interactions, because it is through their potentials that the two species first ‘‘see’’ each other.51 The negative electrostatic potential regions (red color) are related to electrophilic attack while the positive (blue color) ones to nucleophilic attack. As can be seen from the figure, there are two possible sites for electrophilic attack. These sites are spread around the oxygen atom O6 attached with sulphur and nitrogen N7 atom of benzimidazole ring having the values −0.062 a.u. and −0.051 a.u. respectively. The nucleophilic sites are around the hydrogen atoms with values in the range 0.011–0.062 a.u. The maximum value 0.062 a.u. corresponds to the hydrogen atoms of methylene group attached with oxygen. These sites provide information concerning the regions from where the compound can have covalent bonding and intermolecular interactions.
 |
| | Fig. 6 Molecular electrostatic potential mapped on to isodensity surface along the molecular plane for LSP. | |
5. Conclusion
In the present work, we have analyzed the ground state geometric structure of LSP together with conformational stability, spectroscopic signatures, electronic properties, biological activities and chemical reactivity predictions using quantum chemical computations. The experimental spectral properties of LSP are successfully studied by FT-IR, FT-Raman, UV-vis, NMR spectroscopic techniques. The lowest energy conformer of LSP has been identified using PES scan as a function of the selected torsion angle. This conformer reproduces the experimentally reported structure. As spectroscopic methods probe the molecular level, changes in the molecular environment (e.g., H-bonding or π–π interactions) due to solid state changes are detected as a result of altered vibrations of functional groups involved in these interactions. The various modes of vibrations are explicitly assigned on the results of PED output obtained from the normal coordinate analysis. The mismatch between the observed and the calculated wavenumbers in NH and SO stretching vibrations is due to the involvement of these groups in hydrogen bonding. TD-DFT calculations predict a weak transition (oscillator strength = 0.0794) at 287.10 nm which can be correlated to the experimental band at 284.0 nm in ethanol. This is likely due to vanishing overlapping between the MOs involved in the corresponding electronic transition, which usually results in small oscillator strength. The calculated HOMO–LUMO energy gap is 4.7436 eV. It reveals the significant degree of charge transfer interactions taking place in the molecule and is responsible for the enhanced chemical as well as biological activity of the molecule. The nuclear magnetic resonance spectrum shows that the hydrogen atom attached to or near to an electronegative atom gives the most downfield region. In the present compound the largest chemical shift value 11.882 ppm is for the hydrogen atom H26 attached to the nitrogen atom N8. The hydrogen atom H34 in the pyridine ring is also near to the nitrogen atom N9 and has higher chemical shift value than other atom H33. Pass software predicts the mucositis treatment effect with high probability and some other effects as ankylosing spondylitis, phobic disorders, alopecia, proliferative diseases treatment, kidney function stimulant and antimutagenic with decreasing probabilities. Pass predicted effects of this drug are important and may promote quite a number of investigations in the health area. The values of lipophilicity and aqueous solubility are 2.84 and −3.2 respectively. These values indicate tendency of the compound to partition into lipid vs. aqueous environments. Such insights are useful for correlating important pharmaceutical processes such as gastrointestinal absorption, membrane permeation, solubility, volume of distribution, and protein binding. The results of molecular docking studies speculate that this biologically active molecule is metabolized by cytochrome CYP3A4 enzyme thereby indicating its possible pharmacological importance. Under reactivity predictions molecular electrostatic potential map clearly demonstrates that the possible sites for electrophilic attack are spread around the oxygen atom O6 and the nitrogen atom N7, while the nucleophilic sites are around the hydrogen atoms. This shows that the electrophilic ability strengthens and nucleophilic ability weakens as one move outwards from the inner core of the molecule. LSP has negative chemical potential (μ = −4.1689 eV). This implies that it has a weaker tendency to change into the elements it is produced from and does not decompose. It can be produced voluntarily from the elements. The chemical hardness is 2.3718 eV which advocates its relative kinetic stability. Global reactivity descriptors based on structural properties eventually obtained legitimacy within density functional theory. On the basis of Fukui function the possible molecular sites for the nucleophilic attack are O6, C16, N8, C11, C13, C20, C19, N7, C14, C25, C18, C23, C24 and for the electrophilic attack are S1, C21, C17, N9, C10, C12, C22, O5, F4, F2, C15, F3. Locally, a molecular site is more prone to electrophilic attack or another more prone to nucleophilic attack, but the overall philicity of the whole molecule remains conserved. Thus, the present investigation provides a complete and reliable structural, spectral, biological and structure–activity relation of the compound. This investigation not only presents these features of LSP for the first time, but also prepares the ground for future investigations.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
Authors are thankful to Central Drug Research Institute, Lucknow for providing instrumentation facilities for recording NMR and UV-vis spectra of the sample. The authors are grateful to Dr Shreeram Oak, Bruker Optik GmbH, Mumbai for help in recording FT-IR spectra.
References
- R. Marquis, J. Sheng, T. Nguyen, J. Baldeck and J. Olsson, Arch. Oral Biol., 2006, 51, 1015–1023 CrossRef PubMed.
- A. T. Mavrova, K. Anichina, D. Vuchev, J. Tsenov, P. Denkova, M. Kondeva and M. Micheva, Eur. J. Med. Chem., 2006, 41, 1412–1420 CrossRef CAS PubMed.
- M. Boiani and M. Gonzalez, Mini-Rev. Med. Chem., 2005, 5, 409–424 CrossRef CAS PubMed.
- X. Li, T. B. Andersson, M. Ahlstrom and L. Weidolf, Drug Metab. Dispos., 2004, 32, 821–827 CrossRef CAS.
- T. Furuta, N. Shirai, F. Watanabe, S. Honda, K. Takeuchi, T. Iida, Y. Sato, M. Kajimura, H. Futami, S. Takayanagi, M. Yamada, K. Ohashi, T. Ishizaki and H. Hanai, Clin. Pharmacol. Ther., 2002, 72, 453–460 CrossRef CAS PubMed.
- J. T. Rives and W. L. Jorgensen, J. Chem. Theory Comput., 2008, 4, 297–306 CrossRef PubMed.
- M. Orio, D. A. Pantazis and F. Neese, Photosynth. Res., 2009, 102, 443–453 CrossRef CAS PubMed.
- F. Neese, Coord. Chem. Rev., 2009, 253, 526–563 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- H. Yoshida, K. Takeda, J. Okamura, A. Ehara and H. Matsurra, J. Phys. Chem. A, 2002, 106, 3580–3586 CrossRef CAS.
- N. Choudhary, S. Bee, A. Gupta and P. Tandon, Comput. Theor. Chem., 2013, 1016, 8–21 CrossRef CAS.
- V. Deval, A. Kumar, V. Gupta, A. Sharma, A. Gupta, P. Tandon and K. Kunimoto, Spectrochim. Acta, Part A, 2014, 132, 15–26 CrossRef CAS PubMed.
- M. Agrawal, V. Deval, A. Gupta, B. R. Sangala and S. S. Prabhu, Spectrochim. Acta, Part A, 2016, 167, 142–156 CrossRef CAS PubMed.
- G. Onida, L. Reining and A. Rubio, Rev. Mod. Phys., 2002, 74, 601–659 CrossRef CAS.
- M. Cossi, G. Scalmani, N. Rega and V. Barone, J. Chem. Phys., 2002, 117, 43–54 CrossRef CAS.
- D. Jacquemin, V. Wathelet, E. A. Perpete and C. Adamo, J. Chem. Theory Comput., 2009, 5, 2420–2435 CrossRef CAS PubMed.
- R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- D. A. Filimonov, V. V. Poroikov, E. I. Karaicheva, R. K. Kazaryan, A. P. Boudunova, E. M. Mikhailovsky, A. V. Rudnitskih, L. V. Goncharenko and Y. V. Burov, Exp. Clin. Psychopharmacol., 1995, 58, 56–62 CAS.
- I. V. Tetko, J. Gasteiger, R. Todeschini, A. Mauri, D. Livingstone, P. Ertl, V. A. Palyulin, E. V. Radchenko, N. S. Zefirov, A. S. Makarenko, V. Y. Tanchuk and V. V. Prokopenko, J. Comput.-Aided Mol. Des., 2005, 19, 453–463 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- http://accelrys.com/products/collaborative-science/biovia-discovery-studio/visualization-download.php.
- K. Vyas, A. Sivalakshmidevi and G. O. Reddy, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, e572–e573 Search PubMed.
- J. M. L. Martin and C. V. Alsenoy, Gar2ped, University of Antwerp, Antwerp, 1995 Search PubMed.
- P. Pulay, G. Fogarasi, F. Pang and J. E. Boggs, J. Am. Chem. Soc., 1979, 101, 2550–2560 CrossRef CAS.
- G. Fogarasi, X. Zhou, P. W. Taylor and P. Pulay, J. Am. Chem. Soc., 1992, 114, 8191–8201 CrossRef CAS.
- S. Sudha, M. Karabacak, M. Kurt, M. Cinar and N. Sundaraganesan, Spectrochim. Acta, Part A, 2011, 84, 184–195 CrossRef CAS PubMed.
- L. Dyminska, A. Gagor, M. Maczka, Z. Weglinskia and J. Hanuza, J. Raman Spectrosc., 2010, 41, 1021–1029 CrossRef CAS.
- M. Karnan, V. Balachandran and M. Murugan, Spectrochim. Acta, Part A, 2012, 96, 51–62 CrossRef CAS PubMed.
- R. Meenakshi, RSC Adv., 2016, 6, 21822–21831 RSC.
- V. Arjunan, P. S. Balamourougane, S. T. Govindaraja and S. Mohan, J. Mol. Struct., 2012, 1018, 156–170 CrossRef CAS.
- S. Premkumar, T. N. Rekha, R. M. Asath, T. Mathavan, A. Milton and F. Benial, Eur. J. Pharm. Sci., 2016, 82, 115–125 CrossRef CAS PubMed.
- J. Wang, X. Q. Xie, T. J. Hou and X. Xu, J. Phys. Chem. A, 2007, 111(20), 4443–4448 CrossRef CAS PubMed.
- S. Pinchas, D. Samuel and M. Weiss-Broday, J. Chem. Soc., 1961, 1688 RSC.
- L. Kahovec and K. W. F. Kohlreusch, Monatsh. Chem., 1941, 74, 333 CrossRef.
- M. W. Wong, Y. Steudel and R. Steudel, Chem.–Eur. J., 2007, 13, 502–514 CrossRef CAS PubMed.
- N. Sundaraganesan, C. Meganathan and M. Kurt, J. Mol. Struct., 2008, 891(1–3), 284–291 CrossRef CAS.
- T. S. Xavier, N. Rashid and I. H. Joe, Spectrochim. Acta, Part A, 2011, 78(1), 319–326 CrossRef CAS PubMed.
- D. D. Eley, H. Pines and P. B. Weisz, Advances in Catalysis, Academic Press, United States, 1966, vol. 16 Search PubMed.
- V. V. Poroikov, D. A. Filimonov, W. Ihlenfeldt, T. A. Gloriozova, A. A. Lagunin, Y. V. Borodina, A. V. Stepanchikova and M. C. Nicklaus, J. Chem. Inf. Comput. Sci., 2003, 43, 228–236 CrossRef CAS PubMed.
- J. A. Arnott, R. Kumar and S. L. Planey, J. Appl. Biopharm. Pharmacokinet., 2013, 1, 31–36 Search PubMed.
- S. O. Podunavac-Kuzmanovic, D. D. Cvetkovic and D. J. Barna, J. Serb. Chem. Soc., 2008, 73(10), 967–978 CrossRef CAS.
- C. Sarbu, T. Djakovic-Sekulic and N. Perisic-Janjic, J. Pharm. Biomed. Anal., 2001, 30, 739–745 CrossRef.
- J. Huuskonen, M. Salo and J. Taskinen, J. Chem. Inf. Comput. Sci., 1998, 38, 450–456 CrossRef CAS PubMed.
- F. P. Guengerich, Chem. Res. Toxicol., 2008, 21(1), 70–83 CrossRef CAS PubMed.
- J. P. Perdew, R. G. Parr, M. Levy and J. L. Balduz, Phys. Rev. Lett., 1982, 49, 1691–1694 CrossRef CAS.
- M. Levy, J. P. Perdew and V. Sahni, Phys. Rev. A, 1984, 30, 2745–2748 CrossRef.
- J. P. Perdew and M. Levy, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56(24), 16021–16028 CrossRef CAS.
- R. Parthasarathi, J. Padmanabhan, V. Subramanian, B. Maiti and P. K. Chattaraj, J. Phys. Chem. A, 2003, 107(48), 10346–10352 CrossRef CAS.
- C. Morell, A. Grand and A. Toro-Labbe, J. Phys. Chem. A, 2005, 109, 205–212 CrossRef CAS PubMed.
- A. M. Sapse, Molecular Orbital Calculations for Biological Systems, Oxford University Press, Oxford, New York, 1998 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ra00130d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a