
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mutational analysis to identify the residues essential for the acetyltransferase activity of GlmU in Bacillus subtilis
Meng Wang,
Minhua Huang,
Huawei Gu,
Shan Li ,
Yi Ma* and
Jufang Wang*
,
Yi Ma* and
Jufang Wang*
School of Bioscience and Bioengineering, South China University of Technology, 510006 Guangzhou, China. E-mail: bimayikobe@scut.edu.cn; jufwang@scut.edu.cn; Fax: +86-20-39383034; Fax: +86-20-39380626; Tel: +86-20-39383034 Tel: +86-20-39380626
First published on 1st March 2017
Abstract
The novel and attractive antimicrobial drug target, glucosamine-1-phosphate acetyltransferase/N-acetylglucosamine-1-phosphate uridyltransferase (GlmU), is a bifunctional enzyme that catalyzes two sequential steps in the biosynthesis of UDP-GlcNAc, essential for both lipopolysaccharide and peptidoglycan in Gram positive and Gram negative bacteria. Glucosamine-1-phosphate acetyltransferase catalyzes the formation of N-acetylglucosamine-1-phosphate and N-acetylglucosamine-1-phosphate uridylyltransferase catalyzes the formation of UDP-GlcNAc. In this study, glmU genes from Escherichia coli and Bacillus subtilis were cloned and the corresponding proteins were produced in an E. coli BL21 (DE3) expression system. The kinetic parameters and reaction conditions, such as initial reaction rate, optimal temperature and pH, and the effect of Mg2+ ion concentration of GlmU enzymes from E. coli (Ec-GlmU) and B. subtilis (Bs-GlmU) were detected. Compared to Ec-GlmU, Bs-GlmU exhibits much lower glucosamine-1-phosphate acetyltransferase activity. Based on the in silico results of a virtual amino acid mutation and molecular docking of Bs-GlmU, five mutants were successfully constructed to verify the predicted alterations of acetyltransferase activity. The result of point mutations indicated that the C405 and A380 residues played key role in catalytic mechanism of acetyltransferase of Bs-GlmU providing new insight into the reaction mechanism of GlmU enzymes and supporting the further development of antimicrobial drugs.
1 Introduction
Uridine 5′-diphospho N-acetylglucosamine (UDP-GIcNAc), an acetylated nucleotide sugar, plays an important role in the biochemistry of all living organisms.1–3 As an essential substrate to synthesize many biomolecules, including lipopolysaccharide, peptidoglycan and oligosaccharides, UDP-GIcNAc has high potency to exploit pharmaceuticals and functional materials.4,5 However, due to the lack of an efficient method to synthesize this product, UDP-GlcNAc is in great demand and its price is notoriously high.6,7 Infection caused by pathogenic bacteria has become a serious threat to public health in recent years. A sharp rise in bacteria resistant to antibiotics has emerged as a serious issue which renders drugs ineffective for fighting against bacterial infection.8,9 Few new antibiotics to combat Gram-positive organisms and no new drugs for Gram-negative pathogens have become available for clinical research or in the pharmaceutical market for last 40 years.10–12Recent research has suggested that enzymes involved in cell wall biosynthesis could be useful targets for developing new antibiotics.13,14 Peptidoglycan is a major component of the cell wall that provides structural strength and rigidity and is essential for survival and growth of the bacterium.15–17 Peptidoglycan chains are made up of a repeating N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM) units, which are cross linked by peptide bridges.18 Several reactions in different cytoplasmic and membranes steps are involved in the biosynthesis of peptidoglycan. Three enzymes, glutamine fructose-6-phosphate transferase (GlmS), phosphoglucosamine mutase (GlmM) and glucosamine-1-phosphate acetyltransferase/N-acetylglucosamine-1-phosphate uridyl transferase (GlmU), participate in the synthesis of UDP-GlcNAc, which is the main intermediate in the formation of peptidoglycan and lipid A.16,19,20 Both peptidoglycan and lipid A are necessary for cell viability and perturbing UDP-GlcNAc synthesis which catalyzed by GlmU have grave effects on the fitness of the bacteria.21,22
GlmU catalyzes two consecutive reactions: the first is an acetyltransferase reaction with the help of acetyl coenzyme A (Ac-CoA) to convert α-D-glucosamine-1-phosphate (GlcN1P) to N-acetyl α-D-glucosamine-1-phosphate (GlcNAc1P) and produces the by-product coenzyme A (CoA). The second step is a uridyltransferase reaction converting GlcNAc1P to UDP-GlcNAc using UTP as a uridyl group donor and releasing pyrophosphate (PPi).23–25 The first step is widespread in bacteria enzymes but is missing in humans, whereas the uridyltransferase step is present in enzymes of both bacteria and humans.26,27 The acetyltransferase domain could be a suitable target for developing nontoxic antibacterial inhibitors.28
The study of Ec-GlmU have confirmed that the N-terminal portion of the enzyme contains the uridylyltransferase domain and the C-terminus functions as an acetyltransferase.29,30 The crystal structure of Ec-GlmU with multiple substrates revealed that the 2-amino group of GlcN-1-PO4 is positioned in proximity to AcCoA to facilitate direct thioester attack by a ternary complex mechanism.31,32 The cell wall structure of Gram-positive bacteria is quite different to that of Gram-negative bacteria. There is urgent need to explore Gram-positive bacteria such as B. subtilis since understanding Bs-GlmU will provide important information for developing novel inhibitors. However, there are few articles pertaining to Bs-GlmU. The GcaD protein in B. subtilis is the similar to GlmU with uridyltransferase activity but no acetyltransferase activity.33,34 In this study, Bs-GlmU is recombinant expressed and characterized and the structure model of Bs-GlmU is obtained. Based on the in silico results of virtual amino acid mutation and molecular docking of Bs-GlmU, five mutants are constructed to investigate the mechanism and key amino acid residues of acetyltransferase in Bs-GlmU, providing a new template for understanding the structure and function of GlmU in Gram-positive bacteria. Our results will support further GlmU research and facilitate high-throughput inhibitor screening.
2 Materials and methods
2.1 Materials
Reagents related to E. coli and B. subtilis glmU gene cloning and site-directed mutagenesis were purchased from TakaRa Biotechnology Co., Ltd (Dalian, China). Competent cells E. coli DH5α and BL21 (DE3) were obtained from TIANGEN Biotech (Beijing, China). The Ni-NTA His-binding column from QAIGEN (Dusseldorf, Germany) was used for protein purification. The chemicals used for enzyme assays were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA). All chemical reagents used in this study were analytical grade.2.2 Nucleic acid manipulations
Genome DNA from E. coli and B. subtilis were prepared using a genome extraction kit (Sangon, Shanghai, China) as described in the manufacturer's handbook. The GlmU genes of E. coli (Ec-glmU) and B. subtilis (Bs-glmU) were amplified by PCR reaction using Prime STAR HS DNA Polymerase (TaKaRa, Dalian, China) from the genome DNA of E. coli and B. Subtilis strain respectively. The forward primer used for Ec-glmU was 5′-CG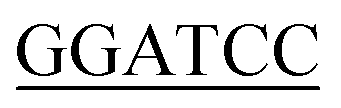 ATGTTGAATAATGCTATGAGCGTAGTGA-3′ (BamH I restriction site underlined), and the reverse primer for Ec-glmU was 5′-CCG
ATGTTGAATAATGCTATGAGCGTAGTGA-3′ (BamH I restriction site underlined), and the reverse primer for Ec-glmU was 5′-CCG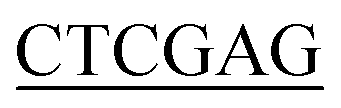 CTTTTTCTTTACCGGACGACGCCAG-3′ (Xho I restriction site underlined). The forward primer used for Bs-glmU was 5′-CG
CTTTTTCTTTACCGGACGACGCCAG-3′ (Xho I restriction site underlined). The forward primer used for Bs-glmU was 5′-CG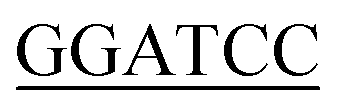 ATGGATAAGCGGTTTGCAGTTGTTT-3′ (BamH I restriction site underlined), and the reverse primer for Bs-glmU was 5′-CCG
ATGGATAAGCGGTTTGCAGTTGTTT-3′ (BamH I restriction site underlined), and the reverse primer for Bs-glmU was 5′-CCG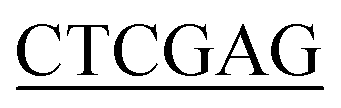 TTTTTTATGAATATTTTTCACATAATCGTC-3′ (Xho I restriction site underlined). Ec-glmU was amplified in 50 μL of reaction buffer by initial denaturation temperature at 98 °C for 2 min followed by 32 cycles of 98 °C for 10 s, 71 °C for 15 s, and 72 °C for 90 s with a final 10 min extension at 72 °C. Bs-glmU was amplified in 50 μL of reaction buffer by initial denaturation temperature at 98 °C for 2 min followed by 32 cycles of 98 °C for 10 s, 65 °C for 15 s, and 72 °C for 90 s with a final 10 min extension at 72 °C. The Ec-glmU and Bs-glmU genes were digested with BamH I and Xho I (Fermentas of Thermo Scientific, Pittsburgh, PA), then ligated with the double digested pET21a vector (Novagen, Wisconsin, USA) yielding the plasmids pET-Ec-glmU and pET-Bs-glmU, respectively. Competent E. coli DH5α cells (Invitrogen, CA, USA) were used as the host for cloning. Transformed cells were spread on Luria-Bertani agar plates containing 100 ng μL−1 of ampicillin and incubated at 37 °C overnight. The positive clones were identified by colony PCR and confirmed by DNA sequence analysis. Sequence similarity searches were performed with the BLAST program (http://www.ncbi.nlm.nih.gov/BLAST/).
TTTTTTATGAATATTTTTCACATAATCGTC-3′ (Xho I restriction site underlined). Ec-glmU was amplified in 50 μL of reaction buffer by initial denaturation temperature at 98 °C for 2 min followed by 32 cycles of 98 °C for 10 s, 71 °C for 15 s, and 72 °C for 90 s with a final 10 min extension at 72 °C. Bs-glmU was amplified in 50 μL of reaction buffer by initial denaturation temperature at 98 °C for 2 min followed by 32 cycles of 98 °C for 10 s, 65 °C for 15 s, and 72 °C for 90 s with a final 10 min extension at 72 °C. The Ec-glmU and Bs-glmU genes were digested with BamH I and Xho I (Fermentas of Thermo Scientific, Pittsburgh, PA), then ligated with the double digested pET21a vector (Novagen, Wisconsin, USA) yielding the plasmids pET-Ec-glmU and pET-Bs-glmU, respectively. Competent E. coli DH5α cells (Invitrogen, CA, USA) were used as the host for cloning. Transformed cells were spread on Luria-Bertani agar plates containing 100 ng μL−1 of ampicillin and incubated at 37 °C overnight. The positive clones were identified by colony PCR and confirmed by DNA sequence analysis. Sequence similarity searches were performed with the BLAST program (http://www.ncbi.nlm.nih.gov/BLAST/).
2.3 Expression and purification of Ec-GlmU and Bs-GlmU
Plasmids pET-Ec-glmU and pET-Bs-glmU were transformed into E. coli BL21 (DE3) (Novagen, Madison, WI, USA). The recombinant bacteria were induced to express recombinant Ec-GlmU and Bs-GlmU by adding isopropyl-β-D-thiogalactopyranoside (IPTG) at a final concentration of 1 mM to a culture with OD600 of approximately 0.6–0.8 and incubated at 37 °C for 3 h. Cell cultures (1 L) were separated by centrifugation at 5000×g for 20 min. The cell pellets were re-suspended in 35 mL of binding buffer (20 mM Tris–HCl, 500 mM NaCl, 20% Glycerin, 50 mM imidazole, pH 8.0) and lysis with 450 sonication pulses (400 W, 3 s with a 5 s interval) cooled in an ice water bath.35–37 The suspension was centrifuged (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×g at 4 °C for 30 min), the supernatant passed through a 0.22 μm filter and applied to a HiTrap™ Chelating HP column (GE Healthcare, Piscataway, NJ, USA). The Ec-GlmU and Bs-GlmU were purified using the standard nickel affinity chromatography procedure with elution buffer (20 mM Tris–HCl, 500 mM NaCl, 20% glycerol, 500 mM imidazole, pH 8.0). The purity of the proteins was evaluated using SDS-PAGE and the concentrations were determined by a BCA Protein Assay Kit (Sangon, Shanghai, China). Elution fractions of the containing proteins of interest were held in storage buffer (20 mM Tris–HCl, 50 mM NaCl, 20% glycerin, pH 8.0) at −80 °C.
000×g at 4 °C for 30 min), the supernatant passed through a 0.22 μm filter and applied to a HiTrap™ Chelating HP column (GE Healthcare, Piscataway, NJ, USA). The Ec-GlmU and Bs-GlmU were purified using the standard nickel affinity chromatography procedure with elution buffer (20 mM Tris–HCl, 500 mM NaCl, 20% glycerol, 500 mM imidazole, pH 8.0). The purity of the proteins was evaluated using SDS-PAGE and the concentrations were determined by a BCA Protein Assay Kit (Sangon, Shanghai, China). Elution fractions of the containing proteins of interest were held in storage buffer (20 mM Tris–HCl, 50 mM NaCl, 20% glycerin, pH 8.0) at −80 °C.
2.4 Enzymatic activity
The glucosamine-1-phosphate acetyltransferase activities of the wild-type and mutant GlmU were measured by a colorimetric microplate assay with 5, 5′-dithiobis (2-nitrobenzoic acid, DTNB).38,39 The 50 μL reaction mixture (50 mM Tris–HCl, pH 8.0, 5 mM MgCl2, 0.5 mM GlcN-1-P, 0.5 mM AcCoA, and 0.5 ng purified GlmU protein) was incubated in a 96 well microtiter plate at 37 °C for different time gradients from 1 min to 10 hours. The reaction was terminated by addition of 50 μL stop solution (50 mM Tris–HCl, 6.4 M guanidine hydrochloride, pH 8.0) and the mixture was incubated for 10 min at 37 °C with 50 μL Ellman's reagent solution (0.2 mM DTNB in buffer [50 mM Tris–HCl, 1 mM EDTA, pH 8.0]).35 The production of TNB2−, generated from the reaction of CoA-SH and DTNB, was monitored at a wavelength of 412 nm. A blank control with GlcN-1-P and AcCoA with inactive GlmU was used to correct the trial errors generated from the –SH group of GlmU protein.2.5 Molecular modeling and docking
The 3D model of full-length Bs-GlmU was obtained by the homology modeling method on I-TASSER server.40 Five prokaryotic GlmU from E. coli K-12 (PDBID: 2OI5), M. tuberculosis Rv37 (PDBID: 3DK5-A), Streptococcus pneumonia (PDBID: 1HM9-A), Yersinia pestis (PDBID: 3FWW-A) and Haemophilus influenza (PDBID: 2VOH-A) were selected as structural models to improve the accuracy of modeling. The initial AcCoA binding mode to Bs-GlmU was modeled through alignment of the Ec-GlmU and Bs-GlmU structures. Clashes between Bs-GlmU and AcCoA were removed by 2000 step energy minimization using NAMD2 program with CHARMM22 force field.41 AcCoA force field parameter was assigned by the CHARMM General Force field (CGenFF) program on ParamChem server.42 The mutated structures were modeled by visual molecular dynamics,43 and 2000 step energy minimization applied to refine the structures. All structural representations were drawn by visual molecular Dynamics Software.2.6 In vitro mutagenesis
Multiple protein sequences, including target protein sequences, were aligned and analyzed. The amino acid sites selected for mutation are shown in the Table 1. In recent years, the method called Restriction Free (RF) cloning has been widely applied to insert desired DNA bases in mutation studies.44,45 This restriction free cloning PCR method was applied to construct the mutated genes from the wild-type Bs-glmU gene template using the primer pairs shown in Table 1. For double mutation in plasmid, this method was used with DNA sequence analysis for site mutations one by one to obtain the required plasmid. The GlmU proteins were expressed with a C-terminal 6 × His tag to allow protein purification that was provided by pET-21a (+) fused with an expression tag.| Mutants plasmid | Name | Sequence | Notes |
|---|---|---|---|
| a Mutated sites are marked in bold and underlined. | |||
| pBS-GlmUC380A | BS-GlmUC380A-F | GGCACTGATGTAAACCTGGGC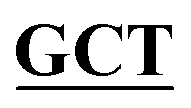 GGTTCAATTACTGTCAATTATGATGGA GGTTCAATTACTGTCAATTATGATGGA |
|
| BS-GlmUC380A-R | TCCATCATAATTGACAGTAATTGAACC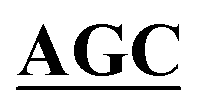 GCCCAGGTTTACATCAGTGCC GCCCAGGTTTACATCAGTGCC |
||
| pBS-GlmUC405S | BS-GlmUC405S-F | AAATTGAAGATGGCGCGTTTATCGGC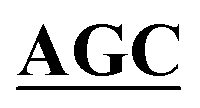 AATTCCAACTTGGTTGCCCCTGTC AATTCCAACTTGGTTGCCCCTGTC |
|
| BS-GlmUC405S-R | GACAGGGGCAACCAAGTTGGAATT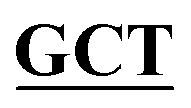 GCCGATAAACGCGCCATCTTCAATTT GCCGATAAACGCGCCATCTTCAATTT |
||
| pBS-GlmUC405A | BS-GlmUC405A-F | TGAAGATGGCGCGTTTATCGGC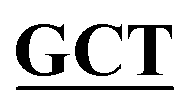 AATTCCAACTTGGTTGCCCCT AATTCCAACTTGGTTGCCCCT |
|
| BS-GlmUC405A-R | AGGGGCAACCAAGTTGGAATT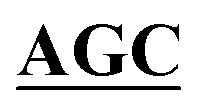 GCCGATAAACGCGCCATCTTCA GCCGATAAACGCGCCATCTTCA |
||
| pBS-GlmUC380A-C405S | BS-GlmUC380A-C405S-F | AAATTGAAGATGGCGCGTTTATCGGC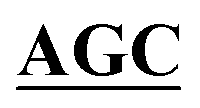 AATTCCAACTTGGTTGCCCCTGTC AATTCCAACTTGGTTGCCCCTGTC |
Gene of pBS-GlmUC380A as a template to construct pBS-GlmUC380A-C405S |
| BS-GlmUC380A-C405S-R | GACAGGGGCAACCAAGTTGGAATT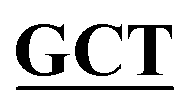 GCCGATAAACGCGCCATCTTCAATTT GCCGATAAACGCGCCATCTTCAATTT |
||
| pBS-GlmUV386C-C405S | BS-GlmUV386C-C405S-F | AAACCTGGGC GGTTCAATTACT GGTTCAATTACT AATTATGATGGAAAGA AATTATGATGGAAAGA |
|
| BS-GlmUV386C-C405S-R | TCTTTCCATCATAATT AGTAATTGAACC AGTAATTGAACC GCCCAGGTTT GCCCAGGTTT |
||
2.7 Kinetic studies of GlmU acetyltransferase
The initial velocity of bifunctional GlmU was closely associated with both the reaction time and concentration curves.46 Conditions to optimize the initial velocity of GlmU were investigated in the ranges of pH 3–11 at 20–80 °C for temperature, and 0–25 mM for Mg2+ concentration. The steady-state kinetic parameters Km and Vmax for two substrates (GlcN1P and AcCoA) were determined at various concentrations (0.05 to 5 mM) of one substrate maintaining the other substrate at fixed value (0.5 mM) for 10 min under optimal reaction conditions. The fixed value (0.5 mM) has been saturated for all GlmU enzymes by the pre-experiment results (data not show). All measurements were performed in triplicate and the data were analyzed by GraphPad software program (GraphPad Software, Inc. CA, USA).3 Results and discussion
3.1 Expression and purification of Ec-G lmU and Bs-GlmU in E. coli
The Ec-GlmU and Bs-GlmU were expressed as C-terminal 6 × His tag fusion proteins using pET21a expression vectors in E. coli BL21 (DE3). The CA tag was fused at the N-terminal of Ec-GlmU and Bs-GlmU to achieve high yield. Ec-GlmU and Bs-GlmU have shown protein bands corresponding to about 52 kDa in SDS-PAGE (Fig. 1a and b). Both Ec-GlmU and Bs-GlmU were expressed in the soluble fraction with a yield of about 85% after 3 h induction with IPTG (Fig. 1a and b). No more target proteins was produced in longer induction time. The proteins then were purified by affinity chromatography and both purity reached 95% detected by SDS-PAGE gel (Fig. 1c and d).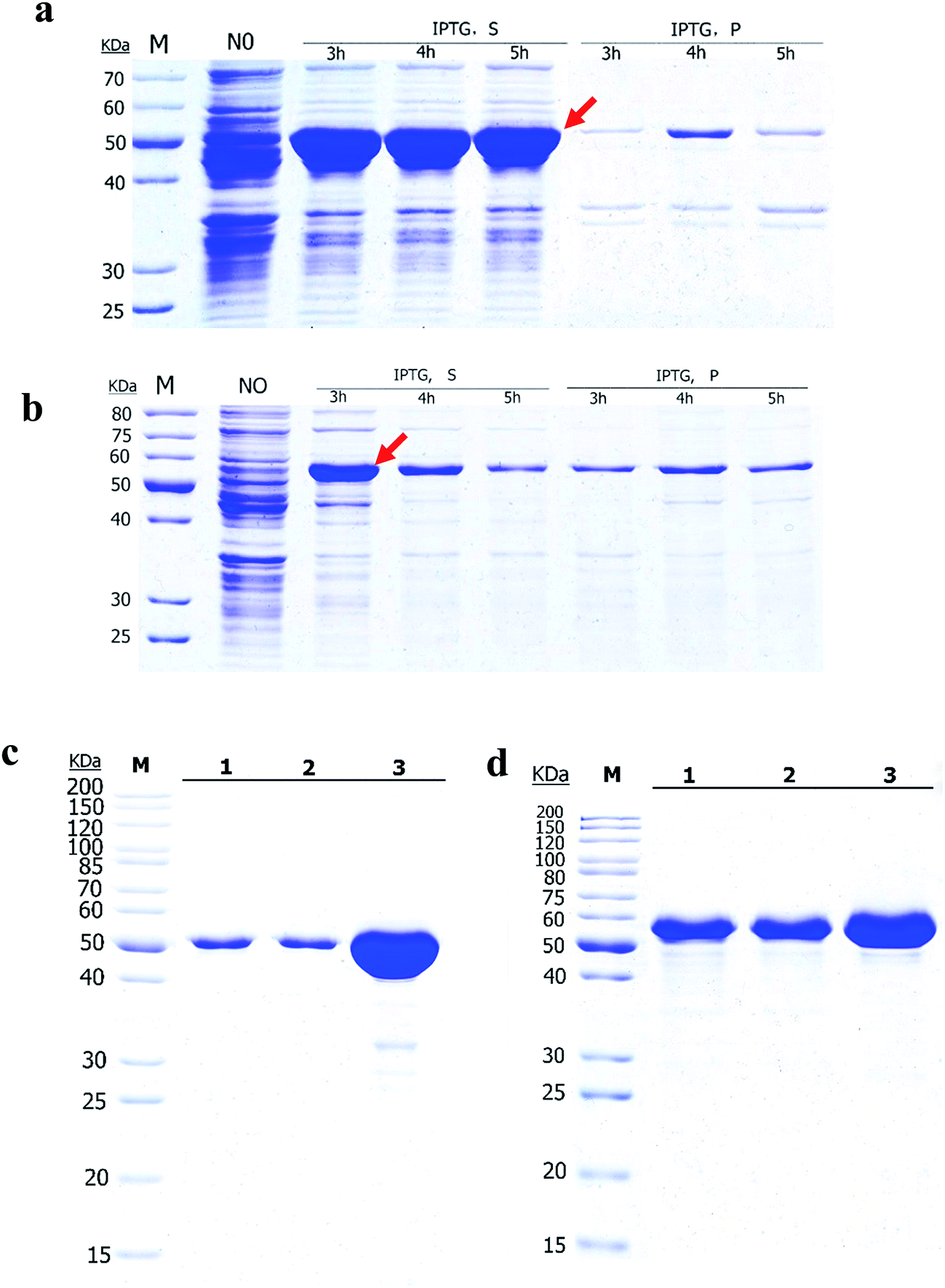 | ||
| Fig. 1 Expression and purification of Bs-GlmU and Ec-GlmU in BL21 (DE3) cells. SDS-PAGE analysis of Ec-GlmU (a and c) and Bs-GlmU (b and d). A total of 1 mM IPTG was added into the cells when OD600 reach to 0.4–0.6. “NO” indicates that the cells were not induced with IPTG. Cells were induced with IPTG for 3, 4, or 5 h respectively. “IPTG S” means supernatant fractions of induced cells after sonication; “IPTG P” indicates the insoluble pellets of induced cells after sonication. “1, 2, 3” in (c) and (d) indicates different concentrations of Ec-GlmU and Bs-GlmU. Positions corresponding to Ec-GlmU and Bs-GlmU are marked by arrows. Optimal conditions of 1 mM IPTG and 3 h expression were used for the purified Ec-GlmU, Bs-GlmU. M, protein marker. | ||
3.2 Characterization and analysis of acetyltransferase activities of Ec-GlmU and Bs-GlmU
Elman's Reagent (DTNB) reacted with materials containing a-SH group and produces yellow TNB2−, which has particularly absorption at 412 nm.38 This assay with high sensitivity was used to detect the –SH produced from AcCoA by Ec-GlmU and Bs-GlmU acetyltransferase catalysis.38 The reaction with Ec-GlmU reaches equilibrium in 30 min however Bs-GlmU need 6 hours. Furthermore, the total –SH produced from AcCoA by Ec-GlmU was 2.5 fold than that produced by Bs-GlmU (Fig. 2). The acetyltransferase activities of Ec-GlmU was similar to other reports.47 Also, the GlmU acetyltransferase activity was tested from pH 3 to 11 and the optimal pH for both enzymes was 8.0 (Fig. 3a). The GlmU acetyltransferase activity was observed from 20 to 80 °C and there was a little difference in the optimal temperature of Ec-GlmU (30 °C) and Bs-GlmU (37 °C) (Fig. 3b). The concentration of Mg2+ had no effect on the enzymatic activity of Ec-GlmU, but Bs-GlmU activity was optimal at 15 mM (Fig. 3c.). According to these data analysis, the huge difference about the acetyltransferase activities between Ec-GlmU and Bs-GlmU illustrated that both catalysis mechanisms was not identical. Further virtual amino acid mutation on Bs-GlmU was implemented to interpret these differences.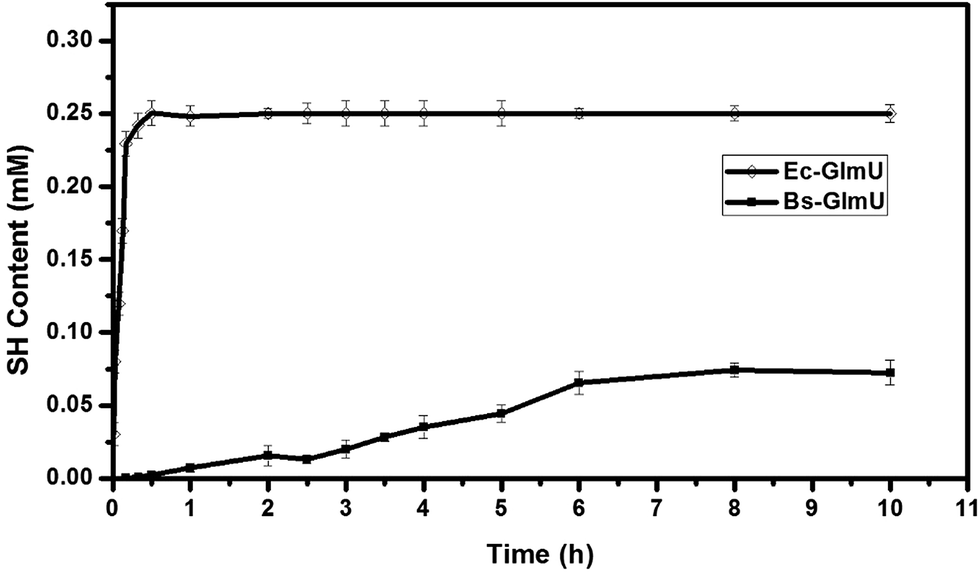 | ||
| Fig. 2 Characterization and analysis of acetyltransferase activities between Ec-GlmU and Bs-GlmU. A colorimetric assay coupled with DTNB was taken for the detection of acetyltransferase activities. The acetyltransferase activities of Ec-GlmU, open diamonds; Bs-GlmU, filled square were measured in triplicate. | ||
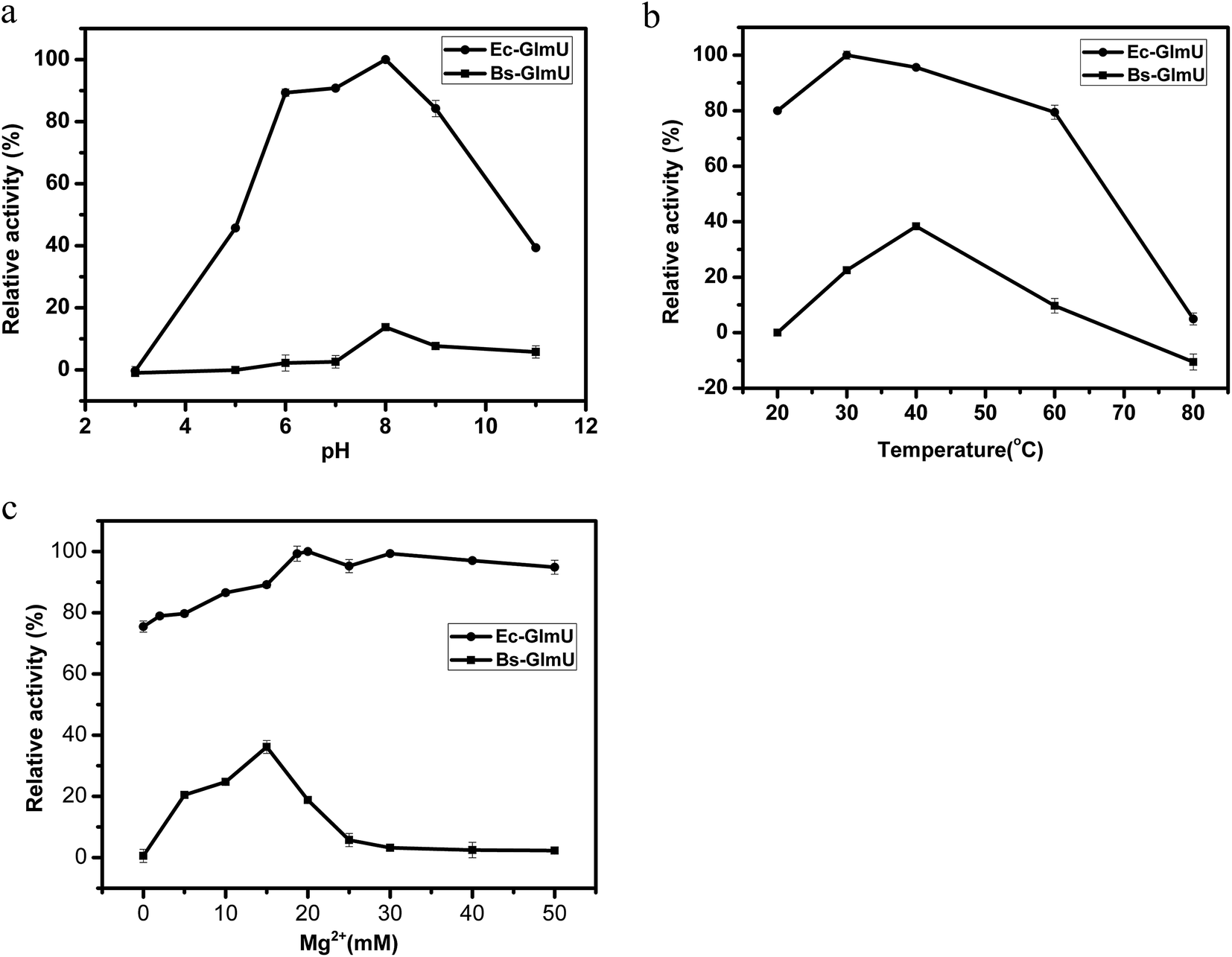 | ||
| Fig. 3 Determination of optimal conditions for Ec-GlmU and Bs-GlmU. (a) The effect of pH (3–11) on Ec-GlmU (solid round) and Bs-GlmU (filled square) acetyl transferase activities. pH buffers with final concentration of 50 mM were as follows: citric acid–sodium citrate (pH 3.0); NaH2PO4–Na2HPO4 (pH 5.0–6.0); Tris–HCl (pH 7.0–9.0); NH4Cl–NH3H2O (pH 11). (b) Different incubation temperatures on the acetyl transferase activities of Ec-GlmU and Bs-GlmU. (c) Different concentrations of Mg2+. | ||
3.3 Structural model of Bs-GlmU and virtual amino acid mutation analysis
GlmU is considered one of the most promising antibacterial drug targets because the first reaction catalyzed by this enzyme is absent in mammals.48 E. coli and B. subtilis are two normal models for Gram-negative and Gram-positive bacteria, respectively. Studies of GlmU form E. coli and B. subtilis are extremely instructive for the development of new antimicrobial agents. The X-ray crystal structure of the Ec-GlmU acetyltransferase active site confirmed the 2-amino group of GlcN-1-PO4 is positioned in proximity to the AcCoA to facilitate direct attack on the thioester by a ternary complex mechanism.31 However, the mechanism of Bs-GlmU has not been reported.The acetyltransferase active site, defined by the binding site for AcCoA, makes use of residues from all three subunits of GlmU and is positioned beneath an open cavity large enough to accommodate the Glc-1-PO4 acetyl acceptor.31 The 2-amino group of GlcN-1-PO4 is positioned in proximity to AcCoA to facilitate direct attack of the thioester by a ternary complex mechanism (based on the structure of E. coli bifunctional GlmU).47 The Protein Data Bank (PDB) was BLAST searched for similar sequences using the target sequence (PDB ID: 3TWD-B, 3DK5-A, 2VOH-A, 3FWW-A, 1HM9-A) and the results were aligned (Fig. 4) to build a tertiary structure of monomeric Bs-GlmU (Fig. 5a) for homology modeling. The quality of the Bs-GlmU model was assessed by Ramachandran statistics49 and Profile-3D.50 93.2% of the total amino acids were located in the optimum region of the Ramachandran plot, with 3.4% in the allowed region and 3.4% in the disallowed region (Fig. 5b). The compatibility score of the majority of the amino acids was greater than zero in the Profile-3D graph (Fig. 5c). These structure evaluation analyses showed that the model represents the Bs-GlmU structure.
 | ||
| Fig. 4 Sequence alignment of Bs-GlmU and other GlmU amino acid sequence. Five GlmU from the following microorganism were selected: Escherichia coli K12, Mycobacterium tuberculosis H37Ra, Haemophilus influenzae, Yersinia pestis pesticides, Streptococcus pneumoniae. The residues in red box are those involved in acetyl transferase activities. The residues marked with asterisk are the key amino acid which was mutated in this study. | ||
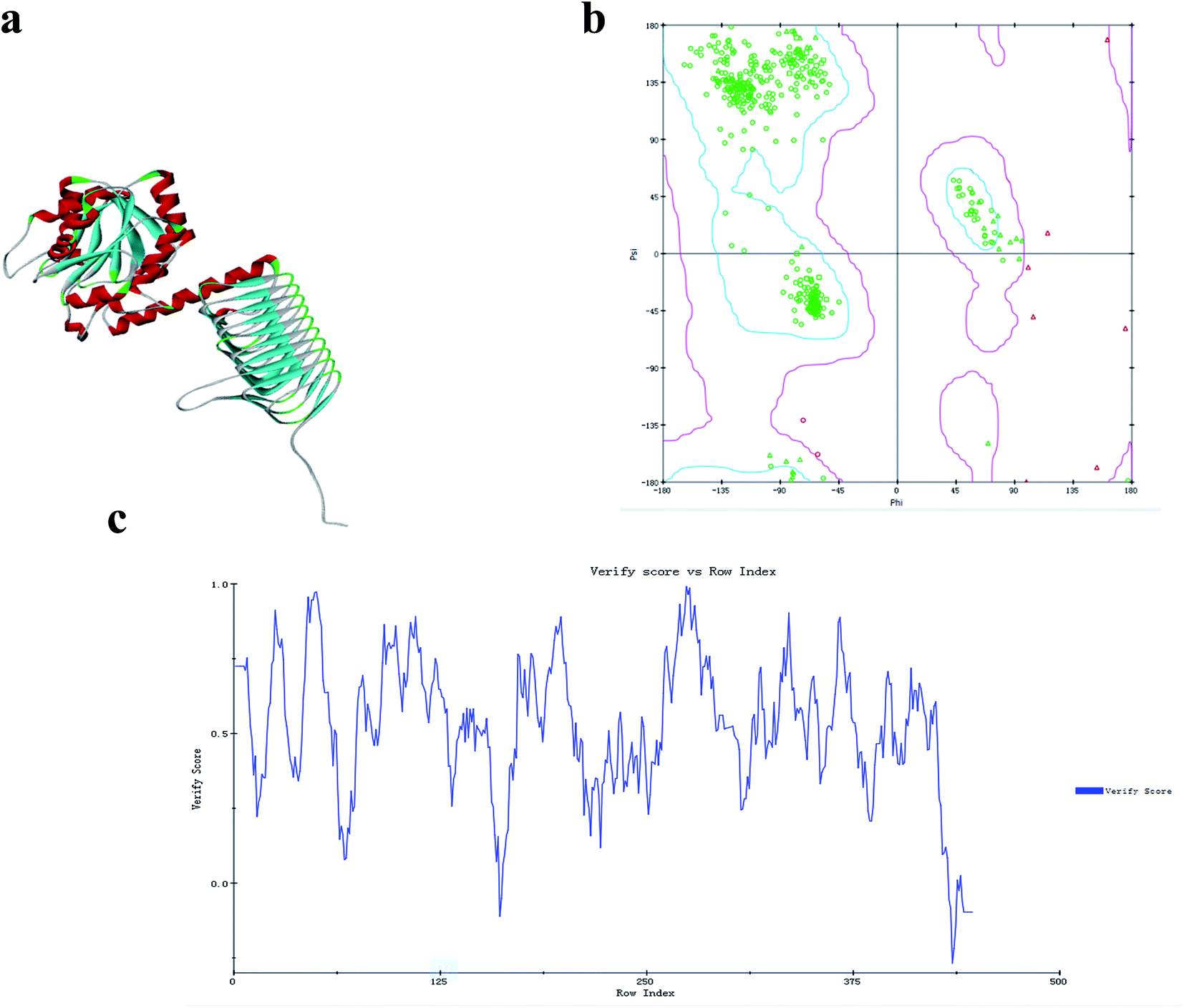 | ||
| Fig. 5 Model of the monomeric structure of Bs-GlmU. (a) The modeled three-dimensional structure of Bs-GlmU. (b) The Ramachandran statistics of the simulated Bs-GlmU structure, showing the residues in the optimum region (blue), the allowed region (magenta), and the disallowed region (red triangle). (c) The structure of Bs-GlmU that was verified using Profile-3D. | ||
The result of the sequence alignment (Fig. 4) has shown that there were several cysteine residues in acetyltransferase region of Bs-GlmU, such as C380, C405 and so on. It is wonder that the disulfide bonds between cysteine residues in this region affected the activity of the acetyltransferase. The model of Bs-GlmU binding a substrate AcCoA was simulated (Fig. 6a). The acetyltransferase catalytic site interacts with AcCoA through a hydrogen bond between residue S362 and carbonyl group of AcCoA and through van der Waals interactions. The close distance between C380 and C405 suggested that these two residues could form a disulfide bond. The resulting disulfide bond has a weak interaction with the carbon atom of acetyl carbonyl group of AcCoA (Fig. 6a). Moreover, the Km value of AcCoA to Bs-GlmUWT was calculated (Fig. 8a) as 1.821 mM. Based on this result, it is speculated that the disulfide bond formed between C380 and C405 could seriously affect acetyltransferase of Bs-GlmU. In order to verify this speculation, the purified Bs-GlmU was dialyzed to buffer S containing DTT from 5 mM to 10 mM to break disulfide bonds formed by cysteine residues. The results showed that the acetyltransferase activity indeed improved in varying degrees when Bs-GlmU was dialyzed to different concentrations of DTT buffer (data not shown). It illustrated that the existence of DTT have interrupted disulfide bonds formed by cysteine residues.
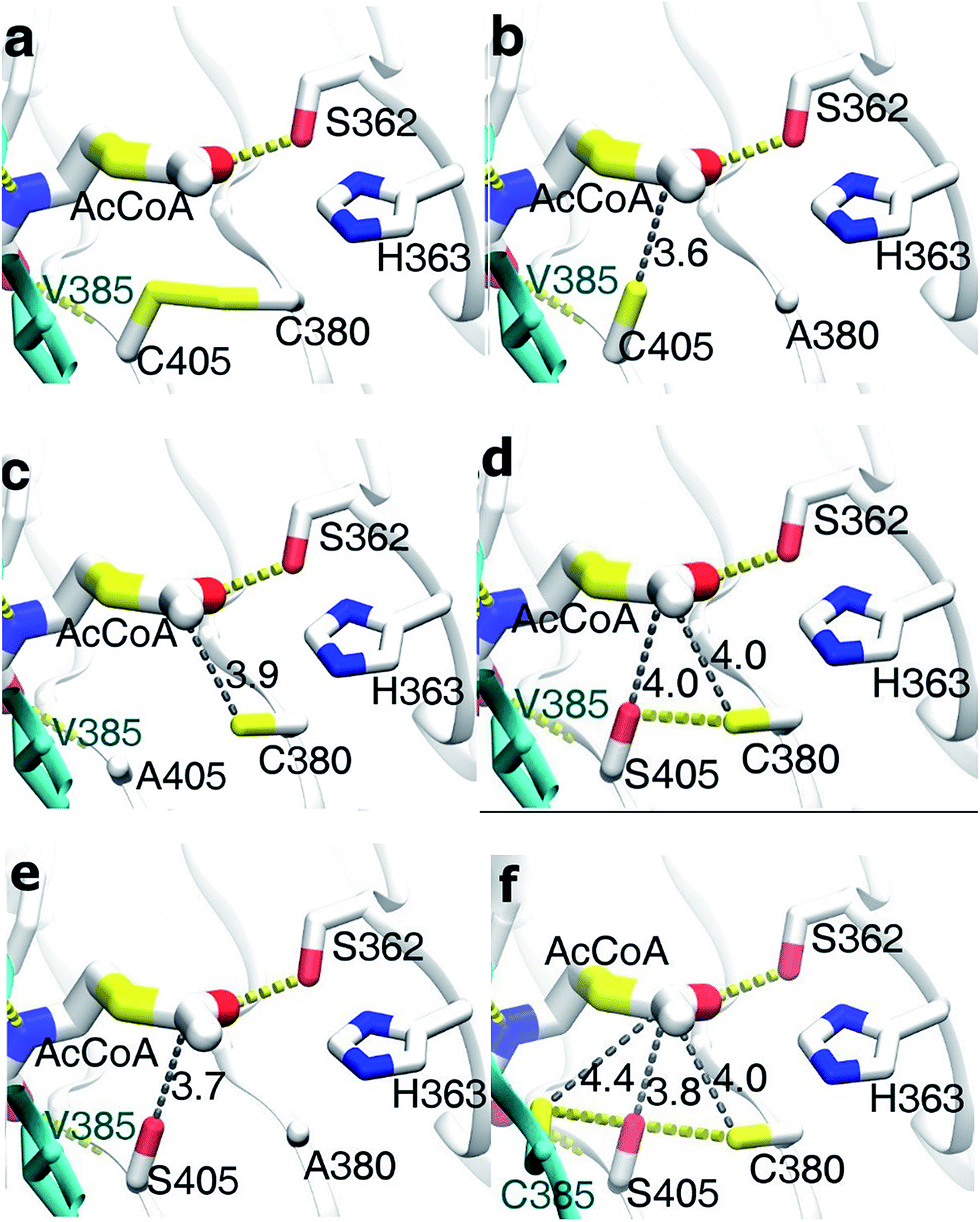 | ||
| Fig. 6 Virtual amino acid mutation analysis and mutant model docking with AcCoA. H-bonds are shown in yellow dotted lines and hydrophobic interaction is indicated as gray dotted line. Arabic numerals next to the gray dotted line represent distance of hydrophobic interaction (Å). (a) Bs-GlmUWT binding AcCoA model (in this model, yellow sticks between C380 and C405 indicated the disulfide bond); (b) Bs-GlmUC380A binding AcCoA model; (c) Bs-GlmUC405A binding AcCoA model; (d) Bs-GlmUC405S binding AcCoA model; (e) Bs-GlmUC380A-C405S binding AcCoA model; (f) Bs-GlmUV385C-C405S binding AcCoA model. | ||
Based on this pre-experimental basis, virtual amino acid mutation analysis was used to design a mutant A (Bs-GlmUC380A) to break the disulfide bond between C380 and C405. The Bs-GlmUC380A mutant binding AcCoA model shows the negative charged sulfhydryl group of C405 interacts with the acetyl carbonyl carbon of AcCoA at distance 3.6 Å (Fig. 6b), which transition state is more stable than the original enzyme. Then the purified mutant protein Bs-GlmUC380A was used to determine the enzyme activity and kinetic parameters (Fig. 7 and 8b). Not surprisingly, the acetyltransferase activity of Bs-GlmUC380A was 2.6 folds high than that of Bs-GlmUWT. Moreover, reaction time of Bs-GlmUC380A to reach equilibrium phase (from 8 h to 30 min) reduced significantly (Table 2). The kcat/Km value of the Bs-GlmUC380A mutant increased 67.1 fold because of the disulfide bond breakdown. The Km of AcCoA to Bs-GlmUC380A also decreased (Table 2). All results indicated that breaking the disulfide bonds formed by C380 and C405 could obviously enhance the acetyltransferase activity.
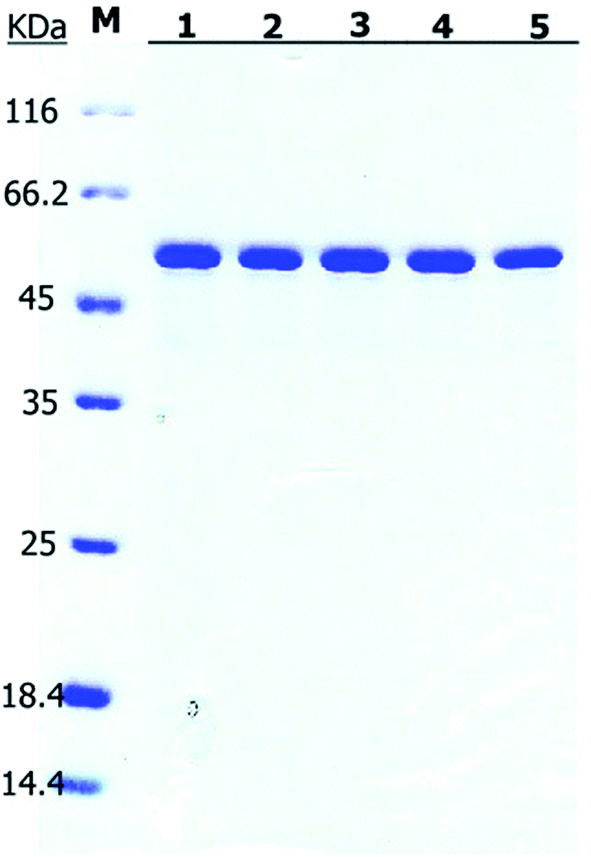 | ||
| Fig. 7 SDS-PAGE of the purified Bs-GlmU mutants. M: protein marker; (1) Bs-GlmUC380A; (2) Bs-GlmUC405S; (3) Bs-GlmUC380A-C405S; (4) Bs-GlmUC405A; (5) Bs-GlmUV385C-C405S. | ||
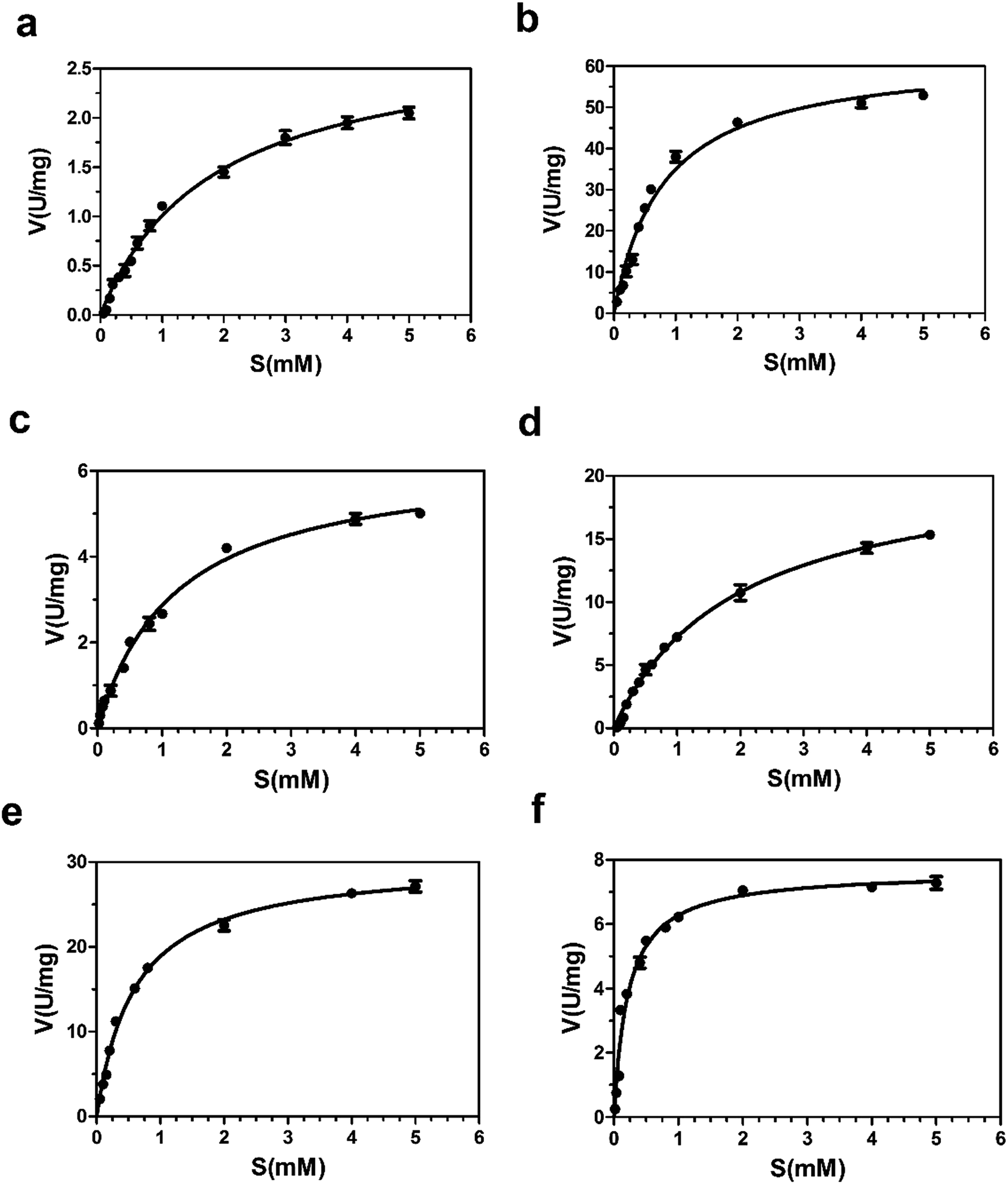 | ||
| Fig. 8 Nonlinear Michaelis–Menten plots of the purified Bs-GlmUWT and other five mutants with AcCoA. The steady-state kinetic parameters Km and Vmax for substrates AcCoA were determined at various concentrations (0.05, 0.1, 0.15, 0.2, 0.3, 0.4, 0.5, 0.6, 0.8, 1.0, 2.0, 4.0, 5.0 mM) of AcCoA maintaining substrate GlcN1P at fixed value (0.5 mM) under optimal reaction conditions. (a) Bs-GlmUWT (b) Bs-GlmUC380A; (c) Bs-GlmUC405A; (d) Bs-GlmUC405S; (e) Bs-GlmUC380A-C405S; (f) Bs-GlmUV385C-C405S; the unit of “U” here is defined as: mmol min−1. | ||
| Mutants | Properties | GlcN-1-P | AcCoA | |||||
|---|---|---|---|---|---|---|---|---|
| Stationary time (h) | Optimal activity (mM) | Km (mM) | kcat (s−1) | kcat/Km (mM s−1) | Km (mM) | kcat (s−1) | kcat/Km (mM s−1) | |
| BS-GlmUWT | 8 | 0.08 ± 0.01 | 0.333 ± 0.012 | 0.810 ± 0.014 | 2.432 ± 1.166 | 1.821 ± 0.341 | 2.459 ± 0.241 | 1.350 ± 0.707 |
| BS-GlmUC380A | 0.5 | 0.21 ± 0.02 | 0.241 ± 0.010 | 25.441 ± 0.051 | 105.564 ± 5.100 | 0.603 ± 0.068 | 54.617 ± 0.074 | 90.546 ± 1.088 |
| BS-GlmUC405S | 2 | 0.10 ± 0.01 | 0.359 ± 0.014 | 2.407 ± 0.009 | 6.705 ± 0.643 | 1.963 ± 0.481 | 18.512 ± 0.472 | 9.430 ± 0.981 |
| BS-GlmUC380A-C405S | 0.5 | 0.18 ± 0.03 | 0.470 ± 0.007 | 30.783 ± 0.010 | 65.496 ± 1.429 | 0.638 ± 0.252 | 26.147 ± 0.347 | 40.977 ± 1.377 |
| BS-GlmUC405A | 6 | 0.11 ± 0.02 | 0.278 ± 0.013 | 3.112 ± 0.007 | 11.194 ± 0.538 | 1.213 ± 0.347 | 5.496 ± 0.294 | 4.531 ± 0.847 |
| BS-GlmUV385C-C405S | 6 | 0.17 ± 0.02 | 0.304 ± 0.005 | 15.756 ± 0.008 | 51.829 ± 1.600 | 0.199 ± 0.024 | 6.629 ± 0.032 | 33.262 ± 1.333 |
To investigate this phenomenon, mutant B (Bs-GlmUC405A) was constructed. Surprisingly, the Bs-GlmUC405A mutant binding AcCoA model showed that the negatively charged sulfhydryl group of C380 released in Bs-GlmUC405A had a little longer distance (3.9 Å) to carbonyl carbon atom of AcCoA (Fig. 6c). This longer distance may not be able to improve stability of the transition state. It is predictable that acetyltransferase activities were not apparently increased. The kinetic assay experiment (Fig. 8c) was also consistent with the modeling assay and showed only a slightly increase in activity (p > 0.05). It suggested that not only the disulfide bonds formed by C380 and C405 can greatly decrease the acetyltransferase activities of Bs-GlmU, but also C405 played a key role in acetyltransferase activities of Bs-GlmU.
In order to validate this conclusion, we designed the mutant C (Bs-GlmUC405S) which substitute cysteine with serine. Both S405 and C380 interact with the acetyl carbonyl carbon of AcCoA and increase stability of the transition state. However, the interaction force was weaker than that of the Bs-GlmUC380A mutant with a longer distance between the protein and substrate (4.0 Å) (Fig. 6d). In addition, there was a hydrogen bond between side chains S405 and C380 that affected the stability of the transition state. In the experiment, the slightly improved stability of the transition state in Bs-GlmUC405S increased slightly (8 fold) in the kcat/Km value (Table 2 and Fig. 8d). The residue C405 replaced by A or S reduced greatly the enzyme activity which demonstrates that the residue C405 is indeed a key amino acid in the acetyltransferase of Bs-GlmU. Additionally, a double mutant D (Bs-GlmUC380A-C405S) was constructed. In the Bs-GlmUC380A-C405S mutant binding AcCoA model, neither disulfide nor hydrogen bonds formed between the 380 and 405 positions (Fig. 6e). The close distance (3.7 Å) between the hydroxy group and carbonyl carbon atom of AcCoA indicated that the C380A-C405S mutation enhanced stability of transition state compared with the C405S mutation. This distance (3.7 Å) was a little longer than Bs-GlmUC380A (3.6 Å). The result of kinetic assay (Fig. 8e) was consistent with the modeling assay and showed that C380A-C405S double mutant has an increased kcat/Km value compared to Bs-GlmUWT (Table 2). Above results indicated that A380 could be an essential amino acid which conferred Bs-GlmU a higher acetyltransferase activity than Ec-GlmU. Finally, a comprehensive analysis conducted to all acetyltransferase activities data, this paper draws such a conclusion that C405 and A380 may act as an important role in acetyltransferase activity of Bs-GlmU.
As reported by Zhang and Olsen, C385 from one monomer of GlmU interact with S405 from another monomer, which could enhance the stability of the Ec-GlmU trimer.23,51 Mutant E (Bs-GlmUV385C-C405S) was constructed to test this interaction between amino acids. The model of Bs-GlmUV385C-C405S binding AcCoA implied that C385, S405 and C380 interact with the pantetheine of AcCoA through hydrophobic interactions. The hydrogen bonds between C385-S405 and C380-S405 also enhance the stability of the transition state (Fig. 6f). The kcat/Km value of the mutant Bs-GlmUV385C-C405S supported the statement (Table 2 and Fig. 8f).
4 Conclusion
As an essential substrate for two important biosynthetic pathways of peptidoglycan and lipid A in bacteria, studies on the GlmU enzyme has attracted increasing interests from both the industrial and the academic in recent decades. In this work, GlmU enzymes from E. coli and B. subtilis were successfully expressed and purified. Compared with Ec-GlmU and GlmU from other microorganisms, Bs-GlmU showed significantly lower acetyltransferase activity. By analyzing the result of the sequence alignment, we predicted the structure of Bs-GlmU and constructed five Bs-GlmU mutants by varying residues close to the acetyltransferase sites. The huge difference in acetyltransferase activity between mutants and Bs-GlmUWT demonstrated that the C405 and A380 residue may play a key role in Bs-GlmU acetyltransferase activity. The studies on amino acid mutation in acetyltransferase of Bs-GlmU provided an important way to comprehensive understand reaction mechanism of Bs-GlmU and set up a platform for the future research on GlmU enzyme in other Gram-positive microorganisms. This work supported the development of GlmU inhibitors to potential new anti-bacterial drugs.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 21306055), Guangdong Natural Science Foundation (Grant no. 2014A030313261), the Ph.D. Programs Foundation of Ministry of Education of China (Grant no. 20130172120041), the Fundamental Research Funds for the Central Universities (Grant no. 2015ZM161) and Special Funds for the Cultivation of Guangdong College Students' Scientific and Technological Innovation (Climbing Program Special Funds, Grant no. pdjh2015b0048). We are grateful to Xuwan Dong and Bihong Li for technical support with the clones.References
- G. Joanny, D. J. Le, D. Bréchemier-Baey, V. Labas, J. Vinh, P. Régnier and E. Hajnsdorf, Nucleic Acids Res., 2007, 35, 2494–2502 CrossRef CAS PubMed.
- J. S. Rush, P. D. Rick and C. J. Waechter, Glycobiology, 1997, 7, 315–322 CrossRef CAS PubMed.
- M. Bardor, R. Louvet, B. Sanit-Jean, C. Burel, B. Baiet, A. Carlier, J. P. Cadoret and P. Lerouge, EP, EP2412815, 2012.
- M. L. Reitman, L. Lang and S. Kornfeld, Methods Enzymol., 1984, 107, 163–172 CAS.
- S. Merello, A. J. Parodi and R. Couso, J. Biol. Chem., 1995, 270, 7281–7287 CrossRef CAS PubMed.
- J. Rodríguezdíaz, A. Rubiodelcampo and M. J. Yebra, Bioengineered, 2012, 3, 339–342 CrossRef PubMed.
- A. H. L. Koenderman, P. L. Loppen, A. M. M. Lian and D. H. V. D. Eijnden, Biomed. Chromatogr., 1986, 1, 104–108 CrossRef CAS PubMed.
- G. L. French, Int. J. Antimicrob. Agents, 2010, 36, S3–S7 CrossRef CAS PubMed.
- I. M. Gould, Int. J. Antimicrob. Agents, 2010, 36, S1–S2 CrossRef CAS PubMed.
- R. E. Isturiz, Int. J. Antimicrob. Agents, 2010, 36(5), S19–S22 CrossRef CAS PubMed.
- R. G. Masterton, Int. J. Antimicrob. Agents, 2010, 36(5), S15–S18 CrossRef CAS PubMed.
- T. R. Walsh, Int. J. Antimicrob. Agents, 2010, 36(3), S8 CrossRef CAS PubMed.
- G. L. Moraes, G. C. Gomes, P. R. Monteiro de Sousa, C. N. Alves, T. Govender, H. G. Kruger, G. E. Maguire, G. Lamichhane and J. Lameira, Tuberculosis, 2015, 95, 95–111 CrossRef CAS PubMed.
- R. Mehra, C. Rani, P. Mahajan, R. A. Vishwakarma, I. A. Khan and A. Nargotra, ACS Comb. Sci., 2016, 18(2), 100–116 CrossRef CAS PubMed.
- P. Doig, P. A. Boriacksjodin, J. Dumas, J. Hu, K. Itoh, K. Johnson, S. Kazmirski, T. Kinoshita, S. Kuroda and T. O. Sato, Bioorg. Med. Chem., 2014, 22, 6256–6269 CrossRef CAS PubMed.
- S. Wang, F. Xuan, Y. Liu, X. W. Liu, W. Lin, J. Fang and G. W. Peng, Carbohydr. Res., 2015, 413, 70–74 CrossRef CAS PubMed.
- N. Vithani and B. Prakash, Encyclopedia of Inorganic and Bioinorganic Chemistry, 2015 Search PubMed.
- C. Rani, R. Mehra, R. Sharma, R. Chib, P. Wazir, A. Nargotra and I. A. Khan, Tuberculosis, 2015, 95(6), 664–677 CrossRef CAS PubMed.
- H. Barreteau, A. Kovac, A. Boniface, M. Sova, S. Gobec and D. Blanot, FEMS Microbiol. Rev., 2008, 32, 168–207 CrossRef CAS PubMed.
- M. A. Pathan, A. S. Jamkhandi and A. K. S. Katti, Int. J. Pharm. Sci. Res., 2014, 5(4), 1493 Search PubMed.
- M. Kotnik, P. S. Anderluh and A. Prezelj, Curr. Pharm. Des., 2007, 13, 2283–2309 CrossRef CAS PubMed.
- C. R. Raetz and C. Whitfield, Biochemistry, 2002, 71, 635–700 CrossRef CAS PubMed.
- Z. Zhang, E. M. M. Bulloch, R. D. Bunker, E. N. Baker and C. J. Squire, Acta Crystallogr., 2009, 65, 275–283 CrossRef CAS PubMed.
- V. K. Singh, K. Das and K. Seshadri, PLoS One, 2012, 7, e43969 CAS.
- J. Min, D. Lin, Q. Zhang, J. Zhang and Z. Yu, Eur. J. Med. Chem., 2012, 53, 150–158 CrossRef CAS PubMed.
- L. R. Olsen, M. W. Vetting and S. L. Roderick, Protein Sci., 2007, 16, 1230–1235 CrossRef CAS PubMed.
- D. Patin, M. Bayliss, D. Menginlecreulx, P. Oyston and D. Blanot, Arch. Microbiol., 2015, 197, 1–8 CrossRef PubMed.
- R. Mehra, R. Sharma, I. A. Khan and A. Nargotra, Eur. J. Med. Chem., 2015, 92, 78–90 CrossRef CAS PubMed.
- A. M. Gehring, W. J. Lees, D. J. Mindiola, C. T. Walsh and E. D. Brown, Biochemistry, 1996, 35, 579–585 CrossRef CAS PubMed.
- R. Sharma, M. R. Lambu, U. Jamwal, C. Rani, R. Chib, P. Wazir, D. Mukherjee, A. Chaubey and I. A. Khan, J. Biomol. Screening, 2016, 21(4), 342–353 CrossRef CAS PubMed.
- L. R. Olsen, M. W. Vetting and S. L. Roderick, Protein Sci., 2007, 16, 1230–1235 CrossRef CAS PubMed.
- X. Li, C. Qi, P. Wei, L. Huang, J. Cai and Z. Xu, Prep. Biochem. Biotechnol., 2016 Search PubMed , accepted.
- B. Hove-Jensen, J. Bacteriol., 1992, 174, 6852–6856 CrossRef CAS PubMed.
- B. Hove-Jensen, Mol. Microbiol., 1989, 3, 1487–1492 CrossRef CAS PubMed.
- Y. Zhou, Y. Xin, S. Sha and Y. Ma, Arch. Microbiol., 2011, 193, 751–757 CrossRef CAS PubMed.
- W. Zhang, V. C. Jones, M. S. Scherman, S. Mahapatra, D. Crick, S. Bhamidi, Y. Xin, M. R. Mcneil and Y. Ma, Int. J. Biochem. Cell Biol., 2008, 40, 2560–2571 CrossRef CAS PubMed.
- J. Zhou, L. Huang, J. Lian, J. Sheng, J. Cai and Z. Xu, Biotechnol. Lett., 2010, 32, 1481–1486 CrossRef CAS PubMed.
- Y. Zhou, Y. Xin, S. Sha and Y. Ma, Arch. Microbiol., 2011, 193, 751–757 CrossRef CAS PubMed.
- Y. Zhai, M. Liang, J. Fang, X. Wang, W. Guan, X.-w. Liu, P. Wang and F. Wang, Biotechnol. Lett., 2012, 34, 1321–1326 CrossRef CAS PubMed.
- C. Meersschaert, E. Thiry and P. P. Pastoret, Rev. Elev. Med. Vet. Pays. Trop., 1988, 41, 235–242 CAS.
- R. C. Troutman and C. Barraquer, Cornea, 1982, 1(3), 251–254 CrossRef.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes and I. Vorobyov, J. Comput. Chem., 2010, 31, 671–690 CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- T. Unger, Y. Jacobovitch, A. Dantes, R. Bernheim and Y. Peleg, J. Struct. Biol., 2010, 172, 34 CrossRef CAS PubMed.
- d. E. F. Van and J. Löwe, J. Biochem. Biophys. Methods, 2006, 67, 67–74 CrossRef PubMed.
- Y. Zhai, M. Liang, J. Fang, X. Wang, W. Guan, X. W. Liu, P. Wang and F. Wang, Biotechnol. Lett., 2012, 34, 1321–1326 CrossRef CAS PubMed.
- L. R. Olsen and S. L. Roderick, Biochemistry, 2001, 40(7), 1913–1921 CrossRef CAS PubMed.
- L. Yongmeng, Z. Yan, M. Yufang and L. Xuebing, Carbohydr. Res., 2011, 346, 1714–1720 CrossRef PubMed.
- R. A. Laskowski, M. W. MacArthur, D. S. Moss and J. M. Thornton, J. Appl. Crystallogr., 1993, 26, 283–291 CrossRef CAS.
- R. Lüthy, J. U. Bowie and D. Eisenberg, Nature, 1992, 356, 83–85 CrossRef PubMed.
- L. R. Olsen and S. L. Roderick, Biochemistry, 2001, 40, 1913–1921 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |