
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of pearl necklace-like ZnO–ZnWO4 heterojunctions with enhanced photocatalytic degradation of Rhodamine B
Yanyan Hao†
a,
Liyun Zhang†b,
Ying Zhang*a,
Lin Zhaoa and
Bingsen Zhang *b
*b
aCollege of Chemistry, Chemical Engineering and Environmental Engineering, Liaoning Shihua University, West No 1 Dandong Road, Wanghua District, Fushun 113001, China. E-mail: zhangying@lnpu.edu.cn
bShenyang National Laboratory for Materials Science, Institute of Metal Research, Chinese Academy of Sciences, 72 Wenhua Road, Shenyang 110016, China. E-mail: bszhang@imr.ac.cn
First published on 16th May 2017
Abstract
Pearl necklace-like ZnO–ZnWO4 heterojunctions composites have been designed and synthesized. The photocatalytic activity of the ZnO–ZnWO4 heterojunctions depended on the calcination temperature and ZnO–ZnWO4 molar ratio. The ZnO–ZnWO4 composites showed higher degradation efficiency than that of ZnO or ZnWO4 individually. The photocatalytic reaction was enhanced due to the heterojunctions construction, which improved charge separation of the photogenerated electron–hole pairs.
1 Introduction
Using semiconductor photocatalysts to solve environmental remediation, especially waste water decontamination, has attracted extensive attention.1 Zinc oxide (ZnO) plays an important role in degrading various organic pollutants due to its high activity, low cost, and environmental friendliness.2 In the past decade, various morphological ZnO, such as rods, sheets, flowers, hollow nanostructures, porous structures, and core–shell structures, have been synthesized by hydrothermal, electrospinning, and sol–gel methods.2,3 Despite achieving great success in the controlled synthesis of single-component ZnO, the photocatalytic efficiency still needs to be improved because of the rapid recombination reaction of photogenerated charge carriers for single semiconductor. Recently, heterostructure composite materials have attracted much attention, because it can improve the primary properties of the constituent materials and create novel functions. Some heterostructure composites have been developed by coupling ZnO with other semiconductors, such as ZnO–WO3,1b,4 ZnO–CdS,5 ZnO–TiO2,6 ZnO–BiVO4,7 and ZnO–SnO2.8 Only few works, however, focused on the synthesis of ZnO–ZnWO4 heterojunctions as well as their employment for the photodegradation of organic compounds.9Zinc tungstate (ZnWO4) has received considerable attention as an ultraviolet (UV) light driven photocatalyst due to its relatively high activity for degradation of organic compounds.10 Amouzegar et al. reported cubic-shaped nanocrystalline ZnWO4 prepared by microwave-assisted precipitation technique showed high photocatalytic activity for the degradation of aqueous solution of Rhodamine B (RhB).11 ZnWO4 was also efficient for the decomposition of malachite green,12 4-chlorophenol,13 and methyl orange.14 Herein, ZnO–ZnWO4 heterostructure with pearl necklace-like morphology was prepared by precipitation method followed by calcination process. Compared with single ZnO and ZnWO4 phase, the as-obtained ZnO–ZnWO4 heterostructure shows an enhanced photocatalytic activity in the photodegradation of RhB.
2 Experimental
2.1 Sample synthesis
For preparing the Zn(OH)2 precursor, NH4OH solution (25%) was slowly dropped into the vigorously stirred Zn(CH3COO)2·2H2O solution with 0.06 mol L−1 concentration until the pH value reached 10.6. The mixed solution was then heated at 80 °C for 2 h to produce Zn(OH)2 precipitate. The precipitate was collected by pump filter and rinsed three times with deionized water and ethanol, respectively. Subsequently, the washed precipitate was dried at 80 °C overnight. For preparing tungstic acid (H2WO4) precursor, 15 g ammonium tungstate was dissolved in 150 mL deionized water under vigorous stirring for 30 min. Following, 30 mL nitric acid (3 mol L−1) solution was slowly added into the above solution and the mixed solution was further stirred for 1 h at 80 °C. The precipitate produced was collected and washed three times with deionized water and ethanol, respectively. The washed sediment was dried at 80 °C overnight to form the H2WO4 precursor.In order to obtain powders with ZnO/ZnWO4 molar ratios equalling to 1/0.04, 1/0.08, 1/0.12, the amounts of the precursors were opportunely determined. The corresponding samples were denoted as Z-ZW0.04, Z-ZW0.08 and Z-ZW0.12, respectively. A certain amount of Zn(OH)2 and H2WO4 with an equal number of Zn and W atoms were used to prepare pure ZnWO4. Pure ZnO was synthesized only using Zn(OH)2 as precursor. Finally, the photocatalysts were obtained by calcining the precursors for 2 h at different temperatures.
2.2 Characterization techniques
The phase composition and crystallinity of the prepared samples were analysed by X-ray diffraction (XRD) using a Rigaku-D diffractometer with Cu Kα radiation. The morphology was investigated by a scanning electron microscope (SEM), equipped with an energy dispersive X-ray spectrometer (EDS) suitable for element identification. Transmission electron microscopy (TEM) observation and scanning TEM (STEM)-EDS elemental mapping were performed by a FEI Tecnai G2 F20. UV-diffuse reflection spectra were recorded by a Perkin Elmer Lambda 900 spectrophotometer. The photoluminescence (PL) spectra of the photocatalysts were obtained by a Hitachi F-4500 spectrophotofluorometer with an excitation wavelength of 325 nm.2.3 Photocatalytic testing
RhB was used as model dye to evaluate the photocatalytic activity of ZnO, ZnWO4 and ZnO–ZnWO4 samples. Photodegradation of RhB was carried out in a 250 mL Pyrex reactor filled with deionized water (60 mL) containing RhB (10 mg L−1) and the photocatalysts (50 mg). The suspension was stirred for 30 min in the dark to obtain adsorption–desorption equilibrium of the dye before illumination. After irradiation with a 250 W Hg lamp (λ = 365 nm), a 5 mL aliquot was took out at different time intervals and immediately centrifuged. The RhB concentration in the clear solution was analysed by optical characteristic absorption at a wavelength of 553 nm for an RhB solution.3 Results and discussion
Fig. 1 shows the XRD patterns of ZnO, ZnWO4, and ZnO–ZnWO4 composites calcined at 850 °C for 2 h. The standard XRD patterns of ZnO (Fig. 1a) and ZnWO4 (Fig. 1b) are corresponding to the hexagonal wurtzite and monoclinic phases, respectively. The diffraction peaks of ZnO and ZnWO4 samples match well with the standard XRD patterns. A peak related to ZnO was detected at 2θ = 34.42° in Fig. 1b. No other impurity peaks were found in the XRD patterns. Fig. 1c displays the XRD patterns of the serials ZnO–ZnWO4 composites. The intensity of the distinct peak indicates the different percentage content of individual ZnO and ZnWO4, respectively. With increasing ZnWO4 content, the crystallite sizes of ZnO calculated by the Scherre's equation were 43.97 nm, 39.55 nm, 37.74 nm and 37.40 nm, respectively. It indicates that the crystal growth of ZnO is inhibited by ZnWO4.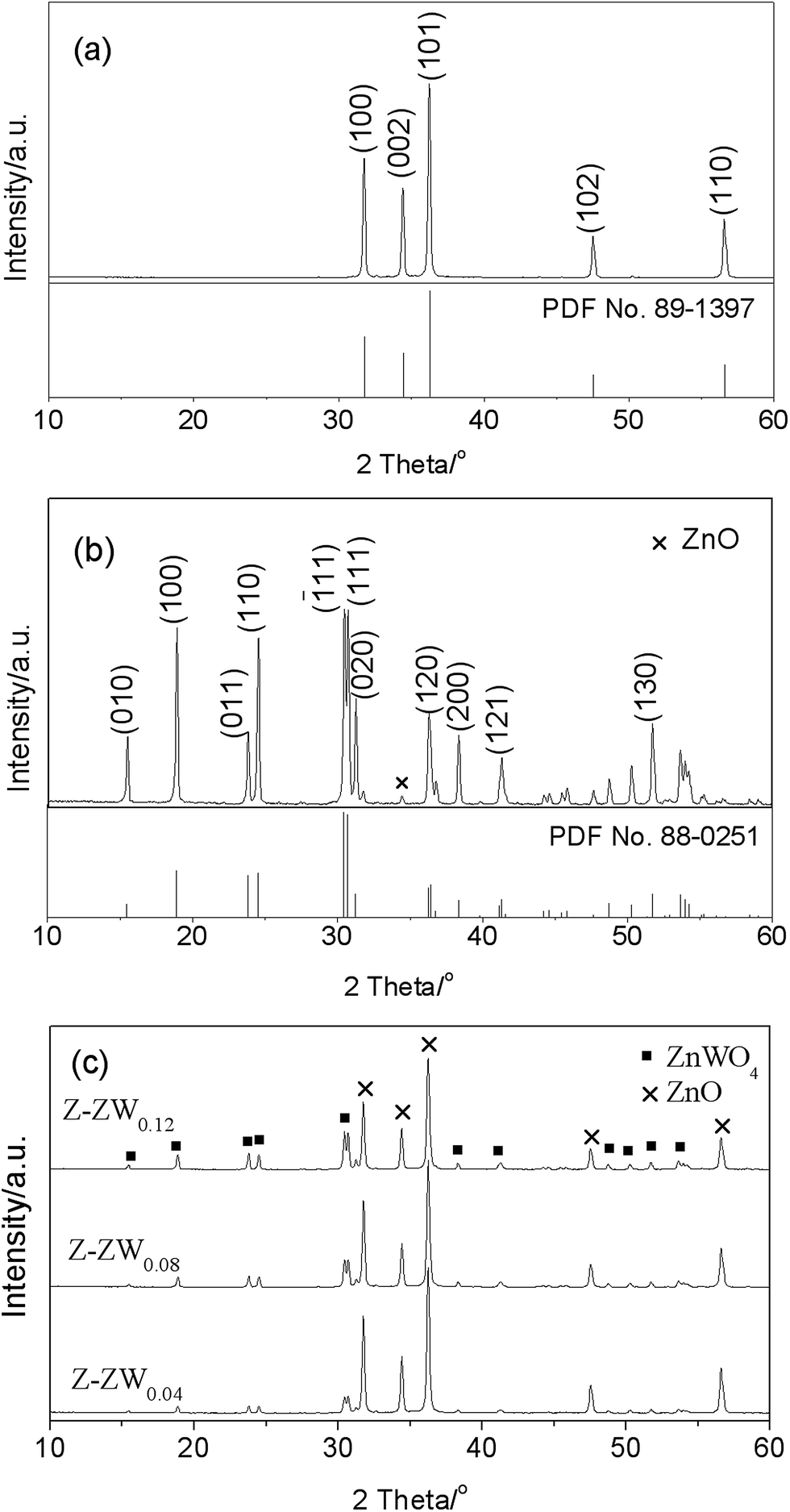 | ||
| Fig. 1 XRD patterns of ZnO (a), ZnWO4 (b), and ZnO–ZnWO4 (c) composites calcined at 850 °C for 2 h. | ||
Fig. 2 shows the representative XRD patterns of Z-ZW0.08 samples calcined for 2 h at 750 °C, 850 °C and 950 °C, respectively. With increasing temperature, the Full Width at Half Maximum (FWHM) of diffraction peaks (101) for ZnO and (111) for ZnWO4 are 0.249°, 0.219° and 0.189°, and 0.250°, 0.204° and 0.198°, respectively, showing the peaks of both phases become sharper. It indicates that the crystallinity can be improved by raising calcining temperature, which is consistent with the published works.3a
 | ||
| Fig. 2 XRD patterns of Z-ZW0.08 calcined for 2 h at different temperatures, 750 °C (a), 850 °C (b), and 950 °C (c). | ||
The morphologies of the samples were observed by SEM. Fig. 3 shows the SEM images of ZnO, ZnWO4, and ZnO–ZnWO4 heterojunctions calcined at 850 °C for 2 h. It displays the rod-like ZnO with average length 3 μm and the needle-like ZnO with average length 4.5 μm. After loading ZnWO4, the pearl necklace-like particles were found. Furthermore, hollow structures were observed at one side of the rod-like/needle-like particles as highlight by circles (Fig. 3b and c). With the increase of the ZnWO4 content, the pearl necklace-like morphology become predominant and the hollow structure diminished gradually, as shown in Fig. 3d. The particle size of ZnO decreased with increasing ZnWO4 content. ZnO with smaller particle size and crystallite size contributes to their influence on charge carrier recombination rate and specific surface area, which may enhance the photocatalytic performance.15 For the ZnWO4 (Fig. 3e), some particles adhere to slantprism-like ZnWO4 surface, while others agglomerated. Clearly, the morphologies of ZnO–ZnWO4 samples depend on the molar ratio of ZnO to ZnWO4.
 | ||
| Fig. 3 SEM micrographs of ZnO (a), Z-ZW0.04 (b), Z-ZW0.08 (c), Z-ZW0.12 (d), ZnWO4 (e), and EDS spectra of ZnWO4 (f and g). | ||
The element composition of the ZnWO4 sample was analyzed by EDS. Zn and W elements were detected in slantprism-like shape ZnWO4 sample and the element ratio of W to Zn was ca. 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (Fig. 3f, carbon derived from the conductive adhesive). However, only Zn and O elements were detected in agglomerated ZnWO4 particles (Fig. 3g). EDS confirmed that there is a small amount of ZnO in the ZnWO4 sample, which is consistent with the XRD result.
:1 (Fig. 3f, carbon derived from the conductive adhesive). However, only Zn and O elements were detected in agglomerated ZnWO4 particles (Fig. 3g). EDS confirmed that there is a small amount of ZnO in the ZnWO4 sample, which is consistent with the XRD result.
As one kind of ZnO–ZnWO4 heterojunctions, Z-ZW0.08 was further characterized by TEM and STEM-EDS elemental mapping. The low-magnification TEM image demonstrates that the Z-ZW0.08 shows pearl necklace-like morphology, as presented in Fig. 4a. The high-resolution TEM (HR-TEM) image (Fig. 4b) clearly displays that the ZnWO4 nanoparticles with a particle size of about 3 nm distribute on the surface of ZnO. The elemental mapping (Fig. 4c) shows that W element is uniform distributed in the sample, which demonstrates ZnWO4 nanoparticles distribute well on the surface of ZnO.
 | ||
| Fig. 4 Low-magnification TEM (a), HR-TEM (b) images and STEM-EDS elemental mapping (c) of Z-ZW0.08. | ||
The ultraviolet-visible (UV-vis) absorption spectra of ZnO, ZnWO4 and ZnO–ZnWO4 composites are shown in Fig. 5. The band gap values (Eg) of the photocatalysts were determined by extrapolating the linear part of the plots of (αhν)2 versus the energy of exciting light.9b The calculated band gap energies of ZnO, Z-ZW0.08, Z-ZW0.12, and ZnWO4 are 3.20 eV, 3.19 eV, 3.18 eV and 3.17 eV, respectively. It can be seen that the presence of ZnWO4 caused the absorption shift towards long wavelength region, which may result from the formation of the energy level of vacancy oxygen since W6+ was doped into the crystal lattice of ZnO.16
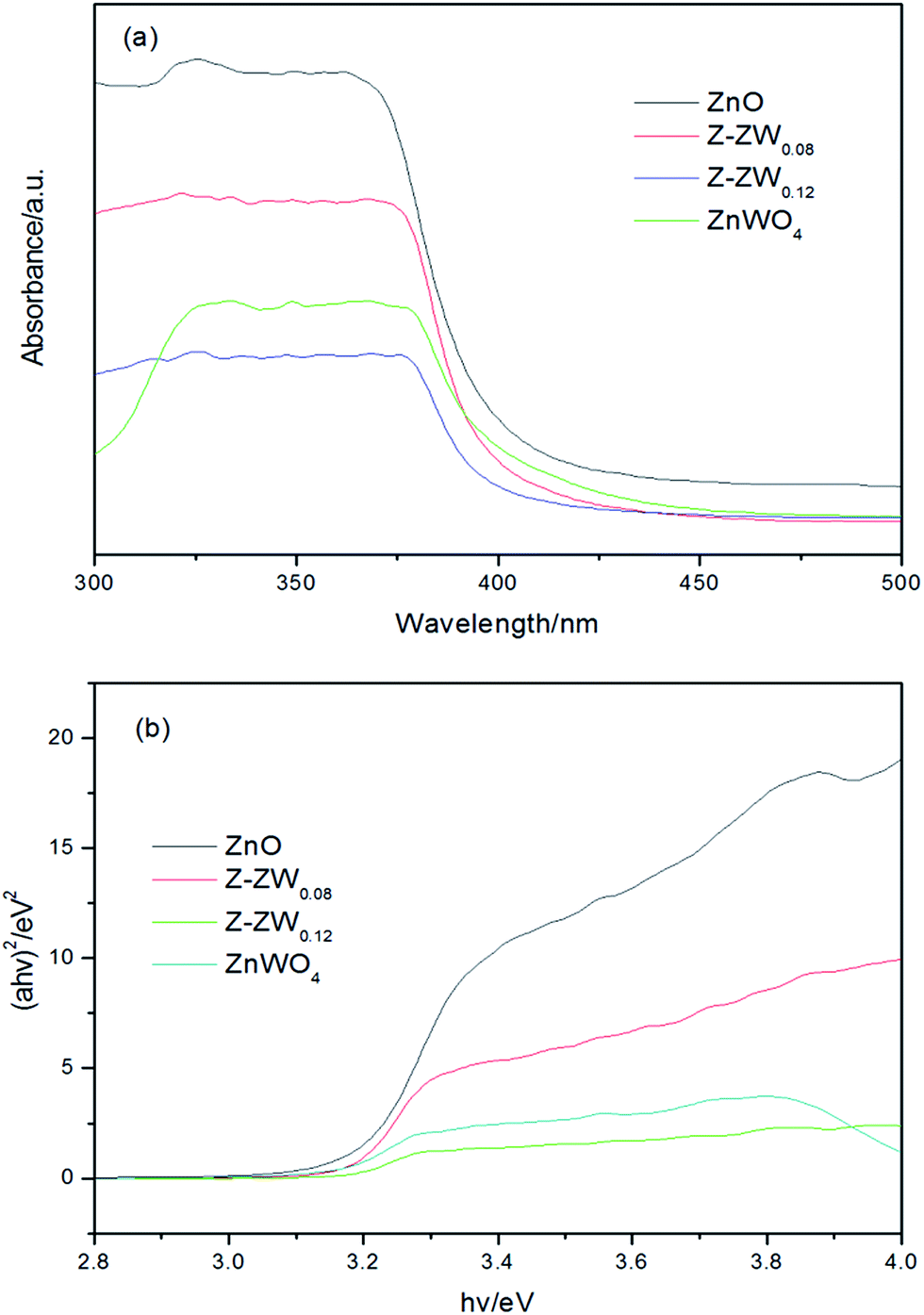 | ||
| Fig. 5 (a) UV-vis diffuse reflectance spectra, (b) (αhν)2 versus energy plots. | ||
The photocatalytic activities of the powders were tested by degradation of RhB solution under UV light irradiation. Fig. 6 shows the effect of calcination temperature on the photocatalytic activity of Z-ZW0.08 sample. The highest efficiency was obtained when the sample was calcined at 850 °C, whereas the powders calcined at 750 °C and 950 °C are less active. The crystallinity of the sample increased with raising the calcination temperature, which reduced the recombination of photo-generated carriers.9b,17 However, the higher calcination temperature resulted in the decrease of surface area, larger particle size, and decrease of dispersion in solution, which caused an adverse effect on the harvest of light and the adsorption of dye, producing a negative effect on activity.
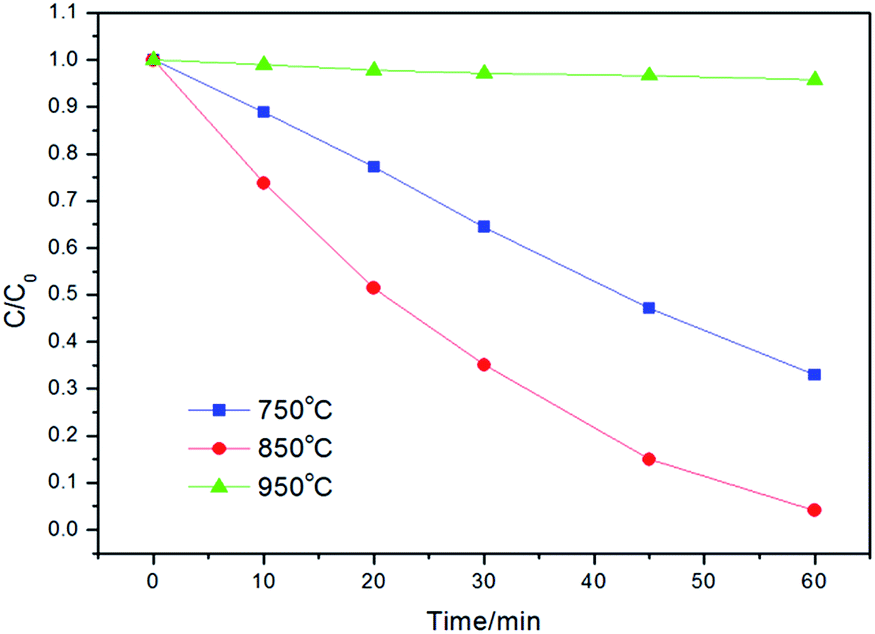 | ||
| Fig. 6 Photodegradation kinetics of RhB over Z-ZW0.08 calcined for 2 h at different temperatures. | ||
Fig. 7a and b show the kinetics of RhB degradation in the presence of ZnO, ZnWO4 and ZnO–ZnWO4 composites calcined at 850 °C. The degradation of RhB could be described by the first-order kinetics of −ln(Ct/C0) = kt, where k is the apparent reaction rate constant, and C0 and Ct are the initial concentration and the concentration at reaction time of RhB, respectively. After 60 min of UV irradiation, 8%, 71%, 91%, 96% and 86% of RhB were degraded by ZnWO4, ZnO, Z-ZW0.04, Z-ZW0.08 and Z-ZW0.12, respectively. The photoactivity of ZnWO4 was very low compared to that of ZnO, but all the ZnO–ZnWO4 samples showed higher photocatalytic activity than single ZnO or ZnWO4. The photocatalytic activities of the composites were found to be related to the ZnO/ZnWO4 molar ratio. The Z-ZW0.08 shows the highest photocatalytic activity. In the Z-ZW0.12 sample, a large amount of free ZnWO4 particles exists, which are less activity, leading to a lower photoactivity.4 Therefore, there is an optimum ZnWO4 content that leads to maximum photocatalytic efficiency. The k values were calculated by the initial slope of the concentration versus time profiles (Fig. 7b). It can be seen that Z-ZW0.08 sample owns the highest rate constant of 0.035 min−1.
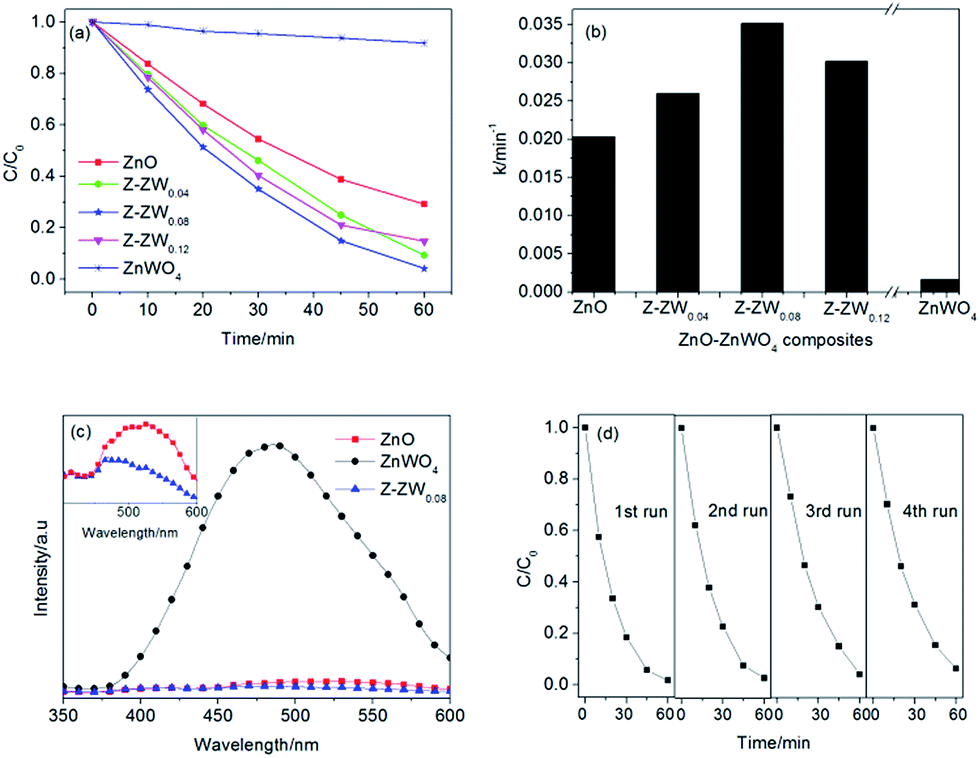 | ||
| Fig. 7 Photodegradation kinetics of RhB over ZnO, ZnWO4, and ZnO–ZnWO4 composites calcined at 850 °C for 2 h (a and b); room temperature PL spectra of as-synthesized samples obtained at the excitation wavelength of 325 nm (c); cycling runs for the photocatalytic degradation of RhB over Z-ZW0.08 (d). | ||
The recombination of electron–hole pairs can release energy, which can be detected by PL emission. Lower photoluminescence intensity indicates a lower recombination of electron–hole pairs, resulting in superior photocatalytic activity.1e The PL spectra of the ZnO, ZnWO4 and Z-ZW0.08 samples obtained at the excitation wavelength of 325 nm are shown in Fig. 7c. The PL intensities of ZnO and Z-ZW0.08 could be ignored compared with that of ZnWO4. The enlarged PL spectra of ZnO and Z-ZW0.08 at the range of 400–600 nm in the inset panel shows that the composites sample has a lower recombination rate of photogenerated electron–hole pairs. The PL results display the importance of the heterostructure of ZnO/ZnWO4 in blocking the recombination of electrons and holes. In order to evaluate the stability of the Z-ZW0.08 photocatalyst, a recycling test was carried out. The result is displayed in Fig. 7d. The degradation rate of RhB still remained over 90% after four cycles.
The enhancement in the photocatalytic activity of Z-ZW0.08 sample arose from the heterojunctions between ZnO and ZnWO4. The formation of the heterojunctions may improve the charge separation efficiency of photoexcited carriers in ZnO–ZnWO4 composites. The potentials of valence band (VB) and conduction band (CB) of a semiconductor can be calculated according to the following empirical equations:
| EVB = X − Ee + 0.5Eg | (1) |
| ECB = EVB − Eg | (2) |
 | ||
| Fig. 8 Degradation schematic mechanism of RhB over ZnO–ZnWO4 composites. | ||
Both ZnO and ZnWO4 are n-type semiconductors and their band gaps match well with each other to form type II heterojunction.18 The CB of ZnO is lower than that of ZnWO4, thus photogenerated electrons transfer easily from CB of ZnO to that of ZnWO4 through the well-developed interface, and the photo-induced holes shift from VB of ZnWO4 to that of ZnO. Therefore, the ZnO–ZnWO4 heterojunctions could act as an active center for hindering the rapid recombination of photogenerated electron–hole pairs. The electrons accumulated in the CB of ZnWO4 can be captured by O2 to produce superoxide radical (˙O2−) and hydrogen peroxide (H2O2), which can interact to produce a powerful oxidant (hydroxyl radical, ˙OH) to decompose RhB. The holes accumulated in the VB of ZnO take part in the oxidation process to make OH− or H2O species to produce reactive hydroxyl radicals.1a,19
To further confirm the heterojunctions effect, the photodegradation of RhB was also studied in the presence of a mechanical mixture of ZnO and ZnWO4, which is labelled as M-Z-ZW0.08. As shown in Fig. 9, the photocatalytic activity of the M-Z-ZW0.08 sample is higher compared to that of ZnO but much lower than that of Z-ZW0.08. This result demonstrates that the Z-ZW0.08 composite can form intimate interface in the heterojunctions, rather than form loose interfaces existing in the mechanically mixed samples.
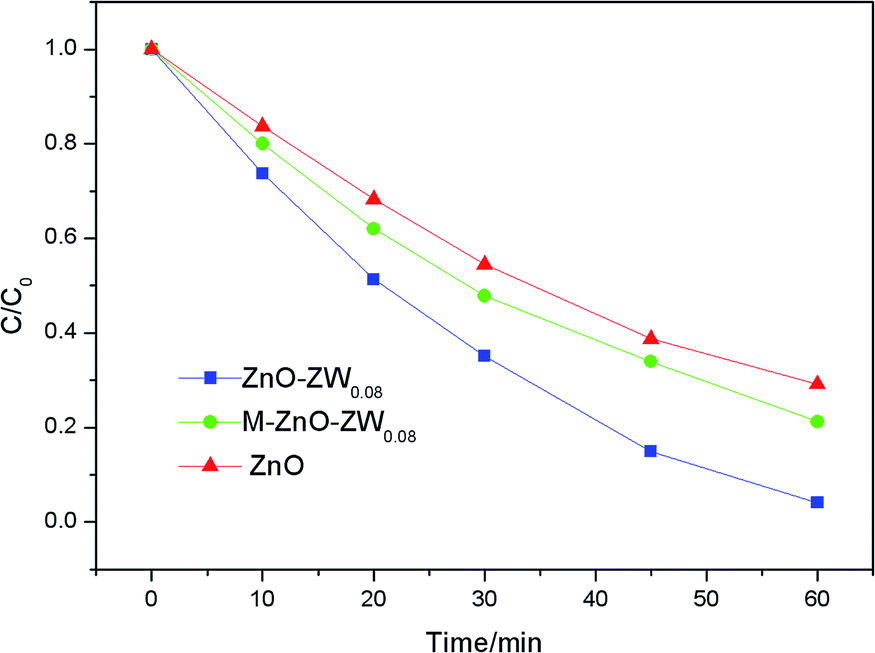 | ||
| Fig. 9 Photocatalytic degradation of RhB over ZnO, Z-ZW0.08, and M-Z-ZW0.08. | ||
4 Conclusions
The pearl necklace-like ZnO–ZnWO4 heterojunctions were successfully synthesized by precipitation method followed by calcination. The photocatalytic activity of ZnO–ZnWO4 composites was found to be dependent on the calcination temperature and ZnO/ZnWO4 mol ratio. The photoefficiency of the ZnO–ZnWO4 heterojunctions was better than that of ZnO and ZnWO4. Z-ZW0.08 composite powders show the best photocatalytic degradation performance for degradation of RhB with degradation efficiency about 96% under UV irradiation in 60 min. The enhanced photoactivity is due to the reduction in the recombination of photo-generated electrons and holes. This work provides a facile method to design and fabricate ZnO–ZnWO4 heterojunctions with special morphology.Acknowledgements
We gratefully acknowledge the financial support provided by the Foundation of Liaoning Educational Committee (L2016004). Dr B. Zhang acknowledges the financial support provided by the National Natural Science Foundation of China (91545119), Youth Innovation Promotion Association CAS (2015152) and the Joint Foundation of Liaoning Province National Science Foundation and Shenyang National Laboratory for Materials Science (2015021011).Notes and references
-
(a) W. Li, L. Y. Cao, X. G. Kong, J. F. Huang, C. Y. Yao, J. Fei and J. Y. Li, RSC Adv., 2016, 6, 23783 RSC
; (b) S. Adhikari, D. Sarkar and G. Madras, RSC Adv., 2015, 5, 11895 RSC
-
(a) C. Q. Zhu, B. G. Lu, Q. Su, E. Q. Xie and W. Lan, Nanoscale, 2012, 4, 3060 RSC
-
(a) Y. Hong, C. G. Tian, B. J. Jiang, A. P. Wu, Q. Zhang, G. H. Tian and H. G. Fu, J. Mater. Chem. A, 2013, 1, 5700 RSC
- X. W. Jiang, X. R. Zhao, L. B. Duan, H. Shen, H. N. Liu, T. Hou and F. G. Wang, Ceram. Int., 2016, 42, 15160 CrossRef CAS
- B. X. Li and Y. F. Wang, J. Phys. Chem. Solids, 2011, 72, 1165 CrossRef CAS
-
(a) S. F. Chen, W. Zhao, W. Liu and S. J. Zhang, Appl. Surf. Sci., 2008, 255, 2478 CrossRef CAS
- S. Balachandran, N. Prakash, K. Thirumalai, M. Muruganandham, M. Sillanpää and M. Swaminathan, Ind. Eng. Chem. Res., 2014, 53, 8346 CrossRef CAS
- A. Hamrouni, N. Moussa, F. Parrino, A. Di Paola, A. Houas and L. Palmisano, J. Mol. Catal. A: Chem., 2014, 390, 133 CrossRef CAS
-
(a) I. Validzic, T. Savic, R. Krsmanovic, D. Jovanovic, M. Novakovic, M. Popovic and M. Comor, Mater. Sci. Eng., B, 2012, 177, 645 CrossRef CAS
- Y. Wang, Q. S. Wang, X. Y. Zhan, F. M. Wang, M. Safdar and J. He, Nanoscale, 2013, 5, 8326 RSC
- Z. Amouzegar, R. Naghizadeh, H. R. Rezaie, M. Ghahari and M. Aminzare, Ceram. Int., 2015, 41, 1743 CrossRef CAS
- M. Mancheva, R. Iordanova and Y. Dimitriev, J. Alloys Compd., 2011, 509, 15 CrossRef CAS
- G. L. Huang, S. C. Zhang, T. G. Xu and Y. F. Zhu, Environ. Sci. Technol., 2008, 42, 8516 CrossRef CAS PubMed
- T. Montim, V. Gombac, A. Hameed, L. Felisan, G. Adami and P. Fornasiero, Chem. Phys. Lett., 2010, 498, 113 CrossRef
- J. Becker, K. R. Raghupathi, J. S. Pierre, D. Zhao and R. T. Koodali, J. Phys. Chem. C, 2011, 115, 13844 CAS
- C. L. Yu, K. Yang, Q. Shu, J. C. Yu, F. F. Cao and X. Li, Chin. J. Catal., 2011, 32, 555 CrossRef CAS
- B. Ohtani, Y. Ogawa and S. Nishimoto, J. Phys. Chem. B, 1997, 101, 3746 CrossRef CAS
- R. Marschall, Adv. Funct. Mater., 2014, 24, 2421 CrossRef CAS
-
(a) A. Di Paola, M. Bellardita, R. Ceccato, L. Palmisano and F. Parrino, J. Phys. Chem. C, 2009, 113, 15166 CrossRef CAS
Footnote |
| † The authors contribute equally to this paper. |
| This journal is © The Royal Society of Chemistry 2017 |