DOI:
10.1039/C6RA28734D
(Paper)
RSC Adv., 2017,
7, 12455-12463
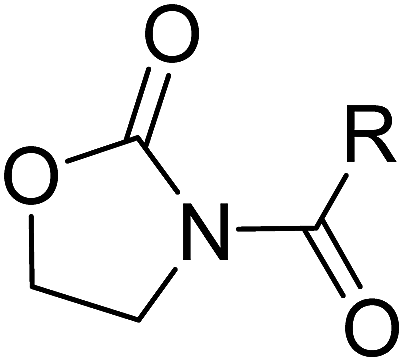
Design, synthesis, and biological evaluation of oxazolidone derivatives as highly potent N-acylethanolamine acid amidase (NAAA) inhibitors†
Received
27th December 2016
, Accepted 16th February 2017
First published on 21st February 2017
Abstract
N-Acylethanolamine-hydrolyzing acid amidase (NAAA) is a lysosomal enzyme that catalyzes the hydrolysis of endogenous fatty acid ethanolamides (FAEs), such as N-palmitoylethanolamide (PEA). PEA exhibits anti-inflammatory and analgesic activities by engaging peroxisome proliferator-activated receptor α (PPAR-α). Preventing PEA degradation by inhibition of NAAA has been proposed as a novel strategy for the treatment of inflammation and pain. In the present study, we reported the discovery of the oxazolidone derivative as a novel scaffold for NAAA inhibitors, and studied the structure–activity relationship (SAR) by modification of the side chain and terminal lipophilic substituents. The results showed that the link chain length of C5, straight and saturated linkages were the preferred shape patterns for NAAA inhibition. Several nanomolar NAAA inhibitors were described, including 2f, 3h, 3i and 3j with IC50 values of 270 nM, 150 nM, 100 nM and 190 nM, respectively. Enzymatic degradation studies suggested that 2f inhibited NAAA in a selective, noncompetitive and reversible pattern. Moreover, 2f showed high anti-inflammatory and analgesic activities after systemic and oral administration.
1 Introduction
N-Acylethanolamine-hydrolyzing acid amidase (NAAA) is a lysosomal cysteine hydrolase widely expressed in many tissues, especially those associated with immune responses, such as the lungs, spleen, and small intestine. NAAA participates in the control of inflammatory and pain1 via involving in the hydrolysis of endogenous palmitoylethanolamide (PEA). PEA is a multifunctional lipid mediator that has been shown to inhibit peripheral inflammation2–4 and cell degranulation, and exhibits antinociceptive activities in rat and mouse models of acute and chronic pain.5–7 Moreover, PEA also could alleviate skin inflammation8 and neuropathic pain in humans.9 Those effects are mainly attribute to the ability of PEA to engage nuclear receptor peroxisome proliferator-activated receptor-alpha (PPAR-α). In recent years, PEA has attracted increasing attention because of its potential therapeutic effects in inflammation and pain. Preventing PEA degradation by inhibition of NAAA10 may provide a novel strategy for the treatment of inflammatory and pain.4,11 For this reason, NAAA has been pursued as a potential drug target.
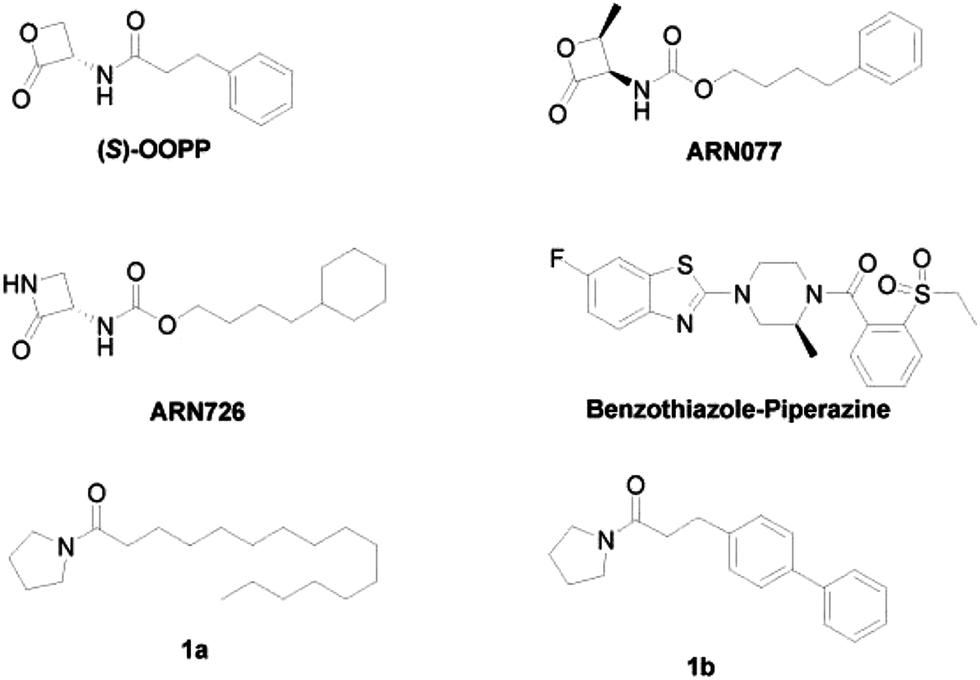
Despite the increasing interest in the design of NAAA inhibitors as anti-inflammatory and analgesic agents, only few potent NAAA inhibitors have been identified so far.12,13 There are two main categories of NAAA inhibitors reported, irreversible covalent inhibitors, such as β-lactone derivatives14–18 and non-covalent reversible inhibitors, such as benzothiazole-piperazine derivatives.19 Although covalent inhibitors, such as (S)-OOPP,4 ARN077,11 ARN726 (ref. 20) (Fig. 1) demonstrated profound anti-inflammatory effects in several animal models, their application is limited due to the poor chemical and plasma stability. These compounds degraded in phosphate buffered saline (PBS) within 120 min and were rapidly cleaved in plasma.16 Non-covalent reversible inhibitors, such as, benzothiazole-piperazine derivatives (Fig. 1),19 are selective, reversible, and competitive NAAA inhibitors. However, only few potent reversible NAAA inhibitors with drug-like properties have been identified so far. Highly potent, stable and reversible NAAA inhibitors are still highly desired for the treatment of inflammation and pain.
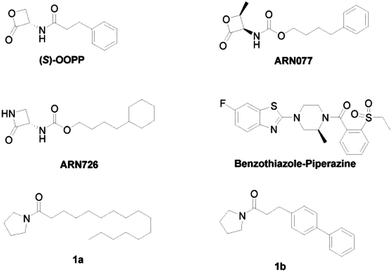 |
| | Fig. 1 The chemical structures of classic NAAA inhibitors. | |
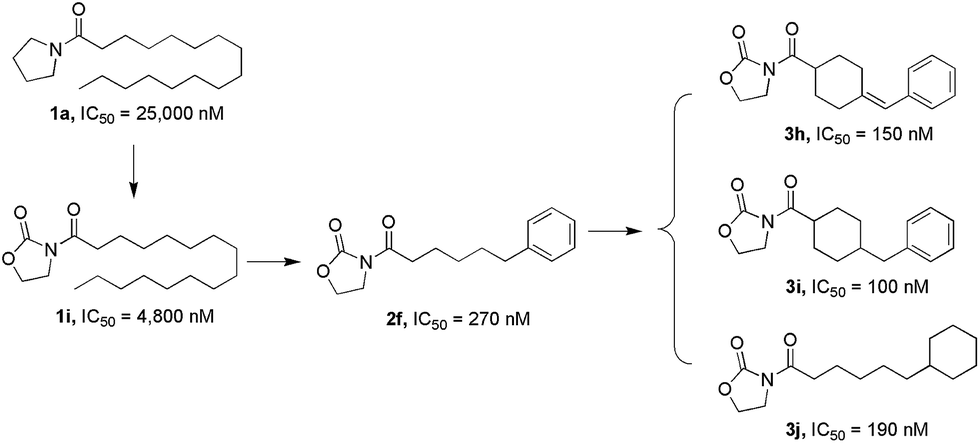
In our previous study, we designed pyrrolidine-based derivatives 1a and 1b (Fig. 1)21 as NAAA inhibitors. Substitution of the saturated alkyl chain of 1a with an aryl-containing lipophilic moiety leaded to a potent NAAA inhibitor 1b (IC50 = 2.1 μM). In vivo pharmacological studies indicated that 1b reduced the mRNA expression levels of inducible nitric oxide synthase and interleukin-6, as well as increased the intracellular PEA levels in mouse macrophages with lipopolysaccharide-induced inflammation.21 Moreover, 1b exhibited excellent chemical stability and plasma stability (t½ > 16 h).
In the present study, we further modified 1a to obtain more potent and biologically stable NAAA inhibitors. A nanomolar NAAA inhibitor 2f was discovered (IC50 = 270 nM), which exerted profound anti-inflammatory and anti-nociceptive effects in rats.22 We then carried out structure–activity relationship (SAR) studies on 2f by modification of the pyrrolidine ring and the lipophilic chain, which led to the identification of several more potent nanomolar NAAA inhibitors, such as 3h, 3i and 3j (Fig. 2). Compound 2f was then selected as a model molecule to study the NAAA inhibition mechanisms and in vivo anti-inflammatory and analgesic activities of oxazolidone derivatives. The results showed that 2f inhibited NAAA in a selective and highly reversible pattern, and efficiently reduced inflammatory and pain after systemic and oral administration. Oral administration of 2f (30 mg kg−1) exhibited the same efficacy with commercial nonsteroidal anti-inflammatory drug ibuprofen at a 6.7-fold higher dose (200 mg kg−1).
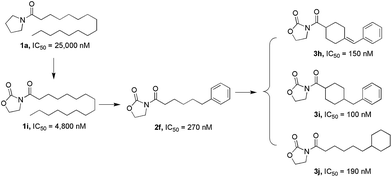 |
| | Fig. 2 Design strategy for oxazolidone derivatives as NAAA inhibitors. | |
2 Results and discussion
2.1 Synthesis of oxazolidone derivatives
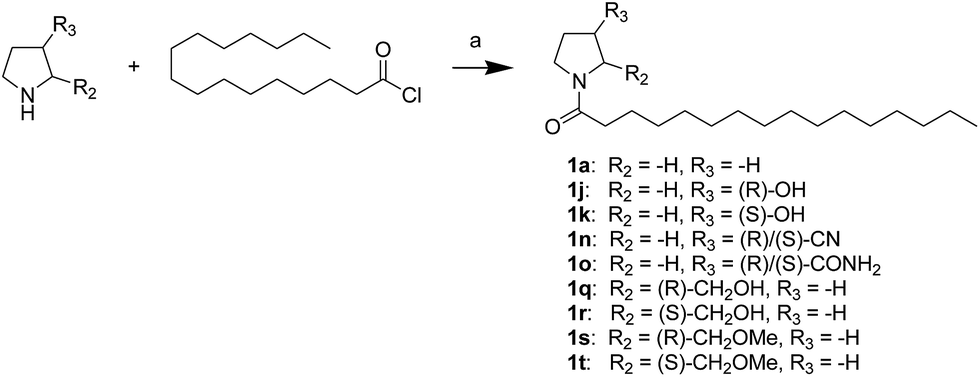
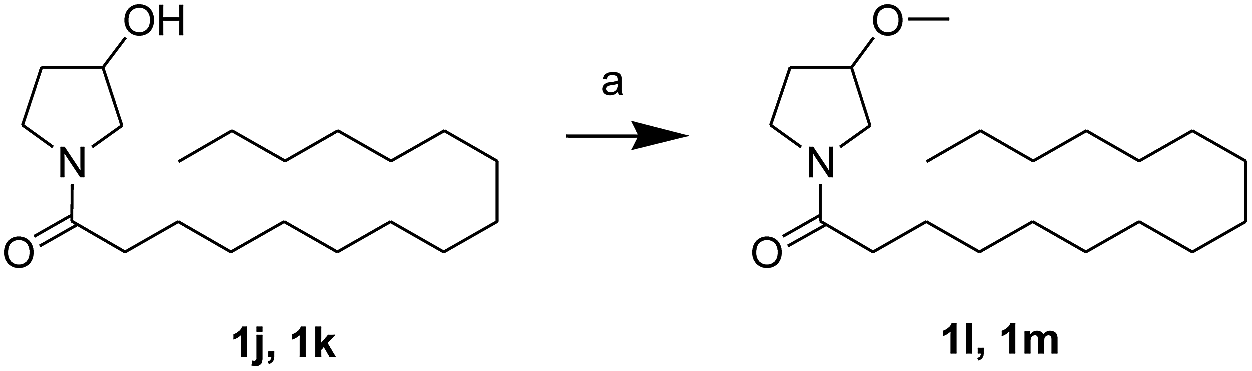
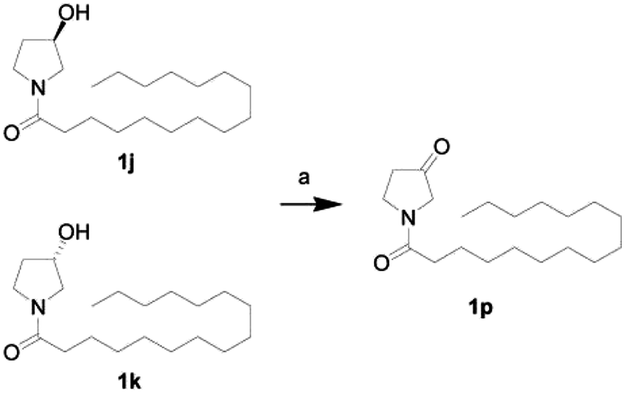
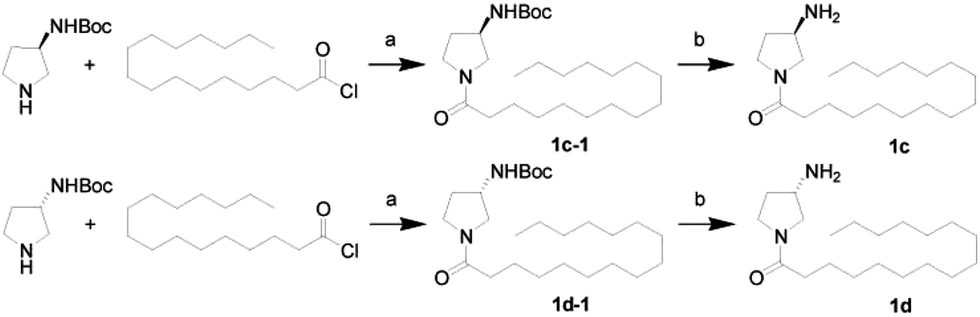
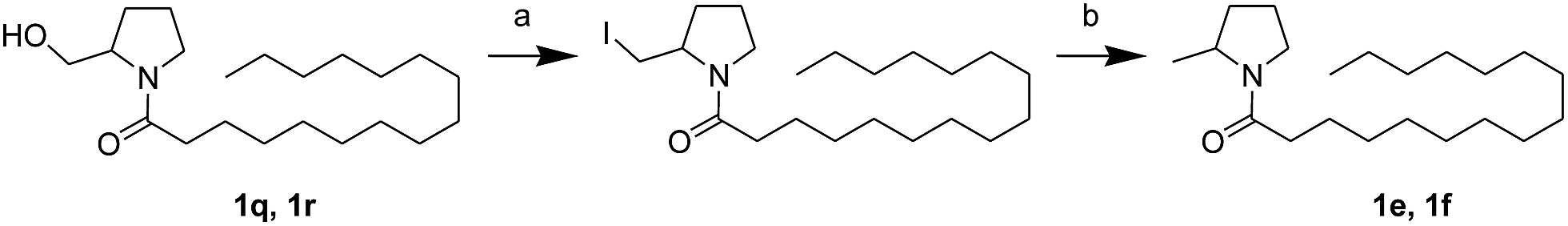
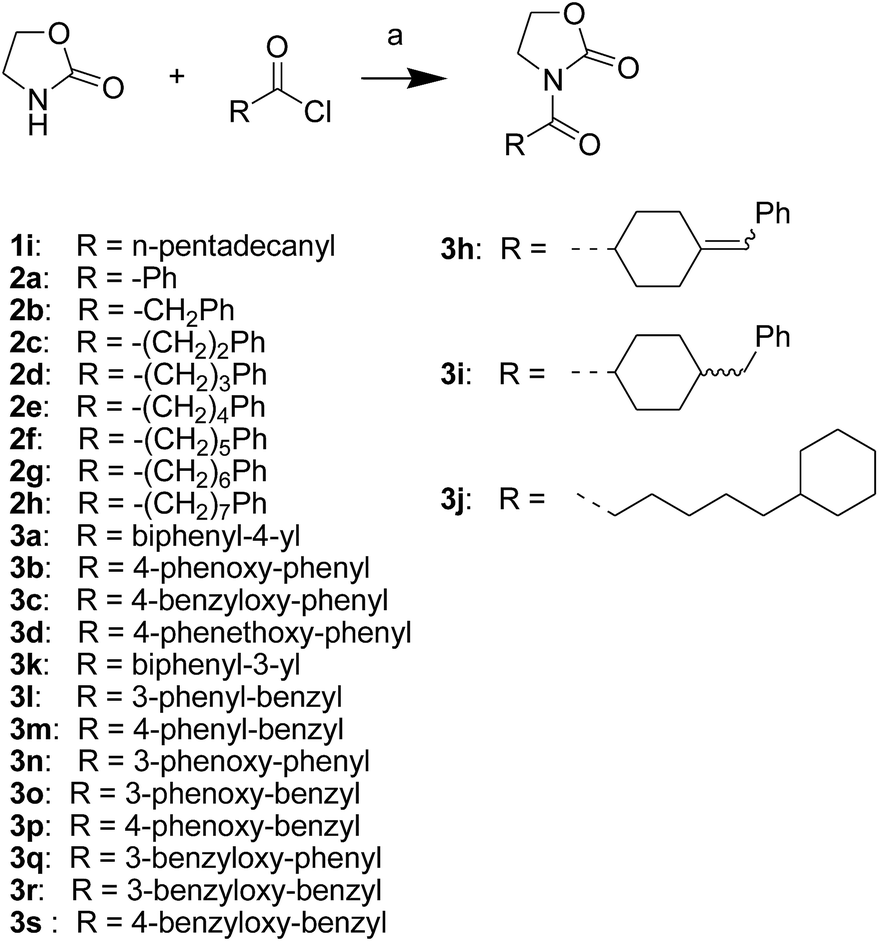
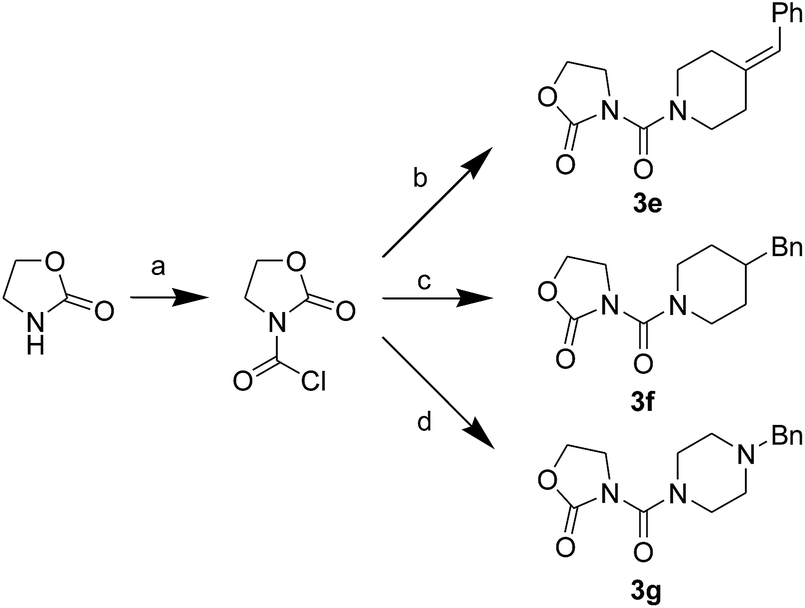
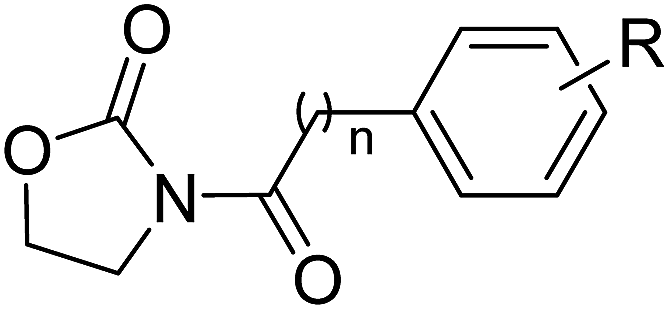
First, we synthesized a series of oxazolidone derivatives with different side chains and terminal lipophilic substituents. Compounds 1a, 1j–1o, and 1q–1t were prepared by the amidation of palmitoyl chloride with the corresponding amine in the presence of triethylamine (Scheme 1).21 1l and 1m were prepared by the methylation of 1q and 1r with iodomethane in the presence of sodium hydride, respectively (Scheme 2).23 1p was prepared by Dess–Martin periodinane oxidation of 1j or 1k (Scheme 3).24 1c and 1d were prepared by the acidic deprotection of pyrrolidine derivatives 1c-1 and 1d-1, which were obtained by the amidation of (R)- or (S)-3-(tert-butoxycarbonylamino)pyrrolidine with palmitoyl chloride, respectively (Scheme 4).25,26 1e and 1f were synthesized by a two-step reaction starting from 1q and 1r, which were converted to iodide derivatives by I2 and PPh3, respectively, followed by hydrogenation with H2 (Scheme 5).27,28 1g, 2a–2h, 3a–3d and 3k–3s were synthesized by the imidate reaction, employing the corresponding acyl chloride with 2-pyrrolidone, 2-imidazolidone, or 2-oxazolidone in the presence of n-BuLi, respectively (Scheme 6).29 3e–3g were synthesized by the amidation of 2-oxooxazolidine-3-carbonyl chloride, generated by reaction of 2-oxazolidone and bis(trichloromethyl) carbonate, with the corresponding substituted piperazine or piperidine (Scheme 7).30 HPLC analysis indicated >99% purity for compound 1g, 1e–1i, 2b, 2d, 2f–2h, 3c, 3e, and 3g, >98% purity for 1c–1d, 1f, 2a, 2e, 3a, 3b and 3g–3i, and >97% purity for 2c and 3d (Table S3†).
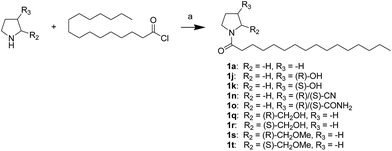 |
| | Scheme 1 The synthesis of compounds 1a, 1j, 1k, 1n, 1o, and 1q–1t. Reagents and conditions: (a) Et3N, CH2Cl2, room temperature. | |
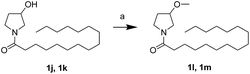 |
| | Scheme 2 The synthesis of compounds 1l and 1m. Reagents and conditions: (a) NaH, MeI, tetrahydrofuran (THF), 0 °C. | |
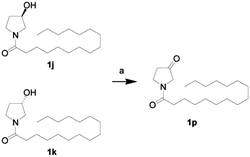 |
| | Scheme 3 The synthesis of compound 1p. Reagents and conditions: (a) Dess–Martin periodinane, CH2Cl2, room temperature. | |
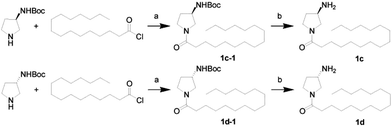 |
| | Scheme 4 The synthesis of compounds 1c and 1d. Reagents and conditions: (a) Et3N, CH2Cl2, room temperature; (b) HCl, dioxane, 0 °C. | |
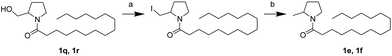 |
| | Scheme 5 The synthesis of compounds 1e and 1f. Reagents and conditions: (a) PPH3, I2, THF, room temperature; (b) H2, Pd/C, CH3OH, room temperature. | |
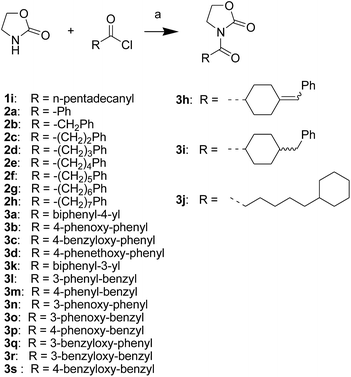 |
| | Scheme 6 The synthesis of compounds 1i, 2a–2h, 3a–3d and 3h–3s. Reagents and conditions: (a) n-BuLi, THF, −78 °C. | |
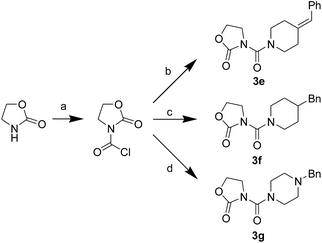 |
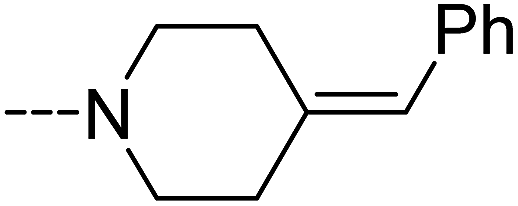
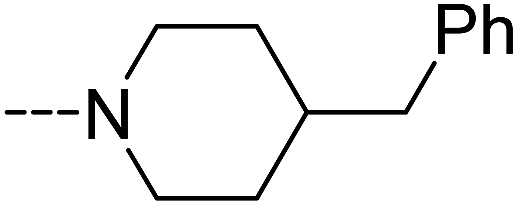
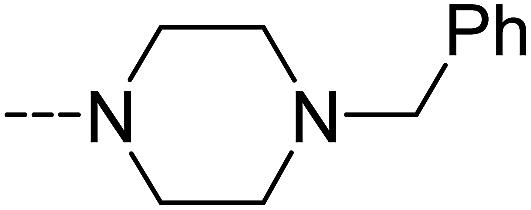
| | Scheme 7 The synthesis of compounds 3e–3g. Reagents and conditions: (a) bis(trichloromethyl) carbonate, Et3N, CH2Cl2, room temperature; (b) 4-benzylidenepiperidine, CH2Cl2, room temperature; (c) 4-benzylpiperidine, CH2Cl2, room temperature; (d) 1-benzylpiperazine, CH2Cl2, room temperature. | |
2.2 NAAA inhibition by oxazolidone derivatives
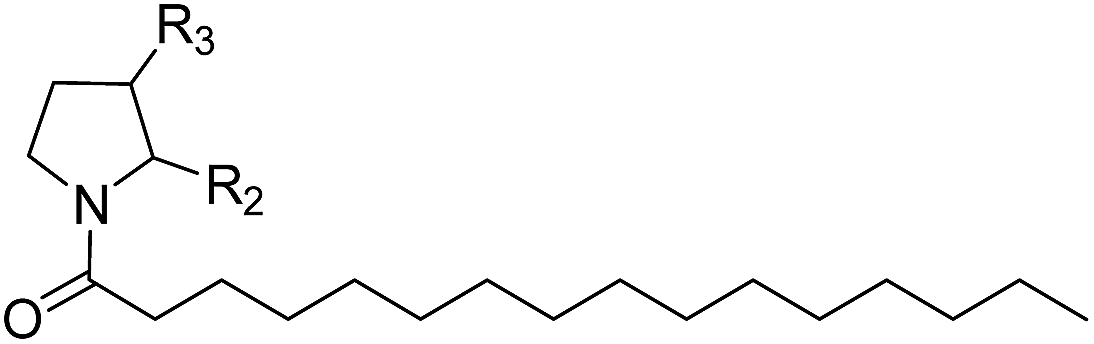
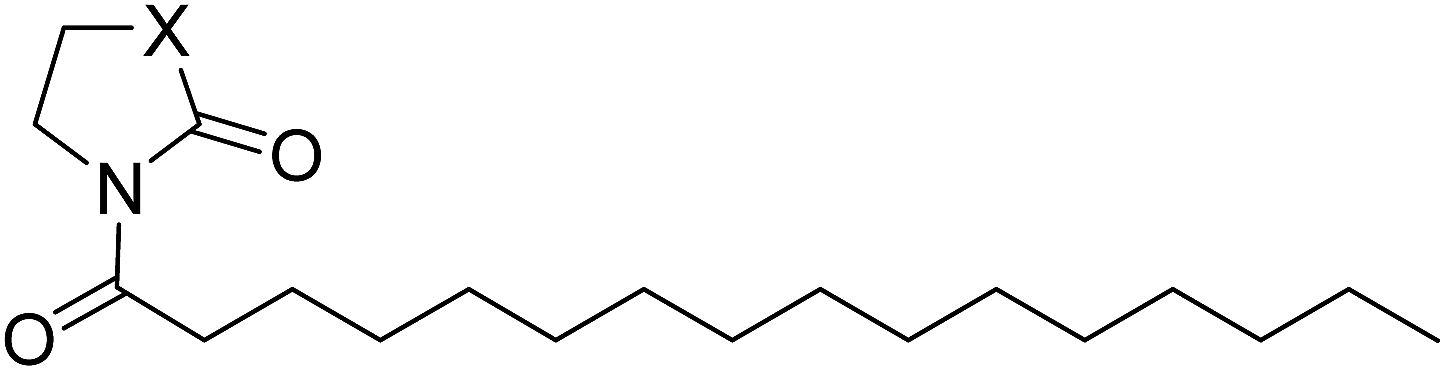
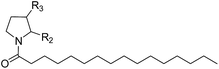
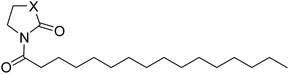
The NAAA inhibition capability of these newly synthesized compounds were tested using heptadecenoylethanolamide as a substrate. Since many NAAA inhibitors also block fatty acid amide hydrolase (FAAH), the FAAH inhibition of these compounds also was examined to obtain NAAA selective inhibitors. (S)-OOPP and well known FAAH inhibitor URB597 were used as standard compounds in the enzymatic assay. The results showed that substitution at 3-position of pyrrolidine with an amino group (1c, 1d) slightly increased NAAA and FAAH inhibition, and substitutions at the 2-position with (S)-methyl (1f) or carbonyl (1g) slightly or moderately enhanced inhibition towards NAAA without changing inhibition against FAAH. However, (R)-methyl (1e), hydroxy (1j, 1k), methoxy (1l, 1m), cyano (1n), formamide (1o), hydroxymethyl (1q, 1r) and methoxymethyl (1s, 1t) substituents diminished the NAAA and FAAH inhibitory effects (Table S1†). 1g exhibited low-micromolar inhibition on NAAA activity (IC50 = 4.6 ± 0.48 μM), and it was over 5-fold more potent towards NAAA than FAAH (Table 1). To further optimize the hydrophilic moieties, a series of 1g isosteres were synthesized and tested (1h, 1i) (Table 2). Enzymatic assay results showed that 1i with oxazolidone ring had similar effects on NAAA inhibition as 1g, but 1i exhibited dramatically enhanced selectivity towards NAAA. Less than 10% FAAH activity was inhibited by 1i even at a concentration of 100 μM. Therefore, 2-oxazolidone ring was selected as the template for further optimization.
Table 1 Inhibitory effects of compounds 1a–1g, (S)-OOPP and URB597 on NAAA and FAAH activities
|
| Compound |
R2 |
R3 |
IC50 of NAAA (μM) |
IC50 of FAAH (μM) |
| 1a |
–H |
–H |
25 ± 5.7 |
21 ± 4.5 |
| 1c |
–H |
(R)-NH2 |
10 ± 0.95 |
3.5 ± 0.65 |
| 1d |
–H |
(S)-NH2 |
11 ± 1.2 |
4.0 ± 0.81 |
| 1e |
(R)-Me |
–H |
>100 |
28 ± 2.7 |
| 1f |
(S)-Me |
–H |
15 ± 2.3 |
18 ± 3.2 |
| 1g |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
–H |
4.6 ± 0.48 |
25 ± 5.3 |
| (S)-OOPP |
|
|
0.83 ± 0.1 |
>20 |
| URB597 |
|
|
>100 |
0.026 ± 0.005 |
Table 2 Inhibitory effects of compounds 1h–1j on NAAA and FAAH activities
|
| Compound |
X |
IC50 of NAAA (μM) |
IC50 of FAAH (μM) |
| 1g |
C |
4.6 ± 0.48 |
25 ± 5.3 |
| 1h |
NH |
>100 |
27 ± 4.9 |
| 1i |
O |
4.8 ± 0.63 |
>100 |
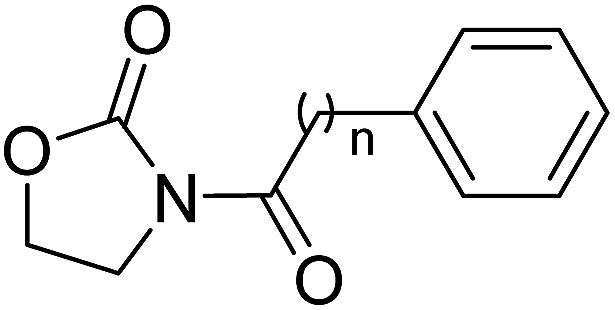
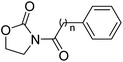
It has been reported that substitutions of the saturated alkyl chain with aryl-containing lipophilic moieties could enhance the inhibitory potency on NAAA and improved the drug-like properties.14,21 Therefore, a series of oxazolidone derivatives incorporating a phenyl ring at the terminus were synthesized and tested. As shown in Table 3, 2a with the chain length of C0 exhibited low-micromolar inhibitory potency toward NAAA (IC50 = 3.4 ± 0.42 μM), while 2f with the chain length of C5 exhibited excellent potency (IC50 = 0.27 ± 0.04 μM), 3-fold more potent than (S)-OOPP tested under the same experimental conditions. Interestingly, sharply reduced or even abolished activity was observed as the chain length decreased from C5 to C1 or increased from C5 to C7. We are not quite sure why their potency dramatically changed when we varied the link chain length, probably because these oxazolidone imides with different chain length are activated to different extent when contacting the NAAA.
Table 3 Inhibitory effects of compounds 2a–2h on NAAA and FAAH activities
|
| Compound |
n |
IC50 of NAAA (μM) |
IC50 of FAAH (μM) |
| 2a |
0 |
3.4 ± 0.42 |
>20 |
| 2b |
1 |
>20 |
>20 |
| 2c |
2 |
>20 |
>20 |
| 2d |
3 |
12.4 ± 2.0 |
>20 |
| 2e |
4 |
8.3 ± 1.8 |
>20 |
| 2f |
5 |
0.27 ± 0.04 |
>20 |
| 2g |
6 |
6.3 ± 1.2 |
>20 |
| 2h |
7 |
>20 |
>20 |
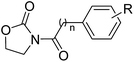
To further optimize the NAAA inhibition activity of 2f, we replaced the rotatable bond of the acyl chain of 2f with an additional phenyl group. As shown in Table 4 and S2,† a series of analogs corresponding to biphenyl (3a, 3k–3m), phenoxyphenyl (3b, 3n–3p), and benzyloxyphenyl (3c, 3q–3s) were obtained and examined. Compared to 2a, 1,4-substituted phenoxyphenyl (3b, IC50 = 0.68 ± 0.11 μM) and benzyloxyphenyl (3c, IC50 = 2.3 ± 0.36 μM) analogs were approximately 10-fold and 2-fold more potent. However, 1,4-substituted bipheny analog 3a (IC50 = 9.4 ± 1.2 μM) was 2-fold less potent (Table 4), and 1,3-substituted isomers (3k, 3l, 3n, 3o, 3q, 3r) were completely inert for NAAA inhibition (Table S2†). The results indicated that straight structures are preferred shape for the hydrophobic channel of NAAA, which is in agreement with previous reports.14,17 The binding pocket of NAAA proximal to the carbonyl moieties seemed to be narrow, as such, only a phenyl moiety is tolerated at this region. Therefore, incorporation of an extra methylene group between the biaryl and carbonyl groups (3m, 3p and 3s) significantly decreased NAAA inhibition.
Table 4 Inhibitory effects of compounds 3a–3d on NAAA and FAAH activities
|
| Compound |
n |
R |
IC50 of NAAA (μM) |
IC50 of FAAH (μM) |
| 3a |
0 |
4-Ph |
9.4 ± 1.2 |
>20 |
| 3b |
0 |
4-OPh |
0.68 ± 0.11 |
>20 |
| 3c |
0 |
4-OBn |
2.4 ± 0.36 |
>20 |
| 3d |
0 |
4-OCH2CH2Ph |
>20 |
>20 |
It's worth noting that the position of the terminal phenyl groups in biaryl series compounds (3a–3d) is corresponding to the location of phenyl moieties in the phenylalkyl series compounds (2e–2h). Increasing the length of the terminal aryl group of 3a by insertion of an oxygen atom or a methyleneoxy unit between the phenyl rings led to 14-fold (3b) or 4-fold (3c) higher potency, whereas the insertion of an additional methylene unit to the aliphatic linker of 3c significantly decreased activity (3d). These results highlighted the importance of the optimal length and connecting pattern of the aryl-containing lipophilic moieties.
It has been reported that NAAA prefer saturated substrates e.g., N-myristoylethanolamine and PEA, rather than unsaturated substrates, e.g., AEA.1 Therefore, we studied whether the replacement of a phenyl ring with a 6-member ring can improve the NAAA inhibition. A series of additional derivatives of 2f were then synthesized and examined (Table 5). The results showed that introducing hexane ring further enhanced the NAAA inhibitory potency. We found two highly potent NAAA inhibitors 3h and 3i with a IC50 value of 150 nM and 100 nM, respectively, around 2-fold more potent than 2f. Moreover, saturated linkages were found to be more beneficial for the activity than unsaturated benzene ring (3h, 3i vs. 3a–3c; 3j vs. 2f). However, nitrogen atoms substitution of the saturated hexane significantly reduced the activity (3e–3g).
Table 5 Inhibitory effects of compounds 3e–3j on NAAA and FAAH activities
2.3 NAAA inhibition pattern
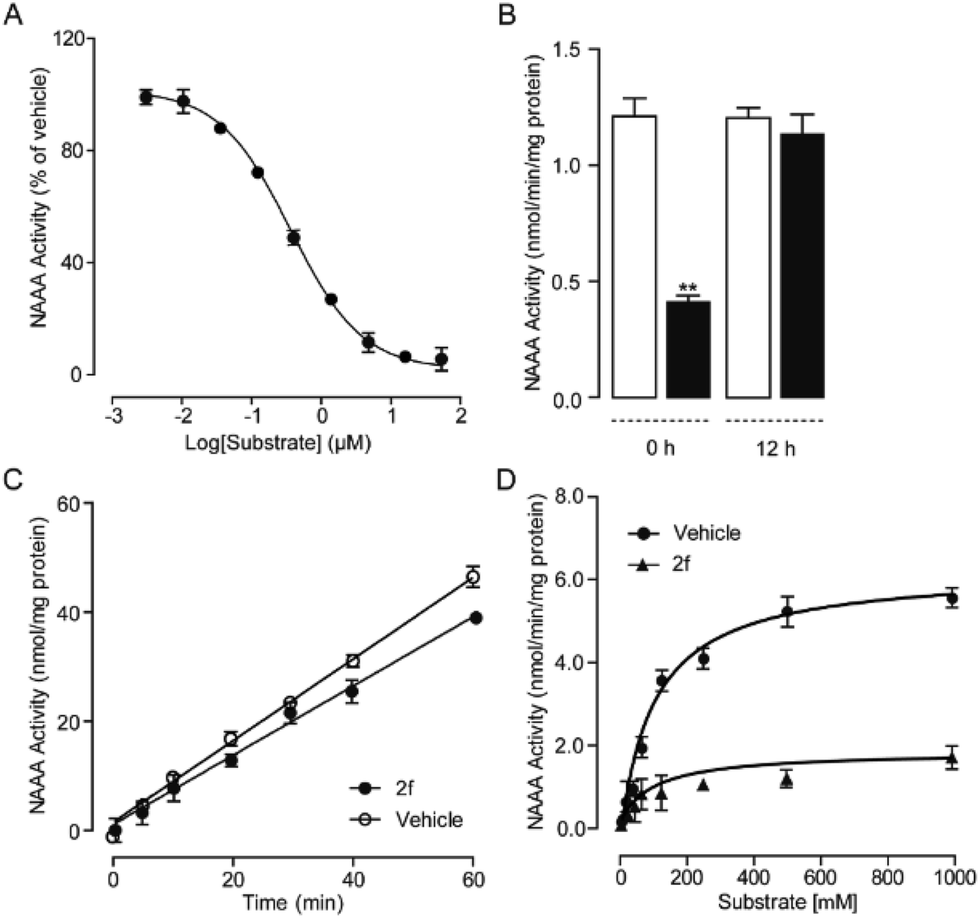
To further understand the NAAA inhibition pattern of these oxazolidone derivatives, compound 2f was selected as a model molecule and its interaction mode with NAAA protein in cells was studied. The results showed that 2f displayed a profound inhibition toward recombinant human NAAA (IC50 = 0.27 ± 0.04 μM, Fig. S1A†) and recombinant rat NAAA in NAAA-transfected HEK293 cells (IC50 = 0.42 ± 0.04 μM, Fig. S1B†), which were similar to the data obtained with the NAAA protein extract. Dialysis (Fig. 3B) and rapid dilution (Fig. 3C) of the 2f–NAAA interaction complex almost completely restored the NAAA activity. Moreover, Michaelis–Menten kinetic analysis revealed that 2f induced changes in the maximal catalytic velocity (Vmax) of NAAA (Vmax in nmol per min per mg protein, vehicle: 6.0 ± 0.51; and 2f, 0.3 μM: 1.9 ± 0.15, Fig. 3D) but did not affect Michaelis–Menten constant Km (Km in μM, vehicle: 174 ± 26; and 2f, 0.3 μM: 212 ± 24). These results indicated that 2f is a reversible and noncompetitive NAAA inhibitor.
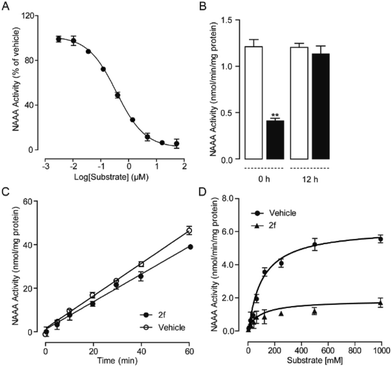 |
| | Fig. 3 Characterization of 2f as a reversible and noncompetitive NAAA inhibitor. (A) Concentration-dependent inhibitory effect of 2f on the rat NAAA. (B) NAAA activity in the presence of vehicle (open columns) or 2f (closed columns) before and 12 h after dialysis. ** P < 0.01 vs. vehicle, n = 3; (C) rapid dilution NAAA assay in the presence of vehicle (1% DMSO, open circles) or 2f (closed circles). (D) Michaelis–Menten analysis of the NAAA reaction in the presence of vehicle (1% DMSO, closed circles) and 2f (0.5 μM, triangles). | |
2.4 In vivo anti-inflammatory and analgesic activities of 2f
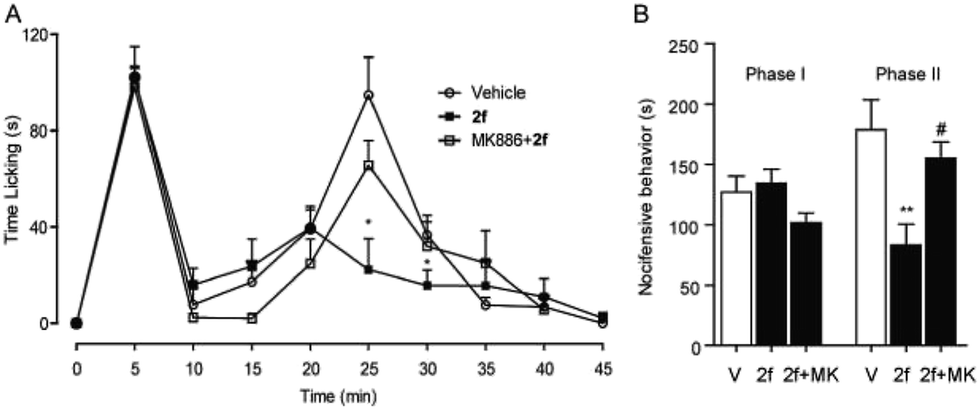
Finally, the in vivo anti-inflammation and analgesic activities of 2f was studied in different pain and inflammation mice models. Experimental mice were treated with 2f via intraperitoneal injection or intragastrical administration. The behavioural response of Mice to formalin stimulation was divided to the first-phase of acute pain (0–10 min) and the second-phase of chronic pain (15–35 min). Single treatment of 2f (10 mg kg−1, i.p.) decreased second-phase but not first-phase (Fig. 4), suggesting a central mechanism of action of 2f. The reduced licking time by 2f at the second-phase was reversed by PPAR-α antagonist MK886 (2 mg kg−1, i.p.), which indicated that 2f relieve pain in a PPAR-α dependent pathway.
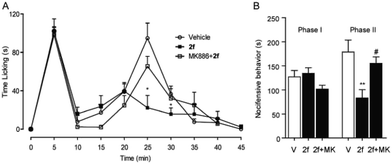 |
| | Fig. 4 2f attenuated formalin-induced pain behaviors. (A) 2f (10 mg kg−1, i.p., open squares), injected 45 min prior to formalin treatment, produced time-dependent changes in composite pain score relative to vehicle (5% PEG/5% Tween-80 in saline, 10 mL kg−1, i.p., open cycles), or a combination of 2f and PPAR-α antagonist MK886 (2 mg kg−1, i.p., close squares). * P < 0.1 vs. vehicle; (B) 2f (close bar) reduced the licking time during phase 2 of the formalin response, ** P < 0.1 vs. vehicle (open bar). MK886 (2 mg kg−1, i.p., close bar) prevented the anti-nociceptive effects of F96, # P < 0.1 vs. 2f. V represents vehicle (5% PEG/5% Tween-80 in saline), and MK represents PPAR-α antagonist MK 886. | |
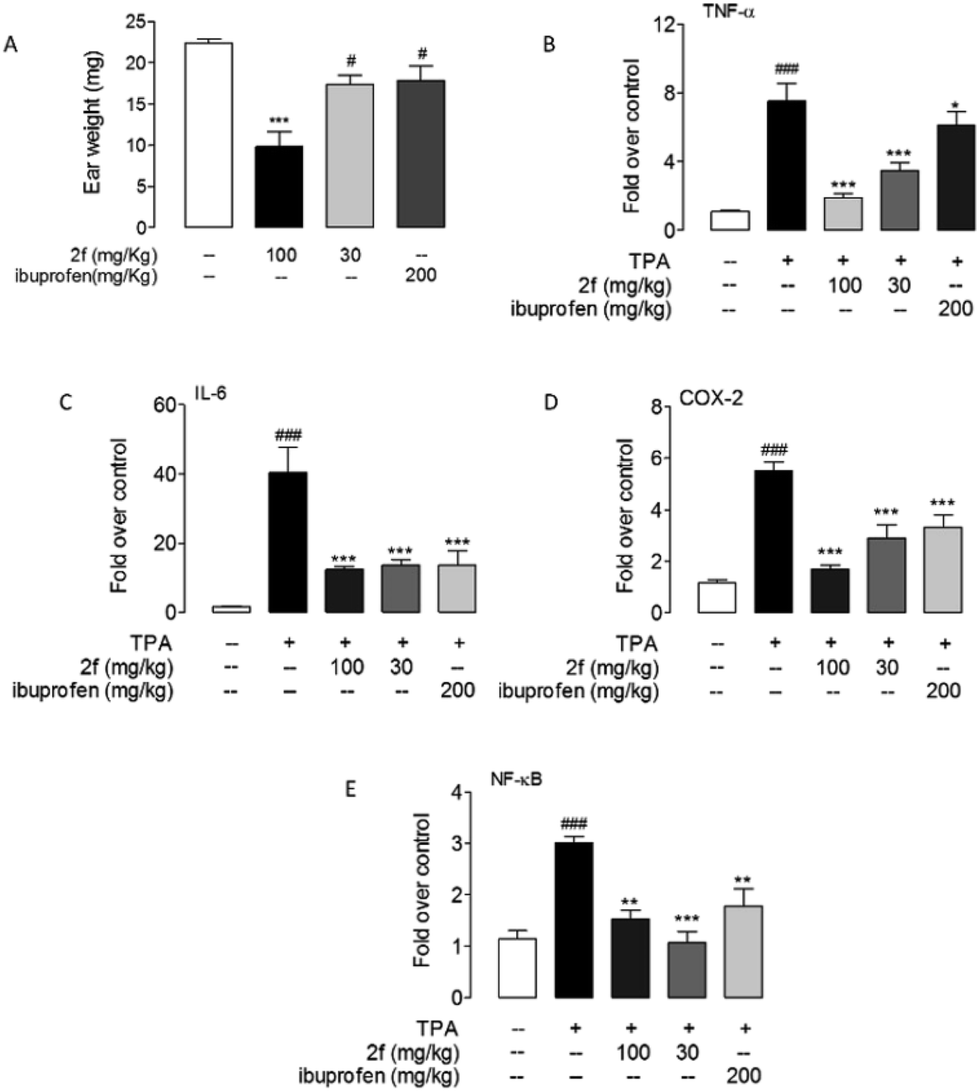
The anti-inflammatory activity of 2f was tested in 12-o-tetradecanoylphorbol-13-acetate (TPA) induced mice ear edema model. A commercial nonsteroidal anti-inflammatory drug, ibuprofen (200 mg kg−1) was intragastrically administrated as a positive control. The results showed that intragastrical administration of 2f (30, 100 mg kg−1) significantly reduce ears edema. Oral administration of compound 2f (30 mg kg−1) presented the same efficacy with ibuprofen (200 mg kg−1). Moreover, 2f significantly prevent the production of proinflammatory cytokines, including TNF-α, IL-6, COX-2 and NF-κB in mRNA level (Fig. 5).
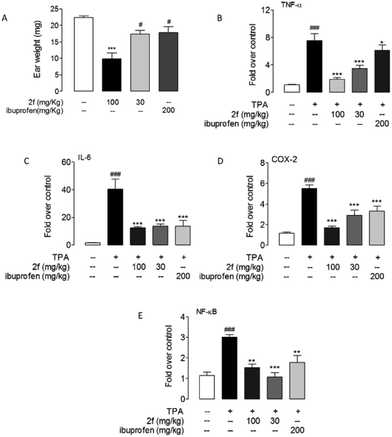 |
| | Fig. 5 Compound 2f alleviated TPA-induced inflammation. Effects of oral administration of 2f and ibuprofen on mice ears edema (A) and mRNA expression levels of TNF-α (B), IL-6 (C), COX-2 (D) and NF-κB (E) in mice ear treated with vehicle (open bars) or TPA (closed bars). Vehicle, 0.1% DMSO; TPA, 0.03% in acetone. ### P < 0.001 vs. vehicle; * P < 0.1, ** P < 0.01, *** P < 0.001 vs. TPA control, n = 8–10. | |
For many nonsteroidal anti-inflammatory drugs, such as COX-2 inhibitors, adverse gastrointestinal effects and increased cardiovascular risks are two main limitations for their clinical application. We tested the toxicity of 2f by monitoring its influence on food intake, body weight and ether-a-go-go-related-gene (hERG) channel activity. Long-term studies suggested that intragastrical administration of 2f (30 mg kg−1 for 30 days, 100 mg kg−1 for 10 days) had no significant effects on food intake and body weight in mice (Fig. S2†). Mice stomach anatomy indicated that 2f did not cause gastrointestinal hemorrhages, while indomethacin (20 mg kg−1 for 3 days, ig) resulted in obvious hemorrhages (Fig. S3†). The low gastrointestinal side effects of 2f may be because it induces analgesia and anti-inflammatory via enhancement of PEA rather than reducing prostaglandins (PGs) in the gut. Moreover, the activity of hERG channels in CHO cells was only inhibited by 38.3% by 40 μM 2f (Fig. S4†), a concentration far above its threshold for NAAA inhibition. However, cisapride, a COX-2 inhibitor, inhibited 50% hERG at a concentration of 0.1 μM. These results suggested that with lower gastrointestinal and cardiovascular side effects, oxazolidone derivatives are promising anti-inflammatory drug candidates in clinical translation.
3 Conclusion
In conclusion, we report the discovery of oxazolidone derivatives as a novel scaffold of NAAA inhibitors. The NAAA inhibition activity of oxazolidone derivatives was modulated by the side chain and terminal lipophilic substituents. The results showed that the link chain length of C5, straight and saturated linkages were the preferred shape patterns. Several nanomolar NAAA inhibitors (2f, 3b, 3h, 3i and 3j) were developed. Compound 2f exhibited highly potent and selective inhibitory activity towards NAAA via a competitive and reversible pattern. Moreover, in vivo study showed that 2f had significant anti-inflammatory and analgesic effects with low side effects. Our study demonstrated that oxazolidone derivatives is a class of potent NAAA inhibitors holds great promise for clinical translation. We anticipate that these oxazolidone derivatives will help better understand the function of NAAA and facilitate the development of improved therapies for inflammation and pain.
4 Materials and methods
4.1 Materials and instruments
All reagents used in the present study were purchased from Sigma-Aldrich (Shanghai, China) unless otherwise indicated. Tetrahydrofuran was distilled prior to use from sodium benzophenone ketyl. Dichloromethane (CH2Cl2) was distilled from phosphorus pentoxide. Dimethylformamide (DMF) was distilled from calcium hydride. Silica gel (300–400 mesh) from Yantai Athy Chemical Technology Co. Ltd. (Zhifu, China) was used for column chromatography, and compounds were eluted with an ethyl acetate/petroleum ether (PE) (60–90 °C) mixture (unless otherwise stated).
The 1H-NMR and 13C-NMR spectra were recorded on a Bruker 400 spectrometer (400 MHz for 1H, 100 MHz for 13C), using CDCl3, d6-DMSO, or CD3OD as solvent and Me4Si as internal substance. IR spectra were recorded on a Nicolet Avatar 360 RT-IR spectrophotometer. Mass spectra were recorded on an Applied Bio systems MDS SCIEX 3200Q-TRAP mass spectrometry (MS) system with electro spray ionization (ESI†) and direct injection. The HRFABMS spectra were recorded on a Bruker APEX-FTMS apparatus. Elemental analysis was performed using a Vario RL analyzer. Melting points were determined on a Yanaco MP-500 melting point apparatus and are uncorrected. HPLC analysis were run on Agilent-1200 series HPLC system equipped with a photodiode array detector, using a Hypersil Gold C18 column (dimensions 250 × 4.6 mm, particle size 5 μm). Photodiode array (PDA) detector range was set as 210–600 nm.
4.2 Synthesis of oxazolidone derivatives
4.2.1 Synthesis of amides 1a, 1j, 1k, 1n, 1o and 1q–1t. Palmitoyl chloride (83 mg, 0.3 mmol) in CH2Cl2 (2 mL) was added dropwise to a mixture of amine (0.3 mmol) and Et3N (0.08 mL, 0.6 mmol) in anhydrous CH2Cl2 (2 mL) under a nitrogen atmosphere. The resulting solution was stirred at room temperature for 4 h, then quenched with saturated aqueous ammonium chloride, and extracted with CH2Cl2 (3 × 10 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel and 1a, 1j, 1k, 1n, 1o and 1q–1t were then obtained.21
4.2.2 Synthesis of amides 1l and 1m. A solution of 1-(3-hydroxypyrrolidin-1-yl)hexadecan-1-one (19 mg, 0.06 mmol) in THF (1 mL) was added slowly to a suspension of NaH (60% in mineral oil, 5 mg) in anhydrous THF (5 mL), under a nitrogen atmosphere at 0 °C. After stirring at the same temperature for 5 min, a solution of CH3I (0.15 mmol) in THF (0.5 mL) was slowly added. The reaction mixture was stirred at 0 °C for 3 h before quenched with saturated aqueous ammonium chloride (5 mL) and extracted with EtOAc (3 × 5 mL). The combined organic layers were treated using the same procedure as 2.2.1 to afford 1l and 1m.23
4.2.3 Synthesis of amides 1c and 1d. A solution of 4 M HCl (1 mmol, 0.25 mL dioxane) was added slowly to 10 mL tert-butyl 1-palmitoylpyrrolidin-3-ylcarbamate (60.0 mg, 0.14 mmol) methanol solution at 0 °C under a nitrogen atmosphere. The resulting solution was stirred at 0 °C for 2 h, quenched with a saturated aqueous solution of Na2CO3, and extracted with EtOAc (3 × 20 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH/CH2Cl2 = 1:15) to afford 1c and 1d.25,26
:MeOH/CH2Cl2 = 1:15) to afford 1c and 1d.25,26
4.2.4 Synthesis of amides 1e and 1f. I2 solution (76 mg, 0.3 mmol) in THF (2 mL) was added slowly to a mixture of imidazole (27 mg, 0.4 mmol) and PPh3 (79 mg, 0.3 mmol) in THF (5 mL), under a nitrogen atmosphere at 0 °C. After stirring at the same temperature for 1 h, a THF solution (2 mL) of 1-(2-(hydroxymethyl) pyrrolidin-1-yl) hexadecan-1-one (71 mg, 0.2 mmol) was slowly added. The reaction mixture was stirred at 0 °C for 0.5 h and allowed to warm slowly to room temperature over 2 h. The reaction was quenched with a saturated solution of Na2S2O3 and extracted with EtOAc (3 × 5 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The mixture in EtOAc (10 mL) was treated with 10% Pd/C (100 mg) and purged with H2. After stirring for 12 h at room temperature, the reaction mixture was filtered through celite and concentrated. The residue was purified by flash chromatography on silica gel (eluent:EtOAc/PE 1:5) to afford 1e and 1f.27,28
4.2.5 Synthesis of oxazolidone imides 1i, 2a–2h, 3a–3d and 3h–3s. A CH2Cl2 (5 mL) solution of oxalyl dichloride (0.06 mL, 0.66 mmol) was added slowly to a solution of the suitable acid (0.55 mmol) and dimethylformamide (0.05 mL) under a nitrogen atmosphere at 0 °C. The reaction mixture was stirred at 0 °C for 1 h and evaporated under reduced pressure. The acid chloride obtained was used as such for the next reaction due to its high instability. A hexane solution (0.2 mL) of 2.5 Mn-BuLi (0.55 mmol, hexane) was added slowly to a THF solution (5 mL) of 2-oxazolidone (44 mg, 0.5 mmol), under a nitrogen atmosphere at −78 °C. After stirring at the same temperature for 10 min, a THF solution (2 mL) of acid chloride (0.55 mmol) was slowly added. The reaction mixture was stirred at −78 °C for 0.5 h and allowed to warm slowly to room temperature over 5 h. The reaction was quenched with saturated aqueous ammonium chloride (1.0 mL) and extracted with EtOAc (3 × 5 mL). The combined organic layers were treated using the same procedure as 2.2.1 to afford 1i, 2a–2h, 3a–3d and 3h–3s.29
4.2.6 Synthesis of acids 3q-1 and 3t-1. Thionyl dichloride (0.2 mL, 3 mmol) was slowly added to 10 mL CH3OH solution of the corresponding carboxylic acid (2.0 mmol) under a nitrogen atmosphere at 0 °C. The reaction mixture was stirred at 0 °C for 2 h and evaporated under reduced pressure. The product was dissolved in acetone (15 mL) and treated with K2CO3 (552 mg, 4 mmol) and alkyl bromide (2.5 mmol). The reaction mixture was filtered and concentrated under reduced pressure after being refluxed for 8 h. The residue was dissolved in a 1:1 mixture of aqueous 2 M NaOH and MeOH (16 mL), stirred at room temperature for 4 h, then treated with 2 M HCl, and extracted with EtOAc (3 × 5 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel to afford 3q-1 and 3t-1.14
4.2.7 Synthesis of carbamates 3e–3g. A CH2Cl2 solution (2 mL) of bis(trichloromethyl) carbonate (98 mg, 0.33 mmol) was added dropwise to a solution of 2-oxazolidone (44 mg, 0.5 mmol) and Et3N (0.16 mL, 1.2 mmol) in CH2Cl2 (5 mL) under a nitrogen atmosphere. After stirring at 0 °C for 1 h and room temperature for 2 h, the amine (0.4 mmol) in CH2Cl2 (1 mL) was added dropwise to the mixture under a nitrogen atmosphere. The resulting mixture was stirred at room temperature for 5 h, quenched with saturated aqueous ammonium chloride, and extracted with CH2Cl2 (3 × 10 mL). The combined organic phases were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography on silica gel to afford 3e–3g.30
4.3 In vitro biological evaluation of oxazolidone derivatives
4.3.1 HPLC-MS/MS analysis. A HPLC-MS/MS method was developed and validated for free fatty acids (FFAs) quantification. An ABI 3200 Q-Trap mass spectrometer equipped with Agilent 1200-HPLC system was used. FFAs were eluted at 1.0 mL min−1 at 40 °C with MeOH:water 95:5, v/v, each containing 0.25% acetic acid and 5 mmol L−1 ammonium acetate, pH = 7.4. The molecular ions were monitored by ESI† negative mode at m/z = 267 for heptadecenoic acid, m/z 303 for arachidonic acid, and m/z 269 for C17:0 FFAs.
4.3.2 Protein preparation and enzymatic assay. HEK293 cells overexpressing rat NAAA (HEK293-rNAAA) or rat FAAH (HEK293-rFAAH) were maintained in Dulbecco's modified Eagle medium (DMEM, Hyclone, Beijing, China) supplemented with 10% fetal bovine serum (FBS, Gibco®, Shanghai, China) containing 0.3 mg mL−1 geneticin G418. HEK293-rNAAA or HEK293-rFAAH cells were harvested, washed with PBS, sonicated in 20 mM Tris–HCl (pH 7.5) containing 0.32 M sucrose, and centrifuged at 800 × g for 15 min at 4 °C. The supernatants were collected, and the protein concentrations were measured by a BCA protein assay kit (Pierce, Shanghai, China). NAAA activity was measured by incubating 30 μg recombinant rNAAA protein with 25 μM heptadecenoylethanolamide at 37 °C for 30 min in 0.2 mL of phosphate buffer (50 mM, pH 5.0) containing 0.1% Triton X-100 and 3 mM dithiothreitol with or without the tested compounds. FAAH activity was measured by incubating 30 μg of protein derived from the HEK293-rFAAH cell extract at 37 °C in Tris–HCl buffer (50 mM, pH 8.0) containing fatty acid-free bovine serum albumin (0.05%). Anandamide (25 μM) was used as the substrate for FAAH. The reactions were terminated by adding 200 μL of methanol containing 1 nmol heptadecanoic acid as internal standard, and the remaining substrates were analysed by HPLC-MS/MS. IC50 of each compound was analysed by GraphPad Prism 5.
4.3.3 Dialysis assay. The dialysis assay was performed using Slide-A-Lyzer dialysis cassettes (Pierce, Shanghai, China). Briefly, 2 mg of NAAA protein was incubated with 2f or dimethyl sulfoxide (DMSO) in 4 mL of Tris–HCl buffer (50 mM, pH 5.0) for 10 min at 37 °C. The mixed reaction solution was loaded into a dialysis cartridge using a syringe and incubated in Tris–HCl buffer (50 mM, pH 5.0) at 4 °C for 8 h. The samples in the dialysis cassettes were collected for the NAAA enzymatic assay.
4.3.4 Rapid dilution assay. The rapid dilution assay was performed following a previous reported method.21 Briefly, samples containing 100-fold concentrated rNAAA protein were pre-incubated with 10-fold of the IC50-equivalent concentration of compounds or vehicle (1% DMSO) for 10 min at 37 °C. The samples were then diluted 100-fold with assay buffer containing substrate to initiate the reactions, and the time course of product formation was determined by HPLC-MS analysis.
4.4 In vivo anti-inflammatory and analgesic activity of compound 2f
All animal experiments were performed in accordance with ‘Guide and Care and Use of Laboratory Animals’ from National Institutes of Health (NIH) and approved by the Animal Care and Use Committees of Xiamen University in China.
4.4.1 In vivo analgesic activity of compound 2f. Formalin-induced pain model was used to evaluate the in vivo analgesic activity of 2f. Male ICR mice (18–20 g) were allowed to acclimate to the Plexiglas chamber for 30 min before testing. Compound 2f (10 mg kg−1, dissolved in 5% polyethylene glycol 400 and 5% Tween-80 in saline) was then injected into the plantar surface of the right hind paw with or without PPAR-α antagonist MK886 (2 mg kg−1). After 30 min, formalin (20 μL of 5% formalin, diluted in saline) was injected into the dorsal surface of the right hind paw and pain responses were recorded by an automated detecting system. The paw licking time was calculated every 5 min over the next 45 min. Nociceptive behavioural response to formalin stimulation was defined as follows: Phase I (0–10 min), acute pain response, Phase II (11–45 min), chronic pain response.
4.4.2 In vivo anti-inflammatory activity of compound 2f. TPA-induced ear edema model was used to test the anti-inflammatory of compound 2f. Male WT 129s mice (20–22 g) were grouped randomly, with 8–10 animals for each group. Compound 2f (30, 100 mg kg−1) was then administered by intragastric gavage 30 min before TPA application. TPA (0.03% in acetone, 10 μL) was applied evenly to both sides of the left ear of each mouse to cause edema and inflammation, while the same amount of acetone alone was smeared to the right ear. Animals were sacrificed 3 h after treatment. Both ears were excised immediately and punched in the same position, with a diameter of 9 mm. The ear swelling was determined by weighing pieces of the ears obtained. The mRNA expression levels of TNF-α, IL-6, COX-2 and NF-κB in mice ear were determined following our previously reported method.21
4.4.3 Long term toxicity of compound 2f. Mice were treat with compound 2f (100 mg kg−1 for 10 d, 30 mg kg−1 for 30 d) or indomethacin (20 mg kg−1 for 3 d) via intragastrical administration with normal diet. Mice body weight and food consumption were measured and recorded every day. The mice were sacrificed 6 h after the last administration and stomachs were harvested, rinsed with distilled water and photographed.
Acknowledgements
We thank Dr Daniele Piomelli at the University of California, Irvine for the kind gifts of the HEK293-rNAAA cells and the HEK293-rFAAH cells. YL also wants to thank William stung for English revision. This work was supported by funds from the National Natural Science Foundation of China (Grant No. 81373273 to QY and 81602974 to YL), Xiamen Southern Ocean Research Center Project (No. 14GYY018NF18) to QY, the Fundamental Research Funds for the Central Universities (No. 20720150054) to QY and China Postdoctoral Science Foundation Funded Project (No. 2016M592876XB) to YL.
References
- K. Tsuboi, Y. X. Sun, Y. Okamoto, N. Araki, T. Tonai and N. Ueda, J. Biol. Chem., 2005, 280, 11082–11092 CrossRef CAS PubMed.
- L. Facci, R. Dal Toso, S. Romanello, A. Buriani, S. D. Skaper and A. Leon, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 3376–3380 CrossRef CAS.
- J. Lo Verme, J. Fu, G. Astarita, G. La Rana, R. Russo, A. Calignano and D. Piomelli, Mol. Pharmacol., 2005, 67, 15–19 CrossRef CAS PubMed.
- C. Solorzano, C. Zhu, N. Battista, G. Astarita, A. Lodola, S. Rivara, M. Mor, R. Russo, M. Maccarrone, F. Antonietti, A. Duranti, A. Tontini, S. Cuzzocrea, G. Tarzia and D. Piomelli, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20966–20971 CrossRef CAS PubMed.
- S. Petrosino, T. Iuvone and V. Di Marzo, Biochimie, 2010, 92, 724–727 CrossRef CAS PubMed.
- A. Calignano, G. La Rana and D. Piomelli, Eur. J. Pharmacol, 2001, 419, 191–198 CrossRef CAS PubMed.
- B. Costa, S. Conti, G. Giagnoni and M. Colleoni, Br. J. Pharmacol., 2002, 137, 413–420 CrossRef CAS PubMed.
- S. Petrosino, L. Cristino, M. Karsak, E. Gaffal, N. Ueda, T. Tuting, T. Bisogno, D. De Filippis, A. D'Amico, C. Saturnino, P. Orlando, A. Zimmer, T. Iuvone and V. Di Marzo, Allergy, 2010, 65, 698–711 CrossRef CAS PubMed.
- J. M. Hesselink and T. A. Hekker, J. Pain Res., 2012, 5, 437–442 CrossRef PubMed.
- B. F. Cravatt, D. K. Giang, S. P. Mayfield, D. L. Boger, R. A. Lerner and N. B. Gilula, Nature, 1996, 384, 83–87 CrossRef CAS PubMed.
- O. Sasso, G. Moreno-Sanz, C. Martucci, N. Realini, M. Dionisi, L. Mengatto, A. Duranti, G. Tarozzo, G. Tarzia, M. Mor, R. Bertorelli, A. Reggiani and D. Piomelli, Pain, 2013, 154, 350–360 CrossRef CAS PubMed.
- J. M. West, N. Zvonok, K. M. Whitten, S. K. Vadivel, A. L. Bowman and A. Makriyannis, PLoS One, 2012, 7, e43877 CAS.
- T. Bandiera, S. Ponzano and D. Piomelli, Pharmacol. Res., 2014, 86, 11–17 CrossRef CAS PubMed.
- C. Solorzano, F. Antonietti, A. Duranti, A. Tontini, S. Rivara, A. Lodola, F. Vacondio, G. Tarzia, D. Piomelli and M. Mor, J. Med. Chem., 2010, 53, 5770–5781 CrossRef CAS PubMed.
- A. Armirotti, E. Romeo, S. Ponzano, L. Mengatto, M. Dionisi, C. Karacsonyi, F. Bertozzi, G. Garau, G. Tarozzo, A. Reggiani, T. Bandiera, G. Tarzia, M. Mor and D. Piomelli, ACS Med. Chem. Lett., 2012, 3, 422–426 CrossRef CAS PubMed.
- A. Duranti, A. Tontini, F. Antonietti, F. Vacondio, A. Fioni, C. Silva, A. Lodola, S. Rivara, C. Solorzano, D. Piomelli, G. Tarzia and M. Mor, J. Med. Chem., 2012, 55, 4824–4836 CrossRef CAS PubMed.
- S. Ponzano, F. Bertozzi, L. Mengatto, M. Dionisi, A. Armirotti, E. Romeo, A. Berteotti, C. Fiorelli, G. Tarozzo, A. Reggiani, A. Duranti, G. Tarzia, M. Mor, A. Cavalli, D. Piomelli and T. Bandiera, J. Med. Chem., 2013, 56, 6917–6934 CrossRef CAS PubMed.
- R. Vitale, G. Ottonello, R. Petracca, S. M. Bertozzi, S. Ponzano, A. Armirotti, A. Berteotti, M. Dionisi, A. Cavalli, D. Piomelli, T. Bandiera and F. Bertozzi, ChemMedChem, 2014, 9, 323–336 CrossRef CAS PubMed.
- M. Migliore, S. Pontis, A. L. Fuentes de Arriba, N. Realini, E. Torrente, A. Armirotti, E. Romeo, S. Di Martino, D. Russo, D. Pizzirani, M. Summa, M. Lanfranco, G. Ottonello, P. Busquet, K. M. Jung, M. Garcia-Guzman, R. Heim, R. Scarpelli and D. Piomelli, Angew. Chem., Int. Ed., 2016, 55, 11193–11197 CrossRef CAS PubMed.
- A. Ribeiro, S. Pontis, L. Mengatto, A. Armirotti, V. Chiurchiu, V. Capurro, A. Fiasella, A. Nuzzi, E. Romeo, G. Moreno-Sanz, M. Maccarrone, A. Reggiani, G. Tarzia, M. Mor, F. Bertozzi, T. Bandiera and D. Piomelli, ACS Chem. Biol., 2015, 2015(10), 1838–1846 CrossRef PubMed.
- Y. Li, L. Yang, L. Chen, C. Zhu, R. Huang, X. Zheng, Y. Qiu and J. Fu, PLoS One, 2012, 7, e43023 CAS.
- L. Yang, L. Li, L. Chen, Y. Li, H. Chen, Y. Li, G. Ji, D. Lin, Z. Liu and Y. Qiu, Sci. Rep., 2015, 5, 13565 CrossRef CAS PubMed.
- E. M. Smith, G. F. Swiss, B. R. Neustadt, E. H. Gold, J. A. Sommer, A. D. Brown, P. J. Chiu, R. Moran, E. J. Sybertz and T. Baum, J. Med. Chem., 1988, 31, 875–885 CrossRef CAS PubMed.
- C. M. Yea, C. E. Allan, D. M. Ashworth, J. Barnett, A. J. Baxter, J. D. Broadbridge, R. J. Franklin, S. L. Hampton, P. Hudson, J. A. Horton, P. D. Jenkins, A. M. Penson, G. R. Pitt, P. Riviere, P. A. Robson, D. P. Rooker, G. Semple, A. Sheppard, R. M. Haigh and M. B. Roe, J. Med. Chem., 2008, 51, 8124–8134 CrossRef CAS PubMed.
- B. J. Backes, K. Longenecker, G. L. Hamilton, K. Stewart, C. Lai, H. Kopecka, T. W. von Geldern, D. J. Madar, Z. Pei, T. H. Lubben, B. A. Zinker, Z. Tian, S. J. Ballaron, M. A. Stashko, A. K. Mika, D. W. Beno, A. J. Kempf-Grote, C. Black-Schaefer, H. L. Sham and J. M. Trevillyan, Bioorg. Med. Chem. Lett., 2007, 17, 2005–2012 CrossRef CAS PubMed.
- S. J. Macdonald, D. J. Belton, D. M. Buckley, J. E. Spooner, M. S. Anson, L. A. Harrison, K. Mills, R. J. Upton, M. D. Dowle, R. A. Smith, C. R. Molloy and C. Risley, J. Med. Chem., 1998, 41, 3919–3922 CrossRef CAS PubMed.
- I. Izquierdo, M. T. Plaza, J. A. Tamayo, F. Franco and F. Sánchez-Cantalejo, Tetrahedron, 2008, 64, 4993–4998 CrossRef CAS.
- S. Mitsumori, H. Zhang, P. Ha-Yeon Cheong, K. N. Houk, F. Tanaka and C. F. Barbas 3rd, J. Am. Chem. Soc, 2006, 128, 1040–1041 CrossRef CAS PubMed.
- L. Huang and W. D. Wulff, J. Am. Chem. Soc, 2011, 133, 8892–8895 CrossRef CAS PubMed.
- J. T. Randolph, C. A. Flentge, P. P. Huang, D. K. Hutchinson, L. L. Klein, H. B. Lim, R. Mondal, T. Reisch, D. A. Montgomery, W. W. Jiang, S. V. Masse, L. E. Hernandez, R. F. Henry, Y. Liu, G. Koev, W. M. Kati, K. D. Stewart, D. W. Beno, A. Molla and D. J. Kempf, J. Med. Chem., 2009, 52, 3174–3183 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details, characterization, and 1H-NMR and 13C-NMR spectra of the intermediate and final compounds. See DOI: 10.1039/c6ra28734d |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence