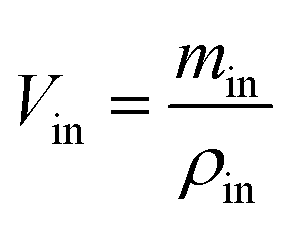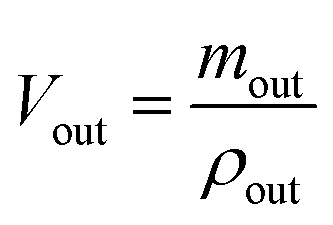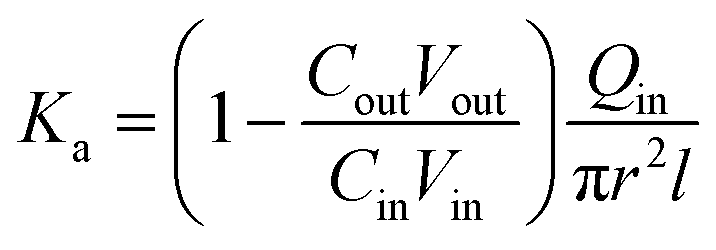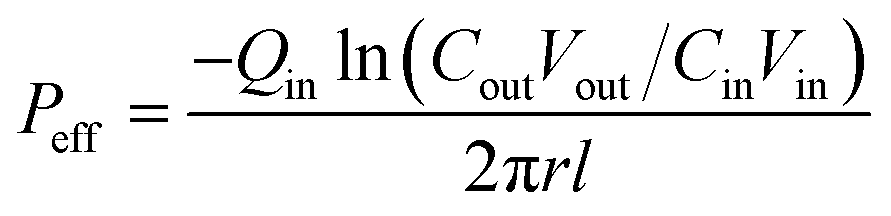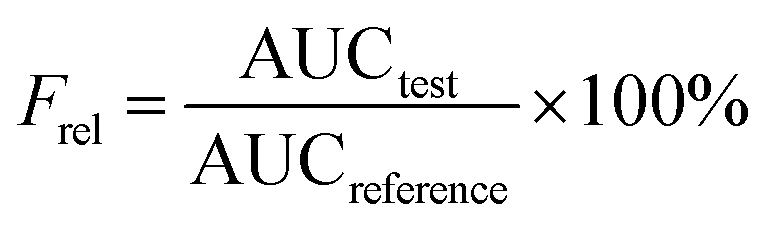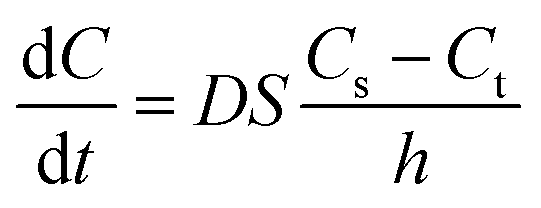DOI:
10.1039/C6RA28676C
(Paper)
RSC Adv., 2017,
7, 13053-13064
Preparation and evaluation of celecoxib nanosuspensions for bioavailability enhancement
Received
25th December 2016
, Accepted 16th February 2017
First published on 24th February 2017
Abstract
Celecoxib (CLX) is a selective cyclooxygenase-2 (COX-2) inhibitor; however, the application of CLX is compromised due to its poor aqueous solubility and low oral bioavailability. This study aims to develop a nanotechnology to overcome these issues. Celecoxib nanosuspensions (CLX-NS) were formulated using D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) as stabilizer via high pressure homogenization (HPH) and then freeze-dried to solid powder. Transmission electron microscopy (TEM), differential scanning calorimetry (DSC), X-ray powder diffraction (XRD) and Fourier transform infrared spectroscopy (FT-IR) were employed to characterize the physicochemical and pharmaceutical properties of CLX-NS lyophilization. In vitro dissolution, an in situ single-pass intestinal perfusion study, and in vivo pharmacokinetic studies were performed for further investigation. Particles of the nanosuspensions were short-rod shaped (∼388 nm) and remained in the same crystalline state as CLX coarse powder. In the test of in vitro dissolution, CLX-NS displayed a higher dissolution amount (90.8% during 60 minutes) compared to groups of CLX coarse powder (47.9%) and physical mixture (52.9%). Moreover, in situ single-pass intestinal perfusion study indicated that CLX-NS could be well absorbed in the whole intestine with the main absorption site being the duodenum. Significant improvements in Cmax and AUC0–t of CLX-NS were obtained in pharmacokinetic analyses with a 245.8% increase in relative bioavailability compared to CLX coarse powder. This study showed that nanosuspension preparations could be a promising strategy in influencing CLX absorption in gastrointestinal tract, and improving dissolution and oral bioavailability of poorly water-soluble drugs.
Introduction
Celecoxib (CLX), a selective cyclooxygenase-2 (COX-2) inhibitor, has been successfully used in the treatment of osteoarthritis, rheumatoid arthritis and acute pain. Several preclinical and experimental studies have also demonstrated the preventive and therapeutic efficacy of combining CLX with chemotherapeutic agents in cancer treatments.1 CLX, provided with hydrophobic methyl and trifluoromethyl groups in the structure (pKa = 11.1, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = 3.5), is classified as a BCS class II substance due to its low aqueous solubility (1–3 μg mL−1) and good permeability,2 resulting in high variability in absorption and erratic blood levels after oral administration. Dolenc et al. has demonstrated that the absolute bioavailability after oral administration of CLX capsules in beagle dogs is only 30% and the drug absorption is limited by the dissolution rate.3 Hence, increasing the solubility and dissolution rate of CLX has the potential for oral bioavailability improvement. Numerous classical pharmaceutical strategies have emerged to increase CLX dissolution rate such as β-cyclodextrin complexes,4 solid dispersions,5 microemulsions6 and microspheres.7 Moreover, a variety of novel drug delivery systems have been designed to improve the solubility and dissolution of poorly water-soluble drugs. For instance, fibrous ordered mesoporous carbon (FOMC) was introduced as a new drug carrier for loading CLX via oral administration to improve drug loading capacity and dissolution rate. The results showed that the dissolution rate of CLX from FOMC was much faster than pure crystalline CLX (more than 70% and no more than 27% in 60 min, respectively) using dissolution medium of pH 6.8 buffer and the AUC0–24 h of CLX-FOMC capsules was about 1.68-fold than that of CLX capsules, demonstrating a marked enhancement in the oral absorption of CLX when administered using mesoporouscarbon as carrier.8 Furthermore, Nguyen et al. fabricated novel CLX silica-lipid hybrid (SLH) microparticles; and the CLX-SLH provided a beneficial enhancement in fasted state oral bioavailability of CLX (6.5-fold higher than aqueous suspensions, and more than 2-fold higher relative to the fed state) after oral administration to beagle dogs.9
P = 3.5), is classified as a BCS class II substance due to its low aqueous solubility (1–3 μg mL−1) and good permeability,2 resulting in high variability in absorption and erratic blood levels after oral administration. Dolenc et al. has demonstrated that the absolute bioavailability after oral administration of CLX capsules in beagle dogs is only 30% and the drug absorption is limited by the dissolution rate.3 Hence, increasing the solubility and dissolution rate of CLX has the potential for oral bioavailability improvement. Numerous classical pharmaceutical strategies have emerged to increase CLX dissolution rate such as β-cyclodextrin complexes,4 solid dispersions,5 microemulsions6 and microspheres.7 Moreover, a variety of novel drug delivery systems have been designed to improve the solubility and dissolution of poorly water-soluble drugs. For instance, fibrous ordered mesoporous carbon (FOMC) was introduced as a new drug carrier for loading CLX via oral administration to improve drug loading capacity and dissolution rate. The results showed that the dissolution rate of CLX from FOMC was much faster than pure crystalline CLX (more than 70% and no more than 27% in 60 min, respectively) using dissolution medium of pH 6.8 buffer and the AUC0–24 h of CLX-FOMC capsules was about 1.68-fold than that of CLX capsules, demonstrating a marked enhancement in the oral absorption of CLX when administered using mesoporouscarbon as carrier.8 Furthermore, Nguyen et al. fabricated novel CLX silica-lipid hybrid (SLH) microparticles; and the CLX-SLH provided a beneficial enhancement in fasted state oral bioavailability of CLX (6.5-fold higher than aqueous suspensions, and more than 2-fold higher relative to the fed state) after oral administration to beagle dogs.9
Over the past decades, strategy of size reduction to nanoscale has been proved to be an undoubtedly promising approach as surface-to-volume ratio increase, thus leading to enhancement of dissolution characteristics and system exposure. According to Noyes–Whitney equation, drugs with smaller particle size have larger surface areas for dissolution velocity increase;10 and the resulting higher concentration gradient between gastrointestinal lumen and blood could further increase oral bioavailability of drugs.11 Recently, tremendous advanced nanosystems, such as nanoparticles,12,13 nanoemulsions14 and nanogels,15 were designed to deliver varieties of hydrophobic drugs to different targets. Among these, nanosuspensions have attracted significant attention of researchers in the pharmaceutical field. Nanosuspension, i.e. nanocrystal, is defined as a colloidal dispersion system, which consists of pure drugs and stabilizers with an average particle size less than 1 μm.16 Compared to other systems, nanosuspensions provide unique features including increase of saturation solubility, dissolution velocity and adhesiveness to cell membranes,17 passive targeting and accumulating in liver or spleen after i.v. administration.18 Moreover, in vivo targetability could be achieved by the surface modification of nanosuspensions with special stabilizers.19
Several strategies, including top-down (e.g. wet milling),20 bottom-up (e.g. precipitation) method,21 and the combination of two methods22 have been applied for nanosuspension production. Among these technologies, high pressure homogenization (HPH) has been widely used for its simplicity, ease of large scale production, absence of organic solvents and reduced product contamination. Li et al. utilized high speed homogenizer (HSH) and HPH to obtain paclitaxel nanosuspensions (PTX-NS) for i.v. injection and the bioavailability of PTX-NS increased 14.38 and 3.51 times, respectively, compared with raw PTX and Taxol® formulation.23 The combination of microprecipitation and HPH method was employed to develop nimodipine nanocrystal which could dramatically improve the aqueous solubility, and exhibit about 2.6-fold higher than Nimotop® in terms of oral bioavailability in beagle dogs.24 Considering long-term stability, patient convenience and ease of transportation, it is necessary to transform liquid nanosuspensions into solid products; and this process can be achieved by multiple drying methods such as freeze-drying, spray drying, vacuum drying, pelletization, granulation, and etc.25
Although nanosuspensions possess many merits, they are less stable and prone to cause particle aggregation/agglomeration and crystal growth.26 Therefore, an eligible stabilizer is critical to suppress agglomeration and maintain the physical stability of the nanosuspension during preparation and storage. The stabilization mechanisms of commonly used stabilizers can be classified into two categories, electrostatic repulsion and steric stabilization.27 For instance, hydroxypropylmethyl cellulose (HPMC), povidone (PVP), D-α-tocopheryl polyethylene glycol 1000 succinate (TPGS) and poloxamer (F68 and F127) are usually used as steric stabilizers; while sodium dodecyl sulfate (SDS) is used as an electrostatic stabilizer. TPGS is a water-soluble derivative of natural vitamin E formed by esterification of vitamin E succinate with polyethylene glycol (PEG) 1000. TPGS has an amphiphilic structure with a lipophilic alkyl tail and hydrophilic polar head with a hydrophilic/lipophilic balance value of 13.2.28 As one of the novel non-ionic surfactants, TPGS has significant surface activity and notable effect on the lipid membrane. Hence, it can solubilize a variety of water-insoluble compounds, and therefore has been widely used in functions of wetting, emulsification, solubilization and absorption enhancer.29 According to the reports, the administration of colchicine formulation containing TPGS resulted in significantly higher systemic exposures and 2-fold increase in AUC as compared to the aqueous reference vehicle.30 TPGS was found to markedly enhance intestinal absorption of berberine chloride (BBR), especially at a concentration of 2.5% achieving around 2.9- and 1.9-fold improvement on Cmax and AUC0–36 h of BBR after oral administration respectively.31
Pharmaceutical researchers have developed different formulations to increase the solubility and oral bioavailability of CLX. For example, nanosuspensions containing celecoxib were produced by emulsion-diffusion method using three different stabilizers (Tween 80, PVP K30 and SDS) and the spray-dried nanosuspensions were compressed to tablets.3 When produced by combining the anti-solvent precipitation and HPH approaches in the presence of HPMC E5 and SDS, the obtained celecoxib nanoparticles could markedly enhance the dissolution rate and oral bioavailability in beagle dog.32 Despite the fact that the fabrication and evaluation of celecoxib nanosuspensions have established, there are few preparation process in these investigations without using organic solvents and large amount of stabilizers. The objective of this study was to prepare celecoxib nanosuspensions (CLX-NS) using TPGS as stabilizer via high speed shear as a pre-treatment step, followed by HPH method. The solidification of fresh nanosuspension was carried out by freeze-drying and the obtained nanosuspension lyophilization was characterized in terms of particle size, zeta potential, morphology, thermal properties, and crystallinity for assessing the variation of physicochemical and pharmaceutical properties during preparation process. The short term stability, solubility, in vitro dissolution patterns, intestinal absorption as well as oral bioavailability in rats of CLX-NS were evaluated in details.
Materials and methods
Materials
Celecoxib (CLX) was purchased from Sigma-Aldrich (St. Louis, USA). D-alpha Tocopherol acid polyethylene glycol succinate (TPGS), poloxamer 188 and PVP K30 was gifts from BASF (Ludwigshafen, Germany). HPMC E15 was obtained from Colorcon (Shanghai, China). Lactose was kindly gifted by Meggle (Shanghai, China). Sodiumdodecyl sulfate (SDS) was purchased from Guoyao Chemical Co., Ltd (Shanghai, China). Heparin sodium salt (180 IU per mg) was purchased from Sigma Chemical Co., Ltd (St. Louis, USA). All other reagents were analytical grade commercial products.
Preparation of CLX-NS and physical mixtures
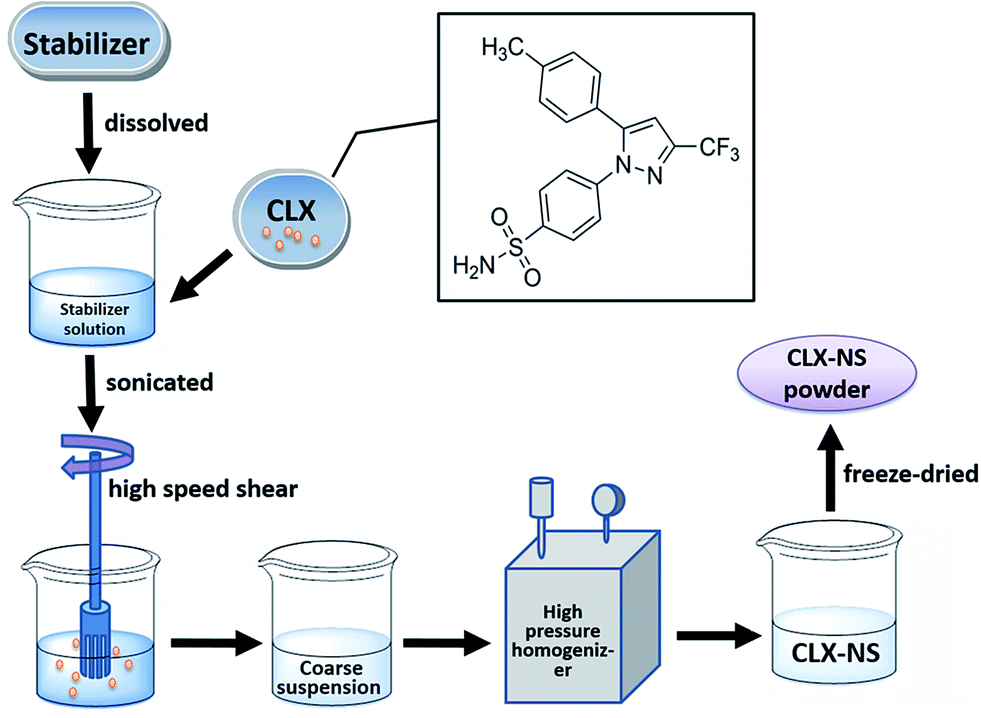
CLX-NS were prepared via HPH by using a homogenize machine (AH-2010, ATS Engineering Inc., Canada). A flow chart of the preparation process was shown in Fig. 1. Briefly, 200 mg CLX coarse powder was added into a 40 mL aqueous solution containing various stabilizers (HPMC E15, poloxamer 188, PVP K30, TPGS, SDS and the combination of SDS and other stabilizers), and then the mixture was placed in an ultrasonic water bath (KQ5200DB, Kunshan Ultrasonic Instrument Co., Ltd, China) for 5 min-sonication. The obtained mixture was stirred at 10000 rpm with high speed shear homogenizer (XHF-D, Scientz, China) for 5 min to obtain a coarse suspension. Then the suspensions were homogenized to nanoscale using a high pressure homogenizer equipped with a heat exchanger. At first, 5 cycles at 200 bar were conducted as pre-milling step and then 20 cycles at 800 bar were run to obtain fresh nanosuspensions. Furthermore, solidification of optimal CLX-NS was achieved by using a laboratory-scale freeze dryer (FD-1A-50, Boyikang Lab Instrument Co., Ltd, China). The freshly prepared CLX-NS solution was rapidly pre-frozen with separate addition of 2.5% (w/v) lactose, sucrose, trehalose or mannitol as cryoprotectant in a −60 °C ultra low temperature freezer (Forma 900, Thermo Fisher Scientific Inc., USA) for 12 h and then freeze-dried at −55 °C for 48 h. The physical mixtures for apparent solubility and dissolution testing were prepared by simply mixing all the powders with the same ratio as CLX-NS until a homogenous mixture was obtained.
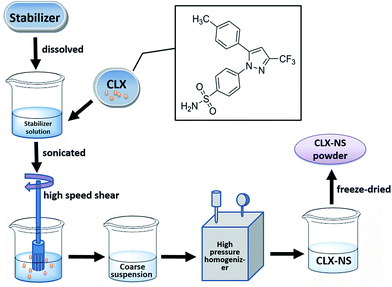 |
| | Fig. 1 The flow chart of the preparation processes for CLX-NS. | |
Characterization of nanosuspensions
Particle size and zeta potential. The particle size distribution of CLX coarse powder was measured using a laser diffractometer equipped with a small sample dispersion unit (Mastersizer 2000, Malvern Instruments Ltd, UK). Using laser diffraction (LD) analysis, it is possible to measure particle size between 0.1–2000 μm. The diameters at 10%, 50%, and 90% of the cumulative volume distribution (d10, d50, and d90, separately) gave the particle size distribution.The mean particle size (Z-ave) and polydispersity index (PDI) of CLX-NS were determined by dynamic light scattering (DLS) in a particle size analyzer (Zetasizer 3000HS, Malvern Instruments Ltd, UK) at room temperature. The nanosuspension was diluted with deionized water to achieve the required particle density for analysis. The zeta potential values were assessed by determination of particle electrophoretic velocity using the same instrument. All measurements were made in triplicate.
Morphology. The morphology of CLX coarse powder was evaluated using scanning electron microscope (SEM, S-3000N, Hitachi, Japan).33 Prior to examination, samples were mounted onto a metal stub using a carbon double sided adhesive tape, and further sputtered with a thin layer of gold palladium. The instrument was operated at a voltage of 10 kV.The morphology of nanoparticles in suspensions was observed by using transmission electron microscopy (TEM, H-7650, Hitachi, Japan). Samples were dropped onto film-coated copper grids and stained with 2% phosphotungstic acid solution for 3 min. The excess was then removed using a filter paper and the copper grids were dried naturally for viewing.
Differential scanning calorimetry (DSC). DSC measurements were carried out using a differential scanning calorimeter (DSC204 F1, Phoenix Netzsch, Germany).34 About 2 mg of the samples (CLX coarse powder, physical mixtures, freeze-dried CLX-NS powder, TPGS, lactose) were weighed and placed in a standard aluminium pan, fitted with a perforated lid for scanning. A heating rate of 10 °C min−1 was employed in the range of 40–280 °C under nitrogen gas. The DSC was pre-calibrated for baseline using empty pans and for temperature and enthalpy using indium.
Powder X-ray diffraction (PXRD). X-ray powder diffraction patterns of the selected samples were obtained using a X-ray powder diffractometer (Bruker D8 Advance, Karlsruhe, Germany) with Cu-Kα radiation (1.5406 Å). The samples were gently consolidated in an aluminium holder and scanned at 40 kV and 40 mA from 3–40° (2θ) using a scanning speed of 1.2° per min and a step size of 0.02°.
Fourier transform infrared spectroscopy (FT-IR). The FT-IR spectra of samples were obtained using FT-IR analyzer (IRTracer-100, Shimadzu, Japan) by KBr method.35 Samples were previously prepared with KBr at 1:5 (sample: KBr, w/w). KBr disks were prepared by compressing the powders at a pressure of 5 tons for 3 min in a hydraulic press and scanned against a blank KBr disk at wave numbers ranging from 500 to 4000 cm−1 with a resolution of 1.0 cm−1.
Apparent solubility determination
The apparent solubility were measured among coarse CLX powder, physical mixture and CLX-NS. Excess of each sample was added into sealed glass vials with 5 mL of deionized water (37 ± 0.5) °C. The vials were shaken at 100 rpm for 48 h in an air bath (HZC-250, BingLab Equipment Co. Ltd, China). After equilibrium was reached, the samples were centrifuged at 10000 rpm for 10 min and the concentration of CLX in the supernatant were determined by high performance liquid chromatography (HPLC, LC-2010, Shimadzu, Japan) equipped with a Hedera C18 column (250 mm × 4.6 mm, 5 μm, Hanbon, China). The mobile phase consisted of methanol and water (78:22, v/v) with a flow rate of 1.0 mL min−1. The detection wavelength was 252 nm and sample injected volume was 20 μL. The method was validated as previously reported.36
In vitro dissolution studies
Dissolution studies were carried out in a dissolution tester (ZRS-8G, Tianjin Tianda Tianfa Technology Co., Ltd, China) by using the USP Apparatus 2 (paddle) method. The bath temperature and paddle speed were set at 37 °C and 50 rpm respectively. The dissolution media included 1000 mL distilled water, hydrochloric acid solution (pH 1.2), acetic acid–sodium acetate buffer (pH 4.5), phosphate buffer (pH 6.8) and tribasic sodium phosphate buffer (pH 12). And each dissolution medium contained 0.3% (w/v) SDS in order to maintain the sink condition during the dissolution process. An accurate weight of CLX-NS powder, physical mixtures and CLX coarse powders (equivalent to 50 mg CLX) were added to the vessels. 5 mL samples were withdrawn at 5, 10, 15, 20, 30, 45 and 60 min, and centrifugated at 10000 rpm for 3 min. The accumulation dissolution amount of CLX was determined by HPLC mentioned above.
Short-term stability study
A short-term stability study was carried out to preliminarily investigate the physical stability of nanosuspensions. The fresh prepared CLX-NS was kept at closed glass vial and stored at two different temperatures (4 °C and room temperature). During a period of two-week-monitoring, particle size and PDI were determined at the time points of 0, 1 and 2 weeks.
In situ single-pass intestinal perfusion study (SPIP)
Animals received care in accordance with the Guidance Suggestions for the Care and Use of Laboratory Animals. All care and procedures of animals were approved by the University Ethics Committee for the use of experimental animals and carried out in strict accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals. SPIP was performed using an established method adapted from the literature.37,38 Briefly, rats were fasted with free access to water for 12 h before the experiment. Anesthesia was induced with an i.p. injection of 20% urethane (1.2 g kg−1) and the rats were fixed on an operation table at 37 °C. The abdomen was opened by a midline incision of 3–4 cm. Aimed intestinal segments of 10 cm were isolated and cannulated with glass tubes, which were connected to a peristaltic pump (BT100-2J, Baoding Longer Precision Pump Co., Ltd, China). The incision area was covered with a pledget wetted by saline (37 °C) to keep moist. The intestine was first washed with normal saline (37 °C), then perfused with Krebs–Ringer's buffer solution (pH 7.4, 7.8 g NaCl, 0.35 g KCl, 1.37 g NaHCO3, 0.02 g MgCl2, 0.22 g NaH2PO4 and 1.48 g glucose in 1000 mL purified water) at 0.2 mL min−1 for 10 min to attain balance. The perfusates (CLX-NS or CLX coarse suspension, at a concentration of 20 μg mL−1 CLX) were then infused at 0.2 mL min−1 and the time was set 0 min with the immediate start of the drug perfusion. After 45 min of saturation, the outflow solution was collected every 15 min up to 120 min in tarred glass tubes and the weight was measured at each time point. Samples were frozen immediately and stored at −20 °C until analysis. At the end of perfusion, the animals were sacrificed by cervical dislocation and the length and inner diameter of segments were accurately measured.
Isolation method of intestinal segment: duodenum segment beginning from 2 cm away from pylorus, jejunum segment beginning from 15 cm away from pylorus, while ileum segment beginning at the site 20 cm upwards cecum, and colon segment beginning at the site downwards cecum.
Sample preparation and analysis. The samples were collected into Eppendorf tube and centrifuged at 12000 rpm for 10 min. The supernatant was accurately diluted with methanol before a 20 μL sample was injected into HPLC (LC 2010C HT, Shimadzu, Japan) equipped with a inertsil ODS-SP C18 column (4.6 mm × 150 mm, 5 μm, Shimadzu, Japan) and detected at 252 nm. The mobile phase consisted of methanol and water (75:25, v/v) with a flow rate of 1.0 mL min−1. The HPLC method was validated as previously reported.39 Gravimetric method with density correction was used to correct the volume change which prevents the results from the interference by water absorption and secretion during the perfusion.| |
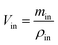 | (1) |
| |
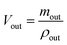 | (2) |
where, min, ρin, Vin are the quality, density, volume of the drug perfusate; mout, ρout, Vout are the quality, density, volume of the sample after correction.The absorption rate constant (Ka) and effective permeability coefficient (Peff) were used to evaluate the ease of drugs absorption in the intestine. These parameters were calculated according to the following equations:
| |
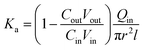 | (3) |
| |
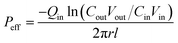 | (4) |
where,
Qin is the perfusion rate (0.2 mL min
−1);
Cin and
Cout are the total concentrations of CLX including CLX molecules and the crystals in the solution entering and exiting the intestine;
r is the inner radius of the intestine, and
l is the length of the intestine.
Pharmacokinetic studies
All animals used in this study were handled in accordance with the University Ethics Committee of Care and Use of Laboratory Animals. Male Sprague-Dawley rats (weighted 200–250 g) purchased from Qinglongshan animal center (Nanjing, China) were used as experiment animals. Twelve rats were divided into two groups (n = 6). Before administration, the rats were fasted over night with free access to water. The next morning, samples (coarse suspension, nanosuspension) were administered orally to rats at a CLX dose of 40 mg kg−1 respectively. Blood samples (0.3 mL) were collected via the orbit vein at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24 and 36 h after administration. The collected blood samples were placed in a dried heparinized Eppendorf tube and then separated immediately by centrifugation at 10000 rpm for 10 min. The supernatant was stored at −20 °C until analysis. Deproteinization method was employed for plasma sample preparation. 5 μL of internal standard solution (10 μg mL−1 diazepam dissolved in methanol) and 300 μL acetonitrile were added to each 100 μL rat blood sample. The mixtures were vortexed for 3 min and centrifugated at 14000 rpm for 10 min. Thereafter 330 μL of the supernatant was transferred into another clean tube, and evaporated under nitrogen gas flow. Then, the residue was redissolved using 100 μL of methanol and vortexed for 2 min. The obtained mixtures were centrifugated at 10000 rpm for 5 min; and 20 μL volume of supernatant was analyzed by the HPLC (LC 2010C HT, Shimadzu, Japan) with the same chromatographic conditions that were applied in SPIP.
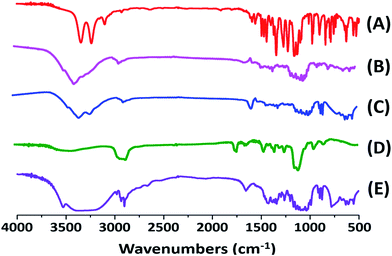
Pharmacokinetic analysis was performed by means of a non-compartmental method using DAS 2.1.1 software (Mathematical Pharmacology Professional Committee of China, China).40 The pharmacokinetic parameters including maximum plasma concentration (Cmax) and the time to reach maximum plasma concentration (Tmax) were directly obtained from plasma data. The area under the plasma concentration–time curves (AUC0–t) was calculated using the trapezoidal method. The relative bioavailability value (Frel) of CLX-NS after oral administration was calculated according to the following formula with the CLX coarse powder as reference:
| |
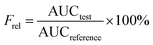 | (5) |
Statistical analysis
All results were expressed as mean ± SD. Student's t-test test was performed using SPSS 19.0 software. The evaluation of data used to assess the significance of differences and *p < 0.05 and **p < 0.01 were regarded as statistically significant.
Results and discussion
Preparation of CLX-NS
In this study, high speed shear combined HPH method was introduced to prepare CLX-NS. As a pretreatment step prior to HPH, the high speed shear is especially important for crushing the big particles and avoiding blocking the homogenizer gap. After pretreatment, homogenization process was conducted to further reduce drug particle size and improve sample homogeneity. It was considered that the pressure and number of homogenization cycles is mainly influenced by the hardness of the drug, the finesse of the starting material and the requirements of the application route or the final dosage form. It has also been shown that increasing the homogenization pressure does not promise a linear reduction in the particle size.41 Considering the above factors and our experimental results, 20 cycles at 800 bar was chosen as final homogeneous condition to obtain the optimal CLX-NS.
Evaluation of the type and concentration of the stabilizers is critical to the successful preparation of nanosuspensions, due to the enormous specific surface area of drug particles in nanosuspension compared to the conventional suspension. In this study, we used both polymeric and surfactant as stabilizers to choose an adequate one which can effectively ensure the stability of CLX-NS. Firstly, 0.25% (w/v) of HPMC E15, poloxamer 188, PVP K30, TPGS, SDS and SDS/other stabilizers (1:4) were used to screen different types of stabilizers with the particle size and PDI value as evaluation index. As shown in Table 1, the particle size of nanosuspensions decreased in the order of SDS > HPMC E15 > HPMC E15/SDS > poloxamer 188/SDS > poloxamer 188 > PVP K30 > TPGS/SDS > PVP K30/SDS > TPGS. Furthermore, the PDI values of CLX-NS increased in the order of HPMC E15 < HPMC E15/SDS < TPGS < PVPK30/SDS < PVP K30 < poloxamer 188 < TPGS/SDS < poloxamer 188/SDS < SDS. These results demonstrated that the smallest particle size and relatively small PDI value were obtained from the CLX-NS stabilized with TPGS. TPGS is a kind of amphiphilic surfactant, which could adsorb on the particle surface of hydrophobic drug through electrostatic interaction and significantly reduce surface tension of drug particles. Moreover, TPGS could also prevent the drug particles from aggregation by steric effect and maintain the stability of the nanosystem.
Table 1 Effect of the type of stabilizer on particle size, and PDI (n = 3)
| Stabilizer |
Size (nm) |
PDI |
| HPMC E15 |
382.2 ± 2.5 |
0.15 ± 0.04 |
| Poloxamer 188 |
291.3 ± 3.3 |
0.30 ± 0.01 |
| PVP K30 |
285.3 ± 2.7 |
0.29 ± 0.03 |
| TPGS |
265.1 ± 2.6 |
0.25 ± 0.01 |
| SDS |
672.7 ± 19.9 |
1.0 |
| HPMC E15/SDS |
360.5 ± 7.3 |
0.22 ± 0.04 |
| Poloxamer 188/SDS |
304.0 ± 2.5 |
0.38 ± 0.03 |
| PVP K30/SDS |
274.4 ± 4.6 |
0.28 ± 0.01 |
| TPGS/SDS |
281.2 ± 4.1 |
0.31 ± 0.02 |
To further optimize the particle size and PDI values of nanosuspension, the ratio of stabilizer to drug was also screened. As shown in Table 2, along with the ratio of TPGS to drug increasing, the trends of the particle size and PDI values showed decrease firstly and then increase. The minimum was obtained when the ratio was 1:4, which indicated that this ratio was more appropriate to the nanosuspension. It could be inferred that when the ratio of TPGS to drug was 1:10, the amount of stabilizer was too little to fully absorb to the surface of drug particles, which led to an instable nanosystem with large particle size and PDI; and when the ratio was more than 1:4, as the amount of stabilizer increase, the excess surfactant would constantly enwrap the drug particles and cause an increase in particle size. In addition, excess surfactant may form micelles spontaneously and result in larger PDI values. Therefore, the ratio of stabilizer to drug was determined to be 1:4.
Table 2 Effect of the stabilizer/drug ratio on the particle size, and PDI (n = 3)
| TPGS:drug |
Particle size (nm) |
PDI |
| 2:1 |
410.6 ± 7.6 |
0.68 ± 0.04 |
| 1:1 |
278.2 ± 5.8 |
0.35 ± 0.03 |
| 1:2 |
265.1 ± 6.3 |
0.25 ± 0.01 |
| 1:4 |
232.5 ± 3.0 |
0.20 ± 0.02 |
| 1:10 |
274.9 ± 7.9 |
0.28 ± 0.04 |
After homogenization process, the fresh CLX-NS was rapidly transformed to solid form by lyophilization to achieve long-term stability. In order to prevent particle aggregation during freeze-drying, several cryoprotectants such as sucrose, lactose, trehalose and mannitol, have been carefully screened. When using 2.5% (m/v) lactose to protect nanosuspensions, a smooth loose white powder with good redispersibility was obtained, which indicated that lactose is the most suitable cryoprotectant for CLX-NS.
Characterizations
Particle size and zeta potential analyses. The coarse CLX was a micronized product and exhibited a fine white appearance with poor flowability. The mean particle size of coarse CLX was 4.37 μm; and the d10, d50, and d90 were 1.18, 3.72 and 8.38 μm, respectively, which indicated a relatively broad distribution. The fresh nanosuspensions were transformed into solid by freeze drying to provide a stable solid form. The obtained CLX-NS powder with smooth loose appearance could be easily redispersed after gently shaking and the particle size was (388.1 ± 20.1) nm measured by DLS. DLS system yields the intensity weighted mean diameter of the bulk population and polydispersity index (PDI) as parameters for size distribution measure. It is reported that PDI value of 0.1–0.3 indicates a narrow size distribution, while that greater than 0.3 indicates a very broad size distribution.42 For lyophilized CLX-NS powder, a low PDI value was found to be (0.21 ± 0.05), indicating a narrow particle size distribution. Generally, the lower PDI value is, the better stability a nanosystem has. Furthermore, a uniform and narrow distribution of particle size is also favorable for improving dissolution rate, promoting intestinal absorption and enhancing oral bioavailability.The zeta potential is another significant index, which has an effect on the stability of dispersion system, because it reflects electrostatic barriers preventing the nanoparticles from aggregation and agglomeration. Particle aggregation is likely to occur when particles possess too low zeta potential to provide sufficient electric or steric repulsion between each other. Generally, a zeta potential of −30 mV at least for electrostatically stabilized systems or −20 mV for sterically stabilized systems was sufficient to obtain a physically stable nanosuspension.43 Therefore, zeta potential of (−21.3 ± 1.1) mV of reconstituted CLX-NS could provide a guarantee for the physical stability of the nanosystem.
Morphology for CLX-NS. SEM-characterized morphology of coarse CLX was shown in Fig. 2A. It can be observed that CLX crystals showed a long-needle shape with particle size ranging from 1 to 15 μm. In order to observe the morphology of CLX-NS powder, TEM imaging was performed. As shown in Fig. 2B, CLX-NS exhibited short-rod shape, with a width ranging from 100 to 200 nm and a length from 200 to 500 nm. This micrograph was in agreement with those measured by DLS.
 |
| | Fig. 2 (A) SEM image of CLX coarse powder and (B) TEM image of CLX-NS. | |
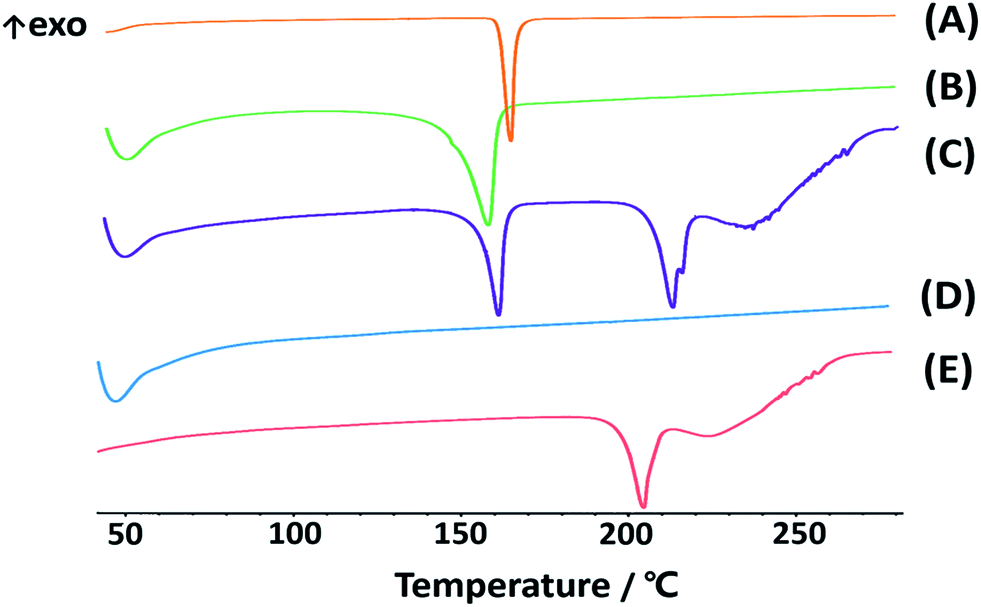
DSC analysis. Crystalline state is another factor influencing dissolution and stability behavior of compounds. The high power density in the piston-gap homogenizer leads to a high energy input during the HPH process.44 Occasionally, this technology may induce changes in the crystalline state, such as increase of amorphous fraction or creation of completely amorphous particles. The extent of such changes depends on the chemical nature and physical hardness of the active ingredients, as well as the applied power density.45,46 To study the differentiations in solid state of CLX in different preparations, thermal analysis of CLX coarse powder, physical mixtures, lyophilized CLX-NS powder, TPGS and lactose were performed. The results of thermal analysis by DSC were shown in Fig. 3, and coarse drug powder displayed a sharp endothermic peak at 161.7 °C which was the characteristic peak of CLX intrinsic melting point,47 while the melting endotherm of the physical mixture and freeze-dried drug nanosuspension powder were recorded at 155.7 °C and 159.1 °C, respectively. In addition, TPGS showed a broad endothermic peak between 45 °C and 55 °C, which might be the glass transition temperature (Tg). Moreover, the melting point of lactose was 206.3 °C. The melting temperatures of CLX in different groups (coarse drug powder, physical mixture and nanosuspension powder) were similar, indicating that neither the stabilizer nor the applied preparation process affected crystalline state of nanosuspensions. In general, an increase in saturation solubility and dissolution rate could be achieved by changing the crystalline state of the drug to amorphous.48 Nevertheless, in terms of long-term storage stability, a change in crystalline is often undesirable. Also, the slight change in melting temperature of CLX may be due to the presence of excipients in the mixtures, which has been confirmed in previous report.49
 |
| | Fig. 3 DSC thermograms of CLX coarse powder (A), physical mixtures (B), lyophilized CLX-NS powder (C), TPGS (D) and lactose (E). | |
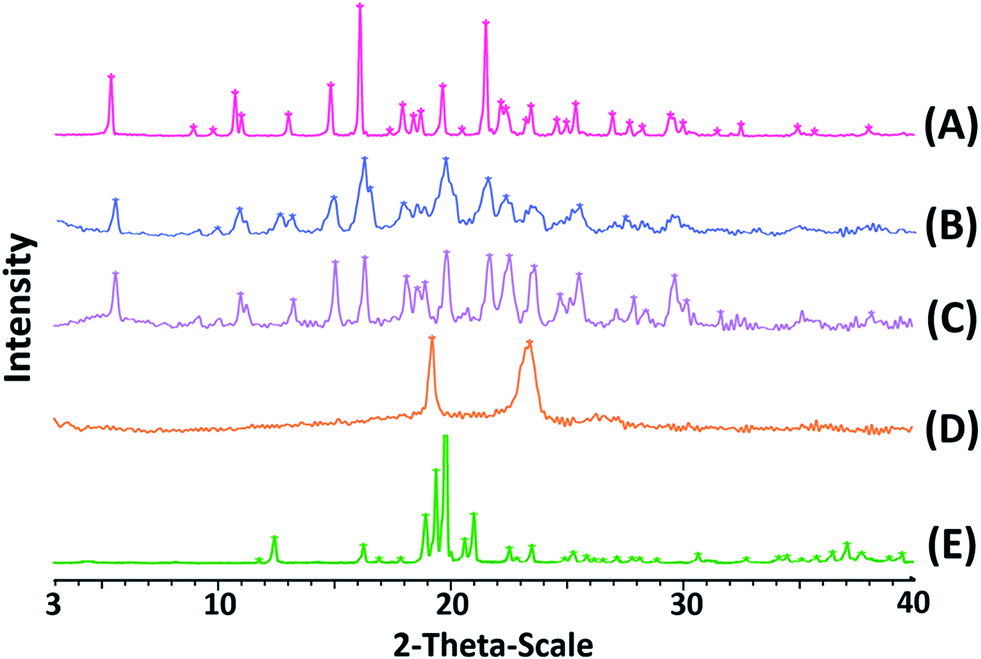
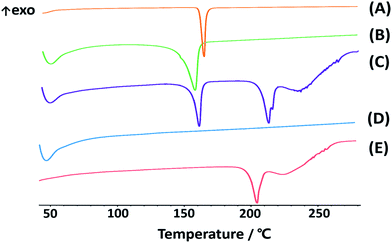
Powder X-ray diffraction (PXRD) analyses. To confirm the crystalline state of lyophilized CLX-NS, PXRD was performed with CLX coarse powder, physical mixtures, lyophilized CLX-NS powder, TPGS and lactose. PXRD patterns are displayed in Fig. 4. It has been reported that X-ray diffraction pattern of raw CLX showed sharp peaks at 2θ values of 5°, 10.5°, 16° and 21.5° which are characteristic peaks for crystalline nature of CLX.50 The diffractograms in figure suggested that there was no significant difference in characteristic peaks position of CLX among these preparations. Accordingly, all kinds of energy input during preparation and the attendance of stabilizer did not alter the crystalline state of lyophilized nanosuspension powder. The only difference detected between the coarse powder and CLX-NS were in the peaks intensity, probably attributed to particle size reduction, stabilizer at the surface and presence of lactose in formulations. Usually, drugs with lower crystallinity and smaller size induce improvement of dissolution rate and oral bioavailability. However, the maintenance of original crystalline structure is beneficial to long-term stability because of its low energy state. Moreover, the same results and induced properties were confirmed by previous studies. For instance, Wang et al. developed a deacety mycoepoxydiene (DM) nanosuspension by HPH process, and demonstrated that DM remained in crystalline state in nanosuspension which is physicochemically and thermodynamically more stable than DM in an amorphous state.42 As well as reported by Ahuja et al., febuxostat nanosuspensions were fabricated by wet media milling technique, in which the drug maintained crystalline state, and could be physically stable within three months.51
 |
| | Fig. 4 PXRD patterns of CLX coarse powder (A), physical mixtures (B), lyophilized CLX-NS powder (C), TPGS (D) and lactose (E). | |
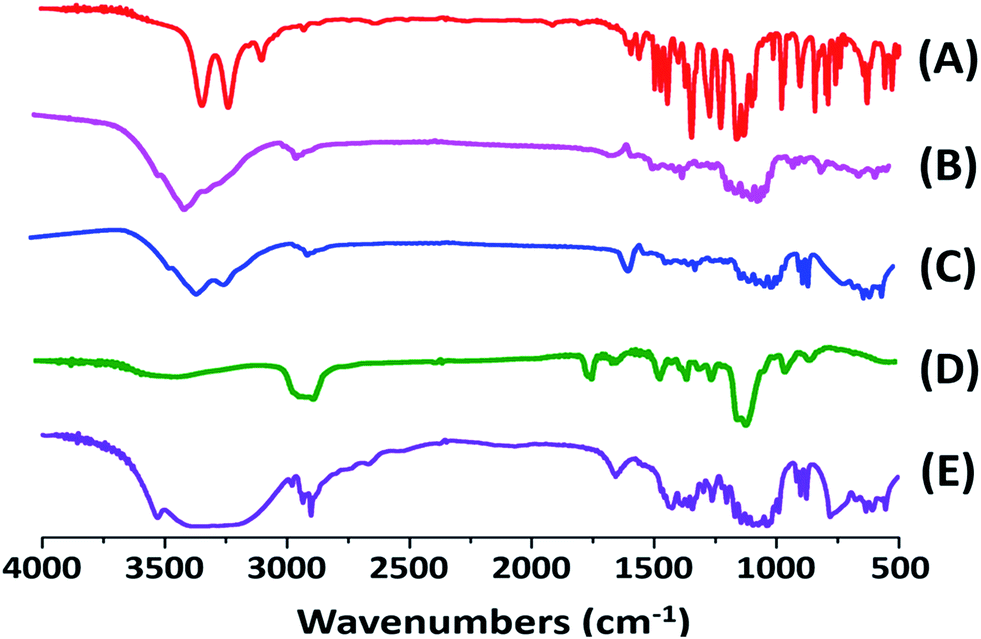
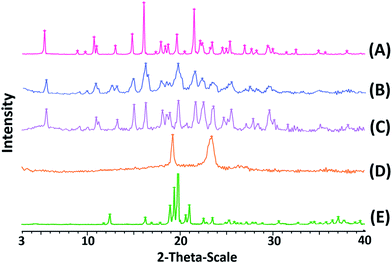
Fourier transform infrared spectroscopy (FT-IR). FT-IR has also been employed to study the possibility of molecular interactions between CLX and TPGS. The FT-IR spectra of CLX coarse powder, physical mixture, CLX-NS, TPGS and lactose samples were shown in Fig. 5. The FT-IR spectra of CLX coarse powder showed characteristic peaks at 3310 and 3202 cm−1 that is attributed to N–H stretching vibration of SO2NH2 group, 1308 and 1124 cm−1 for S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O asymmetric and symmetric stretching, and 1192 cm−1 for C–F stretching. These results were in good agreement with previous data.1 The characteristic peaks of CLX-NS were similar to CLX coarse powder, except the peaks of N–H were less obvious than that of CLX. This may be caused by the hydroxyl peak of lactose which was too broad and the N–H peaks were masked. The spectra of TPGS showed ester CO stretching at 1700 cm−1 and C–H stretching at 2898 cm−1. Regarding the molecular structures of CLX and TPGS, the formation of hydrogen bond is possible, which could be formed between NH2 group of CLX and carbonyl group of TPGS. It has been previously shown that sulfonamide group of CLX can participate as proton donor (NH2) and proton acceptor (SO2), and this can lead to intermolecular hydrogen bonding.47,50 Furthermore, this intermolecular hydrogen bonding has been reported for amorphous form of CLX, which means this intermolecular hydrogen bonding could not occur in crystalline form of CLX because of its spatial conformation.50 It has been previously reported that in amorphous CLX the doublet peak corresponded to NH2 group shifted to 3367 and 3264 cm−1 and the intermolecular hydrogen bonding caused a positive shift of NH2 groups to 3388 and 3264 cm−1.52 However, there is no shift in the wave number according to wave numbers of NH2 groups in CLX-NS. These results suggested that there was no obvious chemical reaction between CLX and TPGS and the CLX maintained its crystalline state in the forming of CLX-NS. These findings also supported the DSC and XRPD results.
O asymmetric and symmetric stretching, and 1192 cm−1 for C–F stretching. These results were in good agreement with previous data.1 The characteristic peaks of CLX-NS were similar to CLX coarse powder, except the peaks of N–H were less obvious than that of CLX. This may be caused by the hydroxyl peak of lactose which was too broad and the N–H peaks were masked. The spectra of TPGS showed ester CO stretching at 1700 cm−1 and C–H stretching at 2898 cm−1. Regarding the molecular structures of CLX and TPGS, the formation of hydrogen bond is possible, which could be formed between NH2 group of CLX and carbonyl group of TPGS. It has been previously shown that sulfonamide group of CLX can participate as proton donor (NH2) and proton acceptor (SO2), and this can lead to intermolecular hydrogen bonding.47,50 Furthermore, this intermolecular hydrogen bonding has been reported for amorphous form of CLX, which means this intermolecular hydrogen bonding could not occur in crystalline form of CLX because of its spatial conformation.50 It has been previously reported that in amorphous CLX the doublet peak corresponded to NH2 group shifted to 3367 and 3264 cm−1 and the intermolecular hydrogen bonding caused a positive shift of NH2 groups to 3388 and 3264 cm−1.52 However, there is no shift in the wave number according to wave numbers of NH2 groups in CLX-NS. These results suggested that there was no obvious chemical reaction between CLX and TPGS and the CLX maintained its crystalline state in the forming of CLX-NS. These findings also supported the DSC and XRPD results.
 |
| | Fig. 5 FT-IR spectra of CLX coarse powder (A), physical mixtures (B), lyophilized CLX-NS powder (C), TPGS (D) and lactose (E). | |
Apparent solubility determination
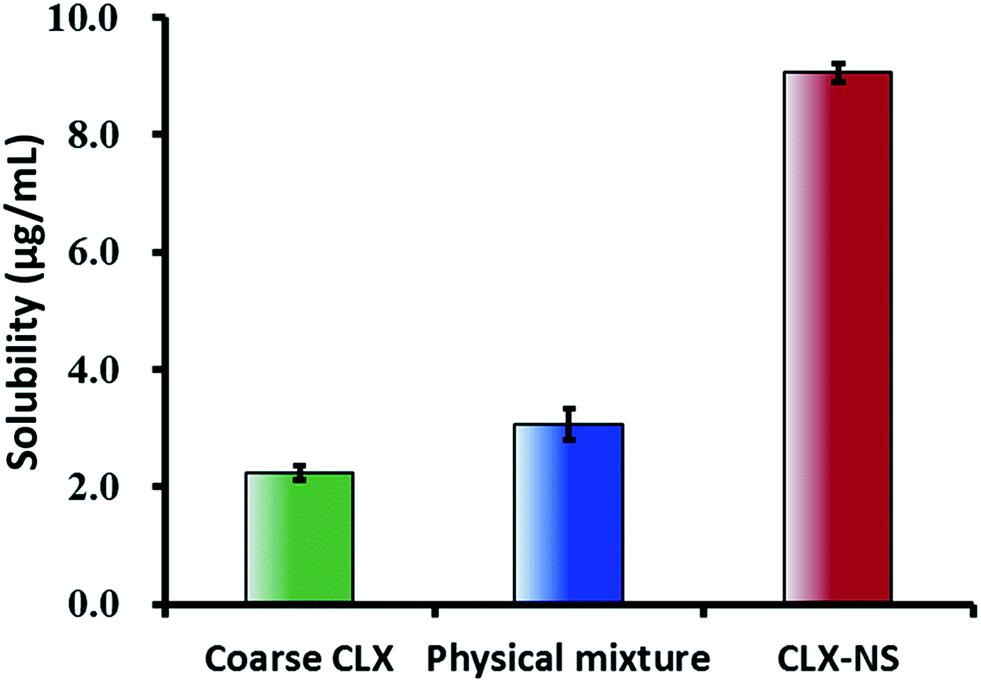
The apparent solubility of the coarse CLX, physical mixture and CLX-NS were shown in Fig. 6. The solubility of the coarse drug and physical mixture were found to be extremely low (2.25 ± 0.12 μg mL−1 and 3.06 ± 0.16 μg mL−1, respectively), which demonstrated that CLX was a poorly water-soluble compound. The apparent solubility of CLX-NS stabilized with TPGS was (9.06 ± 0.27) μg mL−1, 4-fold greater than that of coarse CLX. These results indicated that nanosuspension could be an effective nanotechnology to improve the solubility of CLX. In general, apparent solubility is a compound-specific constant, which depends on the temperature. However, the apparent solubility increases when the particle size below 1 μm.45 In addition, it could be predicted that the enhancement in apparent solubility has the potential to increase the dissolution, and hence improving theoral bioavailability of the BCS class II substance.
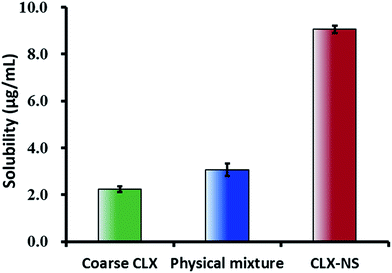 |
| | Fig. 6 Apparent solubility (37 °C) of coarse CLX, physical mixture and CLX-NS in deionized water. Results were expressed as the mean ± SD (n = 3). | |
In vitro dissolution profile
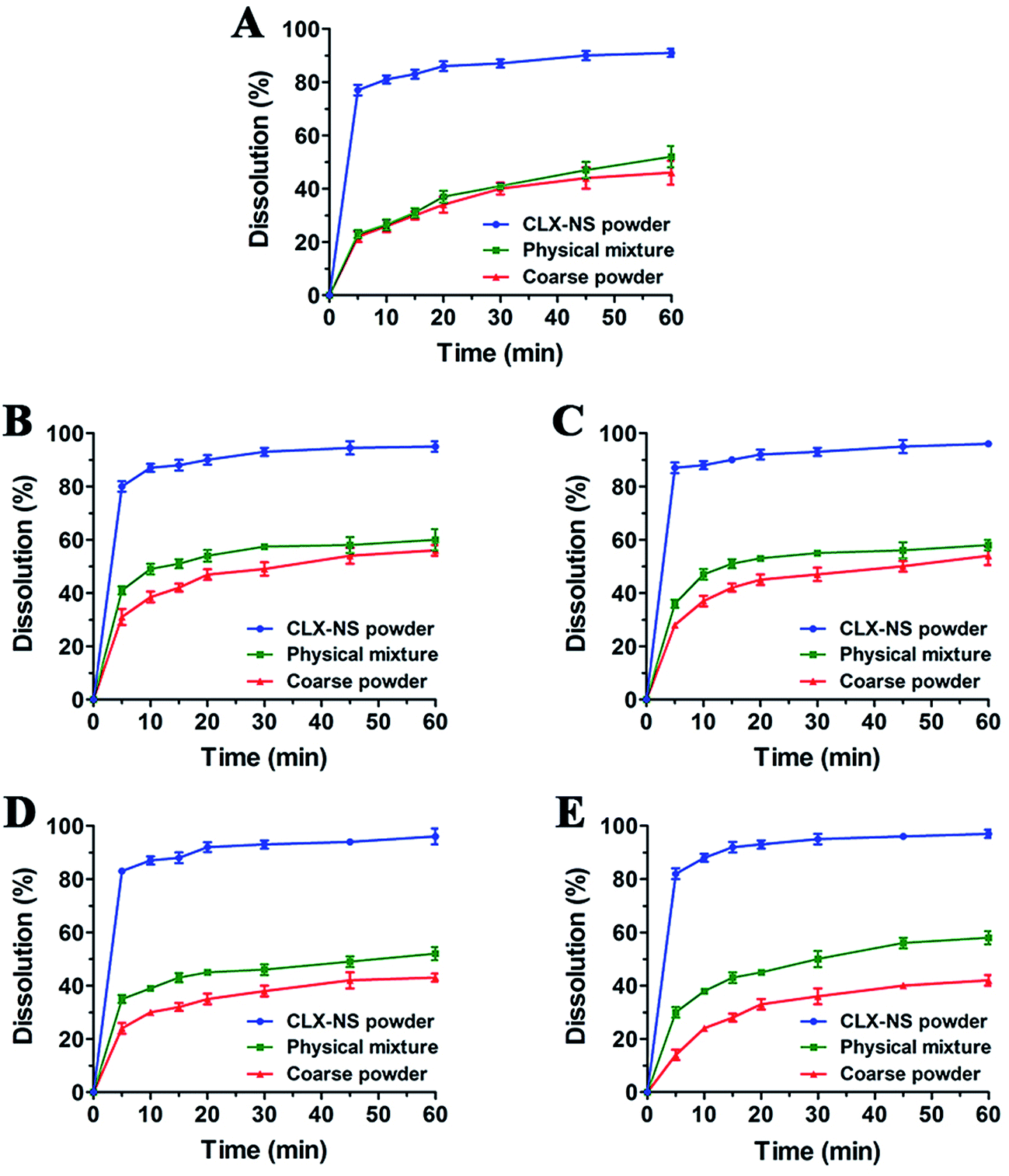
The dissolution behavior of the three samples in solutions with different pH were shown in Fig. 7. CLX coarse powder exhibited similar dissolution behavior with the physical mixture, while CLX-NS displayed a dramatic increase in dissolution rate. The dissolution amounts of CLX coarse powder, physical mixture and CLX-NS powder were 47.9%, 52.9% and 90.8% in 60 min respectively in distilled water containing 0.25% SDS. Moreover, the CLX-NS displayed markedly increased dissolution velocities (more than 80%) compared with the coarse CLX powder and physical mixture (27.9% and 28.3%, respectively) during 10 min. The dissolution profiles of CLX-NS were similar in solutions with different pH including pH 1.2, pH 4.5, pH 6.8 and pH 12, in which more than 80% of CLX was released from CLX-NS within 10 min. These results suggested that dissolution of the freeze-dried CLX-NS powder were distinctly superior compared to the CLX coarse powder and physical mixture. It is widely known that the enhancement in drug dissolution could be attributed to the particle size reduction, especially to the nanometer range which provides a strong increase in surface area available to dissolve.10 This phenomenon can be explained by Noyes–Whitney equation:| |
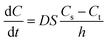 | (6) |
where dC/dt represents the dissolution rate, D is the diffusion coefficient of the solute, h denotes the thickness of the dissolution boundary layer and S represents the surface area, Cs is the saturation solubility and Ct is the bulk concentration. For CLX-NS, their particle size was smaller than that of coarse powder, so they had much more surface area. That might be the reason why they had a higher dissolution rate.
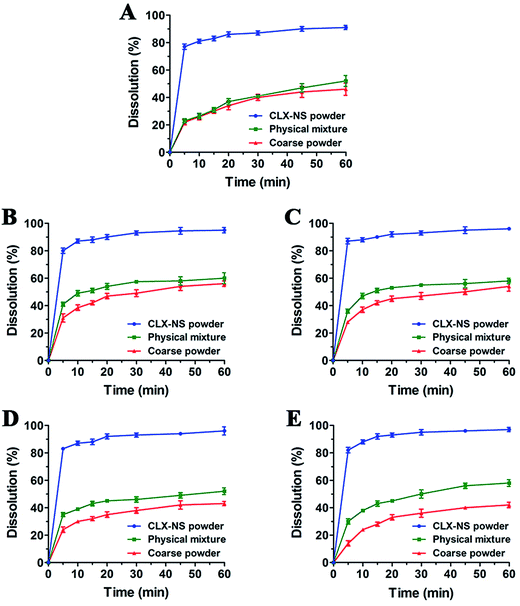 |
| | Fig. 7 Dissolution profiles for CLX coarse powder, physical mixture and CLX-NS powder in different dissolution media ((A) water + 0.25% SDS; (B) pH 1.2 + 0.25% SDS; (C) pH 4.5 + 0.25% SDS; (D) pH 6.8 + 0.25% SDS; (E) pH 12 + 0.25% SDS). Results were expressed as the mean ± SD (n = 3). | |
In fact, besides the size factor, the particle shape also plays an important role in affecting drug dissolution.53 Long, flaky particles with a high degree of irregularity may cause an increase in the average hydrodynamic boundary layer thickness. Thus, the h value would increase and as a result the dissolution rate will decrease. It can be seen from the SEM and TEM images of different sized CLX particles in Fig. 2 that most of them remained rod-like shape. Accordingly, the dissolution enhancement was probably due to their size reduction rather than the alteration of particle shape.
Short term stability
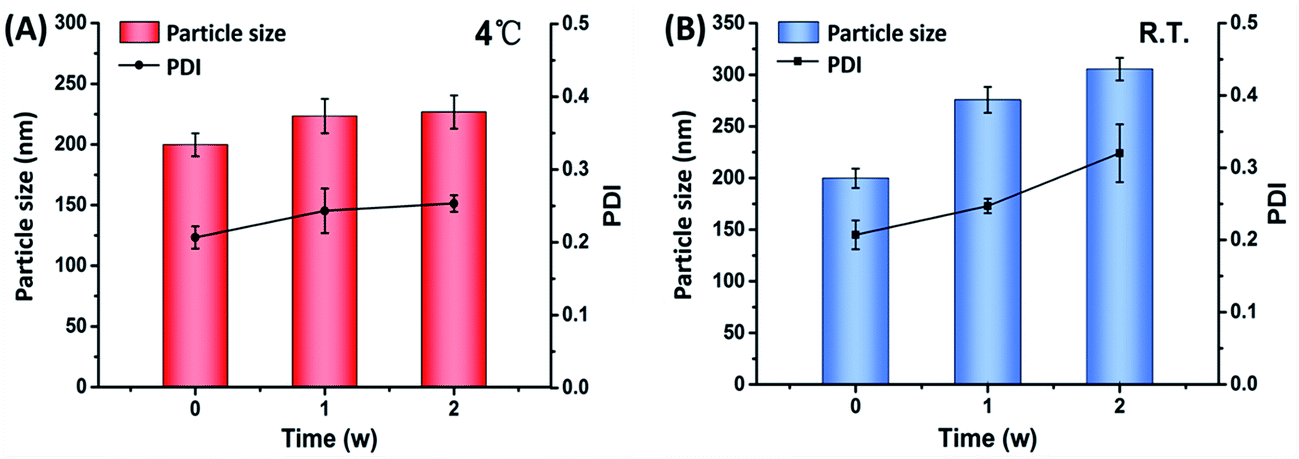
Physical stability issue is crucial in formulating nanosuspensions, mainly caused by Ostwald ripening effect and the aggregation of particles. Short term stability has been performed to evaluate the effect of different temperatures on the physical stability of CLX-NS. Particle size and PDI were detected to show the changes of stability during the storage time. As shown in Fig. 8, a significant increase in particle size of the preparations was observed from 0 to 1 week at all storage temperatures. The particle size of CLX-NS stored at room temperature, continued to increase after one week, while no significant size increase was observed at 4 °C. Obviously, there was some ripening occurring in the nanosuspension system during the storage. Ostwald ripening, a process whereby the larger particles grow by “devouring” smaller particles, might be contributed to the instability of nanoparticles. The driving force of Ostwald ripening arises because the concentration of drug in the vicinity of small particles is higher and that of large particles is lower than the average supersaturation.54 In addition, the nanosuspension system is thermodynamically unstable, and have a tendency to minimize their total energy. Hence, the particle aggregation happens spontaneously.
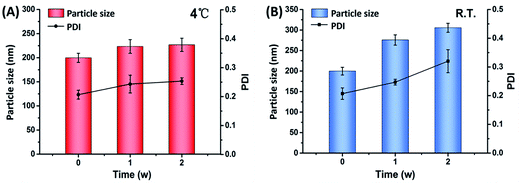 |
| | Fig. 8 Particle size and PDI of CLX-NS during two-weeks storage at different temperatures, such as 4 °C (A) and room temperature (B). Results were expressed as the mean ± SD (n = 3). | |
In situ single-pass intestinal perfusion study (SPIP)
Drug stability and adsorption studies were conducted before commencing the perfusion model. The results proved that CLX was stable in the perfusates and hardly absorbed by intestinal wall. Thus, any loss of CLX from the perfusates was just due to intestinal absorption. It has been reported that phenol red could interfere with the absorption and transport of poor water-soluble drugs in rat intestinal perfusion due to its partial absorption in the intestine.55 Therefore, gravimetric method was used to calibrate the volume change of the perfusates.
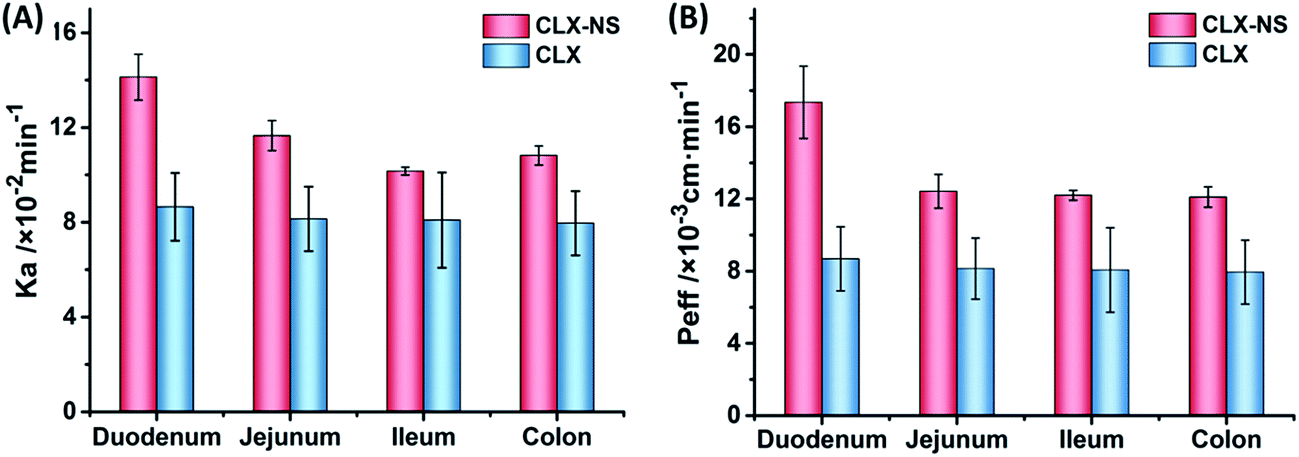
Absorption rate constant (Ka) and apparent effective permeability coefficient (Peff) obtained in SPIP study was illustrated in Fig. 9. It was demonstrated that the order of Ka for CLX-NS was duodenum > jejunum > colon > ileum, and Peff value of CLX-NS in the duodenum were significantly greater than other three intestinal segments (P < 0.05), indicating that CLX-NS could be well absorbed in the whole intestine with the main absorption site at duodenum. Compared with CLX group, Ka value of CLX-NS group was significantly enhanced in duodenum, jejunum and colon; and Peff value was remarkably improved in total intestinal segments (P < 0.05) owing to the nanoscale particle size which significantly increased the apparent solubility and dissolution rate of CLX from CLX-NP. The excellent adhesion of CLX-NS to the mucus or the intestinal villi also attributed to the delivery of CLX across the intestinal wall. Furthermore, the increased absorption of CLX-NS in rat intestine could be explained by the presence of TPGS. As known, TPGS is an excellent solubilizer and absorption enhancer which could be employed to increase the solubility of hydrophobic compounds, or improve the absorption of pharmacologically active drugs. Moreover, TPGS has been demonstrated to inhibit the effect of P-glycoprotein (P-gp), which is extensively distributed and expressed in the intestinal epithelium, hepatocytes, renal proximal tubular cells and so on.29 As shown in Table 3, the absorption parameters of CLX-NS containing TPGS were increased significantly compared with those of CLX group. Hence, TPGS applied as the stabilizer of CLX-NS might be beneficial to further improve intestinal absorption of CLX.
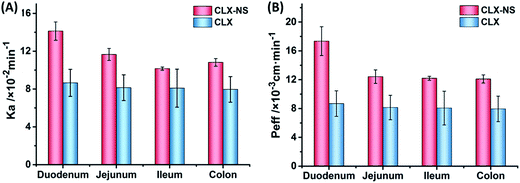 |
| | Fig. 9 The in situ absorption of CLX-NS in rat intestinal segments compared with CLX. The absorption rate (Ka) (A), and the effective permeability coefficient (Peff) (B). Results were expressed as the mean ± SD (n = 5). | |
Table 3 Absorption parameters of CLX and CLX-NS in different intestinal segments with CLX concentration of 20 μg mL−1. Results were expressed as the mean ± SD (n = 5)a
| Samples |
Parameters |
Duodenum |
Jejunum |
Ileum |
Colon |
| *P < 0.05, compared to the corresponding parameters of CLX coarse powder. |
| CLX |
Ka (×10−2 min−1) |
8.65 ± 1.43 |
8.14 ± 1.36 |
8.09 ± 2.01 |
7.96 ± 1.36 |
| Peff (×10−3 cm min−1) |
8.68 ± 1.77 |
8.14 ± 1.69 |
8.06 ± 2.34 |
7.94 ± 1.74 |
| CLX-NS |
Ka (×10−2 min−1) |
14.1 ± 0.97* |
11.7 ± 0.63* |
10.2 ± 0.17 |
10.8 ± 0.40* |
| Peff (×10−3 cm min−1) |
17.4 ± 2.0* |
12.4 ± 0.94* |
12.2 ± 0.27* |
12.1 ± 0.57* |
Pharmacokinetic studies in rats
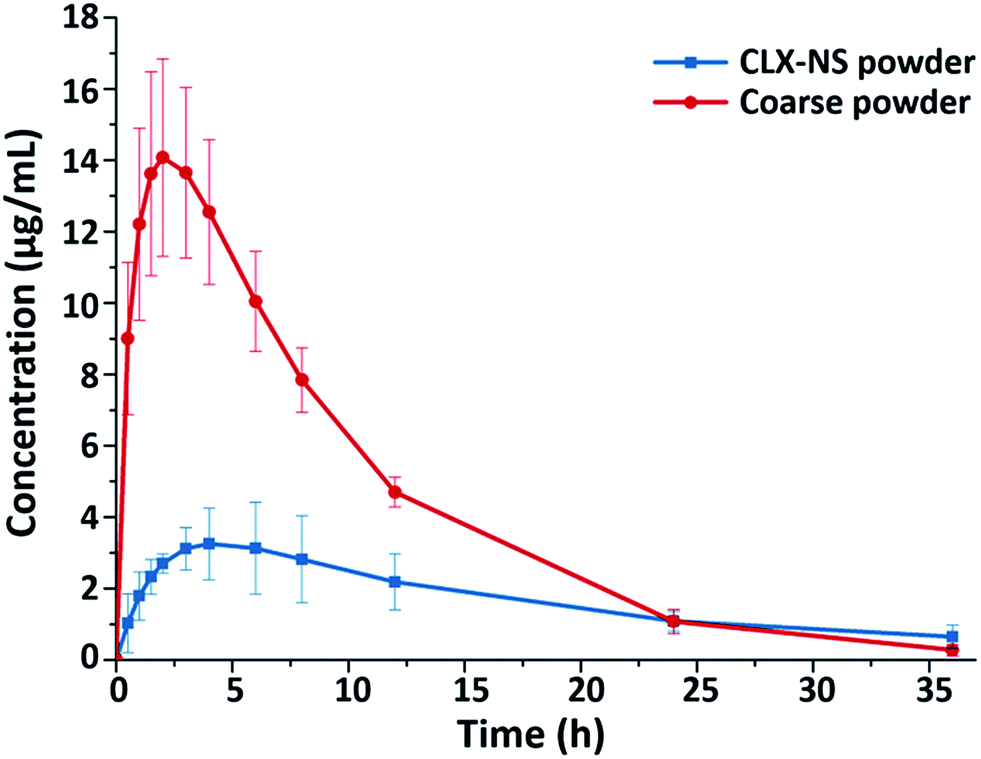
To confirm the positive effects of nanosuspension in enhancing the oral bioavailability of CLX, an in vivo test was carried out in rats; and the pharmacokinetic parameters of the CLX coarse powder and the CLX-NS were compared. The plasma concentration–time curves of two preparations were shown in Fig. 10, and the corresponding pharmacokinetic parameters were summarized in Table 4. As shown, the plasma concentration of CLX in rats after oral administration of CLX-NS was significantly higher than that of CLX coarse powder at nearly every time point except the last. In comparison with CLX coarse powder, CLX-NS exhibited higher Cmax and faster Tmax, indicating a higher absorption amount and more rapid absorption rate. Area under curve (AUC) is an important pharmacokinetic parameter to assess the exposure and circulating time of a drug; and the AUC0–t of CLX-NS and CLX coarse powder were 155.86 μg mL−1 h−1 and 63.40 μg mL−1 h−1, respectively. These results indicated that the oral absorption of CLX in rats was considerably enhanced in nanosuspension preparation.
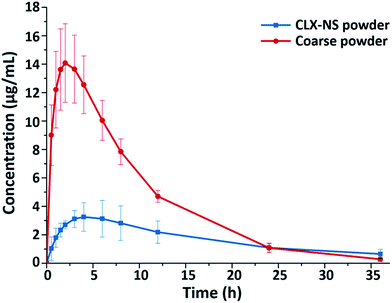 |
| | Fig. 10 Plasma concentration–time profiles of CLX coarse powder and CLX-NS powder. Results were expressed as the mean ± SD (n = 6). | |
Table 4 Pharmacokinetic parameters of CLX coarse suspension and CLX-NS after oral administration in rats. Results were expressed as the mean ± SD (n = 6)a
| Parameters |
CLX coarse powder |
CLX-NS powder |
| **P < 0.01, compared to the corresponding parameters of CLX coarse powder. |
| Cmax (μg mL−1) |
4.82 ± 1.04 |
16.08 ± 3.06** |
| Tmax (h) |
4.30 ± 1.88 |
3.00 ± 0.71 |
| MRT (h) |
12.92 ± 1.49 |
7.95 ± 0.64** |
| AUC0–t (μg mL−1 h−1) |
63.40 ± 9.44 |
155.86 ± 16.49** |
| Frel |
— |
2.46 |
Several factors could be involved in the improvement of the oral bioavailability of CLX, such as particle size decrease, dissolution rate increase, and membrane permeation enhancement. In this research, the particle size of CLX was reduced from micrometer (d50 is 3.72 μm) to nanoscale; this significant decrease may provide tremendous surface area and lead to faster dissolution velocity of the drug as elaborated in the Noyes–Whitney equation. Furthermore, reduction in particle sizes improved drug uptake not only by enhanced dissolution rate but also by other mechanisms, including increasing the mucosal adhesion to gastrointestinal surfaces, which can prolong gastrointestinal transit time and provide efficient oral bioavailability.19 Therefore, the decreased particle size and increased dissolution rates were indeed the determinant reasons for the higher Cmax and AUC0–t of CLX observed after the oral administration of the CLX-NS. Moreover, as a stabilizer, TPGS coated on the surface of nanoparticles may play an important role in enhancing bioavailability of CLX. Firstly, TPGS is one of the amphiphilic surfactant, which could enhance the penetration ability and promotes rapid permeation of the nanoparticles through the intestinal epithelium, and into the circulating blood. Secondly, TPGS is a P-gp inhibitor which could inhibit P-gp activity in the intestinal epithelium cells which subsequently increases the accumulation of the drug in cells.29
Conclusions
CLX-NS was successfully prepared using TPGS as the stabilizer via a combined methodology of high speed shear and HPH. The lyophilized CLX-NS showed small particle size with short-rod surface morphology, and no substantial crystalline change occurred after nanosuspension formation. The obtained CLX-NS containing only traces of stabilizer TPGS exhibited the remarkable increase in dissolution rate and great enhancement in intestinal absorption, owing to the nanoscale particle size, increased solubility and inhibited P-gp efflux by TPGS. The in vivo pharmacokinetic study also demonstrated a significantly improved oral bioavailability in rats by CLX-NS. Collectively, TPGS-stabilized CLX-NS could be a promising strategy to enhance dissolution and oral bioavailability of CLX.
Acknowledgements
The authors gratefully acknowledge financial support from National Natural Science Foundation of China (No. 81501582 and 81573379). This study was also supported Development Funds for Priority Academic Programs in Jiangsu Higher Education Institutions, and Fostering Plan of University Scientific and Technology Innovation Team of Jiangsu Qing Lan Project (2014).
References
- A. Homayouni, F. Sadeghi, J. Varshosaz, H. A. Garekani and A. Nokhodchi, Eur. J. Pharm. Biopharm., 2014, 88, 261–274 CrossRef CAS PubMed.
- A. Homayouni, F. Sadeghi, J. Varshosaz, H. A. Garekani and A. Nokhodchi, Colloids Surf., B, 2014, 122, 591–600 CrossRef CAS PubMed.
- A. Dolenc, J. Kristl, S. Baumgartner and O. Planinšek, Int. J. Pharm., 2009, 376, 204–212 CrossRef CAS PubMed.
- N. Mennini, S. Furlanetto, M. Cirri and P. Mura, Eur. J. Pharm. Biopharm., 2012, 80, 67–75 CrossRef CAS PubMed.
- P. Nkansah, A. Antipas, L. Ying, M. Varma, C. Rotter, B. Rago, A. El-Kattan, G. Taylor, M. Rubio and J. Litchfield, J. Controlled Release, 2013, 169, 150–161 CrossRef CAS PubMed.
- N. Garti, M. Avrahami and A. Aserin, J. Colloid Interface Sci., 2006, 299, 352–365 CrossRef CAS PubMed.
- C. Cannavà, S. Tommasini, R. Stancanelli, V. Cardile, F. Cilurzo, I. Giannone, G. Puglisi and C. A. Ventura, Colloids Surf., B, 2013, 111, 289–296 CrossRef PubMed.
- P. Zhao, H. Jiang, T. Jiang, Z. Zhi, C. Wu, C. Sun, J. Zhang and S. Wang, Eur. J. Pharm. Sci., 2012, 45, 639–647 CrossRef CAS PubMed.
- T. H. Nguyen, A. Tan, L. Santos, D. Ngo, G. A. Edwards, C. J. H. Porter, C. A. Prestidge and B. J. Boyd, J. Controlled Release, 2013, 167, 85–91 CrossRef CAS PubMed.
- A. A. Noyes and W. R. Whitney, J. Am. Chem. Soc., 2002, 19, 930–934 CrossRef.
- W. Li, Y. Yang, Y. Tian, X. Xu, Y. Chen, L. Mu, Y. Zhang and F. Liang, Int. J. Pharm., 2011, 408, 157–162 CrossRef CAS PubMed.
- M. Palamoor and M. M. Jablonski, Colloids Surf., B, 2013, 112, 474–482 CrossRef CAS PubMed.
- R. Wang, X. Gu, J. Zhou, L. Shen, L. Yin, P. Hua and Y. Ding, J. Controlled Release, 2016, 235, 134 CrossRef CAS PubMed.
- R. Chaiittianan and B. Sripanidkulchai, Drug Dev. Ind. Pharm., 2014, 40, 1597–1606 CrossRef CAS PubMed.
- E. S. Lee, D. Kim, Y. S. Youn, K. T. Oh and Y. H. Bae, Angew. Chem., 2008, 47, 2418–2421 CrossRef CAS PubMed.
- X. J. Tang, Y. H. Fu, Q. H. Meng, L. M. Li, X. Y. Ying, M. Han, Q. J. He, B. Yang, S. Zeng and Y. Z. Hu, Int. J. Pharm., 2013, 456, 243–250 CrossRef CAS PubMed.
- V. B. Junyaprasert and B. Morakul, Asian J. Pharm. Sci., 2015, 10, 13–23 CrossRef.
- Y. Wang, L. Wang, Z. Liu, D. Zhang and Q. Zhang, J. Biomed. Nanotechnol., 2012, 8, 760–769 CrossRef CAS PubMed.
- B. E. Rabinow, Nat. Rev. Drug Discovery, 2004, 3, 785–796 CrossRef CAS PubMed.
- X. Zhang, L. C. Li and S. Mao, Curr. Pharm. Des., 2014, 20, 388–407 CrossRef CAS PubMed.
- H. K. Chan and P. C. L. Kwok, Adv. Drug Delivery Rev., 2011, 63, 406–416 CrossRef CAS PubMed.
- H. Xi, C. Xi, Z. Ling, L. Xia, Z. Yu, T. Xing and Y. Wang, Int. J. Pharm., 2014, 472, 130–139 CrossRef PubMed.
- L. Yong, X. Zhao, Y. Zu and Z. Yin, Int. J. Pharm., 2015, 490, 324–333 CrossRef PubMed.
- Q. Fu, J. Sun, D. Zhang, M. Li, Y. Wang, G. Ling, X. Liu, Y. Sun, X. Sui and C. Luo, Colloids Surf., B, 2013, 109, 161–166 CrossRef CAS PubMed.
- B. V. Eerdenbrugh, L. Froyen, J. V. Humbeeck, J. A. Martens, P. Augustijns and G. V. D. Mooter, Eur. J. Pharm. Sci., 2008, 35, 127–135 CrossRef PubMed.
- Y. Wang, Y. Zheng, L. Zhang, Q. Wang and D. Zhang, J. Controlled Release, 2013, 172, 1126–1141 CrossRef CAS PubMed.
- L. Wu, J. Zhang and W. Watanabe, Adv. Drug Delivery Rev., 2011, 63, 456–469 CrossRef CAS PubMed.
- Y. Wu, Q. Chu, S. Tan, X. Zhuang, Y. Bao, T. Wu and Z. Zhang, Int. J. Nanomed., 2015, 10, 5219–5235 CAS.
- Y. Guo, J. Luo, S. Tan, B. O. Otieno and Z. Zhang, Eur. J. Pharm. Sci., 2013, 49, 175–186 CrossRef CAS PubMed.
- B. Bittner, A. Guenzi, P. Fullhardt, G. Zuercher, R. C. González and R. J. Mountfield, Arzneim.-Forsch., 2002, 52, 684–688 CAS.
- W. Chen, Y. Q. Miao, D. J. Fan, S. S. Yang, X. Lin, L. K. Meng and X. Tang, AAPS PharmSciTech, 2011, 12, 705–711 CrossRef CAS PubMed.
- Y. Liu, C. Sun, Y. Hao, T. Jiang, L. Zheng and S. Wang, J. Pharm. Pharm. Sci., 2010, 13, 589–606 CAS.
- Y. Wang, T. Sun, Y. Zhang, B. Chaurasiya, L. Huang, X. Liu, J. Tu, Y. Xiong and C. Sun, RSC Adv., 2016, 6, 37452–37462 RSC.
- X. Ling, Z. Huang, J. Wang, J. Xie, M. Feng, Y. Chen, F. Abbas, J. Tu, J. Wu and C. Sun, J. Mater. Chem. B, 2016, 4, 1787–1796 RSC.
- X. Ling, C. Zhao, L. Huang, Q. Wang, J. Tu, Y. Shen and C. Sun, RSC Adv., 2015, 5, 81668–81681 RSC.
- C. Sun, Y. Shen, D. Sun, T. Hang and J. Tu, J. Chromatogr. Sci., 2012, 50, 343–348 CAS.
- J. S. Kim, S. Mitchell, P. Kijek, Y. Tsume, J. Hilfinger and G. L. Amidon, Mol. Pharm., 2005, 3, 686–694 CrossRef PubMed.
- Z.-M. P., V. H., T. H., A. Y., I. Z., B. S. and B.-J. M., J. Pharm. Pharm. Sci., 2007, 10, 368–379 Search PubMed.
- P. Zakeri-Milani, H. Valizadeh, H. Tajerzadeh, Y. Azarmi, Z. Islambolchilar, S. Barzegar and M. Barzegar-Jalali, J. Pharm. Pharm. Sci., 2007, 10, 368–379 CAS.
- L. Huang, Y. Wang, X. Ling, B. Chaurasiya, C. Yang, Y. Du, J. Tu, Y. Xiong and C. Sun, Carbohydr. Polym., 2017, 159, 178–187 CrossRef CAS PubMed.
- C. M. Keck and R. H. Müller, Eur. J. Pharm. Biopharm., 2006, 62, 3–16 CrossRef CAS PubMed.
- Y. Wang, Z. Liu, D. Zhang, X. Gao, X. Zhang, C. Duan, L. Jia, F. Feng, Y. Huang and Y. Shen, Colloids Surf., B, 2011, 83, 189–197 CrossRef CAS PubMed.
- C. Jacobs, O. Kayser and R. H. Müller, Int. J. Pharm., 2000, 196, 161–164 CrossRef CAS PubMed.
- J. Möschwitzer and R. H. Müller, Eur. J. Pharm. Biopharm., 2006, 62, 282–287 CrossRef PubMed.
- R. H. Müller, C. Jacobs and O. Kayser, Adv. Drug Delivery Rev., 2001, 47, 3–19 CrossRef.
- V. Teeranachaideekul, V. B. Junyaprasert, E. B. Souto and R. H. Müller, Int. J. Pharm., 2008, 354, 227–234 CrossRef CAS PubMed.
- G. P. Andrews, O. Abu-Diak, F. Kusmanto, P. Hornsby, Z. Hui and D. S. Jones, J. Pharm. Pharmacol., 2010, 62, 1580–1590 CrossRef CAS PubMed.
- R. Mauludin, R. H. Müller and C. M. Keck, Int. J. Pharm., 2009, 370, 202–209 CrossRef CAS PubMed.
- C. Hong, Y. Dang, G. Lin, Y. Yao, G. Li, G. Ji, H. Shen and Y. Xie, Int. J. Pharm., 2014, 477, 251–260 CrossRef CAS PubMed.
- G. Chawla, P. Gupta, R. Thilagavathi, A. K. Chakraborti and A. K. Bansal, Eur. J. Pharm. Sci., 2003, 20, 305–317 CrossRef CAS PubMed.
- B. K. Ahuja, S. K. Jena, S. K. Paidi, S. Bagri and S. Suresh, Int. J. Pharm., 2015, 478, 540–552 CrossRef CAS PubMed.
- P. Gupta, A. K. Bansal, R. Thilagavathi and A. K. Chakraborti, J. Pharm. Pharmacol., 2005, 57, 1271–1278 CrossRef CAS PubMed.
- M. Mosharraf and C. Nyström, Int. J. Pharm., 1995, 122, 35–47 CrossRef CAS.
- Y. Hua, T. Fei, P. Wang, B. Tian, L. Xia, H. Xi, Z. Ling, K. Zhang, Z. Yu and T. Xing, Int. J. Pharm., 2014, 477, 88–95 CrossRef PubMed.
- Z. Zhang, H. Bu, Z. Gao, Y. Huang, F. Gao and Y. Li, Int. J. Pharm., 2010, 394, 147–153 CrossRef CAS PubMed.
Footnote |
| † These two authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*