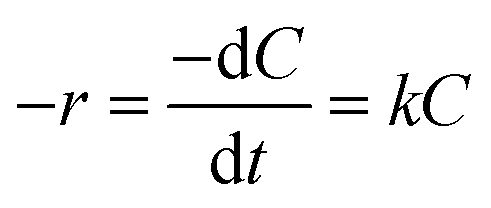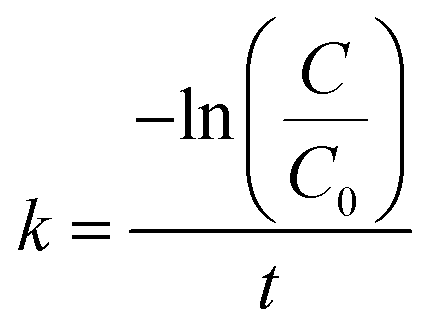DOI:
10.1039/C6RA28673A
(Paper)
RSC Adv., 2017,
7, 15736-15741
Co-assembly of Fmoc-tripeptide and gold nanoparticles as a facile approach to immobilize nanocatalysts†
Received
25th December 2016
, Accepted 6th March 2017
First published on 9th March 2017
Abstract
A facile approach was developed to immobilize gold nanoparticles (AuNPs) onto peptide nanofibers for catalytic applications. In this study, fluorenylmethoxycarbonyl diphenylalanine (Fmoc-FF) was conjuncted with histidine, arginine or cysteine at their C-terminus to provide binding sites for AuNPs. The co-assembly of Fmoc-tripeptide and AuNPs was achieved by dropping the peptide monomer solution directly into the AuNP solution, leading to the formation of nanofibers and the immobilization of AuNPs in one step. Atomic force microscopy, transmission electron microscope and circular dichroism analyses demonstrated that the presence of AuNPs does not significantly change the morphology and secondary structure of the nanofibers. The histidine-containing peptide-immobilized AuNPs were found to display favorable catalytic activity and stability for the reduction of 4-nitrophenol to 4-aminophenol. The present approach to fabricating nanomaterial-supported AuNPs may be extended to the production of other nanoparticle-containing composites in the fields of catalysis, sensing and biomedicine.
Introduction
Over the past few decades, nanocatalysis has become a rapidly growing field that involves the use of nanomaterials as catalysts for a variety of homogeneous and heterogeneous reactions.1–4 Among these materials, gold nanoparticles (AuNPs) have attracted considerable attention because of their excellent applications in carbon monoxide oxidation, hydrogen production and pollutant degradation.5–10 On one hand, the large surface-to-volume ratio endows AuNPs with high activity. On the other hand, the small size of AuNPs inevitably leads to agglomeration, which seriously suppresses their catalytic activity. To conquer this problem, many efforts have been undertaken to immobilize AuNPs onto solid organic or inorganic supports. Dotzauer et al. immobilized AuNPs in a multilayer polyelectrolyte film via layer-by-layer adsorption supported by porous supports, including alumina and polycarbonate.11 Fang et al. reported an approach to simultaneously synthesize and immobilize AuNPs in electrospun polyethyleneimine (PEI)/polyvinyl alcohol (PVA) nanofibers.12 Moreover, natural materials are also advisable for immobilization of nanoparticles. Niu et al. deposited AuNPs on cellulose nanofibers of filter paper to yield bulk cellulose-based catalytic membranes, which exhibited excellent catalytic activities toward the reduction of 4-nitrophenol.13 Lin et al. reported the biological synthesis and binding of AuNPs to the cell surface of yeast Pichia pastoris (P. pastoris), which exhibited high affinity for Au(III) species in aqueous solution.14
Recently, molecular self-assembly has been developed as a popular tool for constructing micro- and nano-structural supports. Low-molecular-weight gels are known as distinct soft materials that are obtained through bottom-up fabrication of small molecules into various types of nano-fibril, strand or tape structures. Therefore, the development of self-assembled architectures from small molecules, including some biological molecules, provides a promising platform for nanoscaled applications. Peptides, especially some aromatic peptides and their derivatives, are well known building blocks for the fabrication of peptide-based nanomaterials because their assembly can be easily conducted under the driving force of π–π stacking between aromatic groups.15 Because these materials combine the simplicity of small molecules with the versatility of peptides, they make excellent nano-carriers or supports in the fields of drug delivery, tissue engineering and chem-biosensing.16–18
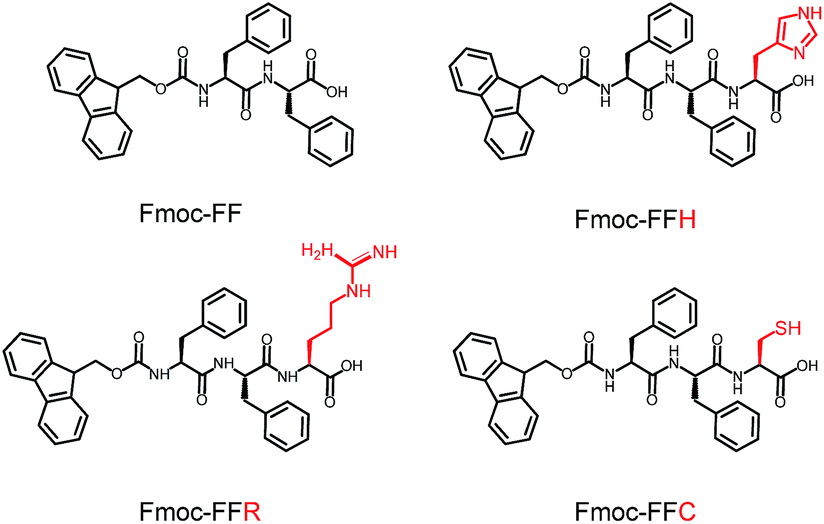
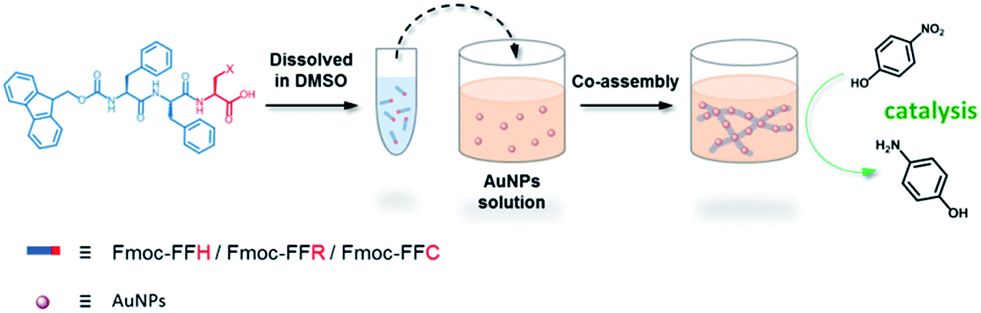
In this study, a peptide-based nanomaterial was developed as a novel support to immobilize AuNPs. Diphenylalanine (FF) is the simplest peptide that can self-assemble into nanotubes in pure water through π–π interactions between the aromatic side chains.19 Fluorenylmethoxycarbonyl (Fmoc) is a commonly used protecting group in peptide synthesis whose aromatic fluorenyl ring can further enhance π–π stacking and promote the formation of supramolecular architectures.15,20 Therefore, Fmoc-FF was chosen as the basic scaffold to build the nano-support. Additionally, it has been reported that many amino acid residues can be attached to metal nanoparticles through different types of interactions. The histidine tag (His6) in biotechnology can directly couple to AuNPs via Nε–Au interactions.21 The positively charged amino acids (arginine and lysine) or the negatively charged amino acids (aspartic acid and glutamic acid) can easily combine with opposite-charged nanoparticles through electrostatic attraction. Moreover, the sulfhydryl group on cysteine can form a strong covalent interaction with AuNPs through the formation of a S–Au bond.22 Herein, Fmoc-FF was further modified to conjugate histidine, arginine and cysteine at their C-terminus to provide a practical binding site for AuNPs (Scheme 1). The co-assembly of AuNPs and peptide (Fmoc-FFH, Fmoc-FFR or Fmoc-FFC) was achieved by dropping the peptide monomer solution directly into the AuNPs solution, resulting in the formation of the nano-support and the immobilization of AuNPs in one step (Scheme 2). The physical properties of the peptide-immobilized AuNPs were characterized and their catalytic activities were evaluated through the reduction of 4-nitrophenol and Congo red.
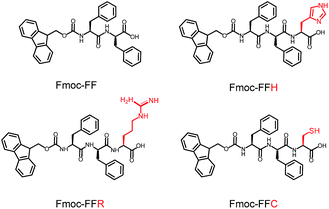 |
| | Scheme 1 Molecular structures of Fmoc-FF, Fmoc-FFH, Fmoc-FFR and Fmoc-FFC. | |
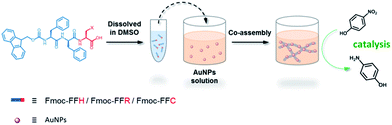 |
| | Scheme 2 One-step immobilization of AuNPs through co-assembly of peptide and AuNPs. | |
Experimental section
Materials
Fmoc-Phe-Phe-His-COOH (Fmoc-FFH), Fmoc-Phe-Phe-Arg-COOH (Fmoc-FFR) and Fmoc-Phe-Phe-Cys-COOH (Fmoc-FFC) were purchased from GL Biochem Ltd. (Shanghai, China). HAuCl4, dimethyl sulfoxide (DMSO), 4-nitrophenol (4-NP) and other chemicals and solvents were commercially purchased.
Synthesis of AuNPs
AuNPs were prepared by citrate reduction of HAuCl4. An aqueous solution of HAuCl4 (1 mL, 1 wt%) was added to ultrapure water (100 mL). After gently stirring for 1 min, an aqueous solution of sodium citrate (1 mL, 1 wt%) was added. Then, 1 mL of NaBH4 solution containing 0.075 wt% sodium citrate and 1 wt% sodium citrate was quickly added to the mixture. Stirring was continued for 5 min and the resulting AuNPs solution was stored at 4 °C.
Co-assembly of peptides and AuNPs
A stock solution was prepared by dissolving 5 μmol of peptides (Fmoc-FFH, Fmoc-FFR or Fmoc-FFC) in 50 μL of DMSO. Then, a certain amount of stock solution was dropped into 1 mL of as-prepared AuNPs solution so that the final concentration of peptide was 5 mM. The mixture was gently shaken and then kept still at room temperature until CoA–H–AuNPs, CoA–R–AuNPs and CoA–C–AuNPs were obtained. For the preparation of self-assembled peptides, the same peptide stock solution was dropped into ultrapure water instead of AuNPs solution. The mixture was gently shaken and then kept still at room temperature to obtain SA-H, SA-R and SA-C. All the samples were washed with ultrapure water so as to remove the free AuNPs.
Characterization
The morphologies of the obtained CoA–X–AuNPs and SA-X were investigated using an Agilent 5500 atomic force microscopy (Agilent, USA). Transmission electron microscope (TEM) were recorded on JEOL 2100F with 200 kV accelerating voltages (JEOL Ltd., Japan). Circular dichroism spectra were obtained on a J-810 CD spectropolarimeter (JASCO, Japan) with a 0.2 mm path length quartz cell at room temperature. The wavelength range was set to 260–190 nm and the scan speed was 100 nm min−1. UV-Vis spectra were obtained on an SpectraMAX 190 ultraviolet-visible spectrophotometer (Molecular device, USA) with a 1 mm path length quartz cell at room temperature. The wavelength range was 220–340 nm. Thermogravimetric analysis (TGA) was performed on a simultaneous TGA-DTA apparatus (PTC-10A, Rigaku, Japan) with the same mass of the dry CoA–X–AuNPs samples. The temperature was programed from 25 °C to 700 °C at the rate of 10 °C min−1.
4-Nitrophenol reduction reaction
4-NP (41.8 mg) and NaBH4 (56.7 mg) were dissolved in ultrapure water (100 and 5 mL, respectively). As a representative example, 750 μL of 4-NP solution, 7 mL of ultrapure water and 100 μL of peptide-immobilized AuNPs solution were pre-heated at 37 °C for 3 min. Then, 1 mL of fresh NaBH4 solution was added to the mixture and stirred continuously. The reaction progress was monitored through determining the concentration of 4-NP on a SpectraMAX 190 spectrophotometer in the wavelength range of 200–500 nm.
Results and discussion
Characterization of peptide-immobilized AuNPs
The self-assembly of peptides with the Fmoc-FF moiety is driven by hydrogen bonding and π–π interactions between the aromatic fluorenyl rings and phenylalanine side chains. Short, aromatic peptides have been typically found to self-assemble into straight nanofibers or nanotubes through π–π stacking, which can form unique 3D scaffolds and appear to be homogeneous hydrogels at the macroscopic scale.23 To determine whether the presence of AuNPs would affect the assembly of Fmoc-FFX (X = H, R or C) peptides, we investigated the gelation of Fmoc-FFH, Fmoc-FFR and Fmoc-FFC with or without AuNPs. As shown in Fig. 1 (top), all peptides can self-assemble into stable hydrogels in water in the absence of AuNPs. When the assembly was conducted in the presence of AuNPs, different phenomena were observed as shown in Fig. 1 (bottom). For the peptide conjuncted with histidine (Fmoc-FFH), the gelation time of CoA–H–AuNPs was prolonged to 60 min, indicating that the interaction between AuNPs and Fmoc-FFH affected the assembly rate of the peptide monomer (Fig. 1, column 1, bottom). In the case of Fmoc-FFR, a reddish brown precipitate was generated immediately upon addition of Fmoc-FFR to the AuNPs solution, which remained insoluble for 24 h instead of forming a stable hydrogel. This result is attributed to the interaction between the positive arginine side chain and the negative AuNPs, which disrupts the electrostatic balance among the Fmoc-FFR molecules (Fig. 1, column 2, bottom). For the peptides conjuncted with neutral cysteine residue (Fmoc-FFC), the combination of AuNPs had little impact on the intrinsic force balance among peptides; hence, the addition of AuNPs did not affect the gelation of Fmoc-FFC (Fig. 1, column 3).
 |
| | Fig. 1 Images of SA-H/CoA–H–AuNPs (column 1), SA-R/CoA–R–AuNPs (column 2) and SA-C/CoA–C–AuNPs (column 3). | |
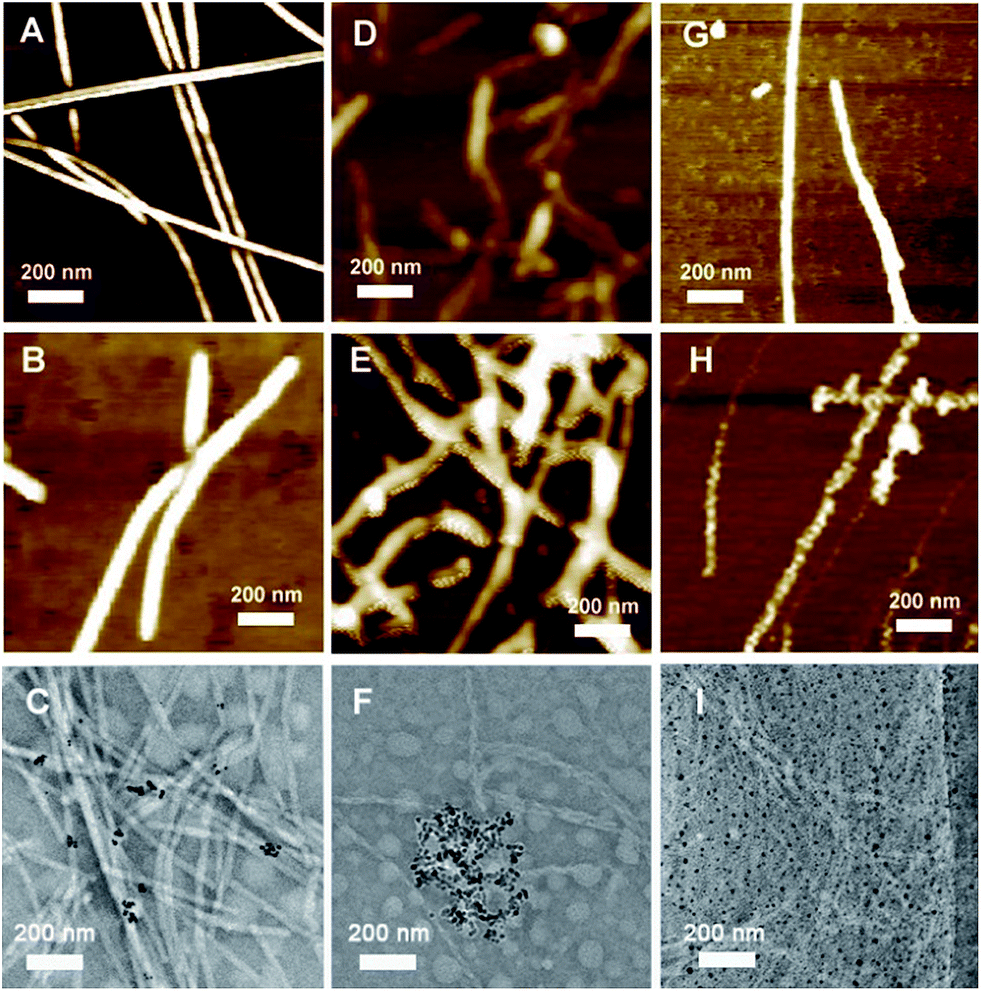
AFM and TEM were used to analyze the morphologies of the as-prepared SA-X and CoA–X–AuNPs, as shown in Fig. 2. Fibrous structures were obtained for all the SA-X and CoA–X–AuNP samples. Compared to SA-H nanofibers (Fig. 2A) with diameters of approximately 20–30 nm, the diameter of the CoA–H–AuNPs increased obviously (Fig. 2B), implying that the addition of AuNPs facilitated the formation of thicker nanofibers. However, the obtained CoA–H–AuNPs fibers were shorter than SA-H, which hinders the formation of 3D scaffolds and may explain the longer gelation time to become a hydrogel. The TEM image of CoA–H–AuNPs (Fig. 2C) demonstrated the association of the AuNPs with the peptide nanofibers. Since the concentration of AuNPs is low, not all fibers were observed to be decorated with them. For SA-R, the peptide monomers assembled into fragile fibrils because of the strong hydrophilicity of the arginine moiety (Fig. 2D). The introduction of AuNPs produced anomalous clusters attributed to the rapid neutralization of the negatively charged AuNPs and the positively charged arginine side chains (Fig. 2E). This result is in agreement with the formation of precipitates observed in Fig. 1 and the TEM result that the agglomeration of AuNPs was generated as shown in Fig. 2F. Unlike the others, the CoA–C–AuNPs display a rougher surface than SA-C (Fig. 2G and H), which implied that AuNPs can be attached to the peptide nanofibers. The TEM image (Fig. 2I) indicated that the AuNPs are distributed uniformly in CoA–C–AuNPs which might due to the strong interaction of S–Au bond.
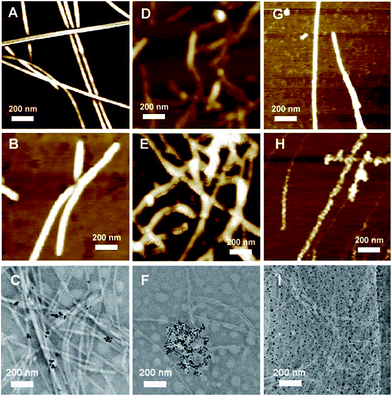 |
| | Fig. 2 AFM or TEM images of SA-H (A), CoA–H–AuNPs (B, C), SA-R (D), CoA–R–AuNPs (E, F), SA-C (G) and CoA–C–AuNPs (H, I). | |
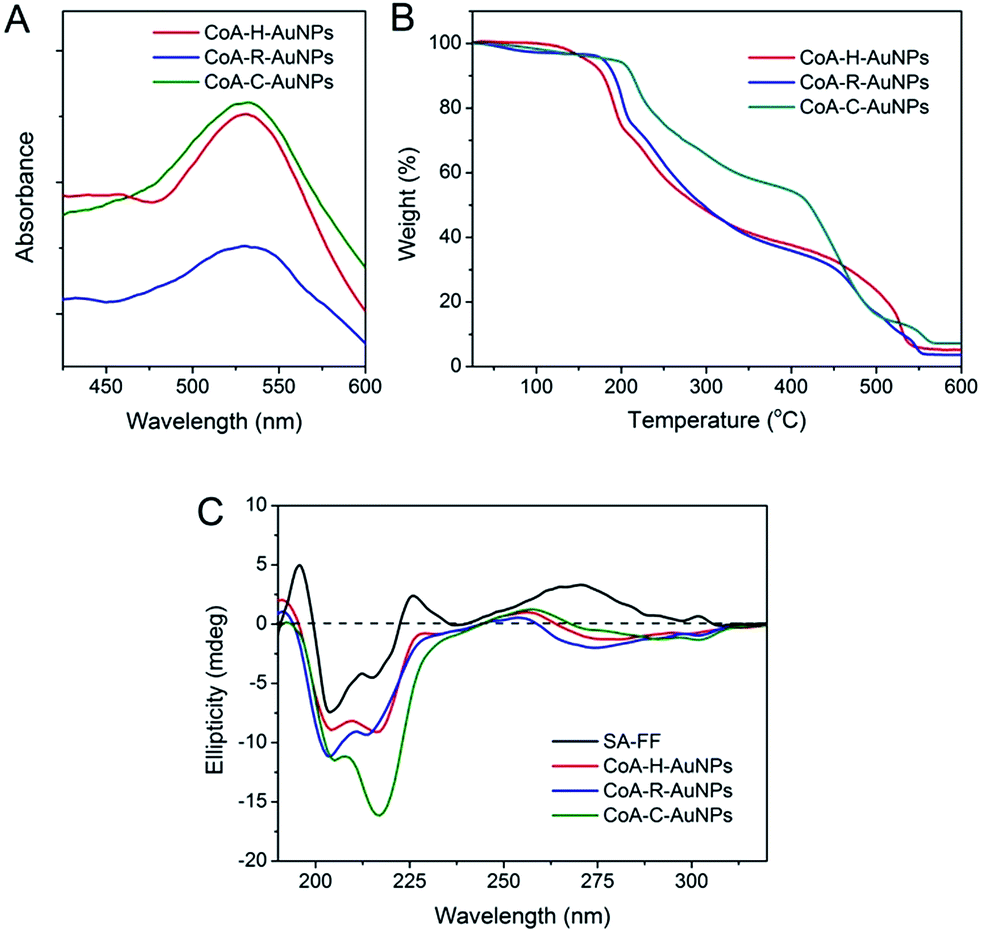
The immobilization of AuNPs on peptide nanofibers can also be determined using UV-Vis spectroscopy (Fig. 3A). For each peptide-immobilized AuNP sample, the spectrum was corrected using the corresponding SA-X nanofibers as a background, thus the observed absorbance at approximately 550 nm arises only from the localized surface plasmon resonance of the AuNPs. For CoA–H–AuNPs and CoA–C–AuNPs, a distinct and relatively sharp peak can be observed at 550 nm; however, when the arginine-containing peptide was used to bind the AuNPs, a broader and red-shifted absorbance was observed. TGA was performed to investigate the relative amount of AuNPs conjuncted to different nanofibers as shown in Fig. 3B. Equal mass of CoA–X–AuNPs samples were gradually heated from room temperature to 700 °C. Due to the loss of bound water, the weight of three samples decreased slowly before 240 °C. The weight began to drop sharply beyond 240 °C, which demonstrated the degradation of peptide nanofibers until 550 °C. Based on the TGA curve, the AuNPs immobilized on CoA–H–AuNPs, CoA–R–AuNPs and CoA–C–AuNPs could be approximately calculated as 4.2%, 3.4% and 7.2%, respectively. The low amount of AuNPs in CoA–R–AuNPs might due to the formation of agglomerations which leads to the weak binding and prone to leaking from the peptide nanofibers. These results suggest that the type of amino acid X not only affect the polydispersity of AuNPs but also the amount of AuNPs immobilized on peptide nanofibers. In addition, the UV-Vis spectrum in the 250–310 nm region is typically assigned to the hydrophobic chromophore groups in the peptide molecules. The absorbance in this range was observed to decrease when AuNPs were attached to the peptide nanofibers (Fig. S1 in ESI†). Compared to the corresponding samples without AuNPs, the maximum absorbance at 263 nm of CoA–H–AuNPs, CoA–R–AuNPs and CoA–C–AuNPs decreased 8.9%, 36.4% and 5.3%, respectively, implying that the binding of AuNPs altered the original hydrophilic–hydrophobic microenvironment of the peptide nanofibers to different extents. Circular dichroism (CD) was utilized to give deep insight into the secondary structure of the peptide-immobilized AuNPs. As shown in Fig. 3C, the structure of all the CoA–X–AuNPs exhibited a mixture of α-helix and β-sheet (positive peak at approximately 185–200 nm and negative peaks at 204 and 216 nm), which is in agreement with Ding's result that Fmoc-FF peptide self-assembled nanofibers (SA-FF) typically display a mixed structure.24 The derivation of Fmoc-FF and the presence of AuNPs did not appear to affect the basic secondary structure of the peptide nanofiber; thus, the Fmoc-FF moiety can be used as an appropriate platform to build nano-scaled supports.
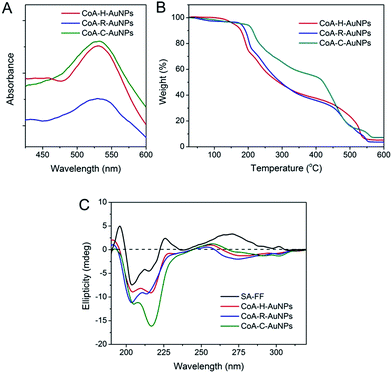 |
| | Fig. 3 UV-Vis spectra (A), TGA analysis (B) and circular dichroism spectra (C) of CoA–H–AuNPs, CoA–R–AuNPs and CoA–C–AuNPs. | |
Catalytic activity of the peptide-immobilized AuNPs
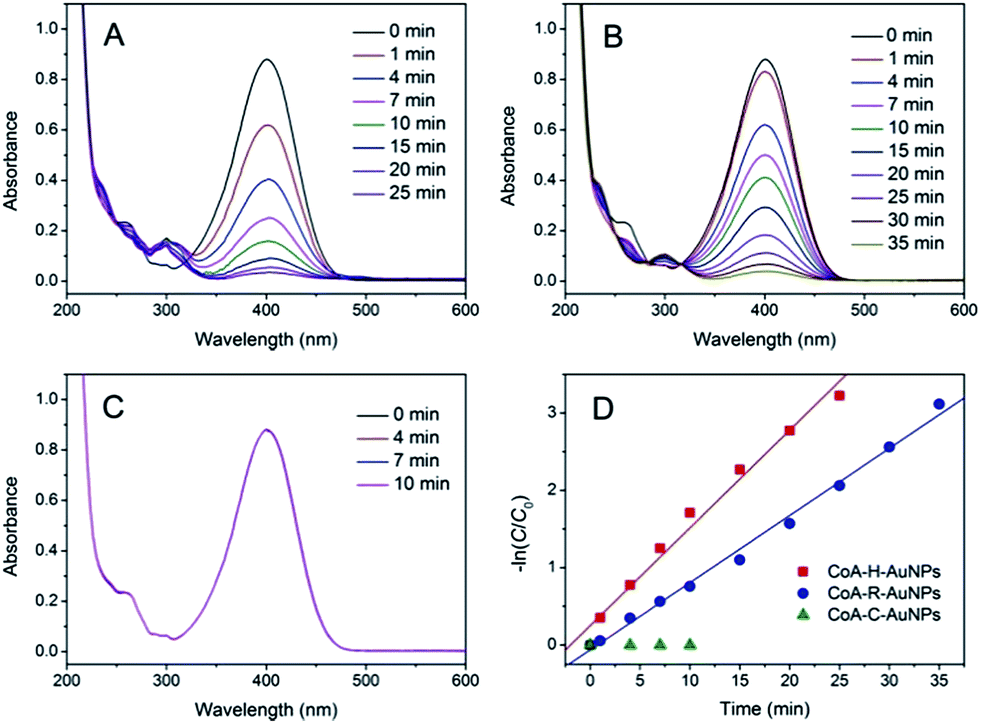
The catalytic activities of the as-prepared CoA–H–AuNPs, CoA–R–AuNPs and CoA–C–AuNPs were evaluated by using 4-NP as a substrate. 4-NP is a commonly used substrate because 4-NP can be catalytically converted to the colourless 4-nitroaniline which can be easily detected on UV-Vis spectrophotometer. Typical time-dependent UV-Vis absorption spectra were obtained during the reaction catalyzed by the peptide-immobilized AuNPs, which showed an obvious decrease in the absorbance peak of 4-NP at 400 nm (Fig. 4A and B) for CoA–H–AuNPs and CoA–R–AuNPs. As a result, the yellow reaction solutions became colorless after 25 and 35 min in the presence of CoA–H–AuNPs and CoA–R–AuNPs, respectively, indicating that the 4-nitrophenol had already been eliminated by the catalysts. However, we also found that the CoA–C–AuNPs lost all of their activity because the absorption of 4-NP remained constant during the reaction (Fig. 4C).
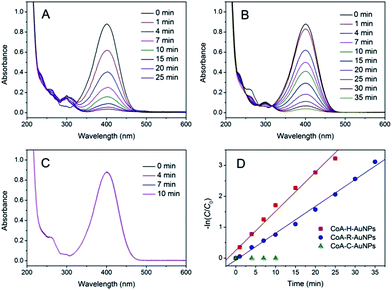 |
| | Fig. 4 Time-dependent UV-Vis spectra for the reduction of 4-nitrophenol catalyzed by CoA–H–AuNPs (A), CoA–R–AuNPs (B), and CoA–C–AuNPs (C) and their corresponding −ln(C/C0) vs. time plots (D). | |
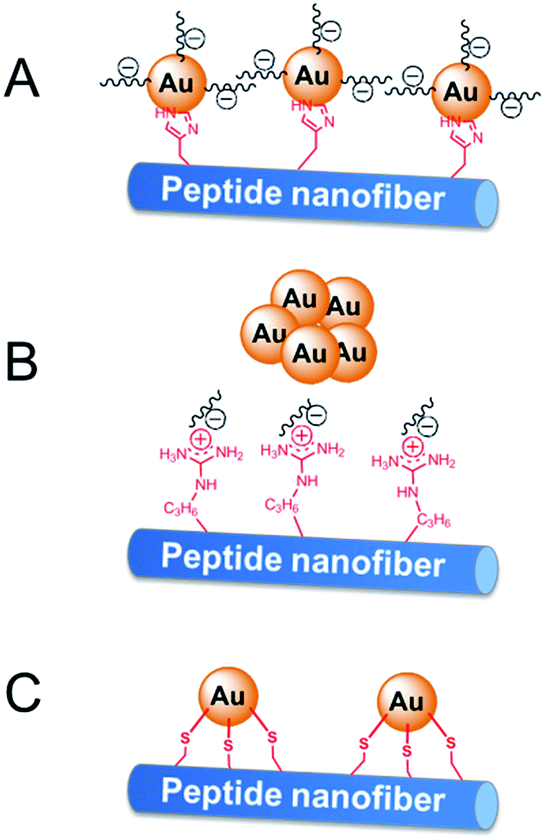
The activity difference of peptide-immobilized AuNPs originates from the different amino acid residues linked to the AuNPs. In this study, histidine, arginine and cysteine were designed as the functional groups to link citrate-capped AuNPs. Histidine-containing peptides have been studied extensively because of their high affinity to metal. When histidine is added to the citrate-capped AuNPs, the citrate molecules can be partly replaced by histidine through ligand exchange. Then, the imidazole group coordinates the AuNPs through Nε–Au interactions,25 which not only fix the AuNPs but also prevent the agglomeration of nanoparticles (Scheme 3A). Therefore, CoA–H–AuNPs display favorable activity in the reduction of 4-NP. In the case of CoA–R–AuNPs, the positive arginine is directly attached to the negative citrate through electrostatic interactions, which severely weakens the electrostatic repulsion between AuNPs, leading to their agglomeration. As a result, the agglomerated AuNPs are more liable to leak from the peptide nanofibers, leading to a low AuNPs amount and the lower catalytic activity (Scheme 3B). The covalent S–Au bond between the thiol group on cysteine and the AuNPs is much stronger than Nε–Au and electrostatic interactions. Many reports have demonstrated that the activity of AuNPs is badly damaged because the active sites on the surface of the AuNPs were irreversibly occupied by the formation of multiple S–Au bonds (Scheme 3C). As a result, the AuNPs on the cysteine-containing peptide nanofibers lost almost all of their catalytic ability.
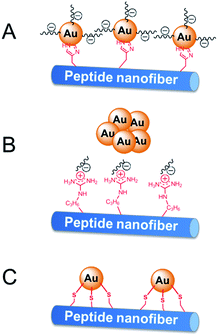 |
| | Scheme 3 Immobilization of AuNPs on peptide nanofibers through different amino acid residuals. | |
Because NaBH4 is present in a significant excess in the reaction, the concentration of NaBH4 is assumed to be constant and the rate of the reaction only depends on the concentration of 4-NP. The reaction rate can be given as:
| |
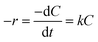 | (1) |
where
C is the concentration of 4-NP at time
t and
k is the reaction rate constant. The rate constant of the pseudo-first-order reaction can be calculated as:
| |
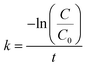 | (2) |
Fig. 4D show the kinetic curves for the degradation of 4-NP by the peptide-immobilized AuNPs. A linear relationship between ln(C/C0) and the reaction time suggests that the reaction is in agreement with the pseudo-first-order kinetics model, and the rate constants are kCoA–H–AuNPs = 0.127 min−1 and kCoA–R–AuNPs = 0.087 min−1. Compared with free AuNPs (kAuNPs = 0.880 min−1) that used for the preparation of peptide-immobilized AuNPs, the activity yield of CoA–H–AuNPs and CoA–R–AuNPs are 14.3% and 10%, respectively. This result indicated that the activity of CoA–X–AuNPs can be further improved if more AuNPs can be attached to peptide nanofibers and favourably using the histidine-containing peptide.
Congo red is a popular used azo dye in textile, paper and plastic industries which usually causes serious damage for environment. In order to investigate the practical performance of the peptide-immobilized AuNPs, we then determined the degradation of Congo red by using peptide-immobilized AuNPs. As shown in Fig. S2,† the linear relationship between ln(C/C0) and the reaction time also indicated that the reaction is in consistent with the pseudo-first-order kinetics model, and the rate constants are kCoA–H–AuNPs = 0.276 min−1 and kCoA–R–AuNPs = 0.089 min−1. Similarly, CoA–C–AuNPs displayed null activity on Congo red. Comparing with AuNPs immobilized on Salmalia malabarica gum (k = 0.236 min−1)26 or GO-G1PAMAM (k = 0.21 min−1),27 the as-prepared CoA–H–AuNPs displayed a better catalytic performance in the degradation of Congo red.
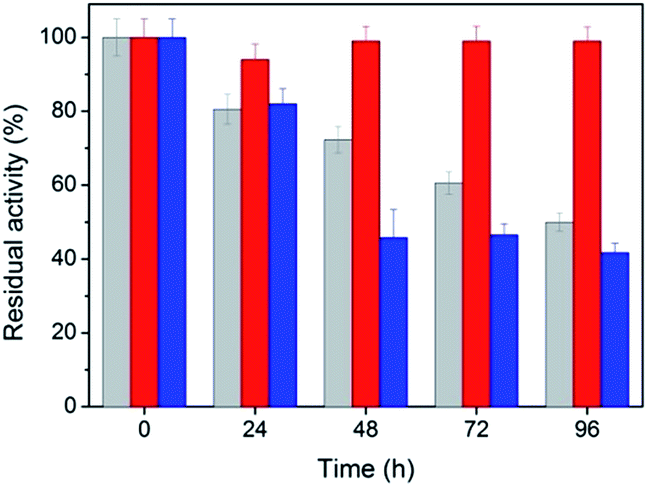
The stability and reusability of peptide-immobilized AuNPs
Stability is an important property for catalysts in industrial applications, especially for nanoscaled catalysts. Agglomeration typically occurs during long reactions, which unfortunately suppresses the activity of the nanocatalyst. Fig. 5 shows the stability of the peptide-immobilized AuNPs under the reaction conditions. CoA–H–AuNPs were found to retain 95% of its activity after 96 h, implying that the AuNPs are stably immobilized on the peptide nanofibers and maintain a high level of activity during the incubation period. This activity is attributed to the fibrous structure, which provides a robust support upon which to fix the AuNPs, preventing them from agglomerating. However, we found that the activity of CoA–R–AuNPs declined rapidly during the incubation period. CoA–R–AuNPs lost nearly 50% of its activity in 96 h, which was similar to that observed for the free AuNPs. Fig. S3† shows the images of CoA–H–AuNPs, CoA–R–AuNPs and free AuNPs before and after 72 h incubation. It can be seen that the CoA–H–AuNPs kept homogeneous hydrogel during the incubation. However, for CoA–R–AuNPs, the reddish brown precipitate became even cloudier which implies the undesired agglomeration of AuNPs during the incubation period. For free AuNPs solution, precipitate can be observed at the bottom of the vessel which implying the low stability of free AuNPs.
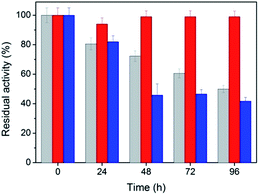 |
| | Fig. 5 Stability of CoA–H–AuNPs (red), CoA–R–AuNPs (blue) and free AuNPs (grey). | |
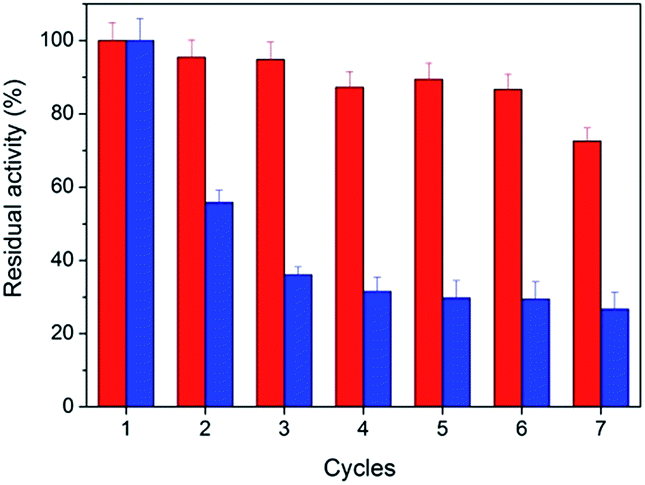
From the point of saving cost and environmental protection, the reusability is another important parameter for catalysts especially in industrial use. The as-prepared peptide-immobilized AuNPs can be separated from the reaction system through centrifugation or filtration. Fig. 6 shows the reusability of the peptide-immobilized AuNPs under reaction condition. CoA–H–AuNPs could be recycled and reused for at least 7 times with a residual activity of 75%. The decline of activity might due to the gradual mass loss after repeated recycles. However, CoA–R–AuNPs lost almost half of its original activity after the first recycle operation, and remained only 29% after the 7 times. This might due to the low stability of the agglomerated AuNPs in CoA–R–AuNPs compared with that of CoA–H–AuNPs. Therefore, we conclude that histidine-conjuncted peptide nanofibers are a preferable support to immobilize AuNPs.
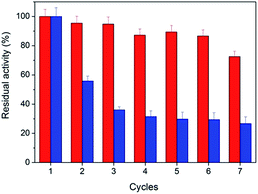 |
| | Fig. 6 Reusability of CoA–H–AuNPs (red) and CoA–R–AuNPs (blue). | |
Conclusions
In this paper, Fmoc-tripeptides conjuncted with different residues (histidine, arginine and cysteine) were used to immobilize AuNPs through co-assembly. AFM, TEM, UV-Vis and CD analyses demonstrated that the AuNPs can be attached to the peptide nanofibers and do not affect the basic secondary structure of the peptide nanofibers. The activity of peptide-immobilized AuNPs mostly depends on the type of residue that the AuNPs are bound to. CoA–H–AuNPs exhibited favorable activity and stability in the reduction of 4-NP and azo dye as a result of the formation of stable Nε–Au interactions, which not only fix the AuNPs but also prevent agglomeration of the nanoparticles.
Acknowledgements
This work was supported by the Ministry of Science and Technology of China (2012YQ090194, 2013AA102204), the Natural Science Foundation of China (21676191, 21476165, 21621004) and the Natural Science Foundation of Tianjin (13JCQNJC09300).
References
- V. Polshettiwar and R. S. Varma, Green Chem., 2010, 12, 743–754 RSC.
- W. Xu, J. S. Kong, Y.-T. E. Yeh and P. Chen, Nat. Mater., 2008, 7, 992–996 CrossRef CAS PubMed.
- R. Schlögl and S. B. Abd Hamid, Angew. Chem., Int. Ed., 2004, 43, 1628–1637 CrossRef PubMed.
- M. Wang, W. Qi, R. Su and Z. He, Chem. Eng. Sci., 2015, 135, 21–32 CrossRef CAS.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- Y. Liu, H. Dai, J. Deng, S. Xie, H. Yang, W. Tan, W. Han, Y. Jiang and G. Guo, J. Catal., 2014, 309, 408–418 CrossRef CAS.
- Q. He, S. J. Freakley, J. K. Edwards, A. F. Carley, A. Y. Borisevich, Y. Mineo, M. Haruta, G. J. Hutchings and C. J. Kiely, Nat. Commun., 2016, 7, 12905 CrossRef CAS PubMed.
- D. Li, J.-F. Wehrung and Y. Zhao, J. Mater. Chem. A, 2015, 3, 5176–5182 CAS.
- Y. Qu, W. Shen, X. Pei, F. Ma, S. You, S. Li, J. Wang and J. Zhou, J. Environ. Sci., 2016 DOI:10.1016/j.jes.2016.09.007.
- H. Wang, Z. Dong and C. Na, ACS Sustainable Chem. Eng., 2013, 1, 746–752 CrossRef CAS.
- D. M. Dotzauer, J. Dai, L. Sun and M. L. Bruening, Nano Lett., 2006, 6, 2268–2272 CrossRef CAS PubMed.
- X. Fang, H. Ma, S. Xiao, M. Shen, R. Guo, X. Cao and X. Shi, J. Mater. Chem., 2011, 21, 4493–4501 RSC.
- T. Niu, J. Xu, W. Xiao and J. Huang, RSC Adv., 2014, 4, 4901–4904 RSC.
- L. Lin, W. Wu, J. Huang, D. Sun, N. u. M. Waithera, Y. Zhou, H. Wang and Q. Li, Chem. Eng. J., 2013, 225, 857–864 CrossRef CAS.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- Q. Li, M. Chen, D. Chen and L. Wu, Chem. Mater., 2016, 28, 6584–6590 CrossRef CAS.
- X. Yan, Y. Cui, Q. He, K. Wang and J. Li, Chem. Mater., 2008, 20, 1522–1526 CrossRef CAS.
- Y. Wang, Z. Zhang, L. Xu, X. Li and H. Chen, Colloids Surf., B, 2013, 104, 163–168 CrossRef CAS PubMed.
- A. N. Rissanou, E. Georgilis, E. Kasotakis, A. Mitraki and V. Harmandaris, J. Phys. Chem. B, 2013, 117, 3962–3975 CrossRef CAS PubMed.
- R. Orbach, L. Adler-Abramovich, S. Zigerson, I. Mironi-Harpaz, D. Seliktar and E. Gazit, Biomacromolecules, 2009, 10, 2646–2651 CrossRef CAS PubMed.
- J. M. Kogot, H. J. England, G. F. Strouse and T. M. Logan, J. Am. Chem. Soc., 2008, 130, 16156–16157 CrossRef CAS PubMed.
- Y. Zhang, J. Jiang, M. Li, P. Gao, Y. Zhou, G. Zhang, S. Shuang and C. Dong, Talanta, 2016, 161, 520–527 CrossRef CAS PubMed.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- B. Ding, Y. Li, M. Qin, Y. Ding, Y. Cao and W. Wang, Soft Matter, 2013, 9, 4672–4680 RSC.
- J. K. Lim, Y. Kim, S. Y. Lee and S.-W. Joo, Spectrochim. Acta, Part A, 2008, 69, 286–289 CrossRef PubMed.
- B. R. Ganapuram, M. Alle, R. Dadigala, A. Dasari, V. Maragoni and V. Guttena, Int. Nano Lett., 2015, 5, 215–222 CrossRef CAS.
- R. Rajesh, S. S. Kumar and R. Venkatesan, New J. Chem., 2014, 38, 1551–1558 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: UV spectra in the range of 250–310 nm, Congo red reduction reaction, images before and after 72 h incubation. See DOI: 10.1039/c6ra28673a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Yanan Zhaoa,
Wei Qi
*ab,
Yanan Zhaoa,
Wei Qi