DOI:
10.1039/C6RA28559G
(Paper)
RSC Adv., 2017,
7, 10345-10352
Synthesis and photophysical properties of phenanthroimidazole–triarylborane dyads: intriguing ‘turn-on’ sensing mediated by fluoride anions†
Received
22nd December 2016
, Accepted 31st January 2017
First published on 8th February 2017
Abstract
Phenanthroimidazole-based triarylborane compounds with an N-phenyl (1Ph, 2Ph) or N-biphenyl (1BP, 2BP) bridge were synthesized and characterized. All four compounds exhibit a dual emission pattern in their photoluminescence (PL) spectra, which can be separated into high- (λem = ca. 380 nm in THF) and low-energy (λem = ca. 480 nm) emissions. While the high-energy emission remains largely unchanged in different organic solvents, the low-energy emission exhibits clear signs of positive solvatochromism. The results of the photophysical analysis and theoretical calculations suggest that the high-energy emission corresponds to a π–π* transition band arising from the phenanthroimidazole, whereas the low-energy emission originates from an intramolecular charge transfer (ICT) transition between phenanthroimidazole and the triarylborane moiety. UV-vis titration experiments examining the association of 1Ph, 2Ph, 1BP, and 2BP with fluoride demonstrate that these compounds associate with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometry in THF and binding constants (Ka) that are estimated to be around 1.0–3.0 × 104 M−1. These compounds show a ratiometrically increased fluorescence response in PL titration experiments upon binding of fluoride to the borane moiety, thereby giving rise to a ‘turn-on’ chemosensor for detection of fluoride anions. The ‘turn-on’ properties can be judged as a result of the reinforcement of π–π* transition on phenanthroimidazole and the restriction of ICT transition to triarylborane.
:1 binding stoichiometry in THF and binding constants (Ka) that are estimated to be around 1.0–3.0 × 104 M−1. These compounds show a ratiometrically increased fluorescence response in PL titration experiments upon binding of fluoride to the borane moiety, thereby giving rise to a ‘turn-on’ chemosensor for detection of fluoride anions. The ‘turn-on’ properties can be judged as a result of the reinforcement of π–π* transition on phenanthroimidazole and the restriction of ICT transition to triarylborane.
Introduction
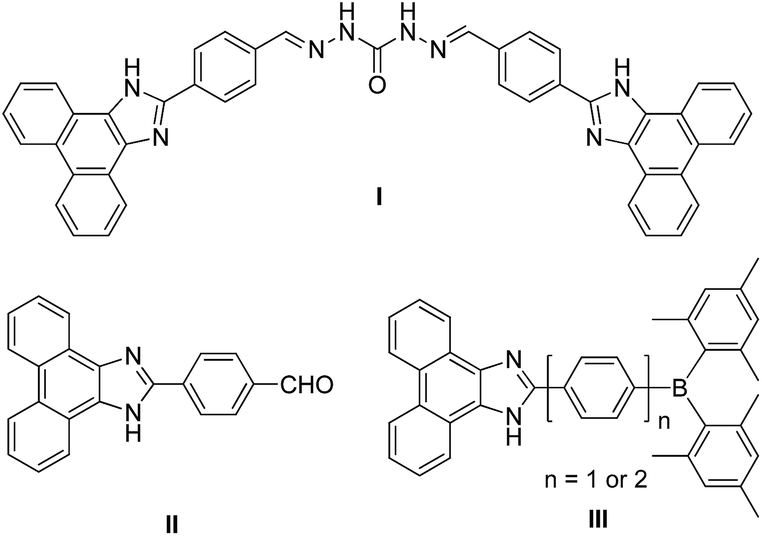
Phenanthroimidazole is a robust, heterocyclic compound, which possesses two nitrogen atoms in the imidazole ring and exhibits high quantum efficiency (Φ = 0.18–0.93)1 and good electron-transporting ability.2 The introduction of various functional groups at the N1- and C2-positions of phenanthroimidazole can lead to the emergence of intriguing properties.3 Accordingly, phenanthroimidazole-based derivatives have received great attention as promising attractive molecular scaffolds for optoelectronic materials of organic light-emitting diodes (OLEDs),1,4 dye-sensitized solar cells,5 and effective mother-luminophores of fluorescent chemosensors for detecting cations.6 In recent years, conjugated phenanthroimidazole-based compounds capable of detecting fluoride anions have been studied thoroughly as naked-eye detectable receptors owing to their remarkable fluorescence changes.7 For example, Mahapatra and co-workers demonstrated that a carbonohydrazone-bridged phenanthroimidazole dimer, driven by hydrogen bonding interactions between –NH group and fluoride, can act as an efficient colorimetric and ratiometric fluorescence sensor that allows detection by naked eye (I in Chart 1).7a Liu and co-workers have also reported an ‘easy-to-prepare’ chemosensor, 2-(4-formyl-phenyl)phenanthroimidazole, suitable as a colorimetric and fluorometric probe for fluoride sensing with good sensitivity and selectivity (II in Chart 1).7b
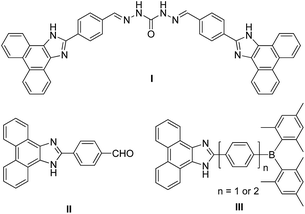 |
| | Chart 1 Examples of phenanthroimidazole-based chemosensors for detecting fluoride. | |
The binding of fluoride to receptors can potentially result in considerable changes in the electronic environment of the phenanthroimidazole moiety. Indeed, Lee group has reported triarylborane–phenanthroimidazole conjugates, where a restricted intramolecular charge transfer (ICT) transition from the phenanthroimidazole moiety to triarylborane group after the binding of fluoride to the boron centre (III in Chart 1) produced a change in the emission colour.7c In this regard, the phenanthroimidazole group can play an important role as an electron or charge donor which mediates ICT transition. These findings indicate that a well-defined molecular design of phenanthroimidazole derivatives equipped with appropriate charge-acceptors or fluoride-receptor units can produce drastic changes in the colour of fluorescence.
Bearing in mind that the potential application of ratiometric and ‘naked-eye’ detectable fluorescence response is worthy of an analytical point of view in chemosensors, we designed and synthesised in this work four phenanthroimidazole–triarylborane-based dyad systems (1Ph, 2Ph, 1BP, 2BP), linked through the nitrogen atom of the phenanthroimidazole unit, which exhibit ratiometric ‘turn-on’ fluorescence upon fluoride binding. The details of synthesis and characterization, including molecular structures in the solid state and the photophysical changes mediated by fluoride sensing, are provided and supplemented with the results of theoretical calculations.
Results and discussion
Synthesis and characterization
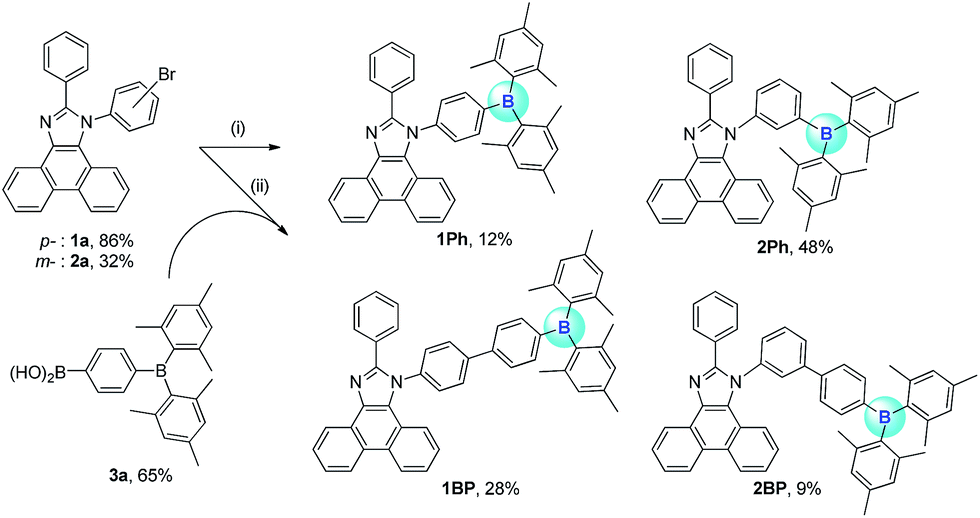
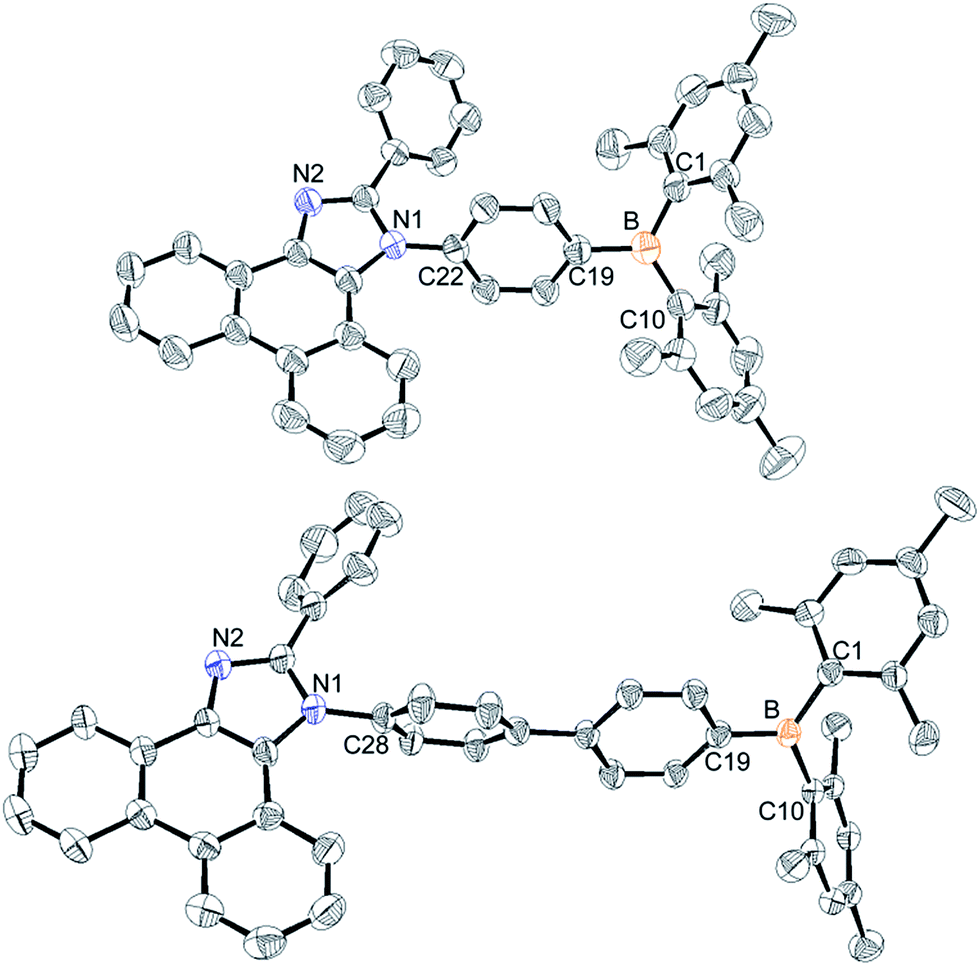
The synthetic procedures for the preparation of N-triarylborane substituted phenanthro[9,10-d]imidazole compounds 1Ph, 2Ph, 1BP, and 2BP are shown in Scheme 1. Lithiation reaction of bromophenyl-substituted phenanthroimidazole (1a and 2a) with dimesitylboron fluoride produced 1Ph (12%) and 2Ph (48%), respectively. Biphenylene-bridged compounds 1BP and 2BP were synthesized using the Suzuki–Miyaura coupling between 1a or 2a and (4-(dimesitylboryl)phenyl)boronic acid (3a) in relatively low yields (28% and 9%, respectively). All the compounds were characterized by multinuclear NMR spectroscopy (Fig. S20–S26†) and elemental analysis (EA). The 1H and 13C NMR spectra of all compounds were in agreement with the predicted structures, and the chemical shifts of the resonances associated with the proton and carbon atoms in these molecules were in expected range. Nevertheless, 11B NMR signals for 1Ph, 2Ph, 1BP, and 2BP could not be observed despite prolonged acquisition times (up to 12 hours). The solid-state structures of 1Ph and 1BP were confirmed by single crystal X-ray diffraction (Fig. 1, Tables S1 and S2†). Single crystals of sufficient quality for structural analysis were obtained by slow evaporation of mixed saturated solution of THF/EA at ambient temperature. The central boron atom observed in both 1Ph and 1BP was found to adopt a perfectly trigonal planar geometry, as evidenced by the sum of the three C–B–C angles (Σ(C–C–C) = 360°, 1Ph: 120.4°, 118.2°, and 121.4° and 1BP: 120.4°, 117.6°, and 121.9°, respectively). Importantly, the X-ray analysis revealed that the phenanthroimidazole plane in both structures is significantly distorted with respect to the N-phenyl group connected to the dimesityl borane, resulting in dihedral angles of 69.7° (1Ph) and 79.8° (1BP), respectively. Although those structural features in solid state of both 1Ph and 1BP would seem that delocalization of electrons between the bridged N-phenyl group and the phenanthroimidazole moiety might be unfavourable, both moieties could be electronically connected by the free rotation of bridged N-phenyl group in solution at least, which is certainly supported by reduction of π–π* transition bands for the phenanthroimidazole part upon the addition of fluoride anions in UV-vis titration experiments (see the fluoride sensing properties below).
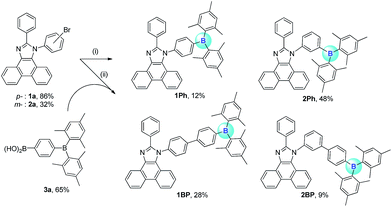 |
| | Scheme 1 Synthetic route of 1Ph–2BP. Reagents and condition: (i) n-BuLi, Mes2BF, THF, −78 °C. (ii) Pd(PPh3)4, K2CO3, THF/H2O, 90 °C. | |
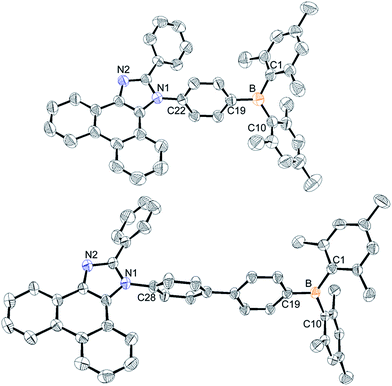 |
| | Fig. 1 X-ray crystal structures of 1Ph (top) and 1BP (down) depicted as 40% thermal ellipsoids. Hydrogen atoms and tetrahydrofuran (for 1BP) are omitted for clarity. | |
UV-vis absorption and emission properties
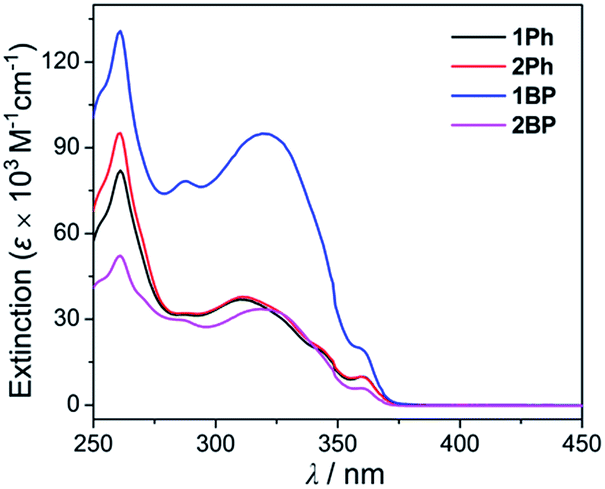
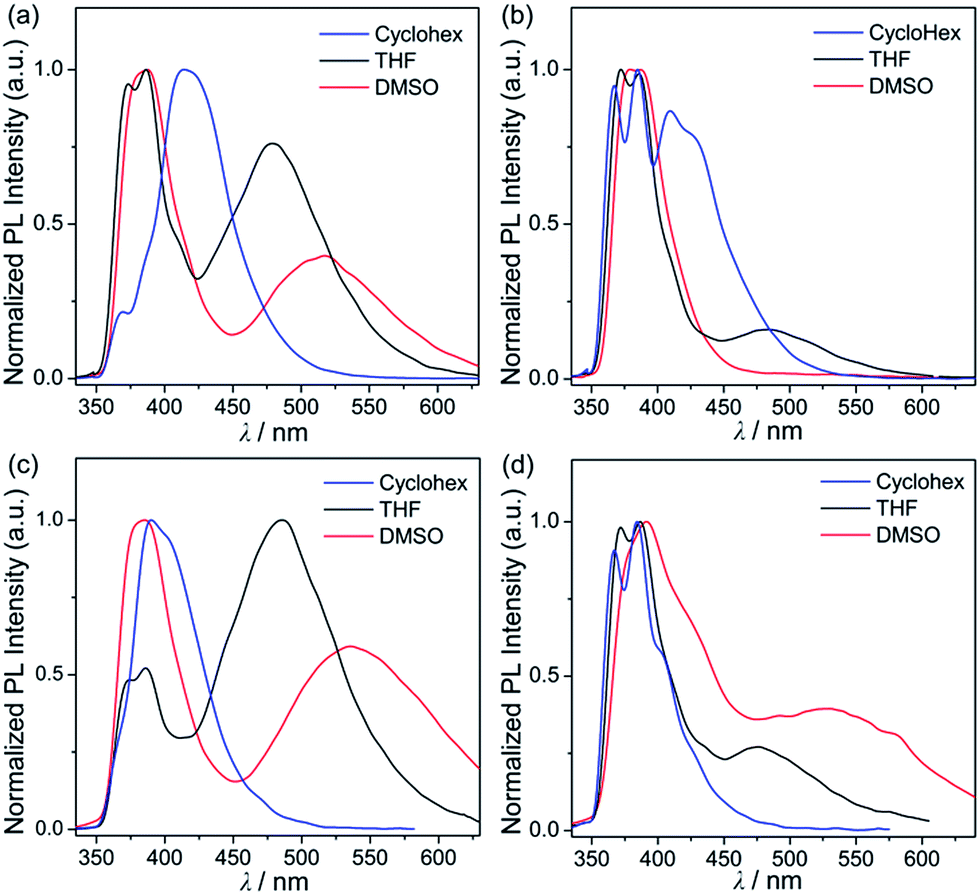
In order to investigate the photophysical properties of all phenanthroimidazole–triarylborane dyads, 1Ph, 2Ph, 1BP and 2BP, UV-vis absorption and photoluminescence (PL) measurements were performed at room temperature (Fig. 1 and Table 1). All compounds exhibited a major absorption band centred at ca. 315 nm, which can be assigned to the π–π* transition of the phenanthroimidazole unit. The spectra also showed a shoulder absorption band in the region above 360 nm, which represents the π(Mes)–pπ(B) charge transfer (CT) transition in the borane moiety (see theoretical calculations for details). The PL spectra of all four compounds in THF exhibited an apparent dual emission band, which could be divided into high- (λem = 386 nm, Table 1) and low-energy (ca. 480 nm) emission regions. In particular, the emission in the low-energy region showed clearly solvent-dependent features. In contrast, the absorption band (Fig. S1†) and the high-energy emission region (Fig. 2) of all the compounds remained practically unaltered in solvents (cyclohexane, THF, and DMSO) with different polarities. With increasing solvent polarity, the low-energy emission band was red-shifted dramatically by over 100 nm for all compounds except for 2Ph (Δλ = 103 nm for 1Ph, 133 nm for 1BP, and 126 nm for 2BP, Table 1). The emission of 2Ph exhibited less significant solvatochromic changes from 409 nm in cyclohexane to 483 nm in THF (Table 1). These solvatochromic changes exhibited by all four compounds suggest that the excited states associated with the low-energy emission are principally polar in character. This polar character, in turn, indicates that these are emissions arising from a charge transfer (CT) state between phenanthroimidazole and the triarylborane moiety. The broad and unstructured shape of the low-energy emission band further supports the CT transition state, which differs from the high-energy emission band originating from the phenanthroimidazole centred π–π* transition. The decay lifetimes of both the high and low-energy emissions for 1Ph–2BP were measured as 2.8–6.5 ns which can be assignable to fluorescence (Fig. S2–S5† and Table 1).
Table 1 UV-vis absorption and fluorescence properties of 1Ph, 2Ph, 1BP, and 2BPa
| |
λabsa/nm (logε) |
λex/nm |
λemb/nm |
Φa,c |
τobsa/ns |
Ka/M−1 |
| Cyclohexane |
THF |
DMSO |
∼385 nm |
∼480 nm |
| c = 2.5 × 10−5 M in THF. c = 2.5 × 10−5 M. Quinine sulfate (Φ = 0.55) was used as a standard. |
| 1Ph |
310 (4.57), 360 (4.00) |
326 |
370, 414 |
386, 479 |
387, 517 |
0.125 |
4.35 |
4.19 |
1.4 × 104 |
| 2Ph |
311 (4.58), 359 (3.99) |
325 |
385, 409 |
386, 483 |
387 |
0.042 |
6.51 |
6.28 |
1.1 × 104 |
| 1BP |
319 (4.98), 360 (4.29) |
296 |
390, 402 |
385, 485 |
385, 535 |
0.033 |
3.45 |
5.44 |
1.0 × 104 |
| 2BP |
318 (4.53), 360 (3.77) |
292 |
384, 403 |
386, 476 |
391, 529 |
0.015 |
2.77 |
5.05 |
3.0 × 104 |
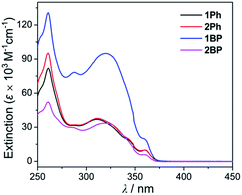 |
| | Fig. 2 UV-vis absorption spectra of 1Ph, 2Ph, 1BP, and 2BP in THF (2.5 × 10−5 M). | |
Fluoride sensing properties
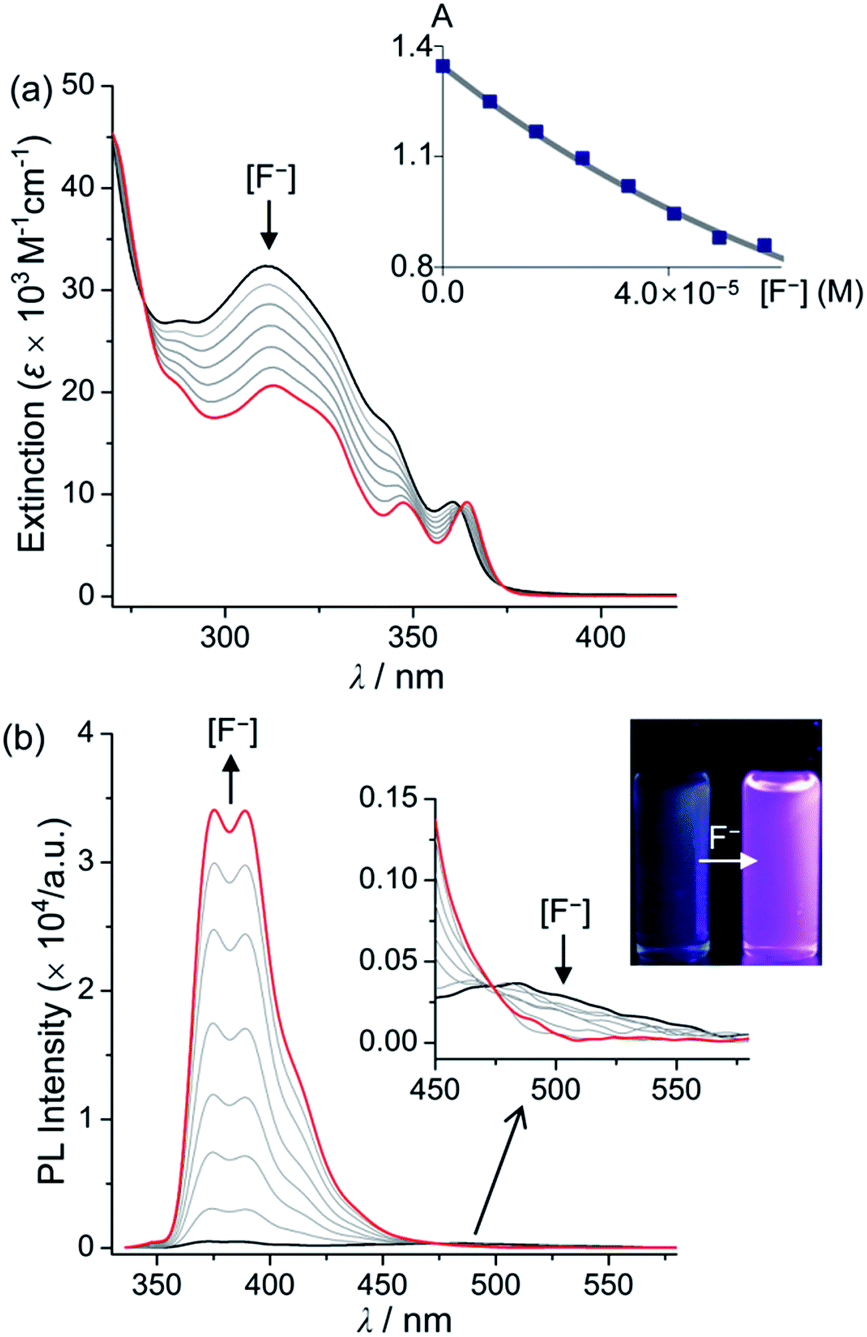
The fluoride anion binding properties of 1Ph, 2Ph, 1BP, and 2BP were investigated using both UV-vis and PL titration experiments in THF as the solvent (Fig. 3 and Table 1). The addition of increasing quantities of fluoride to a solution of 1Ph, 2Ph, 1BP, or 2BP in THF revealed gradual quenching of the major absorption band around 315 nm, arising from phenanthroimidazole centred π–π* transitions in all compounds, and a decrease in the intensity of the absorption region at 360 nm, which can be assigned to π(Mes)–pπ(B) CT band. These spectral changes suggest that the binding of fluoride anion to the boron results in the disruption of the CT transition centred at the triarylborane moiety and also reduction in the absorption arising from phenanthroimidazole centred π–π* transitions. The linear decrease in the absorption bands of all compounds from 310 to 320 nm is indicative of 1:1 binding between the triarylborane and fluoride anion. The binding constants (Ka) for 1Ph, 2Ph, 1BP, and 2BP were estimated as 1.0–3.0 × 104 M−1 (Fig. 4, inset and Table 1) respectively. These values are one order of magnitude lower than that of Mes3B (3.3(±0.5) × 105 M−1).8a This result originates from the fact that the boron atom of amine (NR2)-appended triarylborane compounds would have anionic character due to the resonance structures induced by intramolecular charge transfer from an N (donor) to a B (acceptor) atom.8b Interestingly, the PL titration experiments of all compounds in THF revealed significant changes in their PL spectra following the addition of fluoride (Fig. 4 for 1Ph and Fig. S6–S8† for 2Ph, 1BP, and 2BP, respectively). As shown in Fig. 2, all compounds exhibited dual emission bands: high- (λem = ca. 386 nm) and low-energy (λem = ca. 480 nm) regions, giving rise to a weak blue-green emission colour (Fig. 4 and S6–S8,† inset). Upon addition of incremental amounts of fluoride, the emission band at 386 nm was enhanced tremendously. Importantly, 69-fold enhancement was determined for 1Ph, 13-fold for 2Ph, 40-fold for 1BP, and 67-fold for 2BP, compared to the emission maxima observed prior to the addition of fluoride. In contrast, the low-energy emission was found to decrease only slightly in the presence of fluoride, producing a change in the colour of fluorescence from blue-green to purple. These dramatic changes in emission patterns indicate unambiguously that the fluoride binding interrupts the ICT transition between the triarylborane and phenanthroimidazole moiety, while simultaneously intensifying the π–π* transition arising from the phenanthroimidazole moiety.
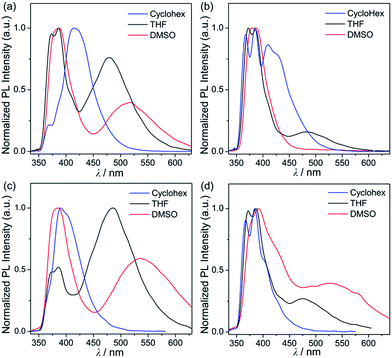 |
| | Fig. 3 Photoluminescence emission spectra of (a) 1Ph (λex = 326 nm), (b) 2Ph (λex = 325 nm), (c) 1BP (λex = 296 nm), and (d) 2BP (λex = 292 nm) in various organic solvents at 298 K. | |
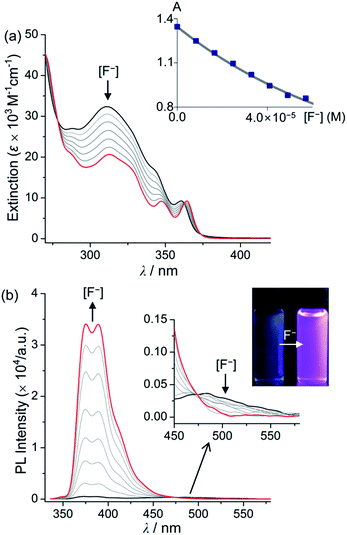 |
| | Fig. 4 Spectral changes in (a) UV-vis absorption and (b) PL intensity of a solution of 1Ph (λex = 326 nm) in THF (4.00 × 10−5 M) following the addition of TBAF (0–5.83 × 10−5 M). The inset shows the changes in the absorbance at 311 nm as a function of [F−]. The line corresponds to the binding isotherm calculated with Ka = 1.4 × 104 M−1. | |
These turn-on features were supported further by the emission patterns of 1Ph and 1BP fluoride adducts. These fluoride adducts were synthesised simply by the addition of 1.5 equiv. of Bu4NF to 1Ph or 1BP in deuterated THF and their structures were characterized by 1H NMR spectroscopy (Fig. S9 and S10†). The PL spectra of the prepared fluoride adducts exhibited only the high-energy emission region (λem = ca. 385 nm) (Fig. S11†). In fact, the shape and intensity of the resulting spectra were quite similar to those obtained at the end of the titration experiments. In particular, the high-energy emission of the fluoride adducts was found to change very little as a function of solvent polarity (adduct of 1Ph: λem = 374, 389 nm in cyclohexane, λem = 389 nm in THF, λem = 388 nm in DMSO; adduct of 1BP: λem = 368, 385 nm in cyclohexane, λem = 387 nm in THF, λem = 387 nm in DMSO, Fig. S11†). Consequently, these results clearly demonstrate that the observed ‘turn-on’ fluorescence phenomena are closely associated with the concentration of π–π* transitions of the phenanthroimidazole moiety and the restricted ICT state between the borane and phenanthroimidazole as a result of fluoride binding.
Theoretical calculations and molecular orbital analyses
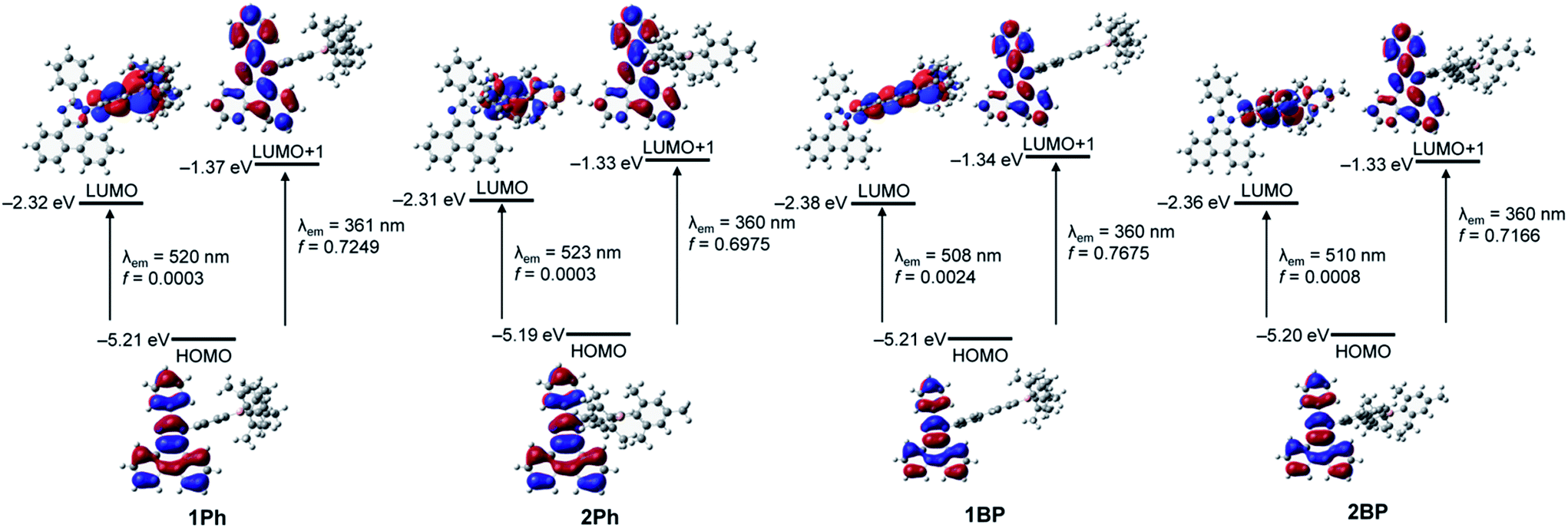
To gain further insight into the origin of the electronic transitions and the ‘turn-on’ emission behaviour of 1Ph–2BP, time-dependent density functional theory (TD-DFT) calculations on the ground state (S0) and the first excited state (S1) optimized structures of four compounds were performed using the B3LYP functional and 6-31G(d) basis sets (Fig. 5, S12–S19,† and Table 2). A conductor-like polarizable continuum model (CPCM) was used in order to account for the solvent effects (THF).9,10 All calculations were performed using the Gaussian 09 Package.11 The wavelengths calculated for 1Ph–2BP in the ground state (S0) exhibited a major low-energy absorption (fcalc > 0.1, Table 2) around 360 nm, which is mainly involved in the transition from HOMO−2 to LUMO. These results are indicative of a π(Mes)–pπ(B) ICT transition, centred at the triarylborane moiety, since the HOMO−2 and LUMO are localised predominantly on the dimesityl borane (>94%, Tables S4, S8, S12, and S16†) or on both the dimesityl borane (∼62%) and the N-bridged phenyl group (∼35%), respectively. On the other hand, the second major low-energy absorption (fcalc > 0.25) determined for 1Ph–2BP involves the HOMO → LUMO+1 transition which can be assigned to the π–π* transition on the phenanthroimidazole moiety only as the molecular occupation of this unit for both the HOMO and LUMO+1 is over 98% (Tables S4, S8, S12, and S16†). These results of computational analysis are consistent with the absorption features observed experimentally and suggest that significant absorptions take place independently of the ICT from phenanthroimidazole to triarylborane and phenanthroimidazole centred π–π* transition.
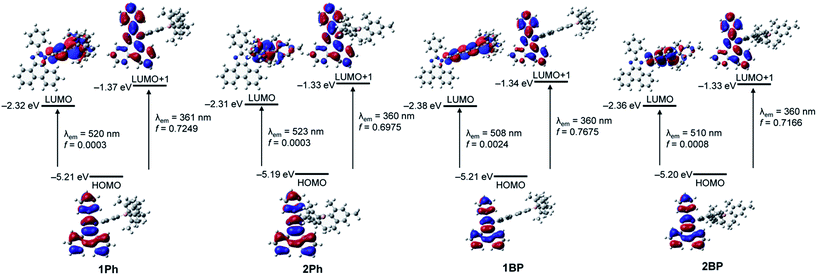 |
| | Fig. 5 Frontier molecular orbitals for 1Ph–2BP at their first excited singlet state (S1) with their relative energies determined using TD-DFT calculations. The transition energies (in nm) were calculated using the TD-B3LYP method with 6-31G(d) basis sets. | |
Table 2 The major low-energy electronic transitions determined for 1Ph, 2Ph, 1BP, and 2BP for the first excited singlet state (S1) using TD-DFT calculationsa
| |
λcalc/nm |
fcalc |
Assignment |
| Singlet energies calculated for the vertical transition in the optimized S1 geometries. |
| 1Ph |
520.27 |
0.0003 |
HOMO → LUMO (99.4%) |
| 361.20 |
0.7249 |
HOMO → LUMO+1 (88.7%) |
| 2Ph |
522.86 |
0.0003 |
HOMO → LUMO (99.7%) |
| 360.37 |
0.6975 |
HOMO → LUMO+1 (88.0%) |
| 1BP |
508.14 |
0.0024 |
HOMO → LUMO (98.9%) |
| 359.61 |
0.7675 |
HOMO → LUMO+1 (89.8%) |
| 2BP |
509.59 |
0.0008 |
HOMO → LUMO (99.3%) |
| 359.76 |
0.7166 |
HOMO → LUMO+1 (90.0%) |
TD-DFT calculations performed on the optimized S1 structures of 1Ph–2BP were used to predict the emission energies and transitions for these molecules. Although the calculated oscillator strengths (fcalc) were not intense, the lowest-energy transition in all compounds could be characterized as the HOMO to LUMO transition (Table 2 and Fig. 5). Each HOMO is located fully on the phenanthroimidazole moiety (99.8%, Tables S6, S10, S14, and S18†), whereas the LUMO levels involve mainly the triarylborane unit (>98%, bridged phenyl rings and dimesityl borane). These results suggest strongly that the experimentally observed low-energy emission, which is turned off upon fluoride binding, originates from the ICT between the phenanthroimidazole donor and triarylborane acceptor. The other major transition observed is that from HOMO to LUMO+1, which is associated with the high-energy emission since the LUMO+1 level is, similarly to HOMO, predominantly located on the phenanthroimidazole moiety. These results also indicate that the emission band around 385 nm arises mostly from the π–π* transition of the phenanthroimidazole moiety. Put together, the results obtained through computational analyses demonstrate that the ratiometric ‘turn-on’ fluorescent behaviour of the phenanthroimidazole-based N-triarylborane system upon fluoride binding originates from the reinforcement of the phenanthroimidazole moiety centred π–π* transition as a result of the restricted ICT transition between the phenanthroimidazole and triarylborane unit.
Experimental
General considerations
All operations were performed under an inert nitrogen atmosphere using standard Schlenk and glove box techniques. Anhydrous grade solvents (Aldrich) were dried by passing them through an activated alumina column and stored over activated molecular sieves (5 Å). Spectrophotometric grade tetrahydrofuran (THF), cyclohexane, dimethyl sulfoxide (DMSO) (Aldrich) was used as received. Commercial reagents were used without any further purification after purchasing from Aldrich (1-bromo-4-iodobenzene, 9,10-phenanthrenequinone, 3-bromoaniline, 4-bromoaniline, benzaldehyde, ammonium acetate, trimethyl borate, tetrakis(triphenylphosphine)palladium(0), potassium carbonate, n-butyllithium (n-BuLi, 1.6 M in n-hexane), acetic acid) and TCI chemicals (dimesitylboron fluoride). Deuterated solvents from Cambridge Isotope Laboratories were used after drying over activated molecular sieves (5 Å). NMR spectra were recorded on a Bruker Avance 400 spectrometer (400.13 MHz for 1H and 100.62 MHz for 13C) at ambient temperature. Chemical shifts are given in ppm and are referenced against external Me4Si (1H and 13C). Elemental analyses were performed on an EA3000 (Eurovector) in the Central Laboratory of Kangwon National University. UV-vis absorption and PL spectra were recorded on a Jasco V-530 and a Spex Fluorog-3 Luminescence spectrophotometer, respectively.
Synthesis of 1-(4-bromophenyl)-2-phenyl-1H-phenanthro[9,10-d]imidazole (1a). To a solution of 9,10-phenanthrenequinone (5.0 mmol) in acetic acid (20 mL) was slowly added a mixture of benzaldehyde (5.0 mmol), ammonium acetate (20.0 mmol) and 4-bromoaniline (25.0 mmol) in acetic acid (30 mL) at ambient temperature. The reaction mixture was heated to 120 °C and stirred overnight. After cooling to room temperature and pouring distilled water (50 mL), a precipitation was obtained by filteration. Washing with water and ethanol gives a pale blue solid (1a, 1.92 g); yield = 86%; 1H NMR (CDCl3): δ = 8.88 (d, 3JHH = 8.0 Hz, phenan-CH, 1H), 8.78 (d, 3JHH = 8.4 Hz, phenan-CH, 1H), 8.71 (d, 3JHH = 8.0 Hz, phenan-CH, 1H), 7.73 (m, 3H), 7.66 (t, 3JHH = 8.4 Hz, 1H), 7.54 (m, 3H), 7.35 (m, 6H), 7.22 (d, J = 9.6 Hz, 1H), 7.66 (t, 3JHH = 8.4 Hz, 1H) ppm; 13C NMR (CDCl3): δ = 151.07 (s, imidazole-NCN), 137.88 (s), 137.63 (s), 133.46 (s), 130.83 (s), 130.33 (s), 129.62 (s), 129.44 (s), 129.16 (s), 128.48 (s), 128.42 (s), 127.97 (s), 127.48 (s), 127.22 (s), 126.52 (s), 125.88 (s), 125.16 (s), 124.34 (s), 123.86 (s), 123.23 (s), 122.92 (s), 120.78 (s) ppm; anal. calcd for C27H17BrN2: C, 72.17; H, 3.81; N, 6.23. Found: C, 71.88; H, 3.49; N, 6.00.
Synthesis of 1-(3-bromophenyl)-2-phenyl-1H-phenanthro[9,10-d]imidazole (2a). This compound was prepared in a manner analogous to the synthesis of 1a using 3-bromoaniline (25.0 mmol). Pale gray solid (0.72 g); yield = 32%; 1H NMR (CDCl3): δ = 8.87 (d, 3JHH = 9.6 Hz, phenan-CH, 1H), 8.78 (d, 3JHH = 8.4 Hz, phenan-CH, 1H), 8.71 (d, 3JHH = 8.0 Hz, phenan-CH, 1H), 7.76 (m, 2H), 7.67 (m, 2H), 7.55 (m, 3H), 7.47 (m, 2H), 7.33 (m, 4H), 7.19 (d, 3JHH = 9.6 Hz, 1H) ppm; 13C NMR (CDCl3): δ = 151.04 (s, imidazole-NCN), 140.11 (s), 137.63 (s), 133.19 (s), 132.31 (s), 131.37 (s), 130.27 (s), 129.59 (s), 129.46 (s), 129.24 (s), 128.52 (s), 128.43 (s), 128.09 (s), 128.04 (s), 127.50 (s), 127.22 (s), 126.59 (s), 125.90 (s), 125.19 (s), 124.37 (s), 123.49 (s), 123.25 (s), 122.92 (s), 122.89 (s), 120.83 (s) ppm; anal. calcd for C27H17BrN2: C, 72.17; H, 3.81; N, 6.23. Found: C, 71.72; H, 3.44; N, 5.88.
Synthesis of 4-(dimesitylboryl)phenyl)boronic acid (3a). A hexane solution of n-BuLi (1.6 M, 4.08 mL, 6.52 mmol) was added at −78 °C to a solution of (4-bromophenyl)dimesityl borane (5.42 mmol) in THF (40 mL) and the mixture was stirred for 1 h at ambient temperature. After cooling to −78 °C, a solution of trimethyl borate (2.2 mL, 19.5 mmol) in THF (15 mL) was added to the mixture. The reaction mixture was then slowly heated at room temperature and stirred overnight. It was quenched by addition of saturated NH4Cl (55 mL) and extracted with dichloromethane (50 mL). The organic layer was dried over anhydrous MgSO4 and passed through silica pad (5 cm). The solvent was removed by rotary evaporation and the residue was washed with n-hexane (100 mL) to give white solid (1.30 g, 65%). 1H NMR (CDCl3): δ = 8.19 (d, 3JHH = 8.0 Hz, 2H), 7.64 (d, 3JHH = 8.0 Hz, 2H), 6.86 (s, 4H), 2.34 (s, Mes-p-CH3, 6H), 2.03 (s, Mes-o-CH3, 12H) ppm; 13C NMR (CDCl3): δ = 150.76 (s, Ar-C-B(OH)2), 141.90 (s), 141.00 (d), 139.06 (d), 135.54 (s), 135.19 (d), 133.16 (d), 128.40 (s), 23.57 (s, Mes-o-CH3), 21.40 (s, Mes-p-CH3) ppm; anal. calcd for C24H28B2O2: C, 77.89; H, 7.63. Found: C, 77.42; H, 7.12.
Synthesis of 1-(4-(dimesitylboryl)phenyl)-2-phenyl-1H-phenanthro[9,10-d]imidazole (1Ph). A hexane solution of n-BuLi (1.6 M, 0.69 mL, 1.1 mmol) was added at −78 °C to a solution of 1a (1.0 mmol, 0.47 g) in THF (10 mL) and the mixture was stirred for 1 h at ambient temperature. After cooling to −78 °C, a solution of dimesitylboron fluoride (1.0 mmol) in THF (5 mL) was added to the mixture. The reaction mixture was then slowly heated to room temperature and stirred overnight. After quenching with water (20 mL), the organic layer was extracted with DCM. The organic layer was dried over anhydrous MgSO4 and filtered. Purification by column chromatography (ethyl acetate:n-hexane = 1:10, v/v) afforded the pale brown solid (72 mg); yield = 12%; 1H NMR (CDCl3): δ = 8.90 (d, 3JHH = 9.2 Hz, phenan-CH, 1H), 8.78 (d, 3JHH = 9.6 Hz, phenan-CH, 1H), 8.71 (d, 3JHH = 9.20 Hz, phenan-CH, 1H), 7.74 (m, 3H), 7.65 (m, 3H), 7.52 (t, 3JHH = 9.8 Hz, 3H), 7.34 (m, 4H), 7.22 (t, 3JHH = 8.4 Hz, 1H), 6.90 (s, 4H), 2.35 (s, Mes-p-CH3, 6H), 2.13 (s, Mes-o-CH3, 12H) ppm; 13C NMR (CDCl3): δ = 150.64 (s, imidazole-NCN), 148.33 (s), 141.47 (s), 141.39 (s), 140.85 (s), 139.44 (s), 137.49 (s), 137.27 (s), 130.37 (s), 129.50 (s), 129.45 (s), 129.33 (s), 128.93 (s), 128.86 (s), 128.79 (s), 128.53 (s), 128.33 (s), 128.28 (s), 128.10 (s), 128.00 (s), 127.33 (s), 127.23 (s), 125.99 (s), 125.65 (s), 124.91 (s), 124.17 (s), 123.13 (s), 122.96 (s), 122.80 (s), 120.95 (s), 23.50 (s, Mes-o-CH3), 21.32 (s, Mes-p-CH3) ppm; anal. calcd for C45H39BN2: C, 87.37; H, 6.35; N, 4.53, found: C, 87.42; H, 6.79; N, 4.25.
Synthesis of 1-(3-(dimesitylboryl)phenyl)-2-phenyl-1H-phenanthro[9,10-d]imidazole (2Ph). This compound was prepared in a manner analogous to the synthesis of 1Ph using 2a (1.5 mmol, 0.71 g). Pale brown solid (0.45 g); yield = 48%; 1H NMR (CDCl3): δ = 8.86 (d, 3JHH = 8.0 Hz, phenan-CH, 1H), 8.76 (d, 3JHH = 8.4 Hz, phenan-CH, 1H), 8.69 (d, 3JHH = 8.4 Hz, phenan-CH, 1H), 7.72 (m, 2H), 7.64 (t, 3JHH = 8.6 Hz, 1H), 7.57 (m, 5H), 7.51 (t, 3JHH = 8.4 Hz, 1H), 7.31 (m, 3H), 7.21 (m, 2H), 6.78 (s, 4H), 2.26 (s, Mes-p-CH3, 6H), 1.99 (s, Mes-o-CH3, 12H) ppm; 13C NMR (CDCl3): δ = 151.21 (s, imidazole-NCN), 149.26 (s), 141.28 (s), 140.83 (s), 139.53 (s), 138.92 (s), 137.38 (s), 136.83 (s), 135.53 (s), 132.12 (s), 130.62 (s), 130.05 (s), 129.86 (s), 129.36 (s), 128.93 (s), 128.53 (s), 128.30 (s), 128.29 (s), 128.25 (s), 127.38 (s), 127.35 (s), 126.06 (s), 125.67 (s), 124.97 (s), 124.25 (s), 123.21 (s), 123.19 (s), 122.90 (s), 121.04 (s), 23.51 (s, Mes-o-CH3), 21.35 (s, Mes-p-CH3) ppm; anal. calcd for C45H39BN2: C, 87.37; H, 6.35; N, 4.53, found: C, 87.50; H, 6.53; N, 4.52.
Synthesis of 1-(4′-(dimesitylboryl)-[1,1′-biphenyl]-4-yl)-2-phenyl-1H-phenanthro[9,10-d]imidazole (1BP). A mixture of 1a (1.36 mmol, 0.64 g), Pd(PPh3)4 (0.068 mmol) and THF (20 mL) was stirred for 10 min. The above boronic acid 3a (1.50 mmol, 0.56 g) in 10 mL of THF and K2CO3 (4.50 mmol) in 10 mL of H2O were subsequently added. The reaction mixture was heated to 80 °C and stirred for 24 h. After cooling to room temperature, the water layer was separated and extracted with DCM (30 mL). The combined organic layers were dried over MgSO4 and the solvents were evaporated under reduced pressure. Purification of the crude product by column chromatography (ethyl acetate:n-hexane = 1:10, v/v) afforded white solid (0.29 g) as a product; yield = 28%; 1H NMR (CDCl3): δ = 8.91 (d, 3JHH = 8.0 Hz, phenan-CH, 1H), 8.79 (d, 3JHH = 8.4 Hz, phenan-CH, 1H), 8.72 (d, 3JHH = 8.4 Hz, phenan-CH, 1H), 7.89 (d, 3JHH = 8.4 Hz, 2H), 7.76 (t, 3JHH = 8.0 Hz, 3H), 7.68 (t, 3JHH = 7.2 Hz, 3H), 7.64 (m, 2H), 7.58 (d, 3JHH = 8.4 Hz, 2H), 7.52 (t, 3JHH = 8.1 Hz, 1H), 7.31 (m, 5H), 6.89 (s, 4H), 2.36 (s, Mes-p-CH3, 6H), 2.09 (s, Mes-o-CH3, 12H) ppm; 13C NMR (CDCl3): δ = 151.17 (s, imidazole-NCN), 145.73 (s), 142.48 (s), 141.94 (s), 141.78 (s), 140.96 (s), 138.39 (s), 138.32 (s), 137.65 (s), 137.27 (s), 131.28 (s), 130.66 (s), 129.61 (s), 129.54 (s), 129.46 (s), 129.40 (s), 128.99 (s), 128.77 (s), 128.41 (s), 128.25 (s), 127.41 (s), 127.37 (s), 126.74 (s), 126.47 (s), 125.76 (s), 125.04 (s), 124.26 (s), 123.24 (s), 123.18 (s), 122.90 (s), 121.02 (s), 23.67 (s, Mes-o-CH3), 21.40 (s, Mes-p-CH3) ppm; anal. calcd for C51H43BN2: C, 88.17; H, 6.24; N, 4.03, found: C, 88.35; H, 6.51; N, 4.45.
Synthesis of 1-(4′-(dimesitylboryl)-[1,1′-biphenyl]-3-yl)-2-phenyl-1H-phenanthro[9,10-d]imidazole (2BP). This compound was prepared in a manner analogous to the synthesis of 1BP using 2a (1.1 mmol, 0.52 g) and 3a (1.2 mmol, 0.45 g). Purification of the crude product by column chromatography (toluene:n-hexane = 10:1, v/v) afforded white solid (0.078 g); yield = 9%; 1H NMR (CDCl3): δ = 8.92 (d, 3JHH = 8.0 Hz, phenan-CH, 1H), 8.80 (d, 3JHH = 8.4 Hz, phenan-CH, 1H), 8.73 (d, 3JHH = 8.4 Hz, phenan-CH, 1H), 7.91 (d, 3JHH = 8.0 Hz, 1H), 7.81 (t, 3JHH = 1.8 Hz, 1H), 7.77 (t, 3JHH = 1.8 Hz, 1H), 7.67 (m, 4H), 7.60 (d, 3JHH = 8.4 Hz, 2H), 7.53 (m, 4H), 7.33 (m, 5H), 6.85 (s, 4H), 2.33 (s, Mes-p-CH3, 6H), 2.04 (s, Mes-o-CH3, 12H) ppm; 13C NMR (CDCl3): δ = 151.13 (s, imidazole-NCN), 145.62 (s), 142.97 (s), 142.48 (s), 141.70 (s), 140.90 (s), 139.34 (s), 138.92 (s), 137.66 (s), 137.23 (s), 130.66 (s), 130.55 (s), 129.64 (s), 129.43 (s), 129.02 (s), 128.56 (s), 128.40 (s), 128.35 (s), 128.21 (s), 128.11 (s), 127.79 (s), 127.42 (s), 127.38 (s), 126.74 (s), 126.46 (s), 125.76 (s), 125.03 (s), 124.29 (s), 123.24 (s), 123.16 (s), 122.92 (s), 121.03 (s), 23.64 (s, Mes-o-CH3), 21.36 (s, Mes-p-CH3) ppm; anal. calcd for C51H43BN2: C, 88.17; H, 6.24; N, 4.03, found: C, 87.72; H, 6.44; N, 4.13.
X-ray crystallography
Single crystals of 1Ph and 1BP were coated with Paratone oil and mounted onto a glass capillary. The crystallographic measurement was performed on a Bruker SMART Apex II CCD area detector diffractometer with a graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The structure was solved by direct methods and all non-hydrogen atoms were subjected to anisotropic refinement by full-matrix least-squares on F2 by using the SHELXTL/PC package, resulting in X-ray crystallographic data of 1Ph and 1BP in CIF format (CCDC 1521689 and 1521688†). Hydrogen atoms were placed at their geometrically calculated positions and refined riding on the corresponding carbon atoms with isotropic thermal parameters. The detailed crystallographic data and selected bond lengths and angles are given in Tables S1–S2,† respectively.
UV-vis absorption and photoluminescence (PL) measurements
UV-vis absorption and PL measurements were performed with a 1 cm quartz cuvette at ambient temperature. Solution PL experiments were performed in degassed solution for all compounds. The solution quantum efficiencies were measured with reference to that of quinine sulfate (0.5 M H2SO4, Φ = 0.55).12
Theoretical calculations
The geometries of the ground state (S0) and first excited state (S1) structures of 1Ph–2BP were optimized using the density functional theory (DFT) method with the B3LYP functional13 and the 6-31G(d)14 basis sets. The structures of the first excited state (S1) of 2 were optimized using the configuration interaction single (CIS) with the 3-21G basis sets. Time-dependent density functional theory (TD-DFT)15 was used with 6-31G(d) to obtain the electronic transition energies which include some accounts of electron correlation. To include the solvation effect of THF, the conductor-like polarizable continuum model (CPCM) was used in the calculations.9,10 All calculations were carried out using the GAUSSIAN 09 program.16 The GaussSum 3.0 was used to calculate the percent contribution of a fragment to each molecular orbital in the molecule.17
Conclusions
We have synthesized and characterized a series of phenanthroimidazole-based N-phenyl (1Ph and 2Ph) and biphenyl (1BP and 2BP) bridged arylborane compounds using lithiation and Suzuki–Miyaura coupling reactions, respectively. The PL spectra of these compounds exhibit a clear dual emission pattern, which can be separated into high- (λem = ca. 380 nm in THF) and low-energy (λem = ca. 480 nm) emission regions. Photophysical data and the results of theoretical calculations indicate that the high-energy emission originates solely from the π–π* transition of phenanthroimidazole unit and the low-energy emission can be assigned to the ICT transition between phenanthroimidazole and triarylborane moiety. The prepared phenanthroimidazole-N-arylborane dyads show ratiometrically increased fluorescence response upon binding of fluoride to the borane moiety, resulting in a ‘turn-on’ chemosensor for the detection of fluoride anions. These ‘turn-on’ properties arise from the reinforcement of π–π* transition of phenanthroimidazole moiety as a result of restricted ICT transition upon fluoride binding to the boron centre.
Acknowledgements
This work was supported by the Nano Material Technology Development Program (NRF-2016M3A7B4909246 for K. M. Lee) and Basic Science Research Program (NRF-2016R1C1B1008452 for M. H. Park) through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning. K. M. Lee is thankful for the Basic Science Research Program (NRF-2015R1D1A1A01057396) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education. This study was further supported by the 2016 Research Grant from Kangwon National University (No. 520160510).
References
-
(a) Y. Yuan, D. Li, X. Zhang, X. Zhao, Y. Liu, J. Zhang and Y. Wang, New J. Chem., 2011, 35, 1534–1540 RSC;
(b) Z. Wang, P. Lu, S. Chen, Z. Gao, F. Shen, W. Zhang, Y. Xu, H. S. Kwok and Y. Ma, J. Mater. Chem., 2011, 21, 5451–5456 RSC;
(c) Y. Zhang, S.-L. Lai, Q.-X. Tong, M.-Y. Chan, T.-W. Ng, Z.-C. Wen, G.-Q. Zhang, S.-T. Lee, H.-L. Kwong and C.-S. Lee, J. Mater. Chem., 2011, 21, 8206–8214 RSC;
(d) Y. Zhang, S.-L. Lai, Q.-X. Tong, M.-F. Lo, T.-W. Ng, M.-Y. Chan, Z.-C. Wen, J. He, K.-S. Jeff, X.-L. Tang, W.-M. Liu, C.-C. Ko, P.-F. Wang and C.-S. Lee, Chem. Mater., 2012, 24, 61–70 CrossRef.
- H. Huang, Y. Wang, S. Zhuang, X. Yang, L. Wang and C. Yang, J. Phys. Chem. C, 2012, 116, 19458–19466 CAS.
-
(a) Z. M. Wang, Y. Feng, X. S. T. Zhang, Y. Cao, G. Zhao, Y. M. Chen, X. J. Zhang, P. Lu, B. Yang, P. Chen, Y. G. Ma and S. Y. Liu, Phys. Chem. Chem. Phys., 2014, 16, 20772–20779 RSC;
(b) W.-C. Chen, Q.-X. Tong and C.-S. Lee, Sci. Adv. Mater., 2015, 7, 2193–2205 CrossRef CAS.
-
(a) C.-J. Kuo, T.-Y. Li, C.-C. Lien, C.-H. Liu, F.-I. Wu and M.-J. Huang, J. Mater. Chem., 2009, 19, 1865–1871 RSC;
(b) Y. Yuan, J.-X. Chen, F. Lu, Q.-X. Tong, Q.-D. Yang, H.-W. Mo, T.-W. Ng, F.-L. Wong, Z.-Q. Guo, J. Ye, Z. Chen, X.-H. Zhang and C.-S. Lee, Chem. Mater., 2013, 25, 4957–4965 CrossRef CAS;
(c) K. Wang, S. Wang, J. Wei, Y. Miao, Y. Liu and Y. Wang, Org. Electron., 2014, 15, 3211–3220 CrossRef CAS;
(d) S. Zhuang, R. Shangguan, H. Huang, G. Tu, L. Wang and X. Zhu, Dyes Pigm., 2014, 101, 93–102 CrossRef CAS;
(e) K. Wang, S. Wang, J. Wei, S. Chen, D. Liu, Y. Liu and Y. Wang, J. Mater. Chem. C, 2014, 2, 6817–6826 RSC;
(f) Y. Yuan, J.-X. Chen, W.-C. Chen, S.-F. Ni, H.-X. Wei, J. Ye, F.-L. Wong, Z.-W. Zhou, Q.-X. Tong and C.-S. Lee, Org. Electron., 2015, 18, 61–69 CrossRef CAS;
(g) X. Tang, Q. Bai, Q. Peng, Y. Gao, J. Li, Y. Liu, L. Yao, P. Lu, B. Yang and Y. Ma, Chem. Mater., 2015, 27, 7050–7057 CrossRef CAS;
(h) B. Wang, G. Mu, J. Tan, Z. Lei, J. Jin and L. Wang, J. Mater. Chem. C, 2015, 3, 7709–7719 RSC;
(i) K. Wang, S. Wang, J. Wei, Y. Miao, Z. Zhang, Z. Zhang, Y. Liu and Y. Wang, RSC Adv., 2015, 5, 73926–73934 RSC;
(j) Z. Wang, X. Li, K. Xue, H. Li, X. Zhang, Y. Liu, Z. Yu, P. Lu and P. Chen, J. Mater. Chem. C, 2016, 4, 1886–1894 RSC;
(k) B. Wang, X. Lv, J. Tan, Q. Zhang, Z. Huang, W. Yi and L. Wang, J. Mater. Chem. C, 2016, 4, 8473–8482 RSC;
(l) B. Liu, Y. Yuan, D. He, D.-Y. Huang, C.-Y. Luo, Z.-L. Zhu, F. Lu, Q.-X. Tong and C.-S. Lee, Chem.–Eur. J., 2016, 22, 12130–12137 CrossRef CAS PubMed;
(m) T. Shan, Y. Liu, X. Tang, Q. Bai, Y. Gao, Z. Gao, J. Li, J. Deng, B. Yang, P. Lu and Y. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 28771–28779 CrossRef CAS PubMed;
(n) Y. Zhang, J.-H. Wang, G. Han, F. Lu and Q.-X. Tong, RSC Adv., 2016, 6, 70800–70809 RSC.
- W. Lee, Y. Yang, N. Cho, J. Ko and J.-I. Hong, Tetrahedron, 2012, 68, 5590–5598 CrossRef CAS.
-
(a) K. C. Song, M. G. Choi, D. H. Ryu, K. N. Kim and S.-K. Chang, Tetrahedron Lett., 2007, 48, 5397–5400 CrossRef CAS;
(b) W. Lin, L. Long, L. Yuan, Z. Cao and J. Feng, Anal. Chim. Acta, 2009, 262–266 CrossRef CAS PubMed;
(c) Y. Guo, Y. Yan, X. Zhi, C. Yang and H. Xu, Bioorg. Med. Chem. Lett., 2013, 23, 3382–3384 CrossRef CAS PubMed.
-
(a) A. K. Mahapatra, P. Karmakar, J. Roy, S. Manna, K. Maiti, P. Sahoob and D. Mandalc, RSC Adv., 2015, 5, 37935–37942 RSC;
(b) F. Kong, Q. Liu, X. Wu, Z. Wang, Q. Chen and L. Chen, J. Fluoresc., 2011, 21, 1331–1335 CrossRef CAS PubMed;
(c) K. C. Song, H. Kim, K. M. Lee, Y. S. Lee, Y. Do and M. H. Lee, Sens. Actuators, B, 2013, 176, 850–857 CrossRef CAS.
-
(a) J. O. Huh, H. Kim, K. M. Lee, Y. S. Lee, Y. Do and M. H. Lee, Chem. Commun., 2010, 46, 1138–1140 RSCS. Solė and F. P. Gabbaï, Chem. Commun., 2004, 1284–1285 RSC;
(b) Z. Yuan, C. D. Entwistle, J. C. Collings, D. Albesa-Jové, A. S. Batsanov, J. A. K. Howard, N. J. Taylor, H. M. Kaiser, D. E. Kaufmann, S.-Y. Poon, W.-Y. Wong, C. Jardin, S. Fathallah, A. Boucekkine, J.-F. Halet and T. B. Marder, Chem.–Eur. J., 2006, 12, 2758–2771 CrossRef CAS PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- M. J. Frischet al., Gaussian 09 Revision D.01, Gaussian. Inc., Wallingford, CT, 2013 Search PubMed.
-
(a) A. M. Brouwer, Pure Appl. Chem., 2011, 83, 2213–2228 CrossRef CAS;
(b) W. H. Melhuish, J. Phys. Chem., 1961, 65, 229–235 CrossRef CAS.
-
(a) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- J. S. Binkley, J. A. Pople and W. J. Hehre, J. Am. Chem. Soc., 1980, 102, 939–947 CrossRef CAS.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision D.01, Gaussian. Inc., Wallingford, CT, 2013 Search PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data of 1Ph and 1BP in CIF format, NMR spectra, and computational details. CCDC 1521689 and 1521688. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra28559g |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a