
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances in the synthesis of acyclic peroxides
H. Gandhiab,
K. O'Reillyab,
M. K. Gupta†
ab,
C. Horgana,
E. M. O'Learyabc and
T. P. O'Sullivan *abc
*abc
aDepartment of Chemistry, University College Cork, Cork, Ireland. E-mail: tim.osullivan@ucc.ie
bAnalytical and Biological Chemistry Research Facility, University College Cork, Cork, Ireland
cSchool of Pharmacy, University College Cork, Cork, Ireland
First published on 31st March 2017
Abstract
Peroxide-containing compounds are an attractive synthetic target, given their widespread abundance in nature, with many displaying potent antimalarial and antimicrobial properties. This review summarises the many developments in the synthesis of acyclic peroxides, with a particular focus on the past 20 years, and seeks to update organic chemists about these new approaches. The synthetic methodologies have been subdivided into metal-catalysed reactions, organocatalytic reactions, direct oxidation reactions, miscellaneous reactions and enzymatic routes to acyclic peroxides.
1 Introduction
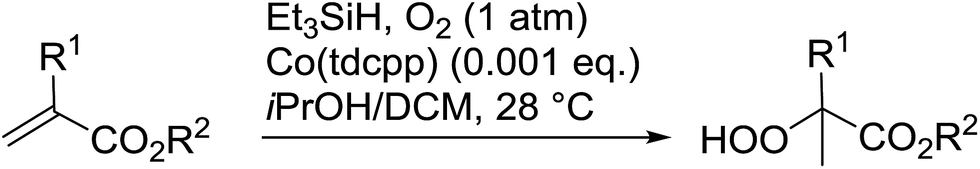
Peroxide-containing compounds are an attractive synthetic target, given their widespread abundance in nature, with many displaying potent antimalarial and antimicrobial properties.1,2 Reliable approaches to peroxide synthesis remain limited, especially for chiral peroxides. Several approaches to the synthesis of peroxide-containing substrates have been established over the years.3 Schenck introduced the preparation of hydroperoxides in 1948 by the radical addition of singlet oxygen to alkenes.4,5 Kharasch discovered that transition metals could be employed in the synthesis of hydroperoxides from alkenes, ketones and amines.6 Lately, various metal-catalysed and organocatalytic routes have been established as highly successful methods for the formation of acyclic chiral and achiral peroxides. Syntheses of cyclic peroxides have been recently reviewed by Terent'ev.7 This review covers the various breakthroughs in the synthesis of acyclic peroxides, with a focus on the past 20 years, and has been subdivided as follows: 2. Metal-mediated reactions; 3. Organocatalysed reactions; 4. Oxygen- or ozone-mediated reactions; 5. Miscellaneous reactions; 6. Enzymatic catalysis.2 Metal-mediated reactions
2.1 Isayama–Mukaiyama reaction
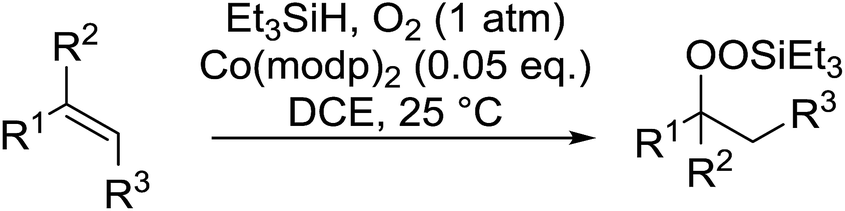
Much attention has been focused on the metal-catalysed, selective oxygenation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bonds by molecular oxygen. Isayama and Mukaiyama pioneered the direct peroxidation of olefins with molecular oxygen and triethylsilane under cobalt catalysis. They were the first to report the smooth and regioselective conversion of alkenes into the corresponding triethylsilylperoxide derivatives in good yields, using a cobalt catalyst in the presence of oxygen and triethylsilane (Table 1).8 Initial screening studies demonstrated that dichloroethane was the best medium for this reaction, with other solvents, such as benzene and ethyl acetate affording lower yields. Further optimization showed that cobalt 1-morpholinocarbonyl-4,4-dimethylpenta-1,3-dionate [Co(modp)2] was the most active among the catalysts tested. Several olefins were subjected to these conditions, and the corresponding triethylsilylperoxides were isolated in high yields (entries 1–5.
C double bonds by molecular oxygen. Isayama and Mukaiyama pioneered the direct peroxidation of olefins with molecular oxygen and triethylsilane under cobalt catalysis. They were the first to report the smooth and regioselective conversion of alkenes into the corresponding triethylsilylperoxide derivatives in good yields, using a cobalt catalyst in the presence of oxygen and triethylsilane (Table 1).8 Initial screening studies demonstrated that dichloroethane was the best medium for this reaction, with other solvents, such as benzene and ethyl acetate affording lower yields. Further optimization showed that cobalt 1-morpholinocarbonyl-4,4-dimethylpenta-1,3-dionate [Co(modp)2] was the most active among the catalysts tested. Several olefins were subjected to these conditions, and the corresponding triethylsilylperoxides were isolated in high yields (entries 1–5.
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Yield |
| 1 | Ph | Me | H | 95% |
| 2 | n-Oct | H | H | 80% |
| 3 | Me | Me | CH2OBz | 99% |
| 4 | CH2NHBz | H | H | 88% |
| 5 | H | Ph | Bz | 85% |
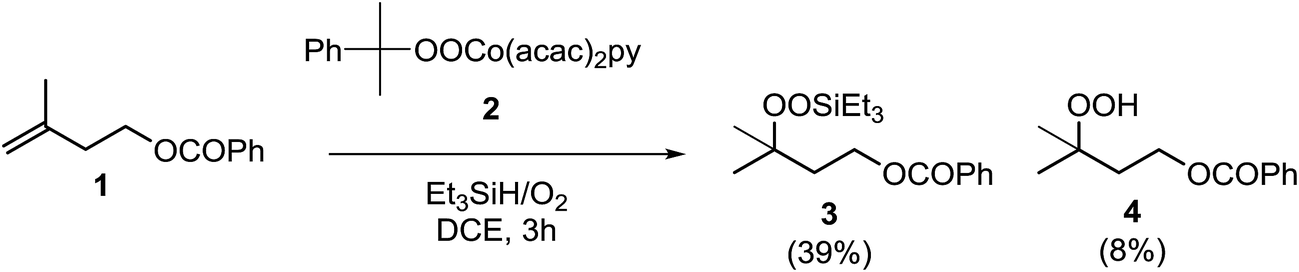
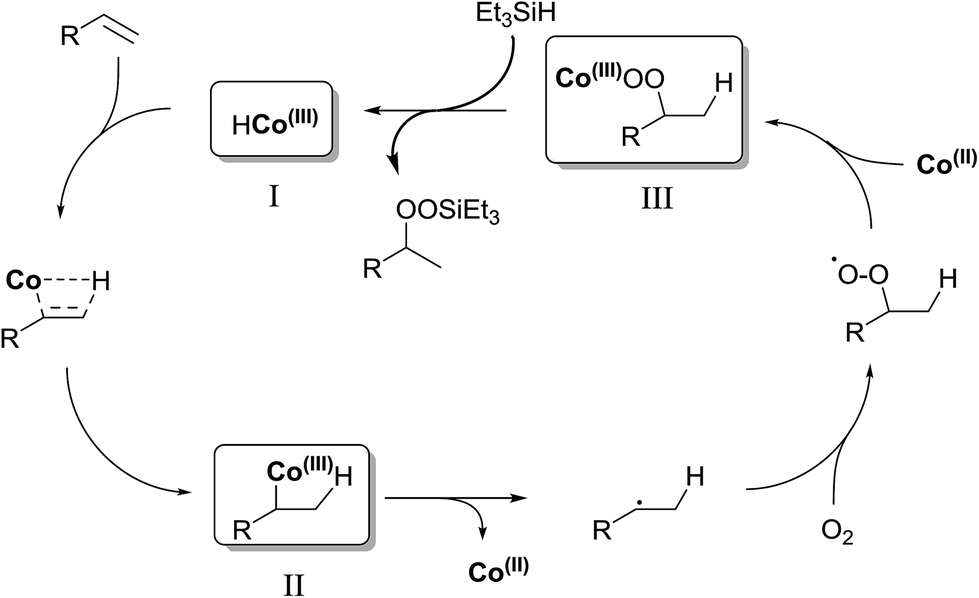
In their efforts to elucidate the exact mechanism at play in the Isayama–Mukaiyama reaction, Nojima et al. investigated whether Co(III)-alkylperoxy complexes could be employed in the peroxidation of alkene substrates.9 They found that a Co(III)–cumylperoxo complex 2 did indeed effect the peroxidation of 1 affording silylperoxide 3 and hydroperoxide 4 in yields of 39% and 8% respectively, along with significant amounts of starting material (45%) (Scheme 1). Accordingly, they proposed a catalytic cycle as outlined in Scheme 2. The first step involves insertion of the alkene into the H–Co bond of Co(III)–hydride complex I to give Co(III)–alkyl complex II. This is followed by homolytic cleavage of the Co–C bond of II to give Co(III)–alkylperoxo complex III. Finally, III undergoes transmetallation with Et3SiH, leading to the formation of triethylsilyl peroxide and regeneration of Co(III)–hydride complex I.
 | ||
| Scheme 1 | ||
 | ||
| Scheme 2 | ||
One of the better catalysts employed in aerobic oxidations is bis(1-morpholinocarbamoyl-4,4-dimethyl-1,3-pentanedio-nato)cobalt(II), Co(modp)2 (Fig. 1).10 However, preparation of this catalyst involves a low yielding, three-step synthesis. O'Neill and co-workers reasoned that a structurally similar catalyst derived from 2,2,6,6-tetramethylheptane-3,5-dione, namely (Co(thd)2), would have comparable electronic properties to Co(modp)2 and would be easier to prepare.11 The β-diketonate was prepared in a simple, one-step process and successfully induced the hydroperoxysilylation of unactivated alkenes, outperforming Co(acac)2 for all substrates tested.
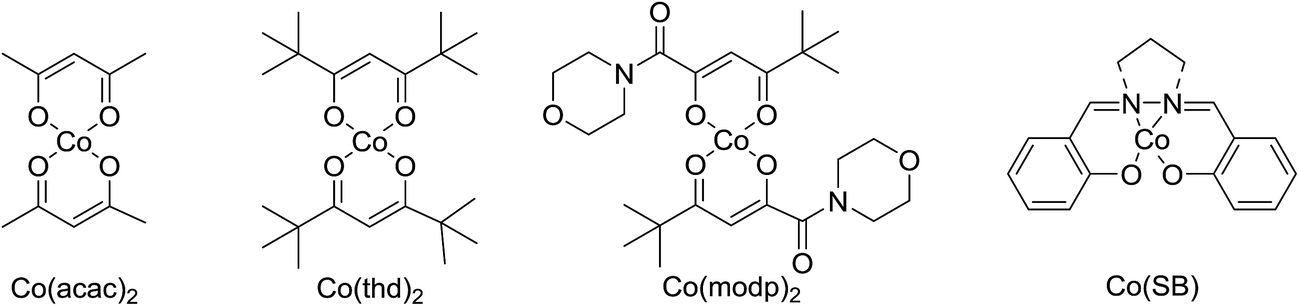 | ||
| Fig. 1 Comparison of cobalt-based catalysts. | ||
The use of Co(thd)2 afforded the desired silylperoxides in excellent yields (68–90%) and with high Markovnikov regioselectivity (Table 2). By contrast, Co(acac)2 was found to be a very poor catalyst for effecting oxidation of alkenes. Utilising different Co(II) complexes, O'Neill attempted to introduce the hydroperoxysilyl group asymmetrically – however, no asymmetric induction was observed. The hydroperoxylsilation of cyclic allylic alcohols was possible using this methodology (entry 7), with the peroxide products serving as useful precursors to 1,2,4-trioxanes.
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Yield |
| 1 | Ph | H | H | 72% |
| 2 | Bu | Me | H | 80% |
| 3 | Me | Ph | H | 79% |
| 4 | Ph | H | CH2OH | 88% |
| 5 | Ph | H | CH2OBz | 85% |
| 6 | Me | CH2OH | H | 90% |
| 7 |  |
72% | ||
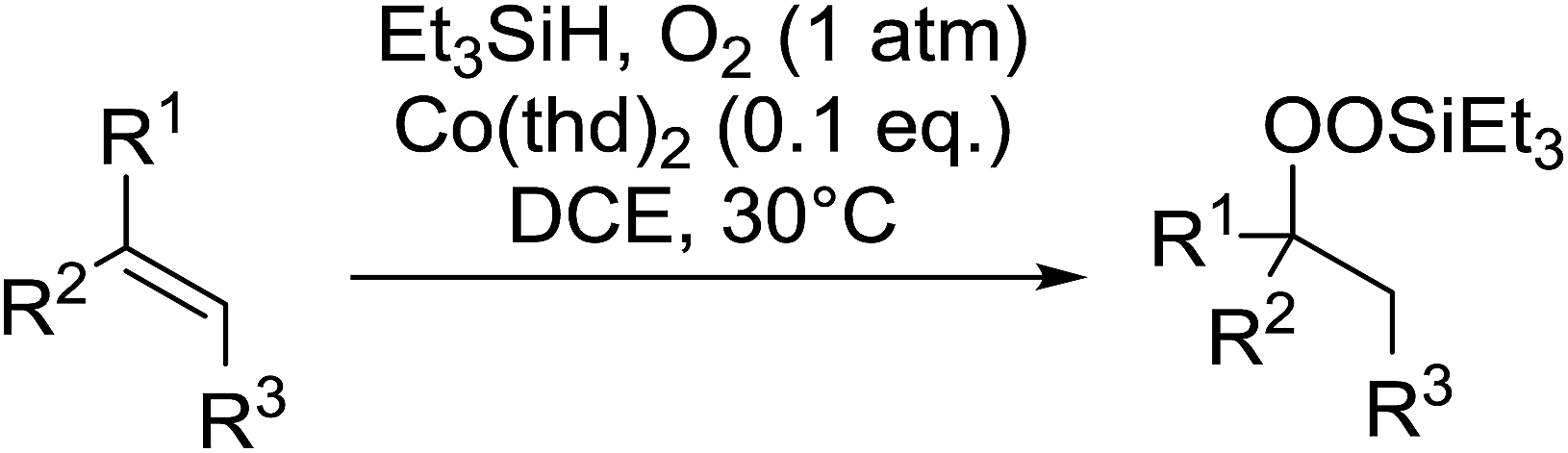
Nojima et al. investigated the competitive peroxidation of various dienes with a cobalt-derived catalyst and discovered that the relative reactivities of the alkene substrates are influenced by three major factors i.e. the stability of the intermediate radical; steric hindrance around the CC bond; electron density in the CC bond.12 Peroxidation of various dienes afforded a mixture of acyclic and cyclic peroxides, with product distribution being dependent on the substitution pattern of the diene substrate (Table 3). Furthermore, by increasing the oxygen concentration, it was found that cyclic 1,2-dioxanes 7 could be formed preferentially (entries 1 vs. 2 and 5 vs. 6).
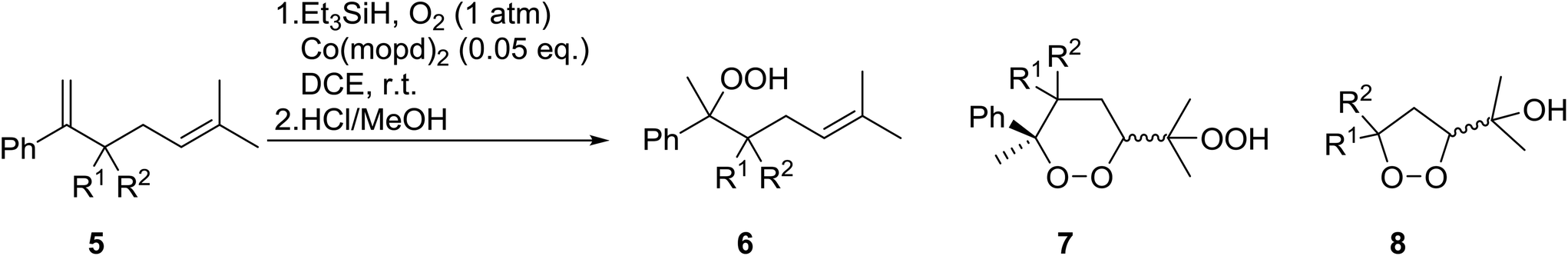
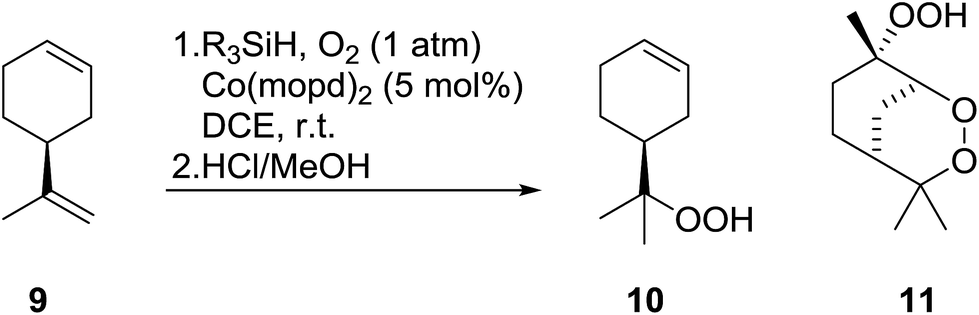
Nojima also used both Co(acac)2 and Co(modp)2 in the synthesis of antimalarial Yingzhaosu A analogues by peroxidation of dienes with atmospheric oxygen and triethylsilane.13 Using (S)-limonene as the test substrate, it was observed that the nature of the solvents, concentration of the substrates and the nature of the catalyst significantly influence the product composition. Further work within the same group outlined the impact of different silane groups on product distribution.14 The ratio of monocyclic hydroperoxide to bicyclic peroxide depends on the steric bulk on the silanes, with the least bulky silane favouring the formation of monocyclic hydroperoxide 10 (Table 4). The yield of 10 increases from 14% to 48% as the silane group decreases in bulk (entries 3 and 1). The effect of the silane group on product distribution is likely due to the change in rate of metal exchange between the Co–alkylperoxo complex and the silane. In the case of the less bulky PhMeSiH2, the transmetallation with the Co-complex, derived from the regioselective oxidation of the less hindered double bond of limonene, occurs rapidly and as a result the monocyclic silyl peroxide is produced exclusively. The metal exchange with the bulky silane group must be slow for steric reasons and as such the reversible formation of the peroxyl radical, followed by intramolecular cyclisation, leads to the formation of the bicyclic product.
|
||||
|---|---|---|---|---|
| Entry | Silane | Reaction time (h) | Yield 10 | Yield 11 |
| 1 | PhMeSiH2 | 2 | 48% | 0% |
| 2 | Et3SiH | 2 | 36% | 22% |
| 3 | Ph2MeSiH | 24 | 14% | 21% |
Dussault has successfully employed Co(thd)2 in the synthesis of spiro-bisperperoxyketals.15 The key step involves the regioselective, cobalt-mediated hydroperoxylsilation of diene 12 (Scheme 3), affording bisperoxide 13 in 52% yield. DIBAL-H reduction of the acetate group, followed by oxidation to the ketone and subsequent intramolecular ketalisation of the carbonyl group under acidic conditions afforded the desired tetracyclic spiroketal. The authors further demonstrated that the Co-mediated bisdioxygenation could be directly executed on dienones for the synthesis of six- and seven-membered spirocycles.
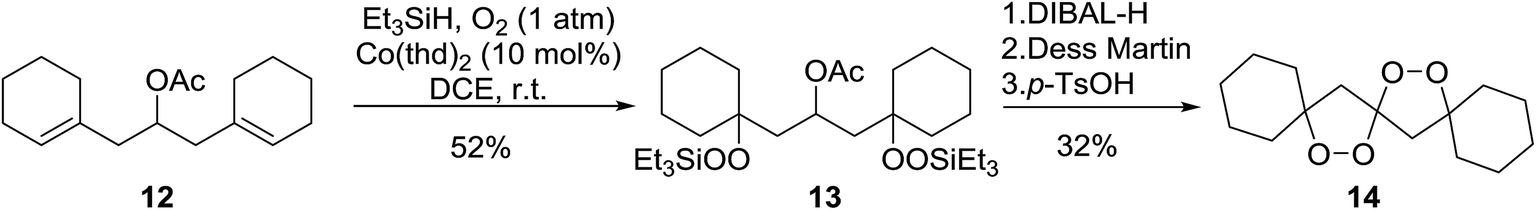 | ||
| Scheme 3 | ||
Gemma et al. synthesised dihydroplakortin and a series of C10-desethyl analogues using a similar approach.16 Catalyst Co(thd)2 was used for the regioselective hydroperoxysilylation of a quaternary olefinic carbon followed by a one-pot intramolecular ring closure to form a 1,2-dioxane ring. Gemma further extended this methodology to the formation of bicyclic cycloperoxides (Scheme 4).17 Hydroperoxysilylation was again accomplished with Co(thd)2 as the catalyst in the presence of oxygen and triethylsilane in moderate to excellent yields. Subsequent DIBAL-mediated reduction of the lactone, followed by addition of excess trimethylsilyl triflate resulted in simultaneous deprotection of the peroxide group and cyclisation to the target cycloperoxides. Similar synthetic strategies have been employed by Vatèle in the first total synthesis of seco-plakortolide E and (−)-ent-plakortide I18 and for the total synthesis of andavadoic acid, a naturally occurring five-membered ring peroxide.19
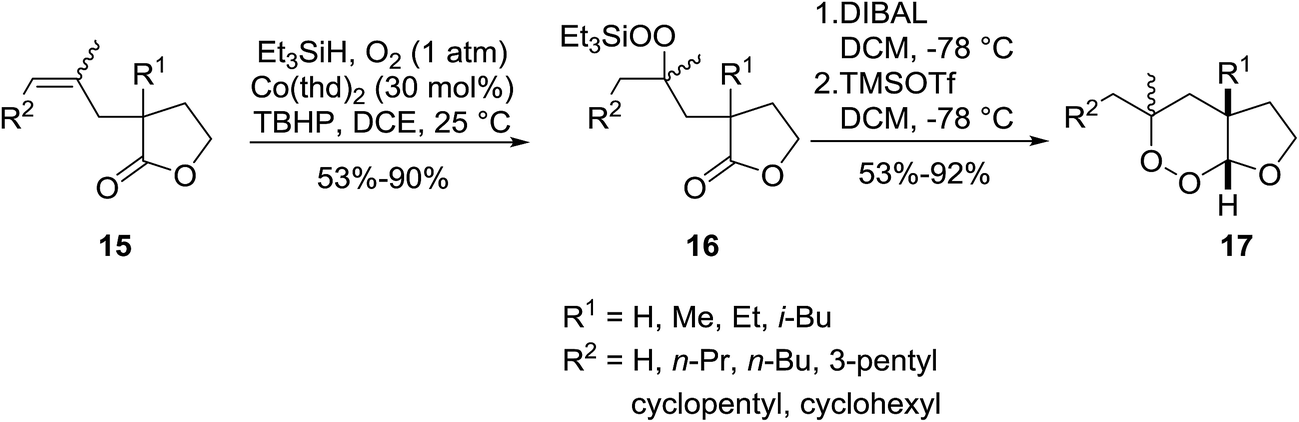 | ||
| Scheme 4 | ||
Isayama demonstrated that the combination of Co(acac)2 and TBHP efficiently promoted the peroxidation of α,β-unsaturated carboxylic acids and esters (Table 5).20 Transformation of the carboxylic acid and ester substrates under mild conditions afforded the corresponding α-triethylsilylperoxy adducts which were then subjected to acidic desilylation to furnish the target α-hydroperoxides in good yields (entries 1–5), along with smaller amount of the β-hydroperoxides. In the case of ethyl cinnamate, however, the observed regioselectivity was reversed (entry 6).
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Time (h) | Yield 19 | Yield 20 |
| 1 | H | Et | 5 | 55% | 0% |
| 2 | CH3 | Et | 21 | 58% | 6% |
| 3 | CH3(CH2)2 | Me | 21 | 70% | 5% |
| 4 | CH3(CH2)5 | Me | 21 | 83% | 3% |
| 5 | CH3(CH2)7 | Me | 21 | 71% | 5% |
| 6 | Ph | Et | 5 | 0% | 82% |
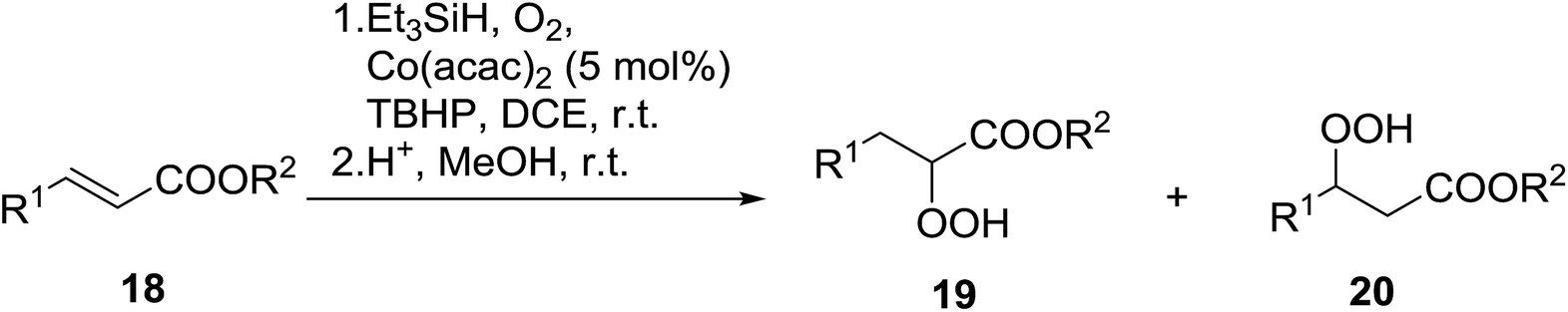
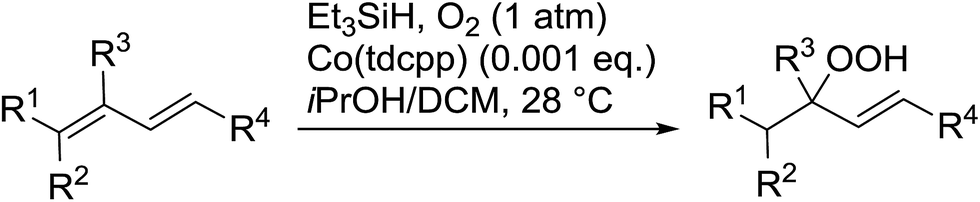
In their studies on the hydroperoxidation of conjugated dienes, Sugamoto et al. discovered that cobalt porphyrins were more effective catalysts than other cobalt-based systems such as Co(acac)2.21,22 From the former group, cobalt 5,10,15,20-tetrakis(2,6-dichlorophenyl)porphine [Co(tdcpp)] proved the most active. Hydroperoxidation of a range of dienes with conjugated electron-withdrawing groups gave the corresponding γ-hydroperoxy-α,β-unsaturated products in moderate to good yields (Table 6). The reaction was highly regioselective for substrates incorporating esters (entries 1–4), amides (entry 5), aldehydes (entries 7–8) and ketones (entry 9). The absence of an electron-withdrawing group led to a dramatic decrease in selectivity and resulted in the formation of an inseparable mixture of products (entry 10). Suitably substituted cyclic dienes were also amenable to hydroperoxidation under these conditions.
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | R4 | Yield |
| 1 | Me | H | H | CO2Et | 77% |
| 2 | n-Pr | H | H | CO2Et | 68% |
| 3 | Me | Me | H | CO2Et | 70% |
| 4 | Me | H | Et | CO2Et | 74% |
| 5 | Me | H | Et | CONHC6H11 | 90% |
| 6 | MeO2C | H | H | CO2Me | 65% |
| 7 | Me | H | H | CHO | 49% |
| 8 | n-Bu | H | H | CHO | 54% |
| 9 | n-Hex | H | H | CO-t-Bu | 55% |
| 10 | Ph(CH2)2 | H | H | H | Mixture |
In the same paper, the authors go on to describe the conversion of acrylic esters to their α-hydroperoxy derivatives in consistently high yields (Table 7).
|
|||
|---|---|---|---|
| Entry | R1 | R2 | Yield |
| 1 | H | Cy | 72% |
| 2 | Me | Cy | 80% |
| 3 | Bu | Et | 78% |
| 4 | CO2Bu | Bu | 81% |
The cobalt-mediated peroxidation of alkenes is well established (vide supra). Woerpel et al. expanded the scope of this reaction to include silyl enol ethers.23 Initial studies indicated that peroxidation of triethylsilyl enols ethers, rather than the more labile trimethyl silyl equivalents, resulted in fewer decomposition products, easier purification and higher yields. Further optimisation revealed that a combination of Co(thd)2 in trifluorotoluene led to an additional increase in yield. Aromatic silyl enol ethers with electron-withdrawing groups afforded higher yields (Table 8, entries 2 and 3) whereas substrates with electron-donating group gave lower yields (entry 1) or complete decomposition (entry 10). This observation may be rationalised on the basis that electron-withdrawing groups stabilise the radical intermediates, thus giving higher yields of the target peroxides. It was also noted that the enolisation step need not be stereoselective, as each component of a mixture of isomeric silyl enol ethers furnished the same product (entries 8 & 9).
|
|||
|---|---|---|---|
| Entry | R1 | R2 | Yield |
| 1 | 4-MeC6H4 | H | 67% |
| 2 | 4-BrC6H4 | H | 92% |
| 3 | 4-NO2C6H4 | H | 96% |
| 4 | 2,4-DichloroC6H4 | H | 54% |
| 5 | CH2Bn | H | 79% |
| 6 | t-Bu | H | 76% |
| 7 | –CHCH2 |
H | 55% |
| 8 | Ph | Ph | 63% |
| 9 | Ph | Me | 69% |
| 10 | 4-MeOC6H4 | H | — |
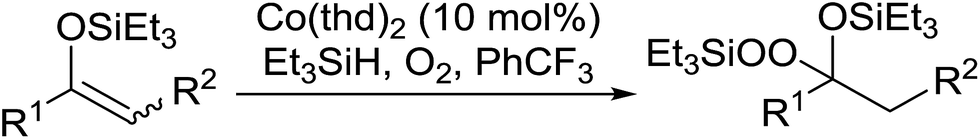
2.2 Kharasch and related reactions
Beginning in the 1950s, Kharasch and co-workers published a number of seminal papers on radical reactions of organic peroxides.24–37 This work culminated in the development of a general methodology for the incorporation of peroxy groups under transition metal catalysis, typically copper, iron or manganese salts. The versatility of this methodology is illustrated by the diverse range of substrates which can be converted to acyclic peroxides in good to excellent yields (Scheme 5).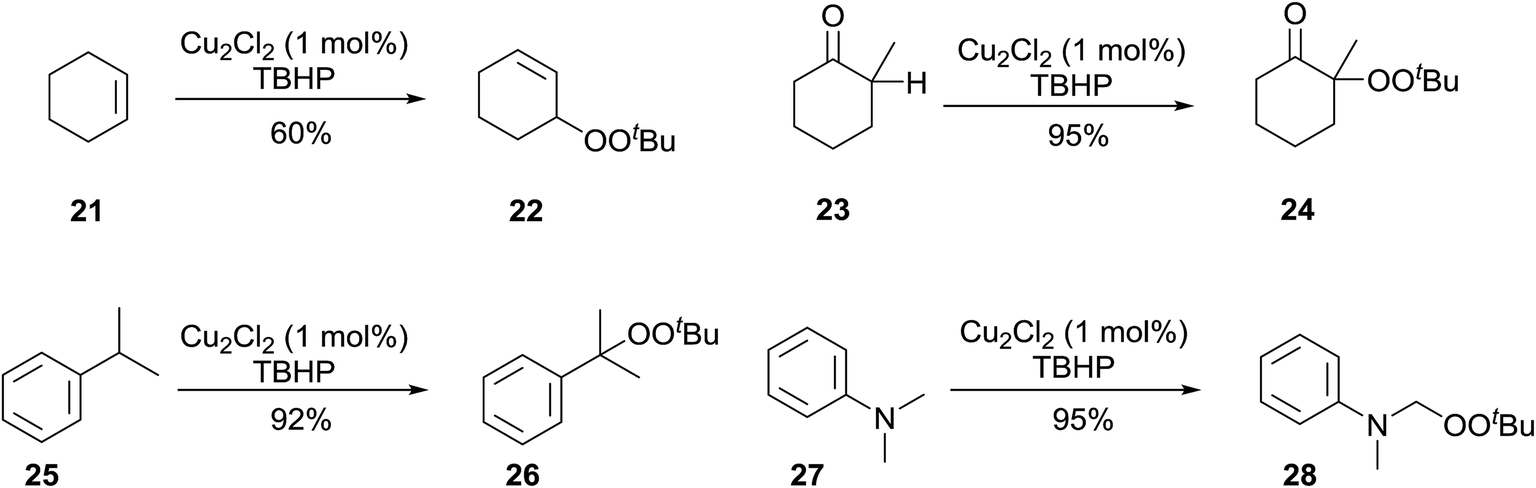 | ||
| Scheme 5 | ||
Gade and Meder studied 1,3-bis(2-pyridylimino)- and 1,3-bis-(2-thiazolylimino)isoindole copper complexes as catalysts for the allylic peroxidation of cyclohexene (Scheme 6).38 Using commercially available 70% aqueous TBHP, they found that catalysts 29 and 30 effected the peroxidation of cyclohexene with conversions in the range of 80–90%. In general, pyridine-based catalyst 29 was found to more active than thiophene-containing 30 with turnover frequencies of 63 h−1 and 18 h−1 respectively. In the absence of the ligands, reactions proceeded very slowly and with poor chemoselectivity.
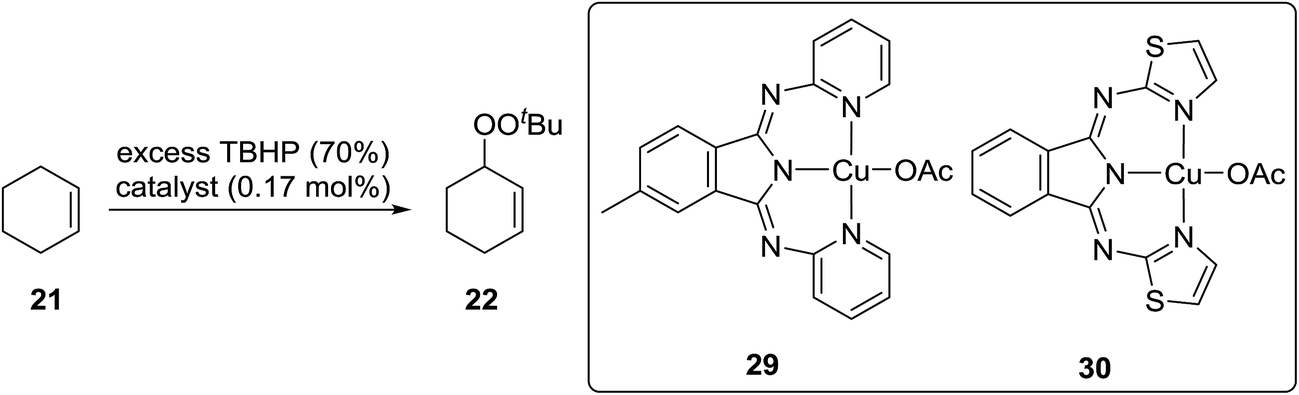 | ||
| Scheme 6 | ||
Schulz et al. reported one of the first examples for the direct asymmetric peroxidation of prochiral allylic and benzylic C–H substrates with tert-butyl hydroperoxide (TBHP) in combination with chiral bisoxazoline–copper complexes (Table 9).39 A mixture of three, regioisomeric, optically active peroxides were isolated in moderate to good yields with minimal side product formation. Peroxidation of 1-methylcyclohexene (31a) with TBHP in the presence of copper(I) triflate and tridentate bisoxazoline ligand 35 led to mixtures of regioisomeric peroxides with (S)-33a being isolated in an enantiomeric excess of 84% under optimised conditions albeit in a low yield of 9% (Table 9, entry 3). It was observed that lower temperatures favoured isomer 32a over 33a while also resulting in higher enantioselectivities (entries 1–3). The reaction was found to be highly substrate dependent, with more sterically hindered substrates affording significantly lower ee's (entries 4 and 5).
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | R | Temp. (°C) | Yield (32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :33:34) :33:34) |
ee (32:33:34) |
| 1 | 31a | Me | 0 | 25%:23%:10% |
19% (R):32% (S):0 |
| 2 | 31a | Me | −36 | 26%:12%:13% |
28% (R):70% (S):0 |
| 3 | 31a | Me | −48 | 31%:9%:14% |
32% (R):84% (S):10% |
| 4 | 31b | Ph | −20 | 27%:54%:16% |
n.d.:8% (S):n.d. |
| 5 | 31c | t-Bu | −20 | 3%:54%:10% |
n.d.:16% (S):n.d. |
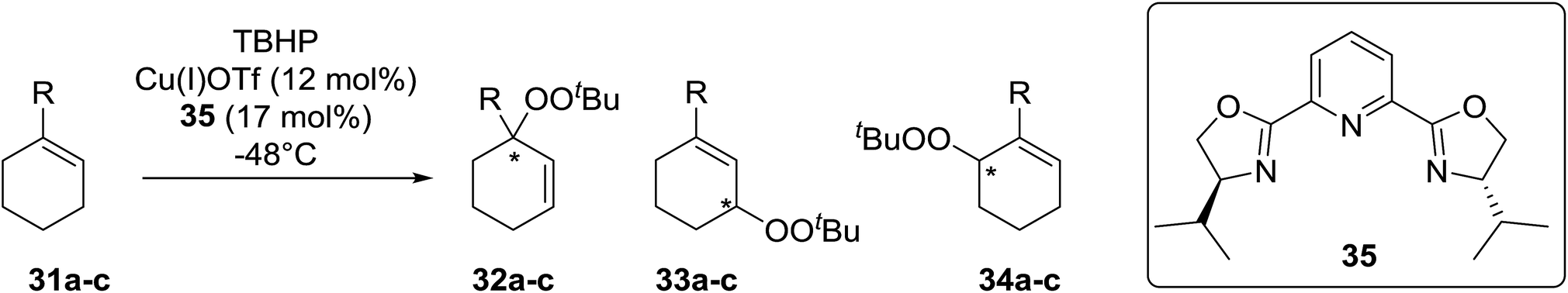
Continuing from their work on the peroxidation of β-dicarbonyl compounds to form 1,2,4,5-tetraoxanes,40 Terent'ev et al. observed that transition metals catalyse the selective peroxidation of β-dicarbonyl compounds at the α-position.41 Following extensive screening studies and reaction optimization, it was discovered that Cu(ClO4)2·6H2O was the most efficient catalyst, outperforming Cu(acac)2, Fe(acac)2 and Mn(OAc)2·4H2O. A selective, gram-scale method was developed for the synthesis of α-peroxidated derivatives of β-diketones, β-ketoesters and diethyl malonate, with the target compounds recovered in high yields from β-diketones (Table 10, entries 1–5), lower yields from β-keto esters (entries 6–10) and lowest yields from malonates (entries 11–14). The mechanism proceeds by initial complexation of the dicarbonyl with Cu(II), followed by reaction with the tert-butylperoxy radical to form the target peroxide and monovalent copper. Subsequent oxidation of Cu(I) by TBHP completes the catalytic cycle.
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Yield |
| 1 | Me | Me | Bn | 73% |
| 2 | Me | Me | 4-NO2–C6H4CH2 | 46% |
| 3 | Me | Me | Allyl | 57% |
| 4 | Me | Me | MeC(O)(CH2)2 | 51% |
| 5 | Me | Me | EtOC(O)(CH2)2 | 75% |
| 6 | Me | OEt | Bn | 89% |
| 7 | Me | OEt | 4-NO2–C6H4CH2 | 53% |
| 8 | Me | OEt | NC(CH2)2 | 66% |
| 9 | Me | OEt | MeC(O)(CH2)2 | 37% |
| 10 | Me | OEt | n-Bu | 78% |
| 11 | OEt | OEt | Bn | 67% |
| 12 | OEt | OEt | Allyl | 52% |
| 13 | OEt | OEt | NC(CH2)2 | 51% |
| 14 | OEt | OEt | MeC(O)(CH2)2 | 37% |
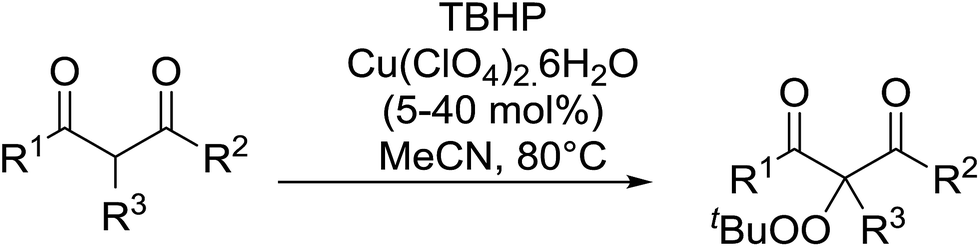
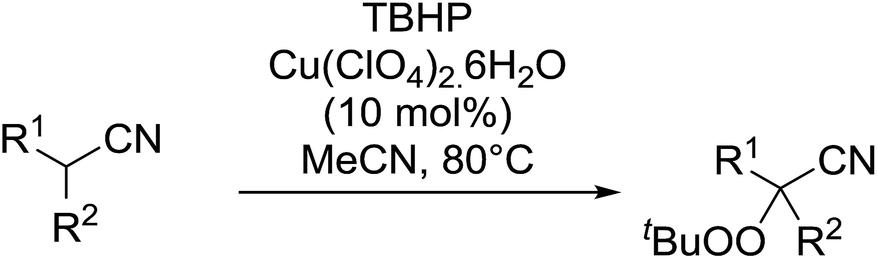
This work was expanded to include aliphatic and aromatic α-substituted malonitriles and β-cyanoesters ester derivatives (Table 11).42 While a 10 mol% loading of the copper catalyst was sufficient for the peroxidation of the malononitrile substrates (entries 7–11), a higher load of 40–80 mol% was required for the complete conversion of the β-cyanoesters (entries 1–6). The synthesis could be easily scaled up to gram quantities without a decrease in yield.
|
|||
|---|---|---|---|
| Entry | R1 | R2 | Yield |
| 1 | CO2Et | Bn | 81% |
| 2 | CO2Et | 4-Cl–C6H4CH2 | 71% |
| 3 | CO2Et | 4-MeO–C6H4CH2 | 63% |
| 4 | CO2Et | EtO2C(CH2)2 | 64% |
| 5 | CO2Et | Allyl | 64% |
| 6 | CO2Et | MeC(O)CH2 | 83% |
| 7 | CN | Bn | 94% |
| 8 | CN | 4-Cl–C6H4CH2 | 89% |
| 9 | CN | 4-MeO–C6H4CH2 | 68% |
| 10 | CN | i-Pr | 78% |
| 11 | CN | Cyclopentyl | 69% |
The Gif-Barton oxidation of several alkylbenzenes was investigated by Minisci et al. as a route to various alkyl peroxides (Table 12, entries 1–4).43,44 The authors conclude that the reaction proceeds via a radical mechanism with the initial formation of tBuO˙, followed by hydrogen abstraction and the generation of a benzylic radical as a key reaction intermediate. This pathway was confirmed by the addition of α-methylstyrene as a radical trap.
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Yield |
| 1 | Ph | H | H | 45% |
| 2 | Ph | Me | H | 62% |
| 3 | Ph | Me | Me | 80% |
| 4 | 4-MeC6H4 | Me | Me | 20% |
These findings were further developed into a free-radical oxidative alkylation, arylation and acylation of acrylonitrile using TBHP under Cu(II) catalysis.45 The reaction is compatible with a variety of substrates including cyclohexane, adamantine, toluene and benzaldehyde (Table 13, entries 1–8). While the alkylation reaction resulted in a mixture of acyclic peroxides (entries 1–3), the corresponding acylation reactions furnished 38 as a single regioisomer (entries 7 and 8).
|
|||
|---|---|---|---|
| Entry | R | Yield 38 | Yield 39 |
| 1 | Cyclohexyl | 36% | 12% |
| 2 | Bn | 16% | 10% |
| 3 | Dioxanyl | 13% | 10% |
| 4 | 1-Admantyl | 44% | — |
| 5 | α-Tetrahydrofuranyl | 80% | — |
| 6 | Ph | 15% | — |
| 7 | Ph–C(O) | 43% | — |
| 8 | n-C3H7–C(O) | 40% | — |
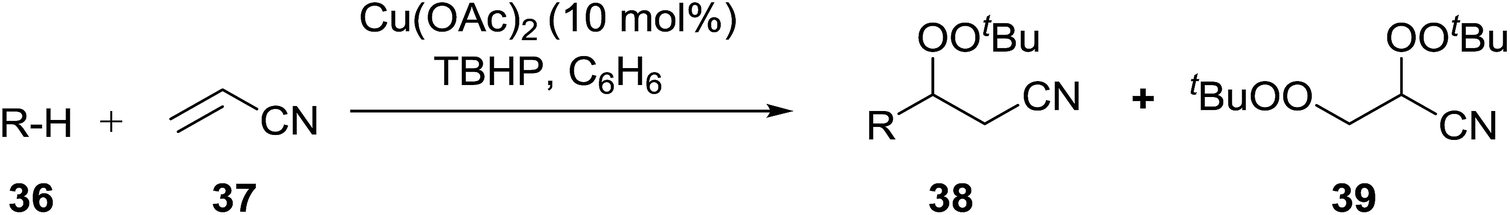
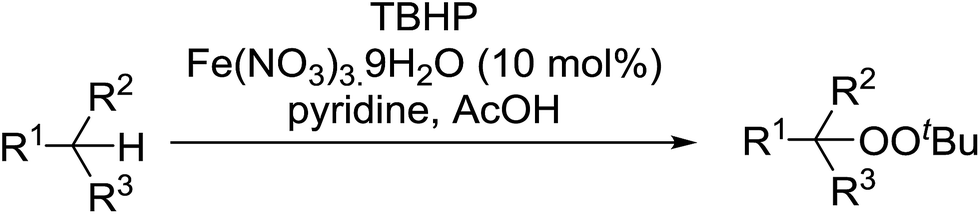
Introduction of a peroxide functionality into an alkane backbone in a synthetically useful manner is a challenging goal. Shul'pin et al. discovered that cyclohexane was susceptible to hydroperoxidation when oxidised with peroxyacetic acid (PAA) and a copper complex – Cu(MeCN)4BF4 – with trace formation of side products cyclohexanol and cyclohexanone (Scheme 7).46 It was proposed that oxidation with PAA is induced via the attack of species HO˙ or CH3C(O)O˙ on the alkane. On switching the oxidising agent to TBHP, a change in the selectivity of the reaction was observed with cyclohexanol being formed as the major product.
 | ||
| Scheme 7 | ||
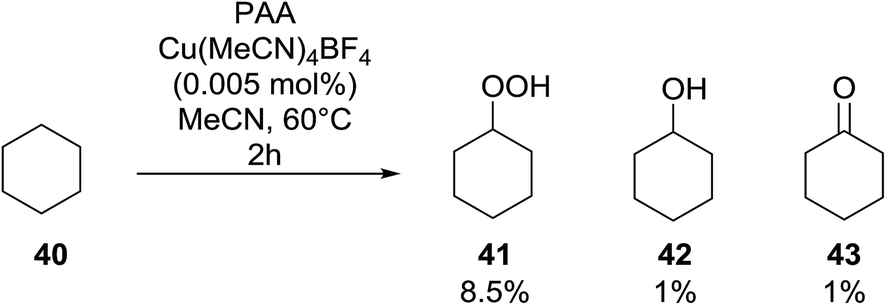
Shul'pin later described the first, non-transition metal-catalysed peroxidation of alkanes using hydrogen peroxide in the presence of a catalytic amount of Al(NO3)3·9H2O.47 Starting with test substrate cyclooctane 44 the corresponding hydroperoxide 45 was isolated in 31% yield after 10 hours at 60 °C along with trace amounts of cyclooctanone and cyclooctanol (Scheme 8). Only negligible amounts of the oxygenated products were recovered in the absence of the aluminium catalyst.
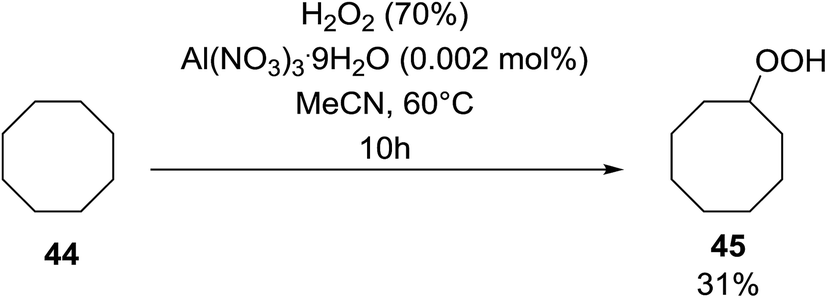 | ||
| Scheme 8 | ||
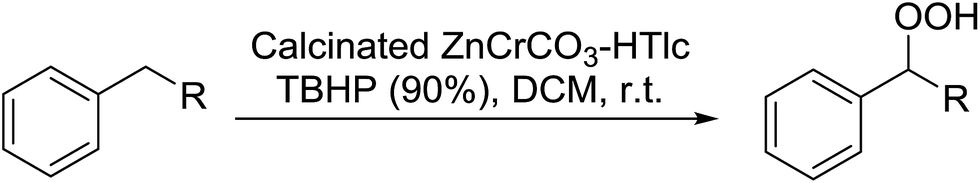
Due to their Bronsted and Lewis acidities, both in their natural and ion-exchanged forms, clays often function as efficient catalysts for organic transformations. Choudary et al. developed a mild and selective hydroperoxidation of aralkanes using calcined ZnCrCO3-HTlc (hydrotalcite-like compounds) with the aid of TBHP.48 The clay catalyst may be recovered and reused after the reaction by simple filtration and the recovered catalyst shows consistent efficiency. The method is quite efficient in the case of tertiary alkanes (Table 14, entry 7) but more moderate yields were obtained in case of primary aralkanes (entries 1 & 2). Use of hydrogen peroxide resulted in poor conversions and selectivity. The presence of an optimum amount of water (90% TBHP) was found to be essential for the higher selectivity at the highest conversion (entry 2 vs. entry 3) which may be attributed to the stabilisation of the hydroperoxide products from further decomposition.
|
|||
|---|---|---|---|
| Entry | Substrate | Product conversion | Hydroperoxide yield (based on NMR) |
| a 70% aqueous TBHP used.b Uncalcined sample used as catalyst.c 30% hydrogen peroxide used as an oxidant and tert-butanol as a solvent. | |||
| 1 | Phenyl methane | 20% | 33% |
| 2 | 1-Phenyl ethane | 40% | 87% |
| 3 | 1-Phenyl ethanea | 10% | 98% |
| 4 | 1-Phenyl ethaneb | 45% | 38% |
| 5 | 1-Phenyl ethanec | 30% | 60% |
| 6 | 1-Phenyl propane | 25% | 79% |
| 7 | 2-Phenyl propane | 60% | 98% |
| 8 | 1-Phenyl butane | 22% | 70% |
| 9 | Fluorene | 40% | 30% |
| 10 | 2-Naphthyl ethane | 37% | 79% |
| 11 | Diphenyl methane | 37% | 33% |
| 12 | Indan | 56% | 34% |
Sudalai and Nikalje have demonstrated that Ru(III)-exchanged montmorillonite K10 clay is a mild and efficient catalyst for benzylic peroxidation.49 Using optimised conditions of two equivalents of TBHP in acetonitrile, Ru-exchanged catalysts exhibited higher activity and selectivity than either Mn- or Cu-exchanged clays. Various alkyl arenes were transformed to the corresponding alkyl peroxides in moderate to good yields (Table 15, entries 1–6). The reaction showed a notable preference for selective oxidation of one methyl group only in the cases of the xylenes (entries 2 and 3) and for methine oxidation over methyl oxidation in the case of ethylbenzene (entry 4). Selective oxidation at the α-positions of tetrahydrofuran and 3,4-dihydropyran was similarly successful (entries 7 and 8).
|
|||
|---|---|---|---|
| Entry | R1 | R2 | Yield |
| 1 | Ph | H | 60% |
| 2 | 2-MeC6H4 | H | 76% |
| 3 | 4-MeC6H4 | H | 47% |
| 4 | Ph | Me | 54% |
| 5 | 2-ClC6H4 | H | 76% |
| 6 | 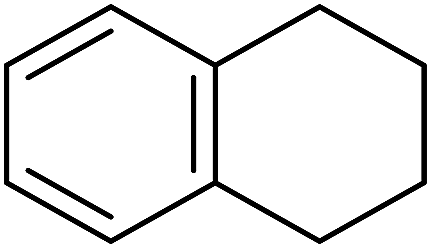 |
50% | |
| 7 | 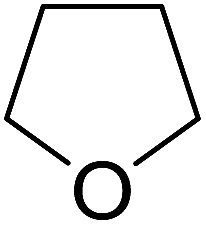 |
45% | |
| 8 | 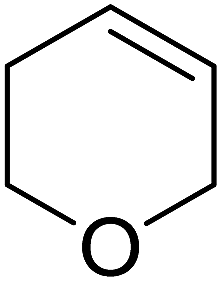 |
70% | |
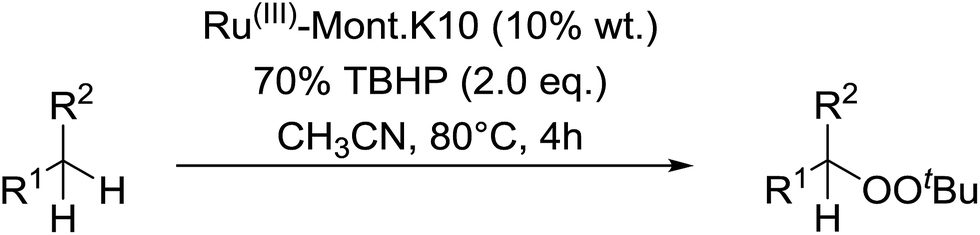
Nishimura and co-workers have investigated the influence of different ruthenium catalysts on the product distribution for the oxidation of aromatic alkenes.50 Oxidation of various styrenes with TBHP in the presence of triethylamine and Ru(II)-bipyridylsilylated montmorillonite (bpy-mont) afforded several products, but with bisperoxide 47 as the major product (Table 16, entries 1–3). Switching to a homogeneous catalyst was accompanied by a significant increase in yield of 47 (entries 4–6). Interestingly, repeating the Ru(II)-(bpy-mont)-catalysed reactions in the absence of triethylamine afforded hydroperoxide 50 almost exclusively (entries 7 and 8). Triethylamine serves to accelerate the generation of the reactive species Ru(IV)O while also enhancing the nucleophilicity of TBHP. In the absence of triethylamine, an intermediate benzyl radical reacts with oxygen forming a peroxy radical and the subsequent hydrogen abstraction from TBHP results in the formation of hydroperoxide 50.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R | Catalyst | Et3N mL | Isolated yields | |||
| 47 | 48 | 49 | 50 | ||||
| 1 | H | Ru(II)-bpy-mont | 0.1 | 30% | 13% | — | — |
| 2 | Me | Ru(II)-bpy-mont | 0.1 | 33% | 11% | 10% | — |
| 3 | Ph | Ru(II)-bpy-mont | 0.1 | 59% | 19% | 8% | Trace |
| 4 | Ph | RuCl2-bpy-(CO2) | 0.1 | 69% | 19% | 5% | Trace |
| 5 | Ph | RuCl2-bpy-2H2O | 0.1 | 71% | 11% | 5% | Trace |
| 6 | Ph | [RuCl2(CO2)]n | 0.1 | 63% | 24% | 5% | 2% |
| 7 | Me | Ru(II)-bpy-mont | — | tr | tr | 1% | 70% |
| 8 | Ph | Ru(II)-bpy-mont | — | tr | — | 18% | 79% |
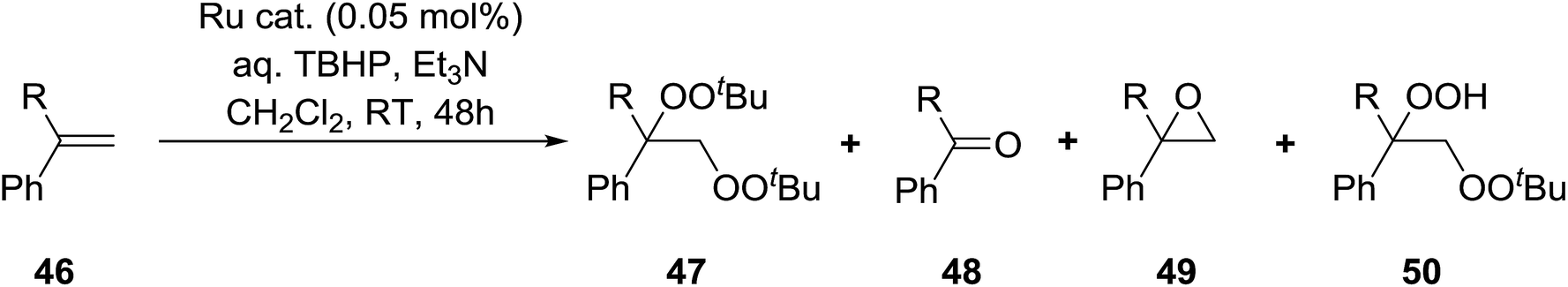
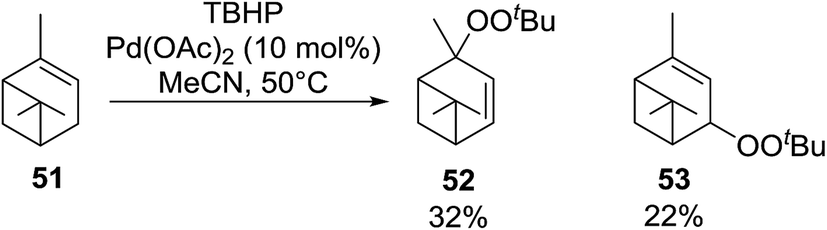
Karim et al. investigated the allylic oxidation of α-pinene 51 with TBHP and found that peroxide products 52 and 53 were obtained in moderate yields of 16% and 28% respectively in the absence of a catalyst (Scheme 9).51 The addition of a catalytic amount of palladium acetate led to significant increase in yields and a switch in preference of products, with 52 and 53 being isolated in yields of 32% and 22% respectively. The mode of addition of TBHP was found to be crucial: the addition of TBHP at once favours formation of the peroxides whereas dropwise addition promotes the formation of the side products verbenol and verbenone.
 | ||
| Scheme 9 | ||
A novel, palladium-catalysed oxidation of olefins under basic conditions was developed by Corey and Yu.52 Numerous olefinic substrates bearing allylic hydrogens were successfully transformed into alkyl peroxide products with only minor competition from the epoxidation side reaction (Table 17, entries 1–5). The addition of a catalytic amount of BINAP prevented unwanted deposition of metallic palladium and facilitated the conversion of Pd(0) to Pd(II) (entries 4 and 5). The choice of palladium ligand was critical to the reaction outcome, with palladium trifluoroacetate affording a regioisomeric peroxide as the major product, most likely as the result of the peroxypalladation of the olfenic bond (entries 6 vs. 1).
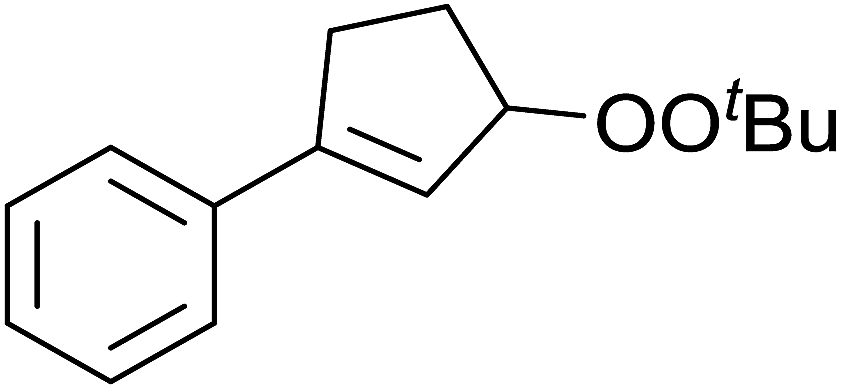
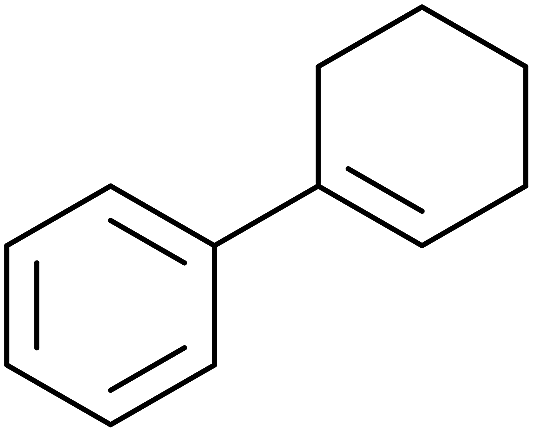
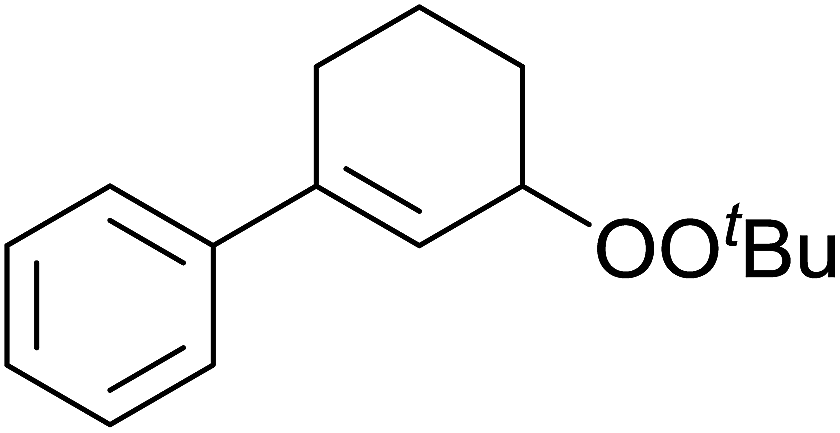
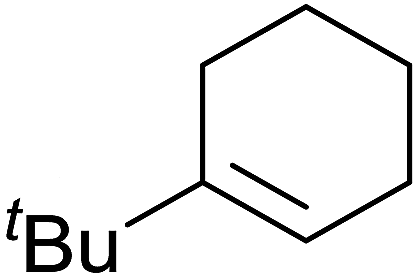
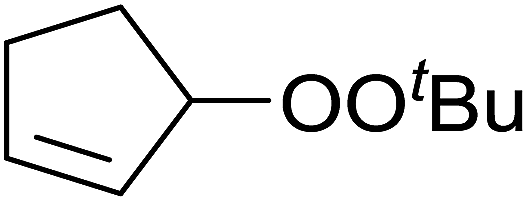
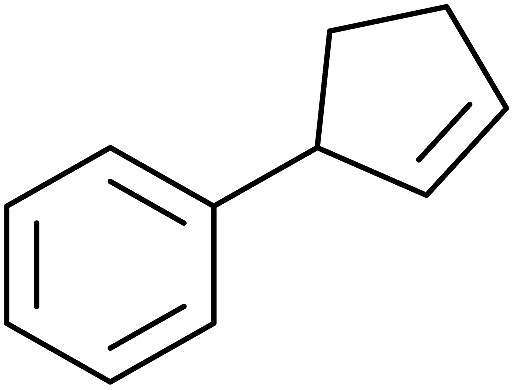
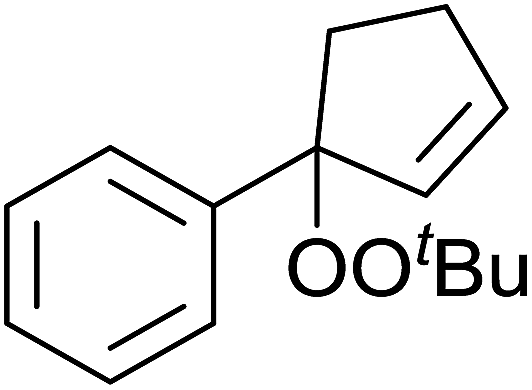
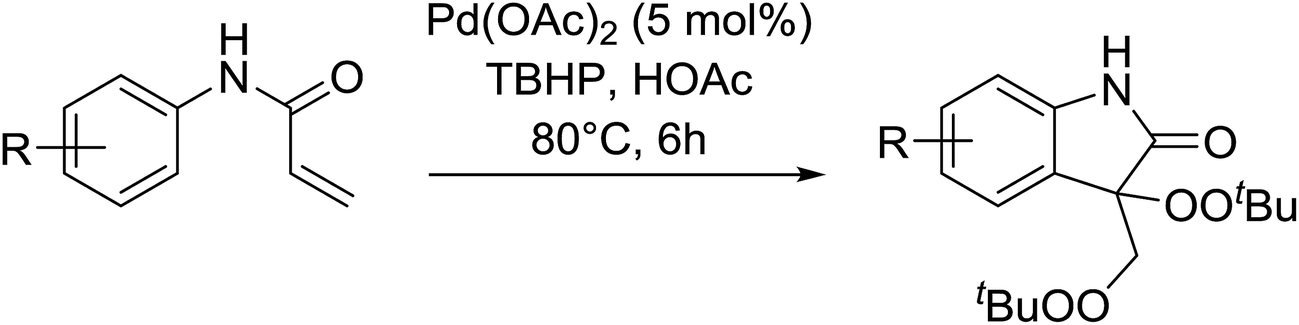
Pan et al. have reported a novel and facile palladium-catalysed tandem bisperoxidation/C–H activation of olefins.53 They found that a combination of palladium acetate and TBHP in acetic acid afforded the highest yields of bisperoxyoxindoles (Table 18, entries 1–13). The reaction tolerated both electron-donating (entries 2–4, 12 and 13) and electron-withdrawing groups (entries 5–11). The latter substrates included ortho-halogenated rings, which indicates that this is a novel palladium-catalysed process which is distinct from the oxindole syntheses described by Hartwig54 and Kündig.55 The authors note, however, that the reaction was unsuccessful with N-methyl substituted substrates.
|
||
|---|---|---|
| Entry | R | Yield |
| 1 | H | 70% |
| 2 | 2-Me | 96% |
| 3 | 3-Me | 90% |
| 4 | 4-Me | 81% |
| 5 | 2-Br | 64% |
| 6 | 3-Br | 54% |
| 7 | 2,4-Dibromo | 46% |
| 8 | 2-Cl | 62% |
| 9 | 4-Cl | 78% |
| 10 | 2,4-Dichloro | 54% |
| 11 | 2,3-Dichloro | 69% |
| 12 | 2-MeO | 86% |
| 13 | 4-MeO | 78% |
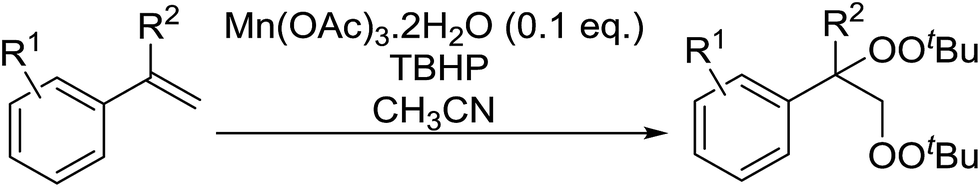
Manganese triacetate has been found to be an effective catalyst for the bisperoxidation of styrenes by Terent'ev and co-workers.56 A range of manganese salts of different oxidation states were initially screened and several were found to catalyse the bisperoxidation of styrene with TBHP. The reaction was optimised using manganese triacetate in acetonitrile which afforded the highest yields. Several styrene substrates were subjected to these optimised conditions and the target bis-peroxides were isolated in yields of 51–75% (Table 19, entries 1–8). Styrenes bearing electron-donating (entries 5 and 6) or electron-withdrawing (entry 8) substituents were equally amenable to peroxidation.
|
|||
|---|---|---|---|
| Entry | R1 | R2 | Yield |
| 1 | H | H | 75% |
| 2 | H | Me | 56% |
| 3 | 4-Me | H | 63% |
| 4 | 4-t-Bu | H | 61% |
| 5 | 4-MeO | H | 46% |
| 6 | 2-MeO | H | 50% |
| 7 | H | Ph | 52% |
| 8 | 4-Cl | H | 51% |
Scettri and co-workers have reported a new method for the racemic peroxidation of furan side chains using a molybdenum catalyst.57 During their studies on the Mo(CO)6-catalysed oxidation of furan derivatives to E- and Z-enediones, they found that formation of E-enediones 55 was preferred over peroxyfurans 56 (Scheme 10). However, with 54b as their substrate and on increasing the equivalents of TBHP from one to three, the authors obtained peroxyfuran 56b as the major product in a yield of 55%. Interestingly, the enediones 55a-b could be directly converted to the corresponding peroxypyranone derivatives 57a-b under similar reactions conditions in yields of 58% and 59% respectively.
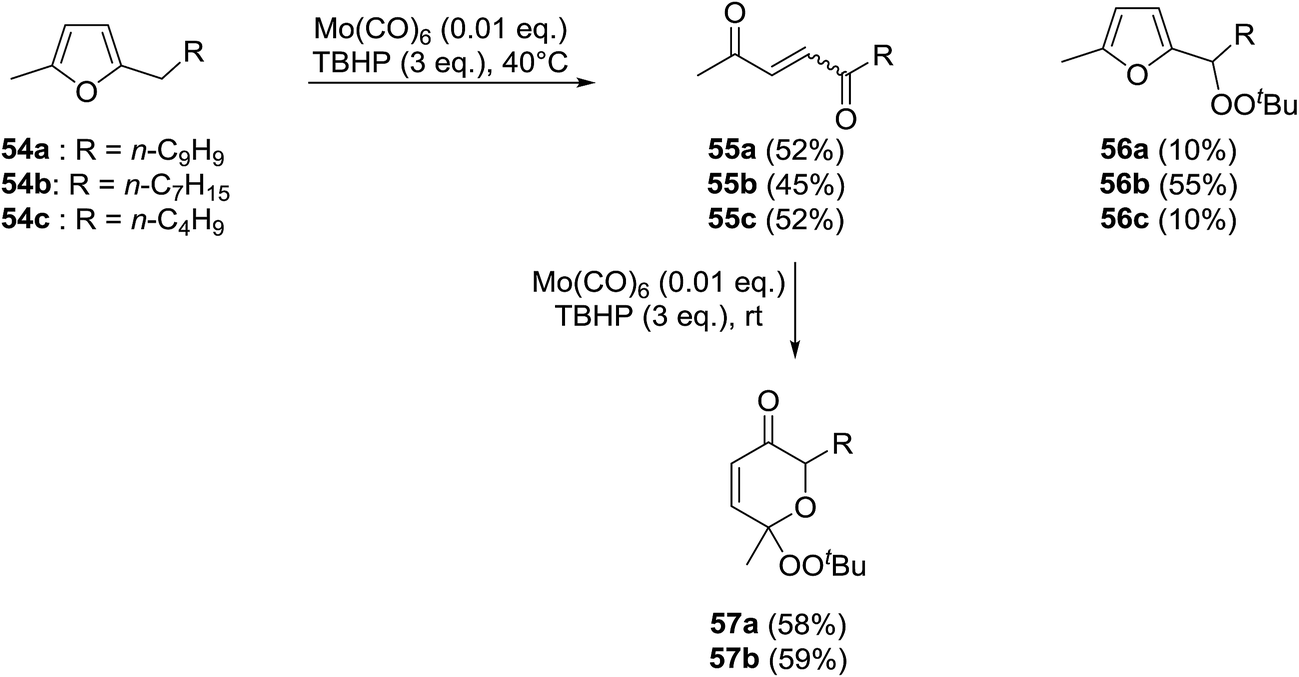 | ||
| Scheme 10 | ||
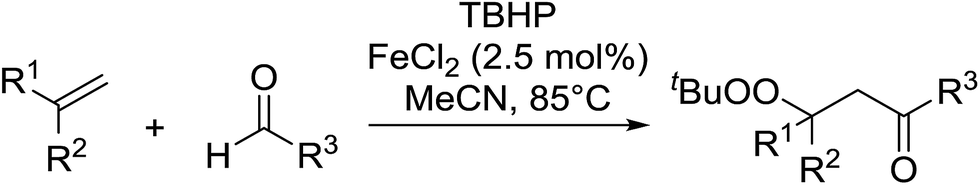
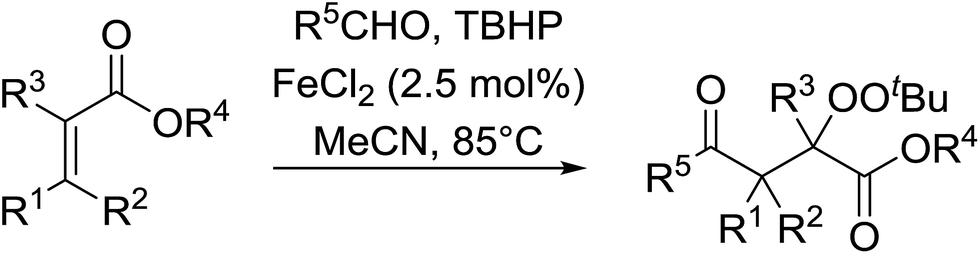
The difunctionalisation of alkenes to introduce a peroxide and another functional group, such as a carbonyl, is a challenging reaction. Li and co-workers published a novel, iron-catalysed, three-component carbonylation–peroxidation reaction of alkenes, aldehydes and hydroperoxides to form β-peroxyketones in moderate to high yields of 39–93% (Table 20).58 Both aromatic aldehydes (entries 1–5) and aliphatic aldehydes (entries 6–13) were coupled to aryl-substituted alkenes. The reaction proceeds by way of a radical mechanism involving initial carbonylation via an acyl radical intermediate, followed by coupling with a t-butylperoxy radical, thereby accounting for the high regioselectively. Interestingly, the addition of base to the reaction leads to the formation of α,β-epoxyketones, thus providing a convenient one-pot method for the preparation of complex epoxides.
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Isolated yield |
| 1 | H | Ph | Ph | 75% |
| 2 | Me | Ph | Ph | 74% |
| 3 | Ph | Ph | Ph | 93% |
| 4 | H | Napthyl | Ph | 52% |
| 5 | H | Pyridyl | Ph | 39% |
| 6 | H | Ph | Cyclopropyl | 75% |
| 7 | Me | Ph | Cyclopropyl | 84% |
| 8 | Ph | Ph | Cyclopropyl | 89% |
| 9 | H | Ph | n-Bu | 75% |
| 10 | H | 4-tBu–C6H4 | n-Bu | 69% |
| 11 | H | 4-Br–C6H4 | n-Bu | 70% |
| 12 | Me | Ph | n-Bu | 83% |
| 13 | Ph | Ph | n-Bu | 83% |
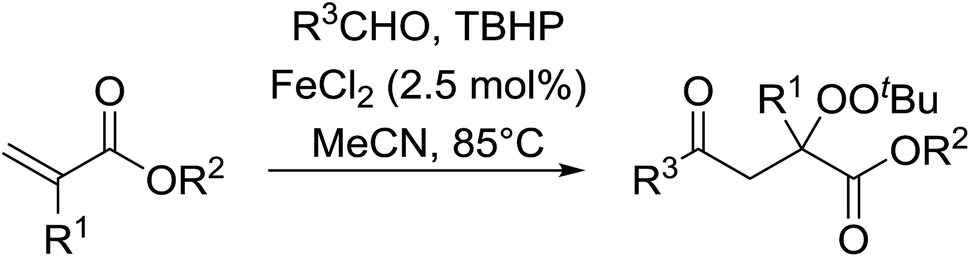
This methodology was also applied to the synthesis of α-ester-β-keto peroxides from α,β-unsaturated esters incorporating a terminal olefinic bond (Table 21).59 The optimised conditions were compatible with a wide variety of unsaturated esters and afforded the target peroxides in good to excellent yields (entries 1–10). This included Baylis–Hillman products bearing O-protected groups (entries 8–10). A range of aldehyde substrates were also tolerated including aromatic (entries 11–15), heteroaromatic (entries 16–18) and aliphatic aldehydes (entry 19). The transformation of formamides (entry 20) was similarly successful.
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Isolated yield |
| 1 | Me | Allyl | Ph | 70% |
| 2 | Me | (CH2)2OH | Ph | 61% |
| 3 | Ph | Et | Ph | 82% |
| 4 | NHAc | Me | Ph | 65% |
| 5 | CH2CO2Me | Me | Ph | 70% |
| 6 | CH2OMe | Me | Ph | 80% |
| 7 | CH2OAc | Me | Ph | 79% |
| 8 | CH(Ph)OAc | Me | Ph | 94% |
| 9 | CH(Ph)OBoc | Me | Ph | 84% |
| 10 | CH(Ph)OTBSMe | Me | Ph | 89% |
| 11 | Me | Bn | 4-MeO–C6H4 | 75% |
| 12 | Me | Bn | 4-Me–C6H4 | 78% |
| 13 | Me | Bn | 4-Br–C6H4 | 72% |
| 14 | Me | Bn | 4-AcNH–C6H4 | 58% |
| 15 | Me | Bn | 4-NC–C6H4 | 46% |
| 16 | Me | Bn | 3-Indolyl | 63% |
| 17 | Me | Bn | 2-Thienyl | 56% |
| 18 | Me | Bn | 2-Furyl | 45% |
| 19 | Me | Bn | n-Bu | 72% |
| 20 | Me | Bn | NMe2 | 45% |
The application of this strategy to tri- and tetra-substituted acrylates constitutes a concise synthetic route to densely functionalised peroxides (Table 22). The target peroxides were efficiently prepared using internal alkenes bearing a β-alkyl (entries 1–3, 7–11), β-phenyl (entries 4–6, 13) or β-ester group (entry 12) in excellent yields. In general, aromatic aldehydes showed lower reactivity than aliphatic aldehydes (entries 1 & 4 vs. entries 2 & 3).
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | R4 | R5 | Isolated yield |
| 1 | H | Me | Me | Me | Ph | 55% |
| 2 | H | Me | Me | Me | n-Bu | 64% |
| 3 | H | Me | Me | Me | Cyclopropyl | 92% |
| 4 | H | Ph | COOMe | Me | Ph | 30% |
| 5 | H | Ph | COOMe | Et | n-Bu | 75% |
| 6 | H | Ph | COOMe | Et | n-Pr | 74% |
| 7 | H | Me | COOEt | Et | n-Bu | 91% |
| 8 | H | Me | COOEt | Et | n-Pr | 87% |
| 9 | H | n-Pr | COOEt | Et | n-Pr | 87% |
| 10 | H | i-Pr | COOEt | Et | n-Pr | 64% |
| 11 | H | Me | C(O)Me | Et | n-Pr | 63% |
| 12 | H | CO2Me | CH2CO2Me | Me | Ph | 90% |
| 13 | H | Ph | CN | Me | Ph | 90% |
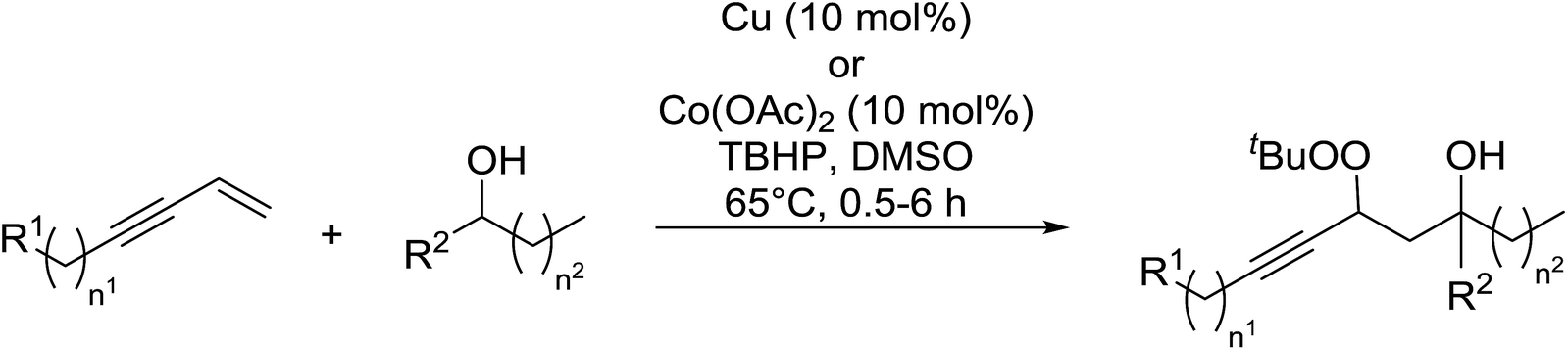
In a similar vein, Loh and Cheng investigated the metal-catalysed coupling reaction of 1,3-enynes with alcohols.60 The highest yields of β-peroxy alcohols were recorded with elemental copper or cobalt acetate as catalysts (Table 23). 1,3-Enynes bearing different silyl protecting groups were well accommodated and furnished the corresponding peroxides in 33–68% yields (entries 1–4) with Cu, whereas reactions with Co(OAc)2 returned better yields (51–73%). Both aromatic (entries 5–9) and aliphatic (10–12) enynes were suitable substrates for this transformation. It was further observed that aliphatic primary (entries 14–16) and secondary (entries 17–21) alcohols were amenable to these conditions, albeit with poorer yields for the former. It was also noted that increasing the chain length of the alcohol was accompanied by decreasing catalyst activity (entries 17 vs. 18 & 20 vs. 21). The proposed mechanism begins with metal-catalysed generation of tert-butyloxy/peroxy radicals followed by hydrogen abstraction from the alcohol to generate an α-hydroxy carbon radical. Attack of this radical on the olefinic double bond is followed by coupling with a tert-butylperoxy radical to afford the β-peroxy alcohol product.
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | n1 | n2 | Yield | |
| Cu | Co(OAc)2 | |||||
| 1 | Ph3Si | Me | 0 | 1 | 68% | 73% |
| 2 | TIPS | Me | 0 | 1 | 36% | 66% |
| 3 | TBDMS | Me | 0 | 1 | 50% | 56% |
| 4 | TES | Me | 0 | 1 | 33% | 51% |
| 5 | Ph | Me | 0 | 1 | 58% | 52% |
| 6 | 4-ClC6H4 | Me | 0 | 1 | 51% | 47% |
| 7 | 4-BrC6H4 | Me | 0 | 1 | 58% | 44% |
| 8 | 4-FC6H4 | Me | 0 | 1 | 51% | 52% |
| 9 | 2-FC6H4 | Me | 0 | 1 | 43% | 61% |
| 10 | Cyclopentyl | Me | 0 | 1 | 35% | 39% |
| 11 | Cyclohexyl | Me | 0 | 1 | 38% | 41% |
| 12 | Cyclohexyl | Me | 1 | 1 | 42% | 33% |
| 13 | Ph | Me | 2 | 1 | 63% | 42% |
| 14 | Ph3Si | H | 0 | 0 | 26% | 44% |
| 15 | Ph3Si | H | 0 | 1 | 55% | 42% |
| 16 | Ph3Si | H | 0 | 2 | 40% | 35% |
| 17 | Ph3Si | Me | 0 | 0 | 62% | 69% |
| 18 | Ph3Si | Me | 0 | 3 | 40% | 55% |
| 19 | Ph3Si | Et | 0 | 1 | 35% | 58% |
| 20 | Ph3Si | i-Pr | 0 | 0 | 55% | 50% |
| 21 | Ph3Si | i-Bu | 0 | 0 | 39% | 32% |
| 22 | Ph3Si | CH2Bn | 0 | 0 | 57% | 57% |
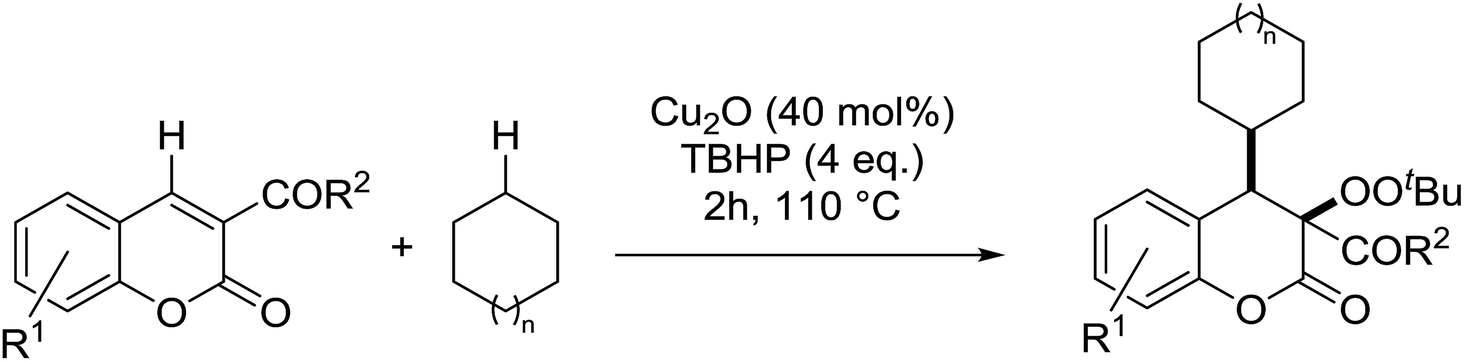
Patel et al. have developed a copper(I)-promoted cycloalkylation–peroxidation strategy involving cycloalkanes, TBHP and coumarins.61 Coumarins bearing both electron-withdrawing (Table 24, entries 2–6) and electron-donating groups (entries 7–8) were converted to the cycloalkylated α-peroxy products in moderate to good yields. The methodology was equally applicable to other 3-acyl/aroyl substituted coumarins (entries 9–11). While cyclopentane was tolerated in place of cyclohexane (entry 12), the use of cyclooctane resulted in the formation of an unwanted cycloalkylated α-hydroxy product. This methodology was further expanded to include ester-substituted coumarins (entries 13–20).
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | n | Yield |
| 1 | H | Me | 1 | 60% |
| 2 | 6-Cl | Me | 1 | 63% |
| 3 | 6-Br | Me | 1 | 59% |
| 4 | 6-NO2 | Me | 1 | 67% |
| 5 | 6,8-Dichloro | Me | 1 | 61% |
| 6 | 6,8-Dibromo | Me | 1 | 58% |
| 7 | 8-MeO | Me | 1 | 65% |
| 8 | 7-MeO | Me | 1 | 42% |
| 9 | H | Et | 1 | 64% |
| 10 | H | n-Bu | 1 | 62% |
| 11 | H | Ph | 1 | 68% |
| 12 | H | Me | 0 | 46% |
| 13 | H | OEt | 1 | 42% |
| 14 | 6-Cl | OEt | 1 | 51% |
| 15 | 6-Br | OEt | 1 | 42% |
| 16 | 6,8-Dichloro | OEt | 1 | 57% |
| 17 | 6,8-Dibromo | OEt | 1 | 50% |
| 18 | H | OEt | 1 | 56% |
| 19 | H | OMe | 1 | 40% |
| 20 | H | OMe | 3 | 38% |
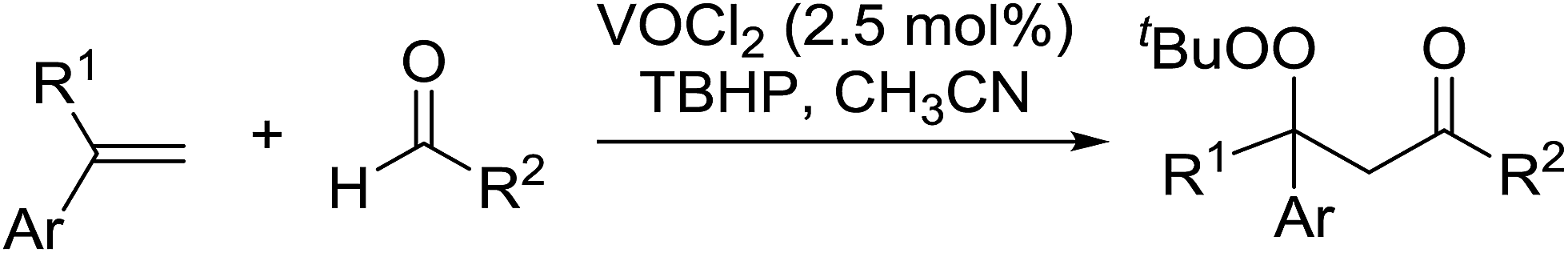
The oxidative carbonylation of styrenes with aldehydes using a vanadium catalyst and TBHP was studied by Weng and co-workers.62 Electron-deficient vanadyl species were found to favour β-peroxidation–carbonylation whereas electron-rich vandyl species resulted in β-hydroxylation–carbonylation. Accordingly, treatment of a range of styrenes and aldehydes with vanadyl dichloride and TBHP furnished β-peroxyketones in good to excellent yields (Table 25, entries 1–12). The lowest yields were recorded for electron rich (entry 3) or hindered (entry 7) aldehydes. Aromatic (entries 1–6), heteroaromatic (entries 8–9) and aliphatic aldehydes (entry 10) were transformed under these conditions. Variation of the styrene precursors, via the ring substituents (entries 11–14) or via the α-substituents (entries 15–17), was likewise tolerated.
|
|||||
|---|---|---|---|---|---|
| Entry | Ar | R1 | R2 | Time (h) | Yield |
| 1 | Ph | H | 4-MeC6H4 | 1 | 74% |
| 2 | Ph | H | 2-MeC6H4 | 1 | 73% |
| 3 | Ph | H | 4-MeOC6H4 | 1.5 | 56% |
| 4 | Ph | H | 4-ClC6H4 | 1 | 75% |
| 5 | Ph | H | 3-ClC6H4 | 1 | 74% |
| 6 | Ph | H | 4-BrC6H4 | 1 | 74% |
| 7 | Ph | H | 1-Naphthyl | 1 | 67% |
| 8 | Ph | H | 2-Thienyl | 1 | 78% |
| 9 | Ph | H | 2-Furyl | 1 | 75% |
| 10 | Ph | H | Cyclohexyl | 1 | 72% |
| 11 | 4-MeC6H4 | H | Ph | 1.5 | 72% |
| 12 | 4-MeOC6H4 | H | Ph | 2 | 68% |
| 13 | 4-BrC6H4 | H | Ph | 1.5 | 70% |
| 14 | 2-Naphthyl | H | Ph | 1.5 | 59% |
| 15 | Ph | CH3 | 4-MeC6H4 | 2 | 81% |
| 16 | Ph | Ph | 4-MeC6H4 | 2 | 72% |
| 17 | Ph | Ph | 4-ClC6H4 | 2 | 93% |
2.3 Peroxidation of alkenes
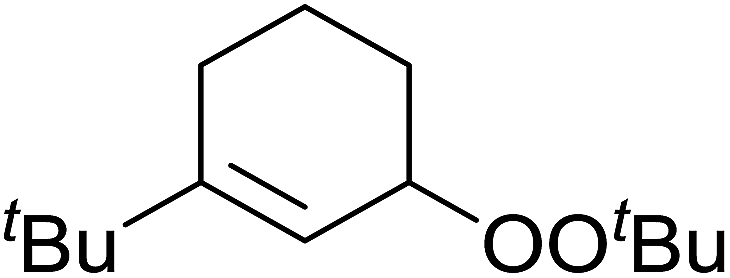
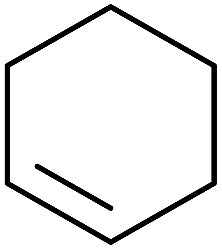
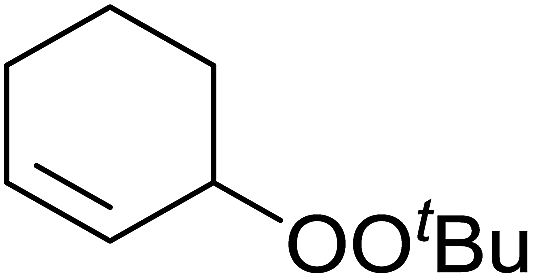
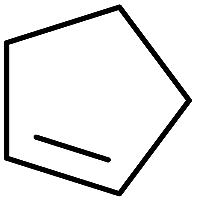
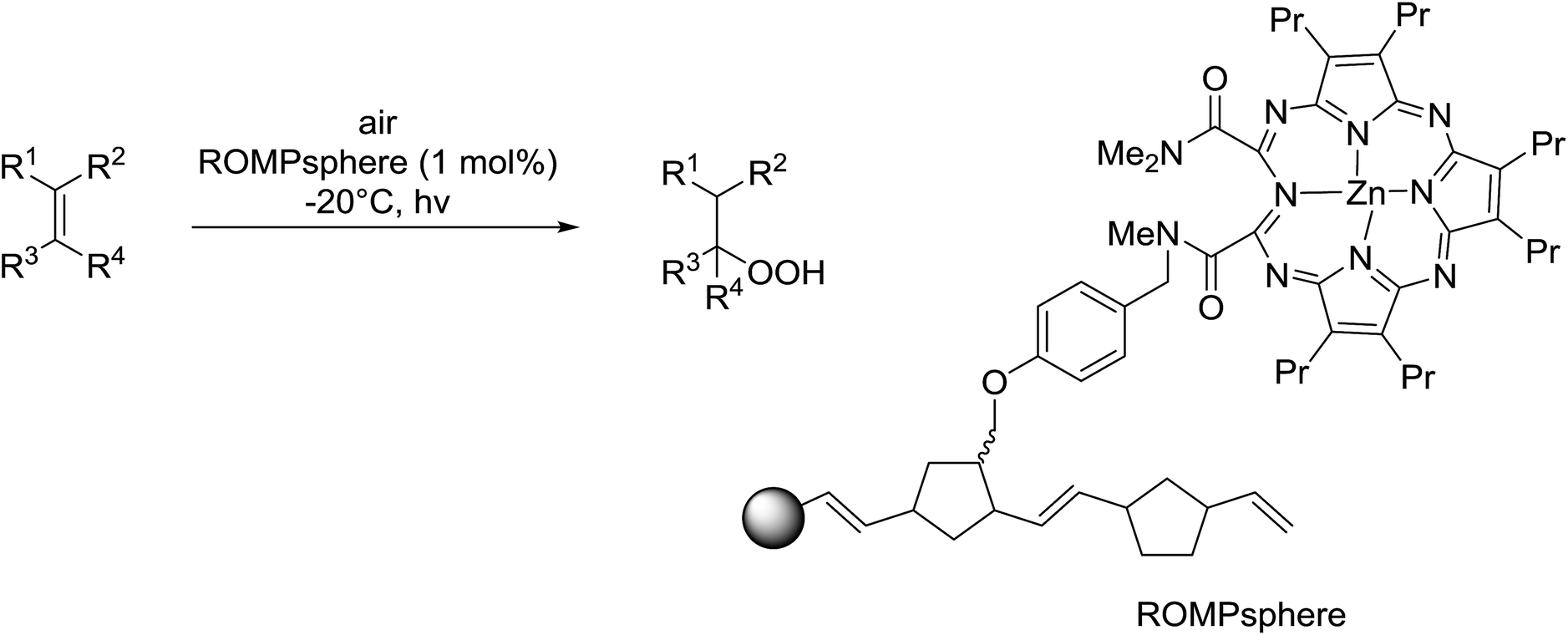
Barrett et al. have developed a novel immobilised, recyclable catalyst for photooxygenation reactions.63 The so-called ROMPsphere (Ring-Opening Metathesis Polymer) catalyst, which is based around polymeric zinc seco-porphyrazines, was screened against several alkenes in the singlet oxygen ene reaction (Table 26). For simple substrates, high yields of hydroperoxides were obtained (entries 1 and 2) whereas for more complex alkenes, extended reaction times were required (entry 3). Longer reaction times were accompanied by decomposition of the catalyst due to degradation of the ROM alkene backbone. The ROMPsphere catalyst was also found to successfully catalyse the conversion of 1,3-dienes into cycloperoxides in good yields.
|
||||
|---|---|---|---|---|
| Entry | Alkene | t (h) | Products | Yield |
| 1 | 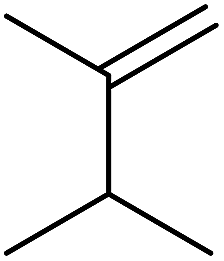 |
20 | 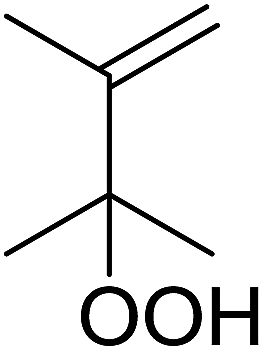 |
>99% |
| 2 | 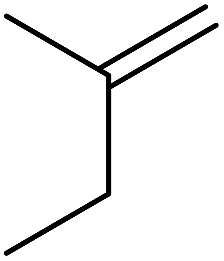 |
20 | 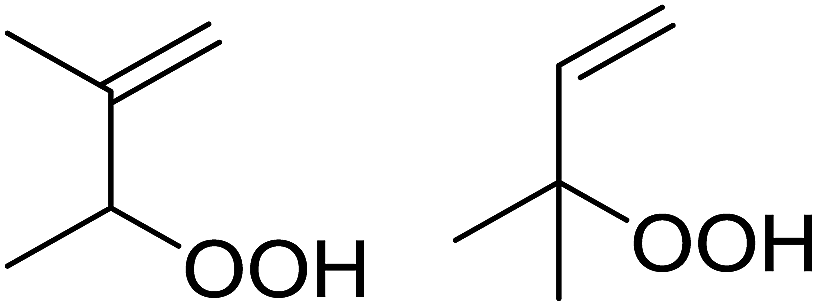 |
43% 57% |
| 3 |  |
44 |  |
68% |
| 4 |  |
16 |  |
30% 28% 8% |
| 5 | 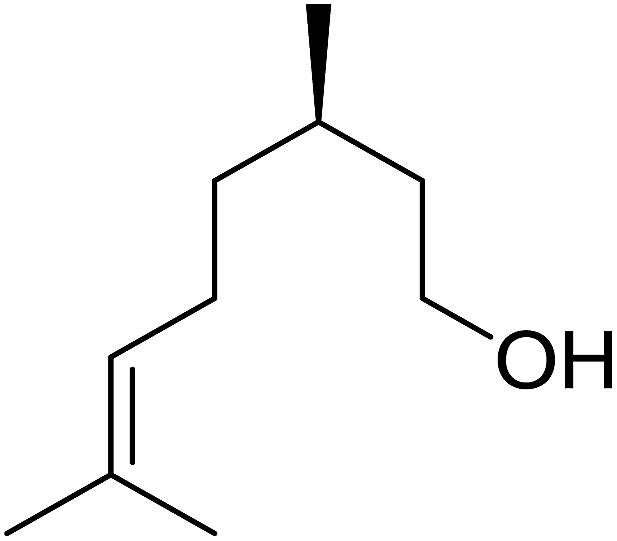 |
16 | 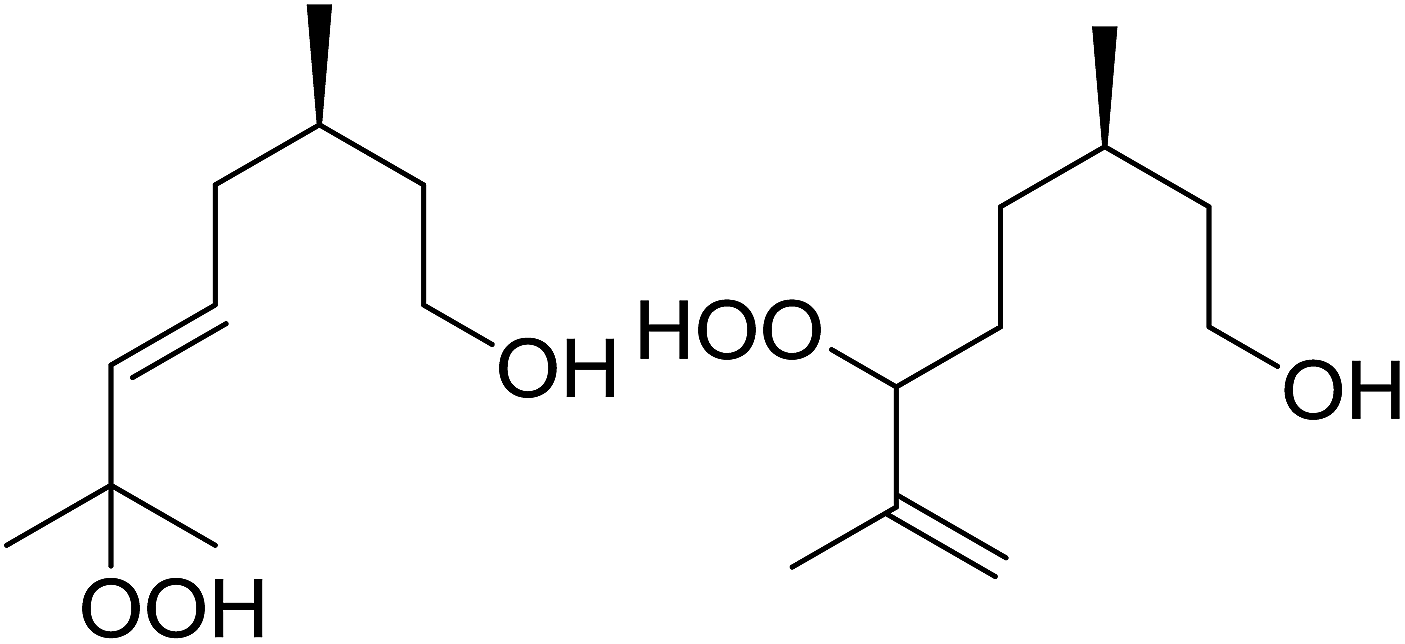 |
37% 19% |
Employing copper nitrates as the catalyst, Johnson et al. have developed a mild and efficient method for the hydroperoxidation of Meldrum's acid.64 The optimised reaction conditions proved tolerant to a variety of potentially vulnerable functional groups including alkenes, terminal and internal alkynes, arenes, tertiary benzylic C–H bonds and esters (Table 27, entries 1–12). In most cases, hydroperoxide products were obtained in over 90% purity after a simple aqueous work up. Both olefins (entries 10 and 11) and barbituric acids (entry 12) were transformed under these conditions.
|
||||
|---|---|---|---|---|
| Entry | Product | R | Time (h) | Yield |
| 1 | 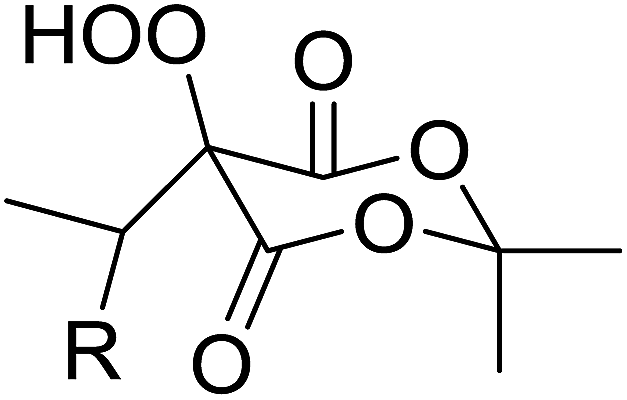 |
CH3 | 6 | 75% |
| 2 | H | 6 | 92% | |
| 3 | Ph | 2 | >95% | |
| 4 | 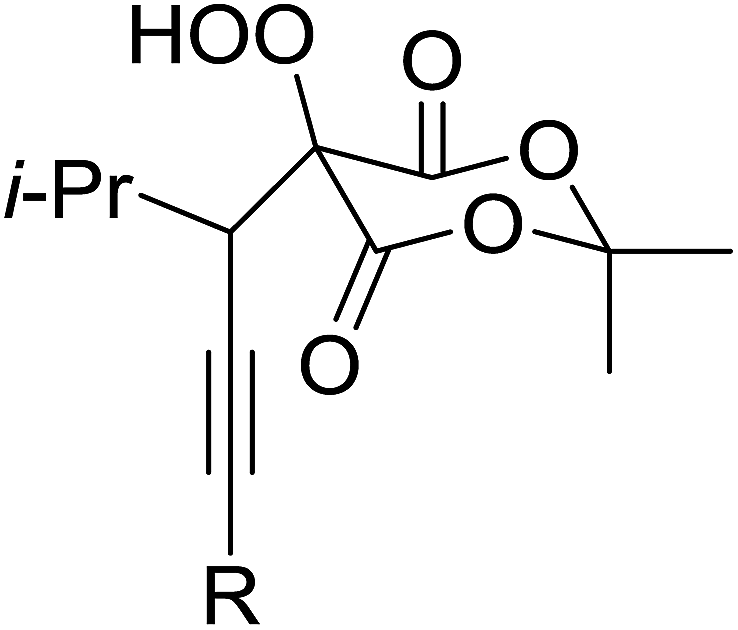 |
H | 2 | 80% |
| 5 | CH3 | 2 | 78% | |
| 6 | C5H11 | 2 | 80% | |
| 7 | C5H11 | 2 | 80% | |
| 8 | CO2Et | 3 | >95% | |
| 9 | Ph | 2 | >95% | |
| 10 | 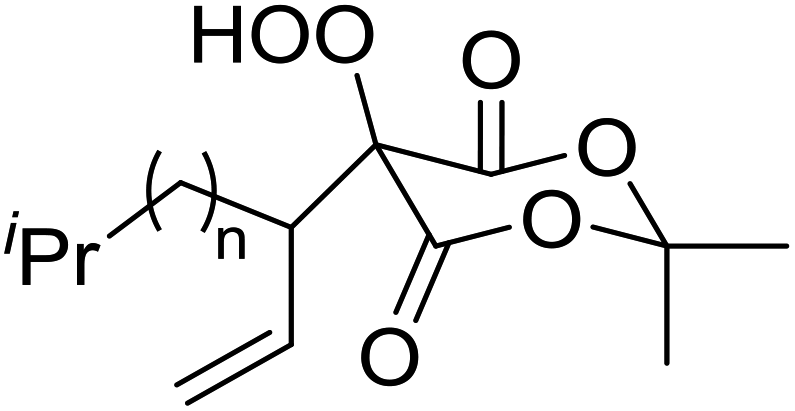 |
n = 0 | 4 | 59% |
| 11 | n = 1 | 4.5 | 86% | |
| 12 |  |
Ph | 8 | 49% |
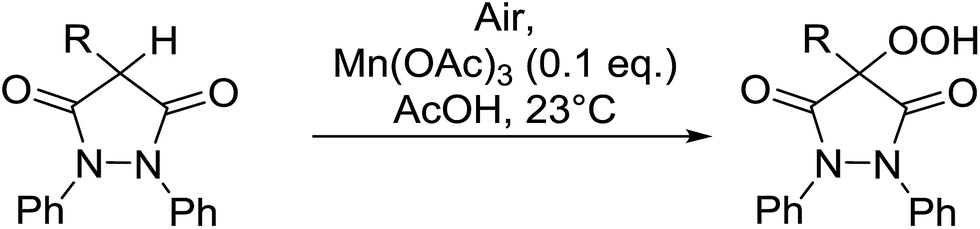
4-Hydroperoxy-1,2-diphenyl-pyrazolidine-3,5-diones are promising candidates as potential nonsteroidal anti-inflammatory agents.65 Nishino and co-workers have successfully demonstrated the aerobic oxidation of 1,2-diphenylpyrazolidine-3,5-diones in the presence of catalytic amount of manganese acetate.66 The introduction of the hydroperoxy group was accomplished for a range of different substrates affording the 4-hydroperoxy adducts in excellent yields (Table 28, entries 2–10). The presence of the catalyst proved crucial, as no product was obtained in its absence (entry 1). Increasing the reaction times or replacing manganese acetate with cerium ammonium nitrate was accompanied by the formation of significant amounts of the 4-hydroxylated by-products.
|
||||
|---|---|---|---|---|
| Entry | R | Catalyst | Time (h) | Yield |
| 1 | i-Pr | None | 3 | — |
| 2 | i-Pr | Mn(OAc)3 | 2 | 97% |
| 3 | Me | Mn(OAc)3 | 2 | 95% |
| 4 | Et | Mn(OAc)3 | 2 | 95% |
| 5 | Pr | Mn(OAc)3 | 2 | 99% |
| 6 | Bu | Mn(OAc)3 | 2 | 99% |
| 7 | t-Bu | Mn(OAc)3 | 10 min | 95% |
| 8 | Bn | Mn(OAc)3 | 2 | 97% |
| 9 | Cp | Mn(OAc)3 | 2 | 99% |
| 10 | i-Pr | Mn(OAc)3 | 12 | 87% |
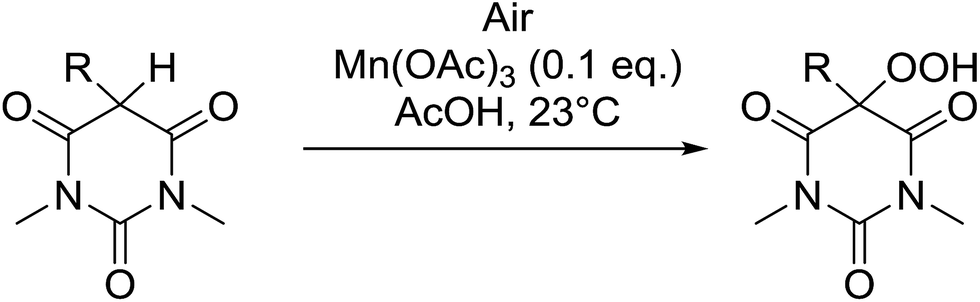
5-Monosubstituted barbituric acids were also amenable to oxidation under these conditions and the corresponding hydroperoxides were isolated in good to excellent yields (Table 29, entries 1–5).67 Prolonged reaction times or the use of cerium in place of manganese were again associated with formation of the alcohol by-product.
|
|||
|---|---|---|---|
| Entry | R | Time (h) | Yield (%) |
| 1 | Bn | 2 | 90% |
| 2 | i-Pr | 4 | 80% |
| 3 | 4-MeOC6H4CH2 | 4 | 94% |
| 4 | 2-MeOC6H4CH2 | 4 | 94% |
| 5 | 2-Napthyl–CH2 | 2 | 88% |
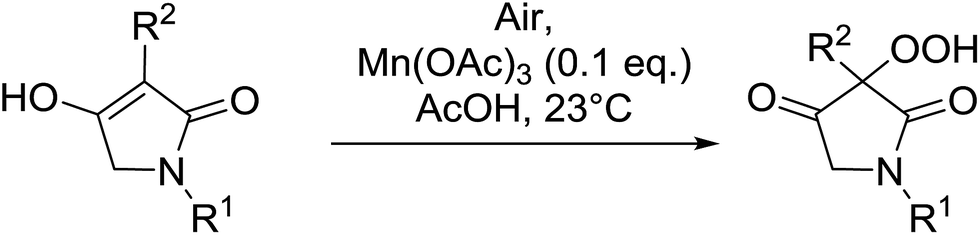
The same methodology was optimised for 3-substituted 2,4-pyrrolidinediones by Nishino and Haque.68 Various 3-alkyl-2,4-pyrrolidinediones were oxidised in the presence of a catalytic amount of manganese acetate, affording the target hydroperoxides in excellent yields (Table 30, entries 1–16). Although the hydroperoxides were mostly isolated as colourless solids, they were generally unstable and gradually decomposed at ambient temperature in air within 2 or 3 days.
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Time (h) | Yield (%) |
| 1 | Bn | Me | 2 | 94% |
| 2 | Bn | Et | 2 | 95% |
| 3 | Bn | Pr | 2 | 98% |
| 4 | Bn | Bu | 2 | 93% |
| 5 | Bn | n-C6H13 | 2 | 93% |
| 6 | Bu | Me | 1.5 | 91% |
| 7 | Bu | Et | 1.5 | 90% |
| 8 | Bu | Bu | 1.5 | 95% |
| 9 | t-Bu | Me | 1.5 | 90% |
| 10 | t-Bu | Et | 1.5 | 96% |
| 11 | t-Bu | Pr | 1.5 | 99% |
| 12 | t-Bu | Bu | 1.5 | 99% |
| 13 | t-Bu | n-C6H13 | 2 | 98% |
| 14 | i-Pr | Me | 1.5 | 95% |
| 15 | i-Pr | Et | 1.5 | 97% |
| 16 | i-Pr | Pr | 1.5 | 98% |
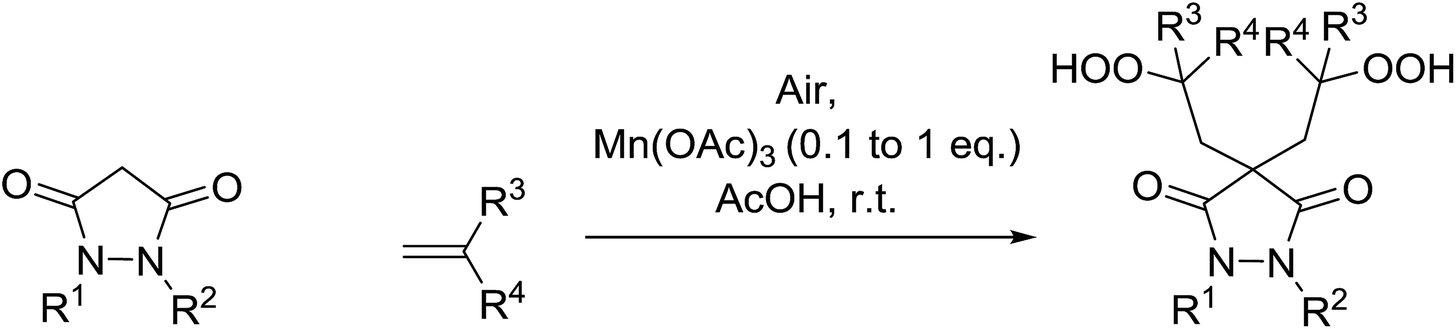
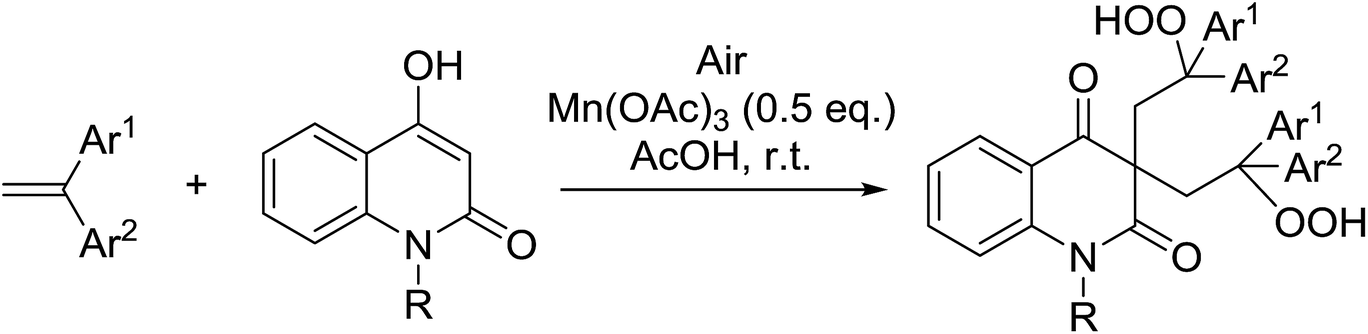
In parallel work, Nishino demonstrated the aerobic oxidation of a two component mixture containing 1,2-disubstituted pyrrazolidine-3,5-diones and alkenes in the presence of catalytic amount of Mn(III) acetate, producing 4,4-bis(2-hydroperoxyalkyl)pyrrazolidine-3,5-diones in good to excellent yields (Table 31).66 When 1,2-diphenyl-3,5-pyrazolidinedione was the substrate, the highest yields were recorded with 0.1 equivalents of the catalyst (entries 1–11). By contrast, when the corresponding 1,2-phenylbenzyl-pyrazolidine-3,5-diones (entries 12–14) or 1,2-bisbenzyl-pyrazolidine-3,5-diones (entries 16–18) were subjected to the same conditions, higher loadings of the catalyst were required for reaction completion.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | R4 | Dione:alkene:cat |
Time | Yield |
| 1 | Ph | Ph | Ph | Ph | 1:1:1 |
12 | 67% |
| 2 | Ph | Ph | Ph | Ph | 1:1:1 |
1 | 80% |
| 3 | Ph | Ph | Ph | Ph | 1:2:0.2 |
3 | 85% |
| 4 | Ph | Ph | Ph | Ph | 1:1:0.1 |
2 | 96% |
| 5 | Ph | Ph | 4-MeC6H4 | 4-MeC6H4 | 1:1:0.1 |
5 | 80% |
| 6 | Ph | Ph | 4-FC6H4 | 4-FC6H4 | 1:1:0.1 |
3 | 95% |
| 7 | Ph | Ph | 4-ClC6H4 | 4-ClC6H4 | 1:1:0.1 |
2 | 98% |
| 8 | Ph | Ph | Me | Me | 1:excess:0.1 |
2 | 91% |
| 9 | Ph | Ph | Et | Et | 1:1:0.1 |
5 | 95% |
| 10 | Ph | Ph | Me | Ph | 1:1:0.1 |
3 | 95% |
| 11 | Ph | Ph | 4-MeC6H4 | Ph | 1:1:0.1 |
14 | 75% |
| 12 | Bn | Ph | Ph | Ph | 1:2:0.2 |
3 | 85% |
| 13 | Bn | Ph | 4-FC6H4 | 4-FC6H4 | 1:2:0.2 |
2 | 90% |
| 14 | Bn | Ph | 4-ClC6H4 | 4-ClC6H4 | 1:2:0.2 |
4 | 87% |
| 15 | –(CH2–)4 | Ph | Ph | 1:2:0.2 |
13 | 95% | |
| 16 | Bn | Bn | Ph | Ph | 1:1:1 |
15 | 93% |
| 17 | Bn | Bn | 4-MeC6H4 | 4-MeC6H4 | 1:1:1 |
10 | 78% |
| 18 | Bn | Bn | 4-FC6H4 | 4-FC6H4 | 1:1:1 |
1 | 98% |
Nishino subsequently expanded this approach to include quinolinone substrates.69 An optimised ratio of 1:2:0.5 of alkene:quinolinone:catalyst afforded the target bisperoxides in moderate to excellent yields (Table 32, entries 1–7). Only trace amounts of the target bisperoxides were recovered when unprotected quinolinones were tested (entry 8).
|
|||||
|---|---|---|---|---|---|
| Entry | Ar1 | Ar2 | R | Time (h) | Yield |
| 1 | Ph | Ph | Me | 4 | 71% |
| 2 | 4-ClC6H4 | 4-ClC6H4 | Me | 12 | 76% |
| 3 | 4-MeC6H4 | 4-MeC6H4 | Me | 4 | 59% |
| 4 | Ph | Ph | Et | 15 | 91% |
| 5 | Ph | Ph | Bn | 12 | 52% |
| 6 | 4-ClC6H4 | 4-ClC6H4 | Bn | 24 | 48% |
| 7 | 4-MeC6H4 | 4-MeC6H4 | Bn | 4 | 44% |
| 8 | Ph | Ph | H | 4 | Trace |
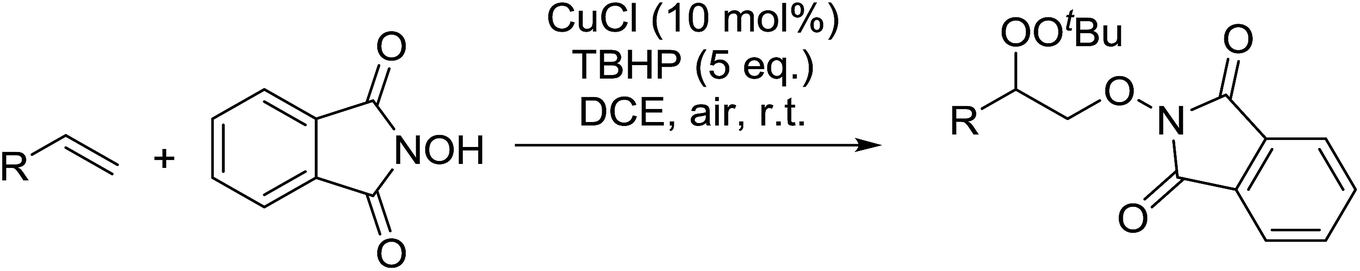
Xia et al. have described an efficient copper-catalysed dioxygenation of olefins under mild reaction conditions.70 An initial catalyst screen revealed that addition of copper(I) chloride to a mixture of styrene, N-hydroxyphthalimide and TBHP furnished the target peroxides in good yields (Table 33). By contrast, when para-toluenesulfonic acid was substituted for CuCl, the corresponding hydroxylated compound was instead isolated. Under the optimised conditions, both electron rich (entries 1 and 2) and electron poor (entries 3–6) substrates performed well irrespective of the substituent position (entries 4–6).
|
||
|---|---|---|
| Entry | R | Yield |
| 1 | Ph | 62% |
| 2 | 4-MeC6H4 | 71% |
| 3 | 4-FC6H4 | 63% |
| 4 | 4-ClC6H4 | 53% |
| 5 | 3-BrC6H4 | 52% |
| 6 | 2-ClC6H4 | 71% |
| 7 | 2-Naphthyl | 44% |
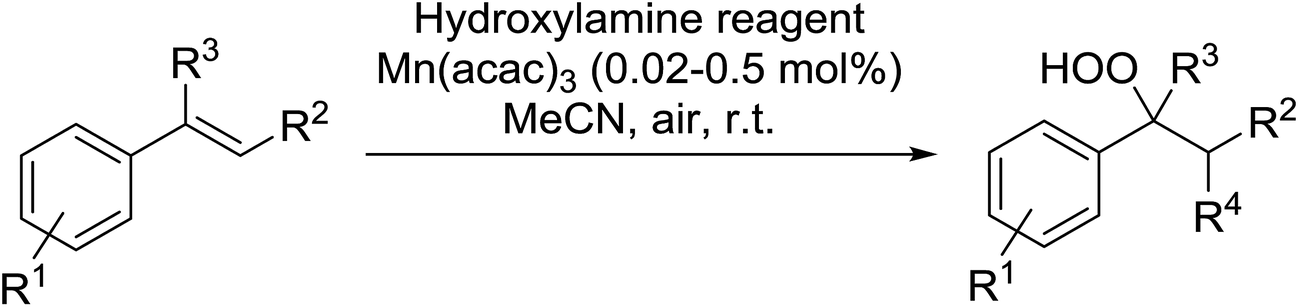
Recently Makino et al. have developed a manganese-catalysed hydroperoxidation method for conjugated alkenes using various hydroxylamine derivatives.71 Reaction of several styrenes with N-hydroxyphthalimide (NHPI) in the presence of 0.02 mol% Mn(acac)2 in air at room temperature furnished the desired hydroperoxides (Table 34, entries 1–10). Yields were highest with styrenes bearing electron-withdrawing groups (entries 4–6), whereas electron-donating substituents returned lower yields (entry 7). More sterically hindered α- and β-substituted substrates were also compatible with these conditions (entries 8–10). This methodology was equally successful when other hydroxylamine reagents were employed, including both N-hydroxybenzotriazole (HOBt) (entries 11–14) or N-hydroxysuccinimide (NHS) (entries 15–18).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Reagent | R4 | Time (h) | Yield (%) |
| 1 | H | H | H | NHPI | –ONPhth | 24 | 64% |
| 2 | 4-Me | H | H | NHPI | –ONPhth | 24 | 96% |
| 3 | 2-Me | H | H | NHPI | –ONPhth | 24 | >99% |
| 4 | 4-F | H | H | NHPI | –ONPhth | 24 | 91% |
| 5 | 4-Cl | H | H | NHPI | –ONPhth | 24 | 94% |
| 6 | 4-Br | H | H | NHPI | –ONPhth | 24 | 88% |
| 7 | 4-MeO | H | H | NHPI | –ONPhth | 48 | 60% |
| 8 | H | H | Me | NHPI | –ONPhth | 24 | >99% |
| 9 | H | Me | H | NHPI | –ONPhth | 36 | 69% |
| 10 | H | Ph | H | NHPI | –ONPhth | 24 | 80% |
| 11 | H | H | H | HOBt | –OBt | 18 | 90% |
| 12 | t-Bu | H | H | HOBt | –OBt | 21 | 52% |
| 13 | H | Me | H | HOBt | –OBt | 20 | 89% |
| 14 | H | H | Me | HOBt | –OBt | 4 | 96% |
| 15 | H | H | H | NHS | –OSu | 24 | 80% |
| 16 | t-Bu | H | H | NHS | –OSu | 24 | 69% |
| 17 | H | Me | H | NHS | –OSu | 48 | 67% |
| 18 | H | H | Me | NHS | –OSu | 28 | 92% |
2.4 Peroxidation of aldehydes and ketones
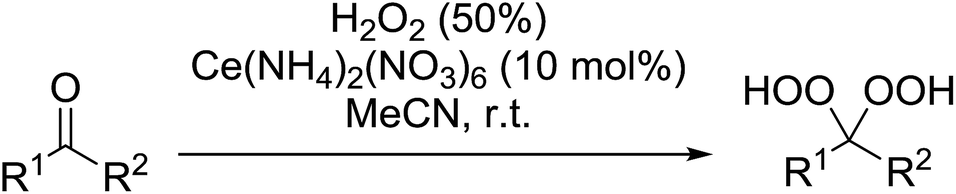
A large number of methods have been developed for the peroxidation of carbonyl-containing substrates. Das et al. found cerium ammonium nitrate (CAN) to be an effective catalyst for the synthesis of gem-dihydroperoxides from ketones and aldehydes.72 CAN acts as a Lewis acid and increases the electrophilic character of the carbonyl. Both acyclic and cyclic aliphatic ketones were transformed in excellent yields (Table 35, entries 1–8). Reactions involving aromatic ketones were generally lower yielding (entries 9–12) while electron-withdrawing substitutents, in particular, were found to have a detrimental effect (entry 12). Under these conditions, aldehydes could likewise be converted to the corresponding gem-dihydroperoxides (entries 13–14). The presence of an electron-withdrawing groups again proved problematic and hydroperoxidation of 4-nitrobenzaldehyde was not successful.
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Time (h) | Yield |
| 1 | Me | n-Nonyl | 1 | 96% |
| 2 | Me | n-Heptyl | 1 | 95% |
| 3 | Me | n-Bu | 2 | 96% |
| 4 | Et | Et | 3 | 89% |
| 5 | Me | iso-Bu | 2 | 90% |
| 6 | Cyclopentanone | 3 | 85% | |
| 7 | Cyclohexanone | 2 | 87% | |
| 8 | Cycloheptanone | 2 | 86% | |
| 9 | Me | Ph | 2 | 48% |
| 10 | Me | 4-MeOC4H6 | 2 | 60% |
| 11 | Me | 4-ClC6H4 | 2 | 62% |
| 12 | Me | 4-NO2C6H4 | 4 | 25% |
| 13 | H | Ph | 4 | 51% |
| 14 | H | 4-MeOC4H6 | 4 | 74% |
In a similar vein, Khosravi used cesium nitrate as a highly efficient, commercially available and non-toxic catalyst for conversion of aldehydes or ketones to the corresponding gem-dihydroperoxides (Table 36).73 In general, cesium-catalysed peroxidations involving aliphatic ketones (entries 7–9) were faster and higher yielding than for aromatic ketones (entries 1–5), while benzophenone did not react under these conditions (entry 6). The peroxidation of aromatic aldehydes was an equally high yielding process (entries 10–14) although aliphatic aldehydes afforded exclusively 1,1-hydroxyhydroperoxides (entries 15 and 16).
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Time (min) | Yield |
| 1 | Ph | Me | OOH | 200 | 67% |
| 2 | 4-MeC6H4 | Me | OOH | 200 | 72% |
| 3 | 4-ClC6H4 | Me | OOH | 205 | 75% |
| 4 | 4-MeOC6H4 | Me | OOH | 190 | 74% |
| 5 | Napthyl | Ph | OOH | 90 | 85% |
| 6 | Ph | Ph | OOH | 300 | — |
| 7 | Et | Et | OOH | 22 | 94% |
| 8 | Et | n-Heptyl | OOH | 19 | 93% |
| 9 | Me | n-Nonyl | OOH | 21 | 96% |
| 10 | Ph | H | OOH | 80 | 81% |
| 11 | 4-ClC6H4 | H | OOH | 90 | 81% |
| 12 | 4-NCC6H4 | H | OOH | 240 | 60% |
| 13 | 4-MeOC6H4 | H | OOH | 80 | 88% |
| 14 | Thienyl | H | OOH | 120 | 81% |
| 15 | n-Heptyl | H | OH | 110 | 90% |
| 16 | Ph(CH2)2 | H | OH | 90 | 92% |
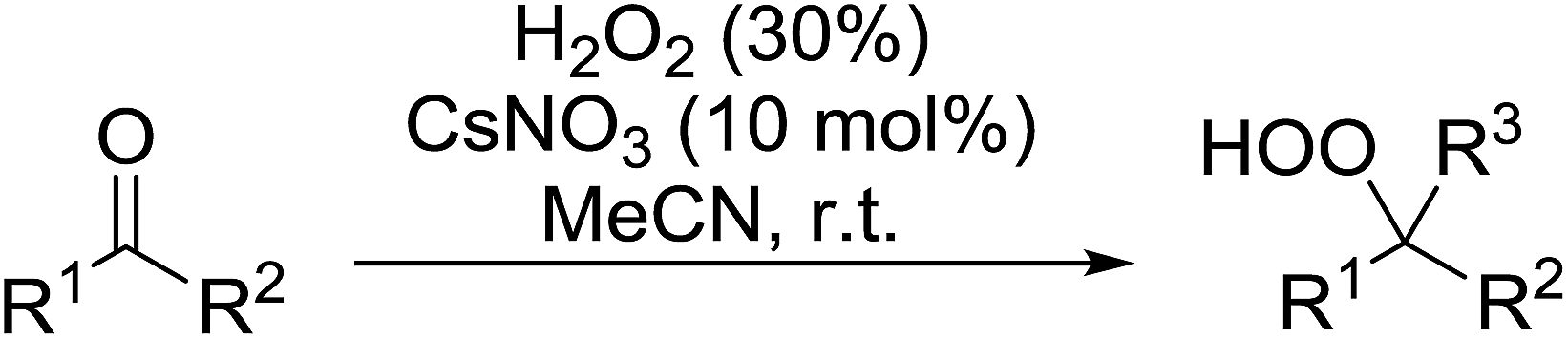
Tin chloride dihydrate was also found to be an efficient catalyst for the synthesis of gem-dihydroperoxides from ketones and aldehydes (Table 37).74 This catalyst system was compatible with acyclic (entries 1–3) and cyclic (entries 4–6) aliphatic ketones in addition to aromatic ketones (entries 7 and 8). The transformation of aromatic aldehydes was similarly successful in yields of 50% to 75% (entries 9–13). Aliphatic aldehydes were converted to 1,1-hydroxyhydroperoxides with this system (entries 14 and 15).
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Time (h) | Yield |
| 1 | Me | n-Heptyl | OOH | 5 | 95% |
| 2 | Et | Et | OOH | 4 | 93% |
| 3 | Me | i-Bu | OOH | 4 | 92% |
| 4 | Cyclohexanone | OOH | 5 | 90% | |
| 5 | Cyclopentanone | OOH | 4 | 94% | |
| 6 | Cycloheptanone | OOH | 4 | 90% | |
| 7 | Ph | Me | OOH | 11 | 45% |
| 8 | 4-MeOC6H4 | Me | OOH | 9 | 62% |
| 9 | Ph | H | OOH | 12 | 50% |
| 10 | 4-MeC6H4 | H | OOH | 12 | 52% |
| 11 | 4-ClC6H4 | H | OOH | 10 | 75% |
| 12 | 4-MeOC6H4 | H | OOH | 10 | 60% |
| 13 | 4-NCC6H4 | H | OOH | 11 | 70% |
| 14 | n-Heptyl | H | OH | 5 | 90% |
| 15 | –CH2Bn | H | OH | 6 | 88% |
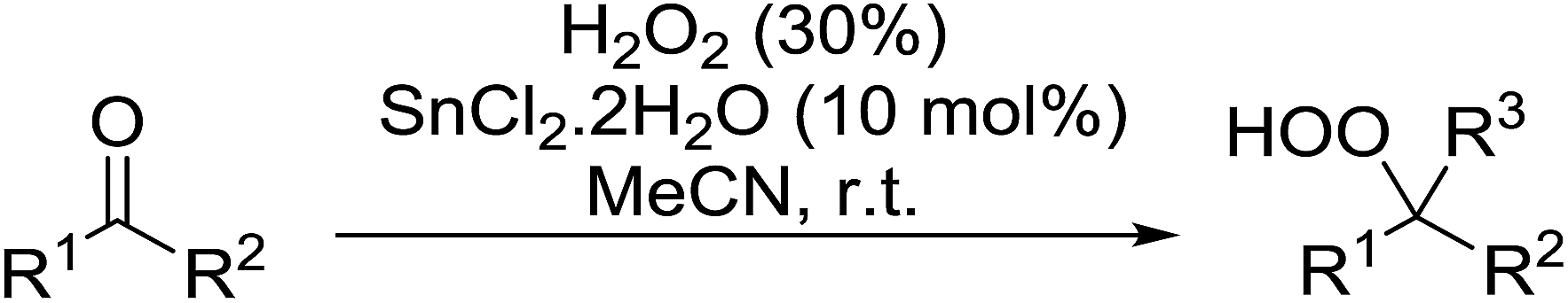
In subsequent work, Azarifar and Khosravi demonstrated that aluminium chloride hexahydrate is a superior catalyst for such transformations, often affording higher yields and reduced reaction times in comparison to tin chloride trihydrate (Table 38).75 As with the tin-catalyst system, aliphatic aldehydes were converted to 1,1-hydroxyhydroperoxides rather than gem-dihydroperoxides (entries 18 and 19).
| Entry | R1 | R2 | R3 | Time (h) | Yield |
|---|---|---|---|---|---|
| 1 | Me | n-Heptyl | OOH | 4 | 97% |
| 2 | Et | Et | OOH | 3 | 96% |
| 3 | Me | i-Bu | OOH | 3.5 | 95% |
| 4 | Cyclohexanone | OOH | 3 | 98% | |
| 5 | Cyclopentanone | OOH | 3 | 97% | |
| 6 | Cycloheptanone | OOH | 3 | 93% | |
| 7 | Ph | Me | OOH | 8 | 56% |
| 8 | 4-MeOC6H4 | Me | OOH | 7.5 | 82% |
| 9 | 2-Naphthyl | Me | OOH | 4 | 95% |
| 10 | Ph | H | OOH | 8 | 67% |
| 11 | 4-MeC6H4 | H | OOH | 10 | 64% |
| 12 | 4-BrC6H4 | H | OOH | 9 | 85% |
| 13 | 4-ClC6H4 | H | OOH | 8 | 80% |
| 14 | 4-FC6H4 | H | OOH | 11 | 84% |
| 15 | 4-MeOC6H4 | H | OOH | 8.5 | 78% |
| 16 | 2-MeOC6H4 | H | OOH | 10 | 75% |
| 17 | 4-NCC6H4 | H | OOH | 10 | 82% |
| 18 | n-Heptyl | H | OH | 4 | 93% |
| 19 | –CH2Bn | H | OH | 5 | 95% |
Rhenium(VII) oxide has been shown to be a mild and efficient catalyst for the peroxyacetalisation of cyclic ketones (Table 39).76 The choice of solvent is important as the reaction proceeds with greater speed and higher yields in acetonitrile than dichloromethane. Dihydroperoxides are produced in high yields from the six- and seven-membered substrates in less than an hour (entries 1–3). Increasing the ring size is accompanied by a decrease in reactivity and extended reaction times (entries 4 and 5).
|
||||
|---|---|---|---|---|
| Entry | n | R | Time (h) | Yield |
| 1 | 1 | H | 0.5 | 94% |
| 2 | 1 | Me | 0.5 | 89% |
| 3 | 2 | H | 1 | 71% |
| 4 | 3 | H | 3 | 96% |
| 5 | 7 | H | 20 | 67% |
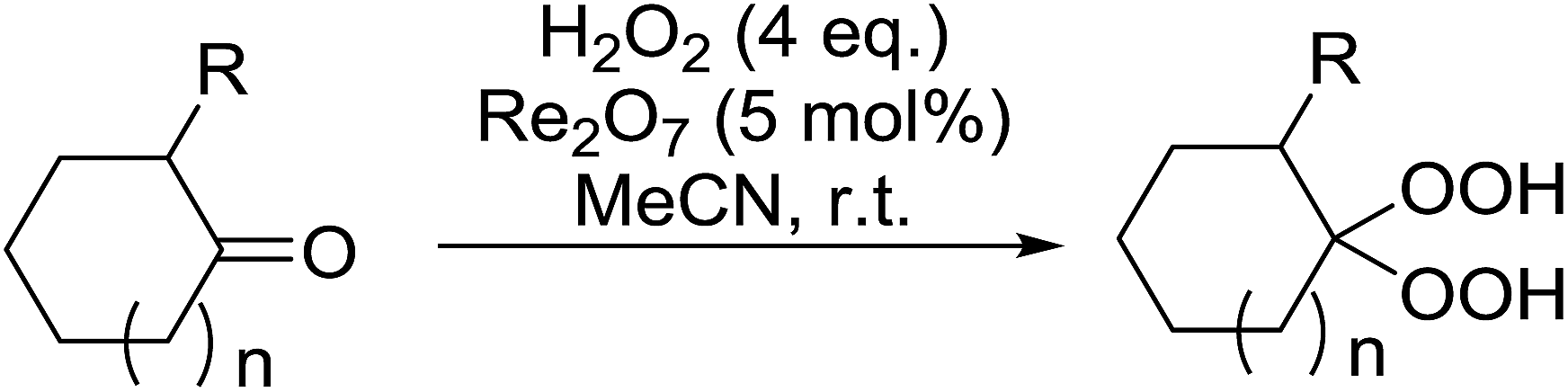
While the preparation of acyclic gem-dihydroperoxides has often proved challenging due to the tendency of the intermediate perhydrates to undergo heterolytic fragmentation, the authors successfully expanded their methodology to encompass both aliphatic and aromatic aldehydes and ketones (Table 40). Aromatic aldehydes and ketones required longer reaction times (entries 4–9). In some cases, lower yields were obtained due to the oligomerisation of the electron-rich products (entries 5 and 9).
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Time (h) | Yield |
| 1 | CH3(CH2)4 | Me | 0.5 | 94% |
| 2 | Ph(CH2)2 | Me | 1 | 89% |
| 3 | Bu | Bu | 1 | 71% |
| 4 | Ph | H | 6 | 96% |
| 5 | 4-MeOC6H4 | H | 2 | 67% |
| 6 | 2-MeOC6H4 | H | 5 | 94% |
| 7 | 3-MeOC6H4 | H | 5 | 92% |
| 8 | 4-ClC6H4 | H | 5 | 96% |
| 9 | Ph | Me | 24 | 69% |
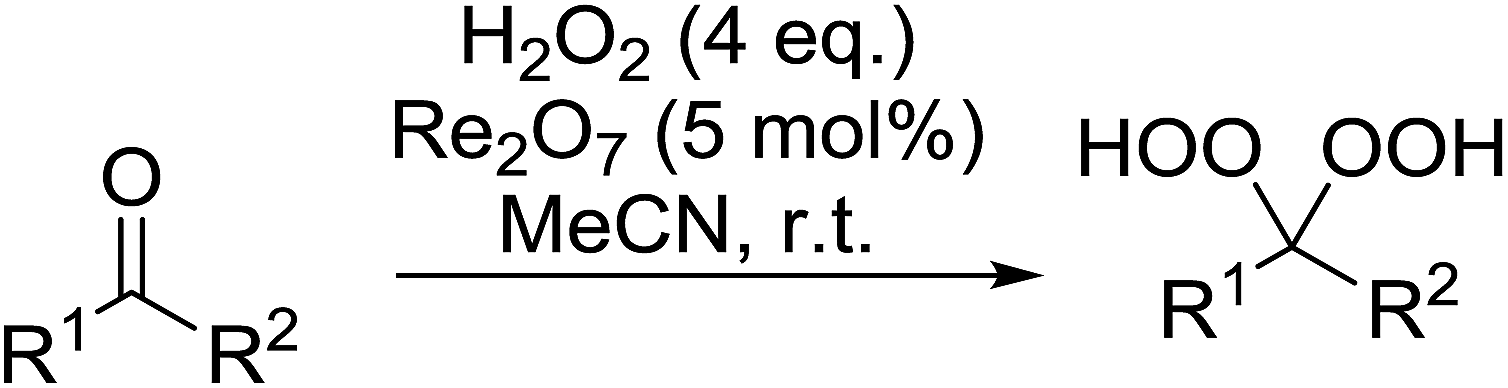
The transformation of gem-dihydroperoxides into alkylhydroperoxides has been outlined by Dussault (Table 41).77 This methodology relies on the observation that bis-hydroperoxyacetals derived from bulky alicyclic or strained cyclic frameworks undergo hydrolysis more rapidly than those based on a strain-free backbone.78 Two different methods were employed in the alkylation of gem-dihydroperoxides. Bisalkylation with primary alkyl iodides or bromides was achieved in good yields using silver oxide (entries 1–4). While no alkylation of the gem-dihydroperoxides was observed with secondary alkyl iodides, the use of primary or secondary triflates and strong base furnished the target compounds in yields of 42–68% (entries 5–7). Subsequent acid hydrolysis furnished the corresponding hydroperoxides in a highly pure form and in moderate to good yields ranging from 48% to 78% (entries 1–7). In related work, Hamada et al. have exploited the silver-mediated alkylation of gem-dihydroperoxides to prepare a library of potential anti-malarial agents.79
|
|||||
|---|---|---|---|---|---|
| Entry | Method | R | Leaving group | Yield 60 | Yield 61 |
| 1 | a | n-C10H21– | –I | 93% | 78% |
| 2 | a | n-C6H13– | –I | 79% | 49% |
| 3 | a | Bn– | –Br | 56% | 60% |
| 4 | a | CH2CH(CH2)8– |
–I | 79% | 62% |
| 5 | b | Ph(CH2)4– | –OTf | 68% | 79% |
| 6 | b | (Me)(n-Hex)CH– | –OTf | 53% | 48% |
| 7 | b | (Me)(n-Dec)CH– | –OTf | 42% | 63% |
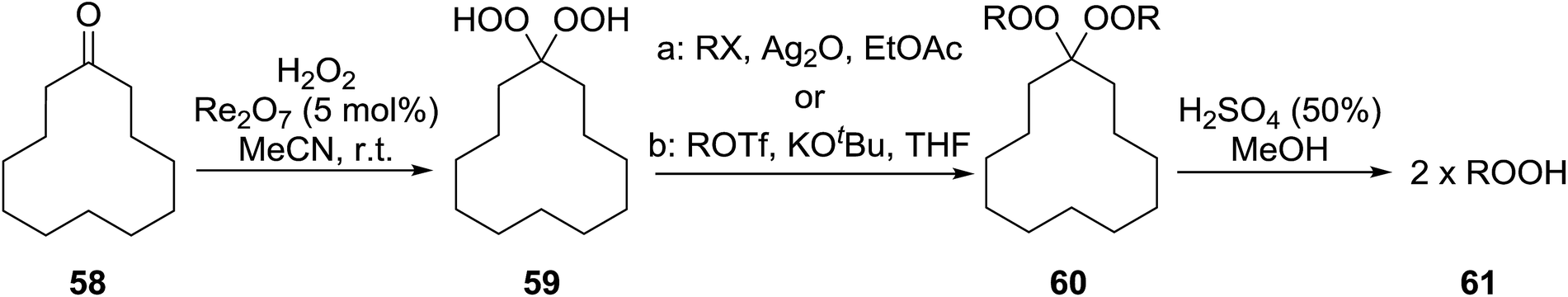
Sung and co-workers have reported the titanium-catalysed addition of TBHP and an alcohol to aldehydes to generate a range of peroxyacetals (Table 42).80 Sterically hindered alcohols such as tert-butyl alcohol resulted in lower yields (entry 4) in comparison to primary and secondary alcohols (entries 1 & 2). Electron withdrawing substituents on the aryl-derived aldehydes gave the peroxyacetal in high yields (entries 7 & 8) whereas electron-donating substituents only provided the target compound when very dry solvent and activated 4 Å molecular sieves were employed (entry 10). This approach is not compatible with ketone substrates as formation of the hemiketal was found to be thermodynamically unfavourable.
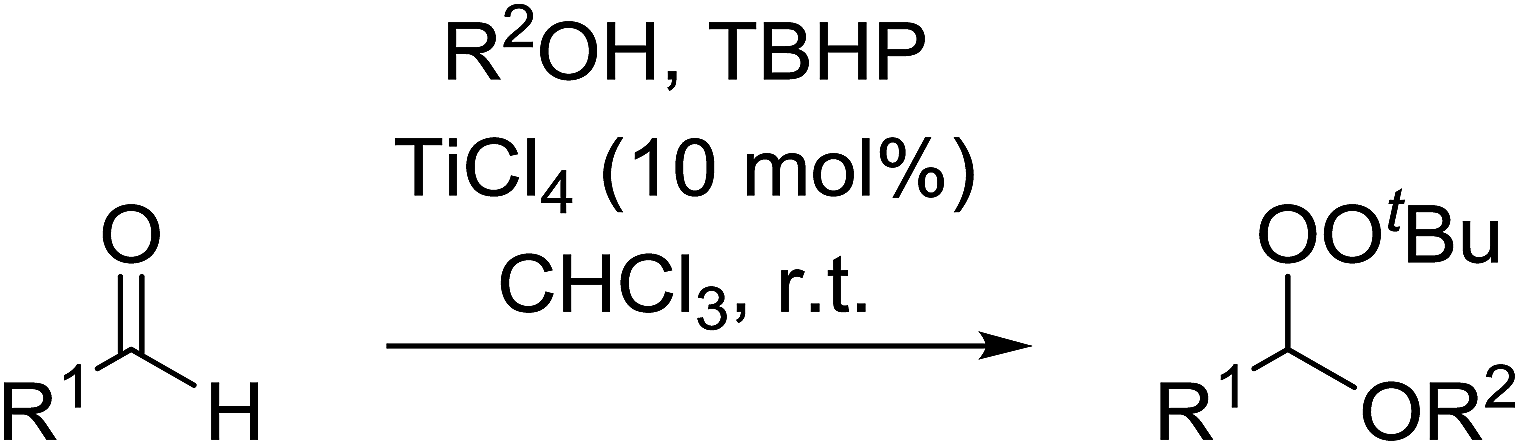
A facile construction of 6-carbomethyloxymethyl-3-methoxy-1,2-dioxane, a central motif in anti-malarial peroxides, has been outlined by Kobayashi and co-workers.81 Key to this approach was a Sc(OTf)3-mediated peroxyhemiacetalisation of an aliphatic ketone (Scheme 11). The nature of the acid greatly affects the formation of the peroxyhemiacetal, with a yield of only 10% observed when Yb(OTf)3 was used. This increased to 83% with Sc(OTf)3. Decreasing the concentration of the hydrogen peroxide–urea adduct from 20 equivalents to 7.5 equivalents saw an increase in product formation from 57% to 83% with minimal side product formation. The high yield may be rationalised by assuming that coordination of the lanthanoid salt to the product likely prevents unwanted acetalisation or an intramolecular Michael addition.
 | ||
| Scheme 11 | ||
Ghorai and co-workers have recently published a protocol for the synthesis of α-azido peroxides, with the azide acting as a useful precursor for the formation of the corresponding primary amines, nitrenes and isocyanates.82 They developed a one-pot, chemoselective methodology for the synthesis of α-azido peroxides from a range of aldehydes utilising trimethylsilyl-azide, tert-butyl hydroperoxide and iron trichloride as the catalyst (Table 43). Aromatic aldehydes with a broad substitution pattern were tolerated although lower yields were recorded with substrates bearing electron-donating substituents (entries 5 and 7) due to their propensity to undergo a competing Baeyer–Villiger rearrangement. The methodology was expanded to include aliphatic aldehydes (entries 11–14) in addition to both cyclic and acyclic ketones (entries 15–19). The authors further demonstrated that secondary α-azido peroxides underwent asymmetric decomposition in the presence of hydroquinine 2,5-diphenyl-4,6-pyrimidinediyl diether [(DHQ)2Pyr] to give enantioenriched α-azido-peroxides in good to excellent enantiomeric excesses (entries 6–10).
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Yield 65 | Kinetic resolution | ||
| t (h) | Yield 66 | ee | ||||
| 1 | Ph | H | 91% | — | — | — |
| 2 | 4-Cl–C6H4 | H | 89% | — | — | — |
| 3 | 2-F–C6H4 | H | 55% | — | — | — |
| 4 | 4-CF3–C6H4 | H | 90% | — | — | — |
| 5 | 4-MeO–C6H4 | H | 58% | — | — | — |
| 6 | 4-Me–C6H4 | H | 87% | 6.5 | 42% | 55% |
| 7 | 4-BnO–C6H4 | H | 52% | 11 | 30% | 82% |
| 8 | 3-Cyclopentyl–O–C6H4 | H | 68% | 14 | 35% | 96% |
| 9 | Napthyl | H | 88% | 36 | 32% | 93% |
| 10 | 4-Me–naphthyl | H | 50% | 26 | 30% | 87% |
| 11 | Bn | H | 80% | — | — | — |
| 12 | BnCH2 | H | 89% | — | — | — |
| 13 | n-Hex | H | 85% | — | — | — |
| 14 | Cyclohexyl | H | 84% | — | — | — |
| 15 |  |
90% | — | — | — | |
| 16 | 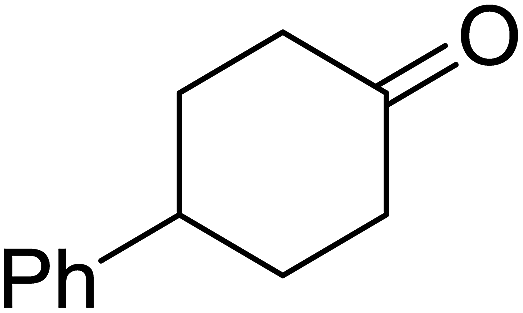 |
85% | — | — | — | |
| 17 | 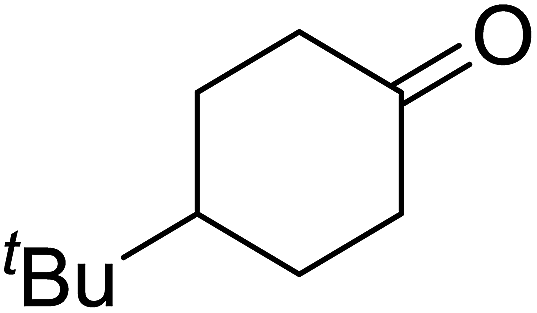 |
84% | — | — | — | |
| 18 | 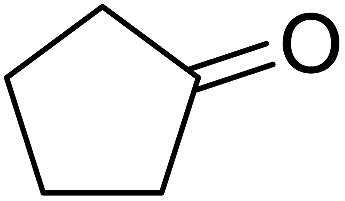 |
60% | — | — | — | |
| 19 | 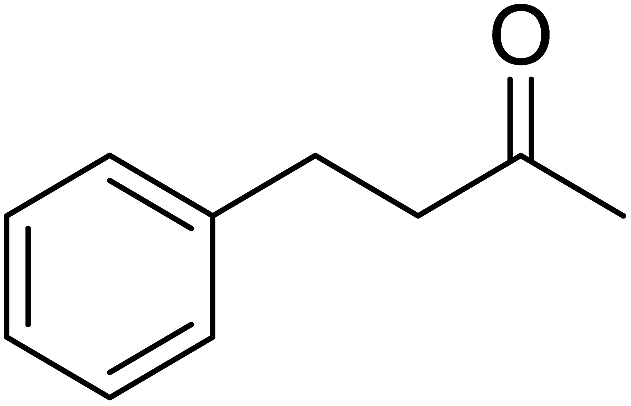 |
51% | — | — | — | |
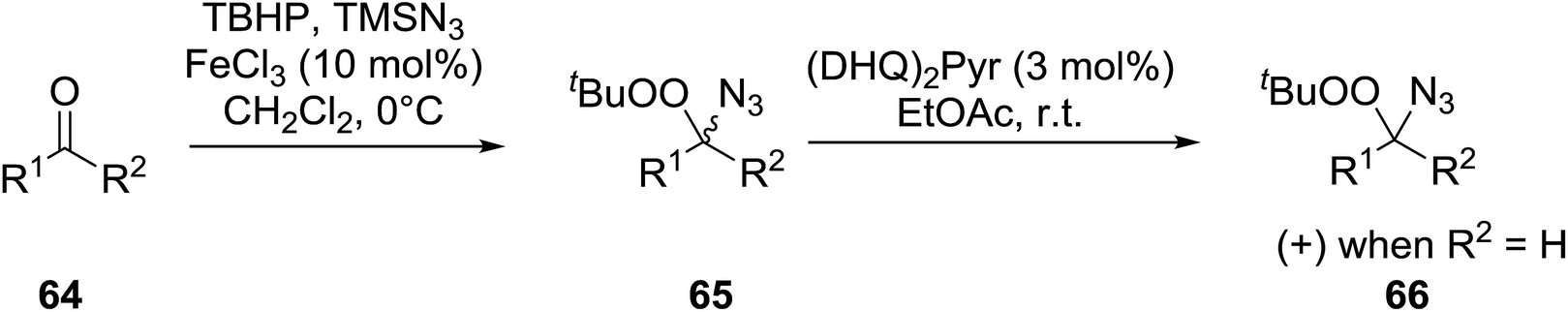
2.5 Peroxidation of phenols and alcohols
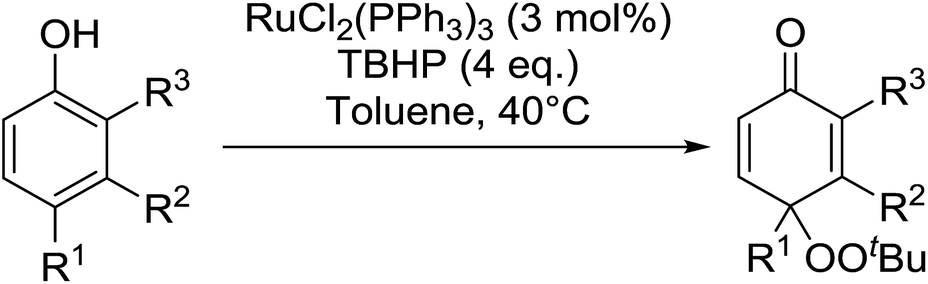
In 1996, Murahashi reported the first example of a selective transformation of phenols to 4-(tert-butylperoxy)cyclohexadienones without the prior requirement for substitution at the 2- or 6-position.83 He demonstrated that TBHP in combination with a low valent ruthenium catalyst, such as RuCl2(PPh3)3, could effectively mimic the enzymatic function of cytochrome P-450. Murahashi expanded the scope of this chemistry to include a wide range of phenolic substrates (Table 44).84 The reaction proceeds chemoselectively even in the presence of easily oxidisable isopropyl and benzyl groups (entries 2 and 5). Both 3,4-disubstituted (entries 7 and 8) and 2,4-disubstituted phenols reacted exclusively at the 4-position. The importance of the choice of metal catalyst is illustrated by entry 11, with peroxydienone formed in 54% yield in the presence of RuCl2(PPh3)3. By contrast, allylic oxidation is the preferred pathway using PdCl2/H2O2.
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Yield |
| 1 | Me | H | H | 85% |
| 2 | i-Pr | H | H | 86% |
| 3 | tBu | H | H | 68% |
| 4 | Ph | H | H | 76% |
| 5 | Bn | H | H | 78% |
| 6 | CH2COOCH3 | H | H | 82% |
| 7 | Me | Me | H | 77% |
| 8 | i-Pr | Me | H | 92% |
| 9 | Bn | H | Cl | 91% |
| 10 | Me | H | COMe | 71% |
| 11 | Allyl | H | OMe | 54% |
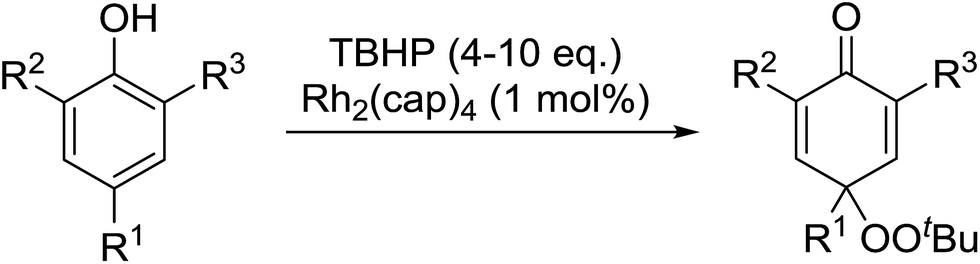
In parallel work, Doyle discovered that dirhodium caprolactamate [Rh2(cap)4] is an efficient catalyst for the generation of tert-butylperoxy radicals, facilitating the peroxidation of para-substituted phenols with TBHP (Table 45).85 Importantly, replacing ruthenium with rhodium allows for the use of aqueous, rather than anhydrous, TBHP. Preliminary screening studies identified a significant rate enhancement in chlorinated solvents and that other metal-containing catalysts (e.g. copper) were inferior to rhodium-based catalysts. A range of para-substituted phenols were transformed to the expected peroxydienones under these optimised conditions (entries 1–11). The use of ten equivalents of TBHP instead of four afforded higher yields of the expected product, most likely a result of the higher flux of tert-butylperoxy radicals (entries 1 vs. 2, 4 vs. 5, 6 vs. 7). The reaction displayed high regioselectivity for the para-position, although increasingly bulky substitutents were accompanied by a fall in yield due to competing oxidation at the ortho-position (entries 8 and 9). The authors propose that the reaction proceeds by abstraction of the phenolic hydrogen by a tert-butylperoxy radical, followed by trapping of the phenolic radical by another tert-butylperoxy radical.
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | eq. TBHP | Yield |
| 1 | Me | t-Bu | t-Bu | 4 | 99% |
| 2 | Me | t-Bu | t-Bu | 10 | 82% |
| 3 | MeO2CH2 | H | H | 10 | 87% |
| 4 | Bn | H | H | 4 | 72% |
| 5 | Bn | H | H | 10 | 91% |
| 6 | Me | OMe | H | 4 | 71% |
| 7 | Me | OMe | H | 10 | 90% |
| 8 | i-Pr | H | H | 4 | 35% |
| 9 | i-Pr | H | H | 10 | 58% |
| 10 | Ph | H | H | 8 | 57% |
| 11 | n-BuO2C | H | H | 10 | 66% |
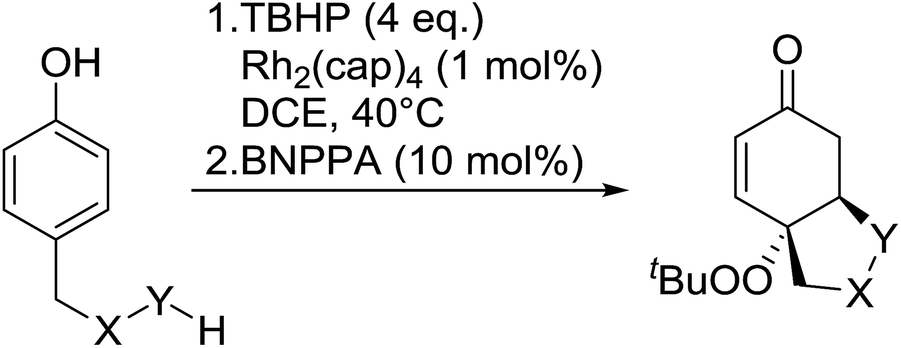
The synthetic utility of this methodology was further illustrated in the development of a one-pot, intramolecular Michael reaction to substituted oxa- and aza-Michael heterocycles.86 Phenols bearing carboxylic acids, alcohols and boc-protected amines at the 4-position were transformed into the corresponding bicyclics in overall yield ranging from 40% to 62% (Table 46). Following initial peroxidation, activation of the intermediate dienone by a chiral phosphoric acid allowed for stereoselective oxo- or aza-Michael additions. The addition of 1,1′-binaphthyl-2,2′-diyl hydrogen phosphate (BNPPA) initiated highly diasteroselective oxo-Michael additions (entries 1–3) while the corresponding aza-Michael reaction proceeded with lower d.r.'s under similar conditions (entries 5 and 6).
|
||||
|---|---|---|---|---|
| Entry | X | Y | Yield | d.r. |
| 1 | –CH2– | –O– | 62% | >20:1 |
| 2 | –(CH2)2– | –O– | 52% | >20:1 |
| 3 | –CH2– | –C(O)O– | 40% | >20:1 |
| 4 | –CH2– | –N(Boc)– | 55% | |
| 5 | –CH(CO2Me)– | –N(Boc)– | 53% | 1.6:1 |
| 6 | –CH(CO2tBu)– | –N(Boc)– | 62% | 1.4:1 |
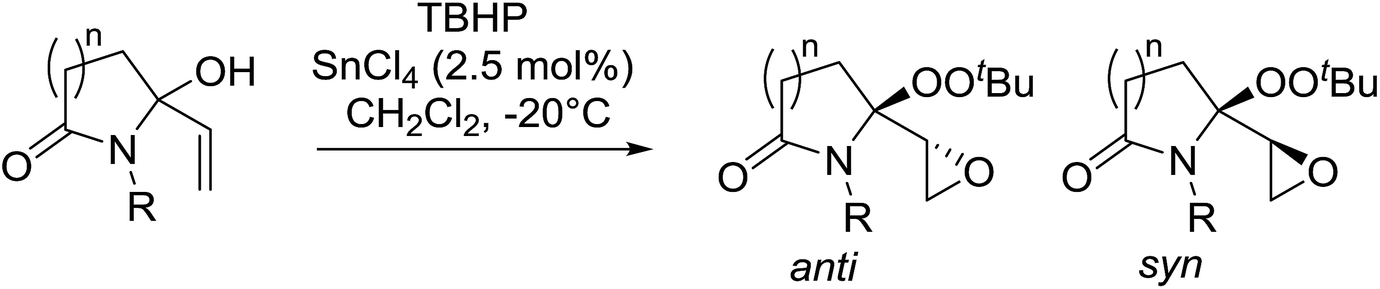
Alcohols offer another route to acyclic peroxides. In their studies on the epoxidation of allylic alcohols, Marson et al. discovered that treatment of lactam-based allylic alcohols with TBHP and tin tetrachloride resulted in diastereoselective epoxidation of the double bond with concomitant conversion of the alcohol to the corresponding alkyl peroxide (Table 47).87 Good yields of epoxy alkyl peroxides were obtained for a variety of N-substituents and for both γ- and δ-lactams (entries 1–4). The reaction outcome was dependent on the concentration of tin chloride. At 0.9 mol% concentration, peroxidation of the alcohol and the double bond was favoured, whereas at a higher concentration of 2.5 mol% the target epoxy alkyl peroxide was the major product. No reaction took place in the absence of tin chloride. The degree of diastereoselectivity was influenced by the size of the lactam ring, with γ-lactams (entries 1–3) displaying higher selectivity than δ-lactams (entry 4).
|
||||
|---|---|---|---|---|
| Entry | R | n | Yield | anti:syn |
| 1 | Me | 1 | 69% | 9:1 |
| 2 | Bn | 1 | 67% | 9:1 |
| 3 | Propargyl | 1 | 80% | 9:1 |
| 4 | Me | 2 | 64% | 7:1 |
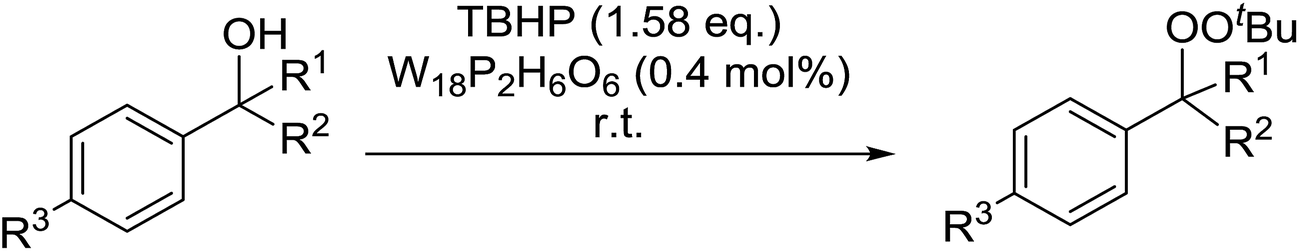
Tarlani and co-workers have investigated the use of the Well–Dawson tungsten heteropolyacid (W18P2H6O62) to promote the peroxidation of benzylic alcohols.88 Treatment of a range of benzylic alcohols with a slight excess of TBHP and a catalytic amount of W18P2H6O62 furnished the desired alkyl peroxides in moderate to good yields (Table 48, entry 1–6). The peroxidation of secondary benzylic alcohols was accompanied by the unwanted formation of the corresponding symmetric ethers (entries 1–3). Switching from secondary to tertiary alcohols blocked this side reaction and the target peroxides were formed exclusively (entries 4 and 5). These conditions were also compatible with other functional groups such as CC (entry 6).
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Yield |
| 1 | Ph | H | H | 69% |
| 2 | Ph | H | Cl | 59% |
| 3 | Ph | H | Me | 80% |
| 4 | Ph | Ph | H | 91% |
| 5 | Me | Me | H | 72% |
| 6 | PhCHCH |
H | H | 90% |
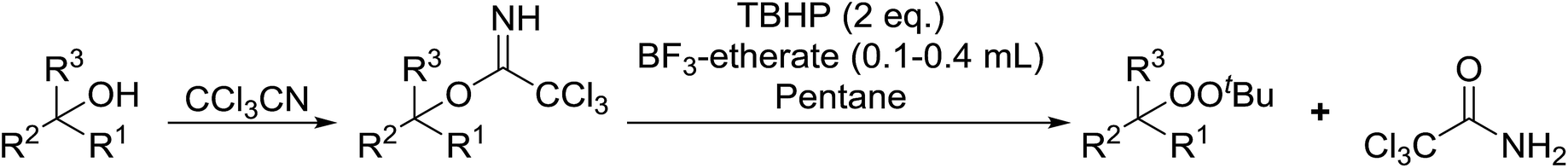
Bourgeois and co-workers have reported that tertiary alcohols may be transformed into alkyl peroxides via a trichloroacetimidate intermediate.89 Reaction of a suitable alcohol with trichloroacetonitrile affords the corresponding trichloroacetimidate. The trichloroacetimidate is then treated with TBHP in the presence of boron trifluoride etherate resulting in attack of the hydroperoxide nucleophile on the stabilised carbocation in an SN1 fashion. Moderate to good yields of the alkyl peroxides were obtained from aliphatic and aromatic tertiary alcohols (Table 49, entries 1–7). A noticeable decrease in yields was recorded when this methodology was applied to primary alcohols (entries 8 and 9).
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Yield |
| 1 | Me | Me | Me | 63% |
| 2 | CH2CH–CH2– |
Me | Me | 55% |
| 3 | Et | Me | Me | 61% |
| 4 | Ph | Me | Me | 45% |
| 5 | Cyclopropyl | Me | Me | 60% |
| 6 | Cyclobutyl | Me | Me | 32% |
| 7 | CH2CH– |
Me | Me | 44% |
| 8 | 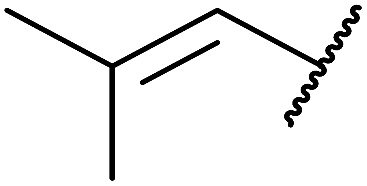 |
H | H | 10% |
| 9 | 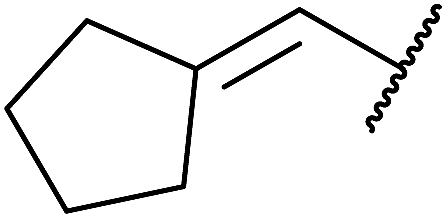 |
H | H | 10% |
2.6 Peroxidation of epoxides and oxetane
Intermolecular reactions of tertiary epoxides are well established.90,91 In a recent example, Vatèle and Barnych described a new route employing bis-epoxide 67 in a tandem epoxide opening-cyclisation reaction using anhydrous hydrogen peroxide and phosphomolybdic acid (PMA) (Scheme 12).92 Hydroperoxide 68 was isolated as the major product in a yield of 40% as a 1:1 mixture of diastereoisomers. The authors suggest that acid co-ordination to the vinyl epoxide occurs, followed by nucleophilic addition of the second epoxide leading to formation of the epoxonium intermediate. Addition of hydrogen peroxide to the most highly substituted carbon of the bridged oxonium ion affords the hydroperoxide.
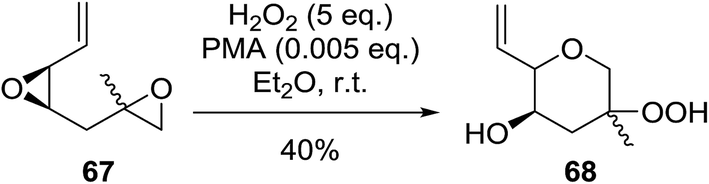 | ||
| Scheme 12 | ||
The perhydrolysis of epoxides has long been used as a route to hydroperoxides.93,94 Phosphomolybdic acid has been demonstrated to be an effective catalyst for the perhydrolysis of various epoxides.95 Unlike previous approaches which required almost neat hydrogen peroxide, Wu and co-workers found that treatment of a range of epoxides with ethereal hydrogen peroxide in the presence of PMA afforded the corresponding hydroperoxides in yields of 60–96% (Table 50, entries 1–11). Substrates containing TBS (entry 4), MOM (entry 5) or allyl (entry 6) protecting groups all underwent successful perhydrolysis. 2,3-Disubstituted epoxides were also amenable to these conditions (entry 8). In the case of 2-monosubstituted oxiranes, the regioselectivity depended significantly on the nature of the substituents with the reaction being controlled by electronic effects in the case of phenyl-substituted epoxides (entry 9) but by steric factors in the case of aliphatic epoxides (entry 10).
|
||||
|---|---|---|---|---|
| Entry | Substrate | Time (h) | Product | Yield |
| 1 |  |
8 |  |
80% |
| 2 | 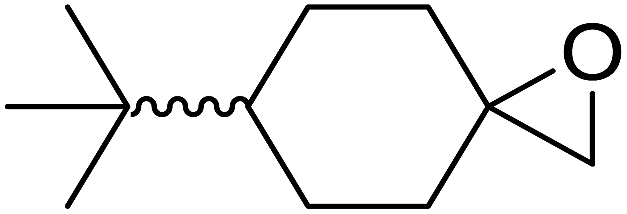 |
10 | 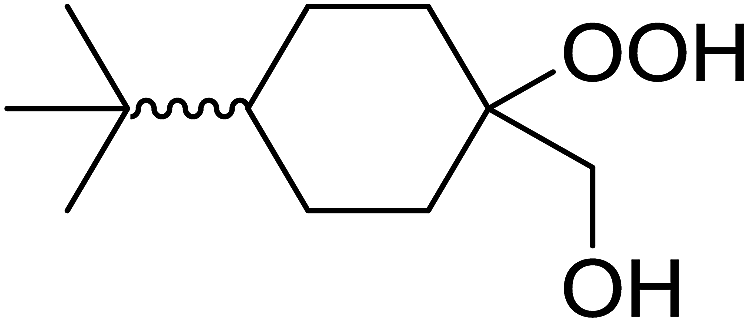 |
76% |
| 3 | 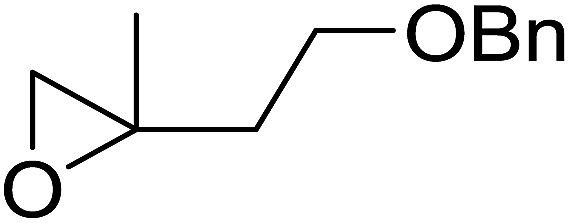 |
5 | 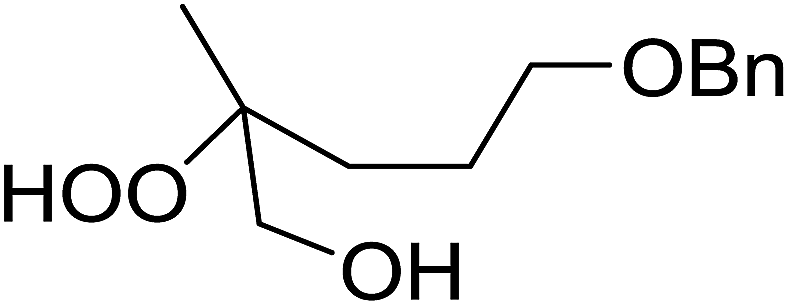 |
87% |
| 4 | 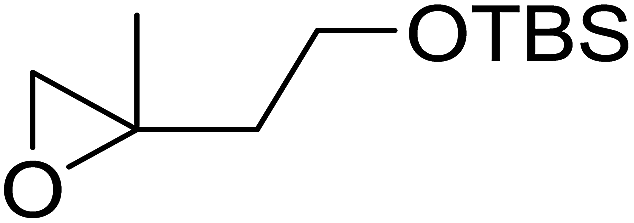 |
4 | 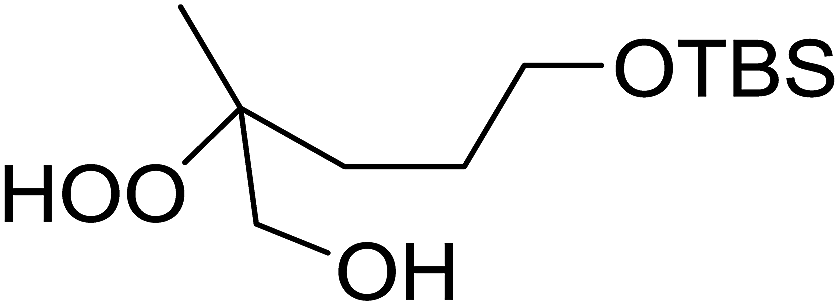 |
66% |
| 5 | 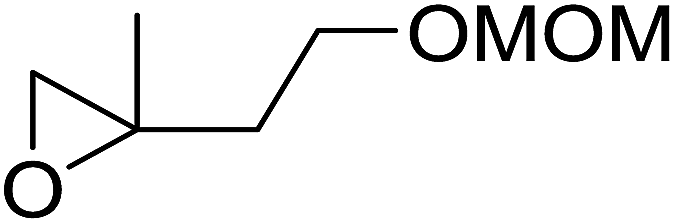 |
7 | 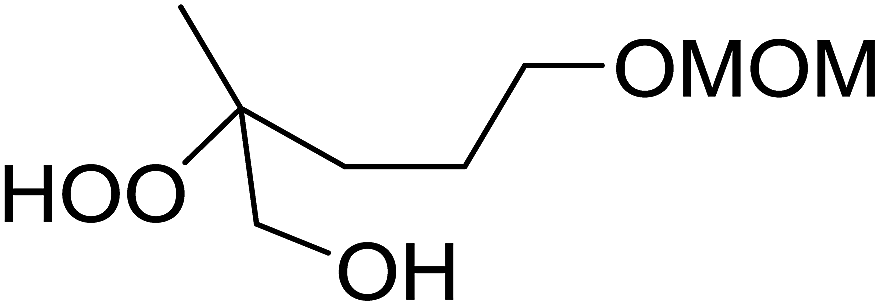 |
60% |
| 6 | 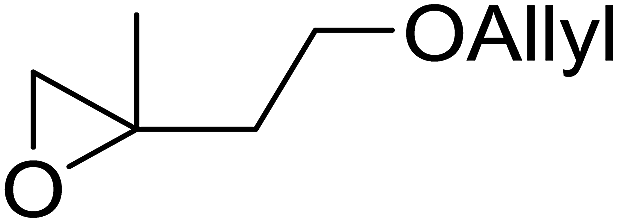 |
8 | 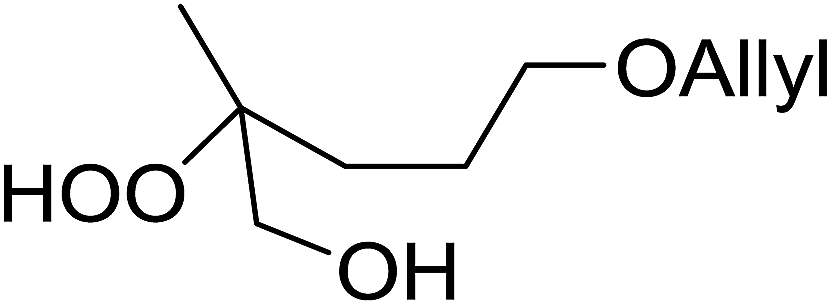 |
62% |
| 7 | 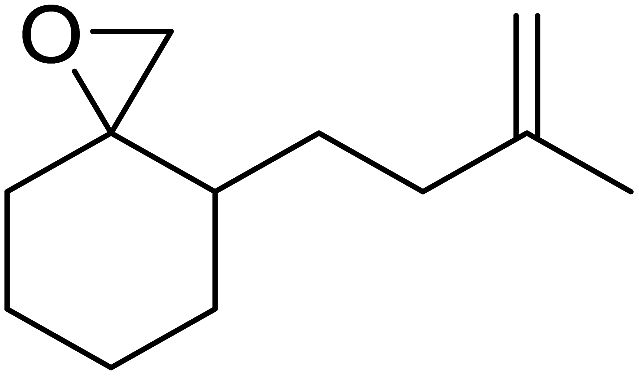 |
8 | 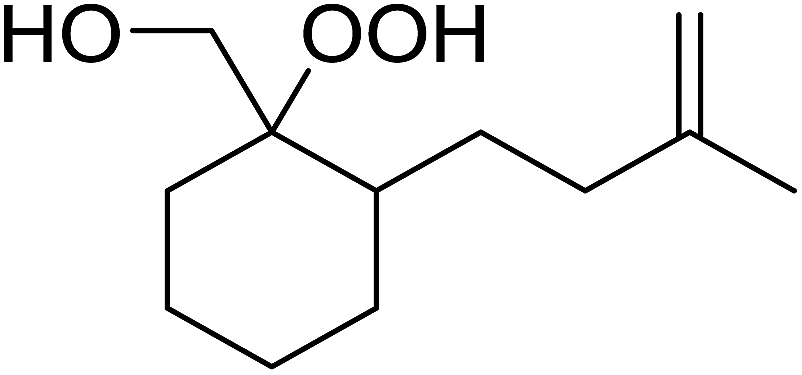 |
81% |
| 8 | 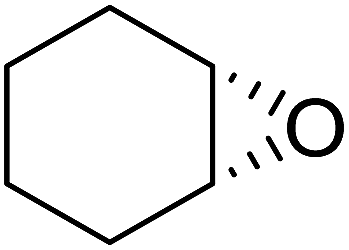 |
6 | 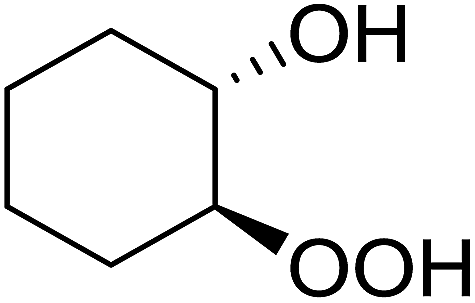 |
95% |
| 9 | 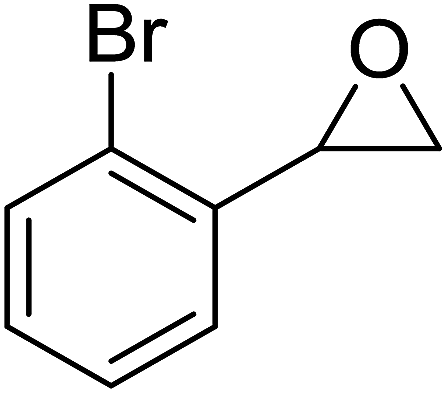 |
10 | 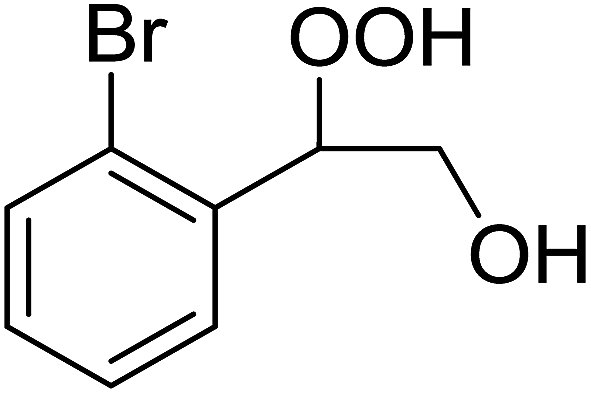 |
96% |
| 10 | 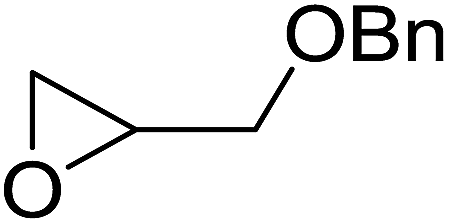 |
12 | 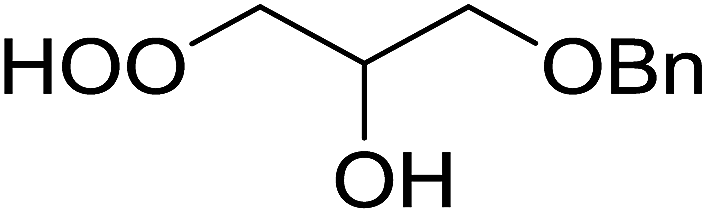 |
82% |
| 11 |  |
2 |  |
91% |
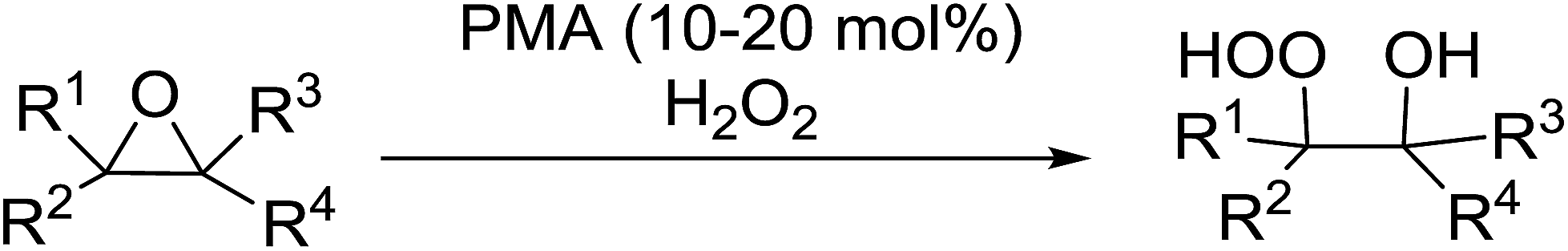
During the course of an investigation into the total synthesis of Artemisinin, Wu et al. encountered a problem with the stereoselective introduction of a hydroperoxyl group via a sterically congested epoxide (Scheme 13).96 They found that a molybdenum species prepared from Na2MoO4 and glycine can effectively catalyse the perhydrolysis of epoxy rings. The perhydrolysis was found to take place with a distinct facial preference and the attack of hydrogen peroxide occurred from the backside of the epoxy ring with inversion of the configuration at C-12a. The reaction was found to proceed in a good yield of 74%.
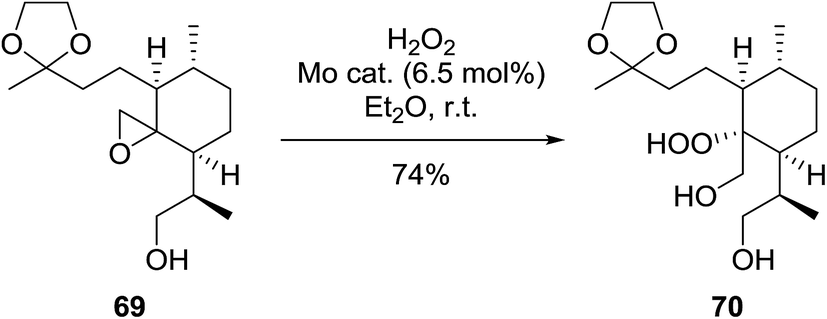 | ||
| Scheme 13 | ||
In a similar vein, Li et al. reported the efficient ring-opening of various epoxides with hydrogen peroxide, with silica-supported antimony trichloride as the catalyst system.97 Perhydrolysis proceeded smoothly across a range of substrates to afford β-hydroperoxyalcohols in a regioselective manner in good to excellent yields (Table 51, entries 1–11). Substituted styrene oxides were found to react at the more substituted carbon (entries 1–8) whereas unsymmetrical alkyl oxiranes were attacked at the terminal position (entries 9 and 10). The reaction was further promoted by ultrasound sonication which significantly reduced reaction times across all substrates.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Without sonication | With sonication | Product | ||
| Time (h) | Yield | Time (h) | Yield | |||
| 1 |  |
9 | 85% | 1.5 | 86% |  |
| 2 | 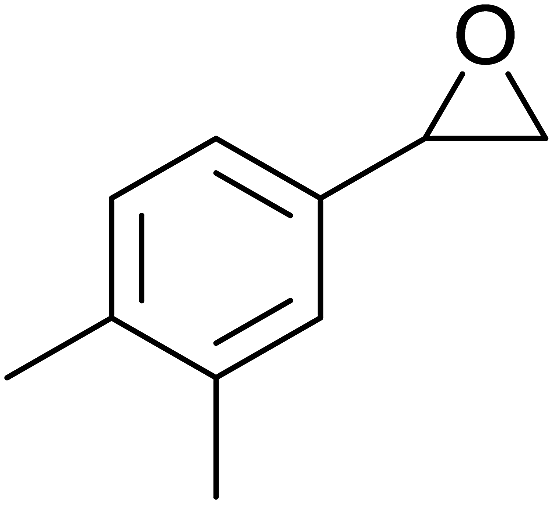 |
12 | 72% | 2 | 75% | 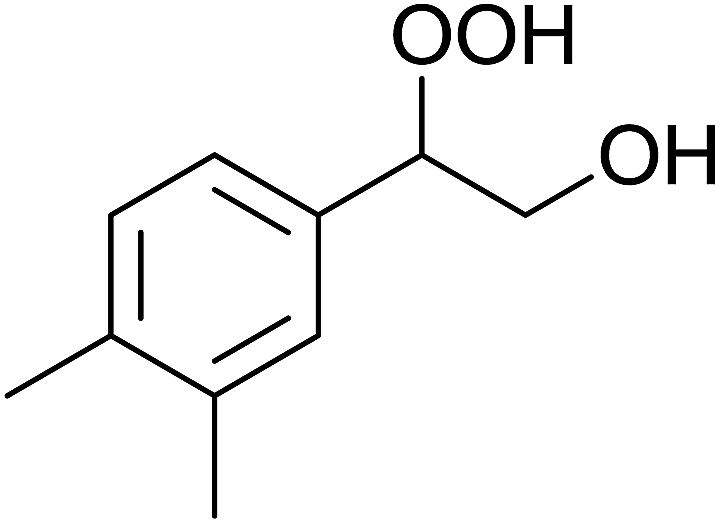 |
| 3 | 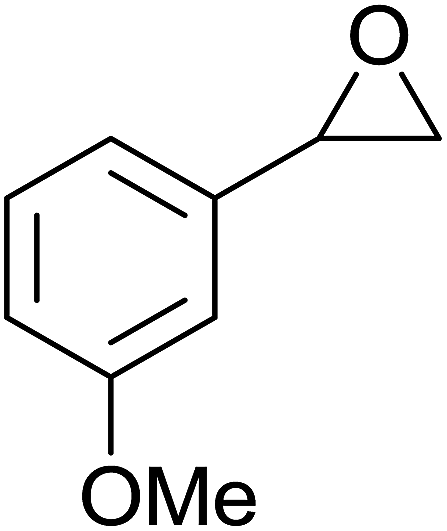 |
6 | 85% | 1 | 85% | 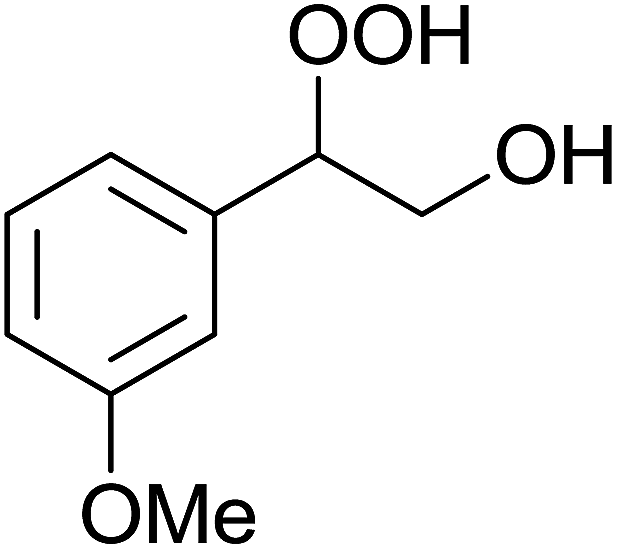 |
| 4 | 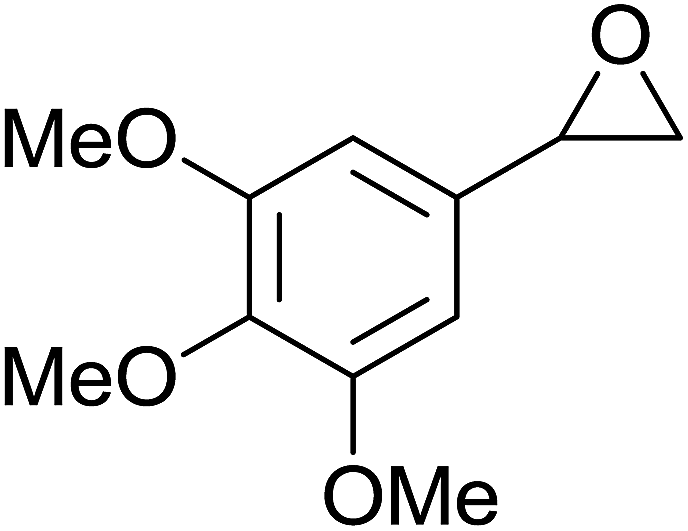 |
2 | 85% | 0.5 | 77% | 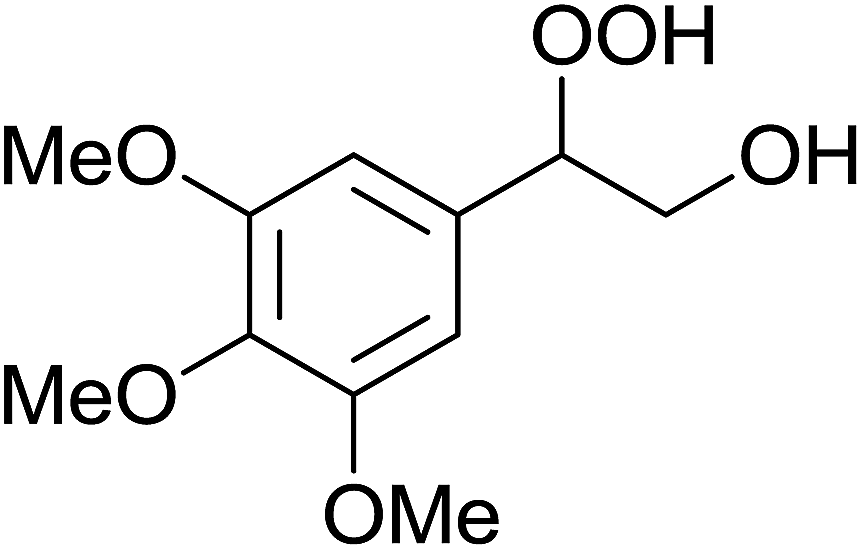 |
| 5 | 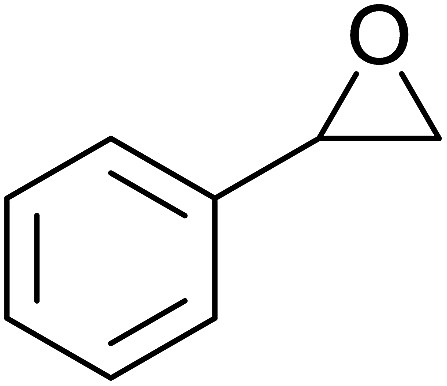 |
5 | 85% | 1.5 | 85% | 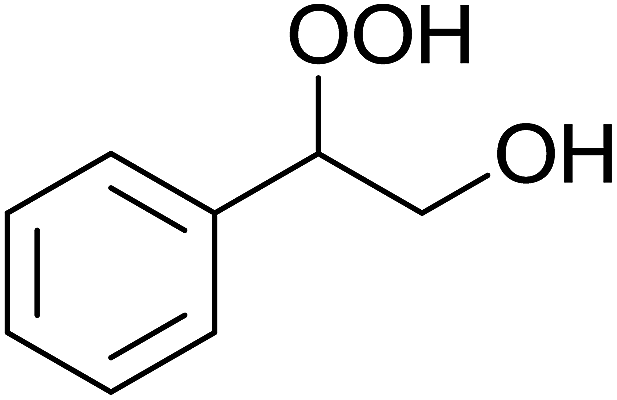 |
| 6 | 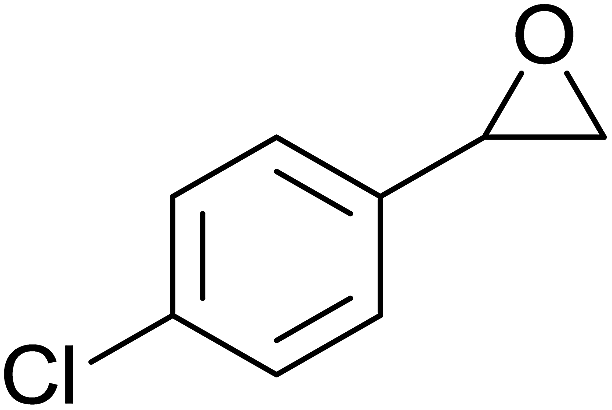 |
5 | 86% | 1 | 85% | 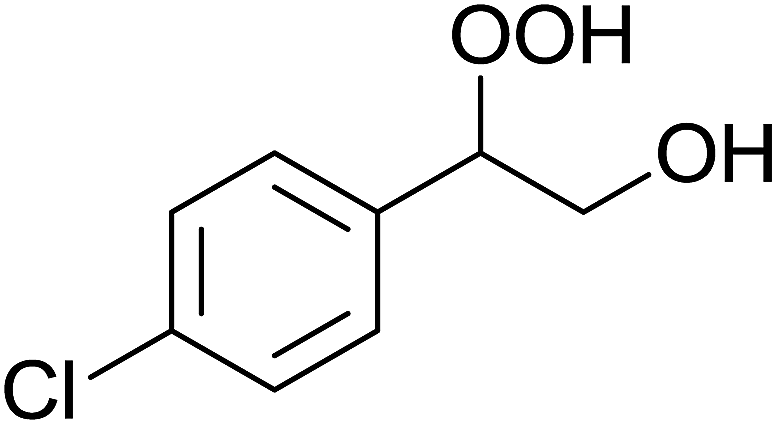 |
| 7 | 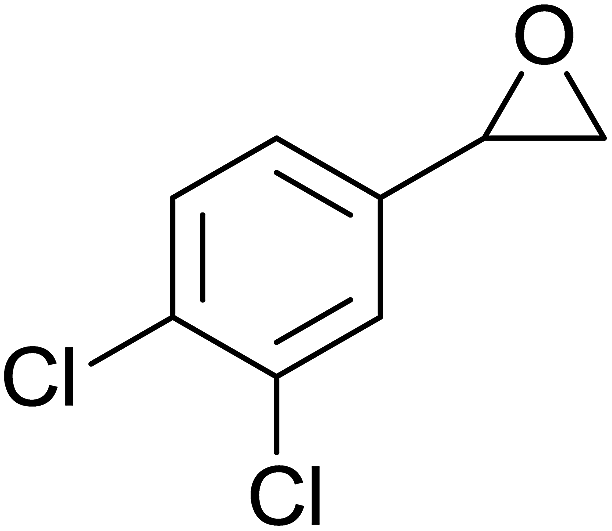 |
5 | 78% | 1 | 78% | 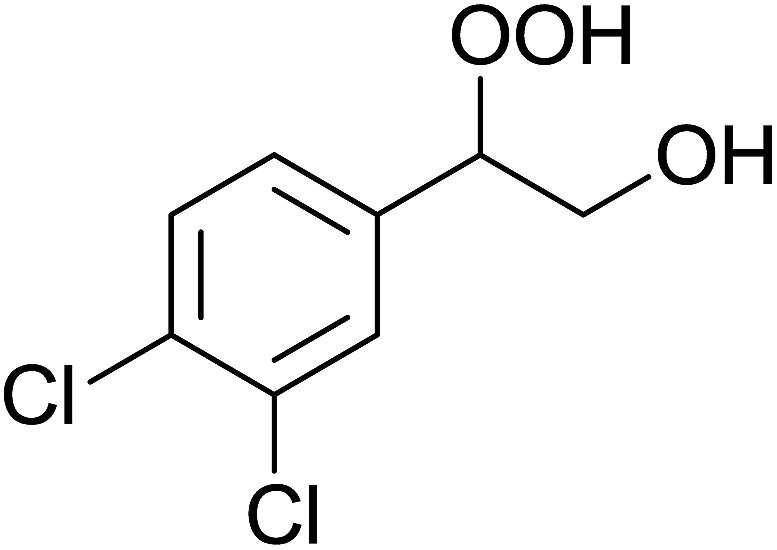 |
| 8 | 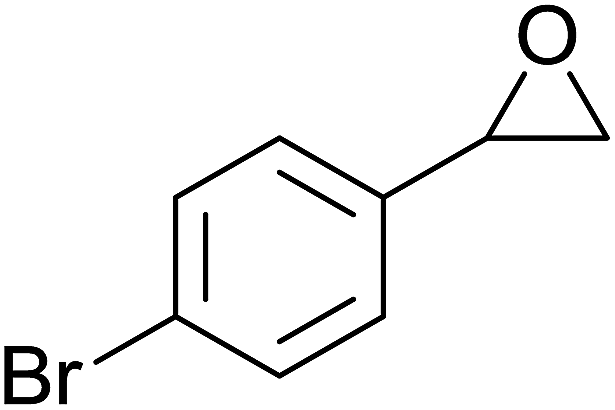 |
10 | 81% | 2 | 85% | 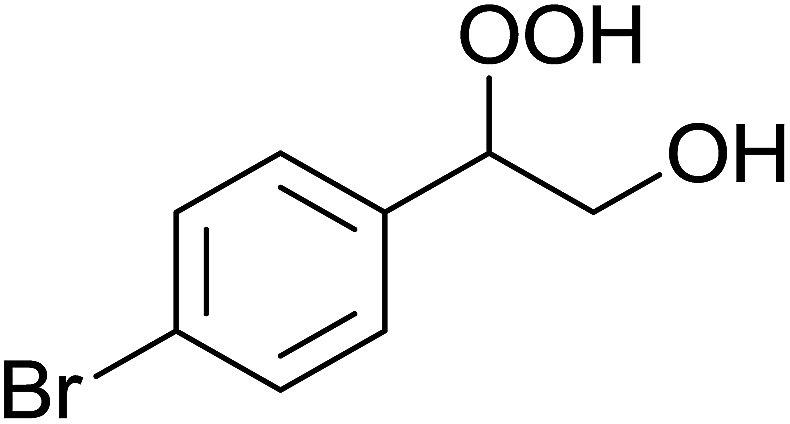 |
| 9 |  |
8 | 82% | 1.5 | 82% |  |
| 10 | 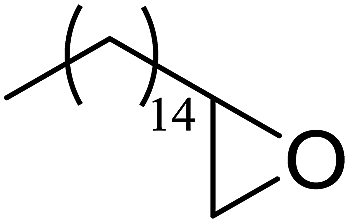 |
12 | 82% | 2.0 | 83% | 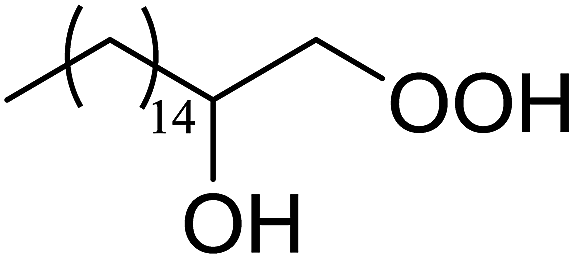 |
| 11 | 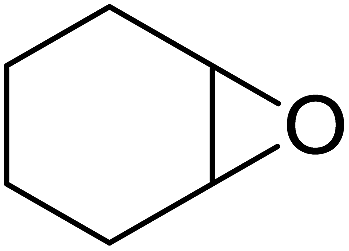 |
12 | 82% | 2.0 | 83% | 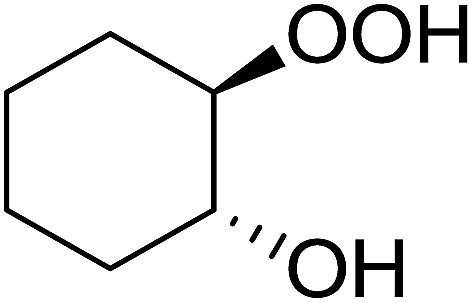 |
Similar reactions involving secondary alkyl epoxides were, until recently, unreported. Dussault and Xu demonstrated the successful Lewis acid-catalysed intermolecular reaction of a hydroperoxide with a secondary epoxide in a 73% yield (Scheme 14).98 The reaction was found to proceed with moderate regioselectivity in favour of the more substituted peroxide.
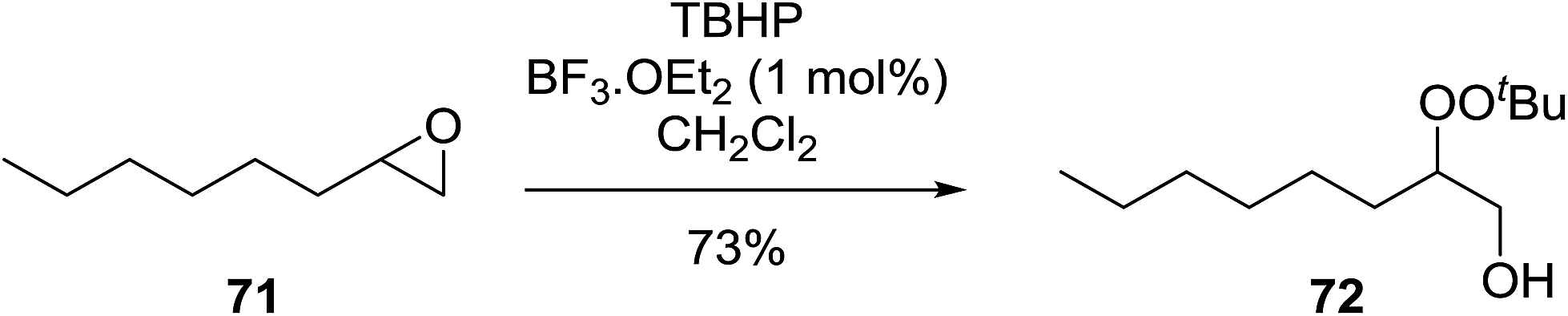 | ||
| Scheme 14 | ||
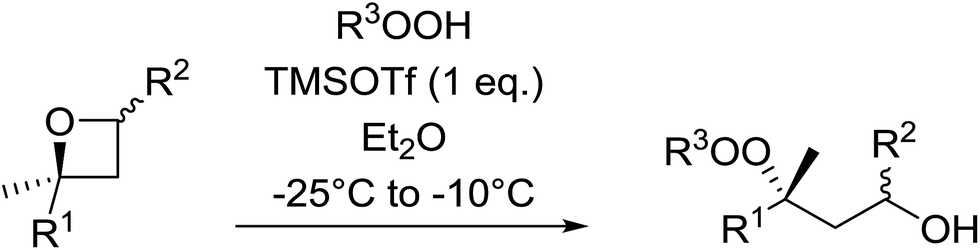
1,3-Peroxyalkanols are useful building blocks for the synthesis of 1,2-dioxalanes. Dussault et al. have reported a novel route to these compounds based on the Lewis acid-catalysed perhydrolysis of substituted oxetanes.99 The oxetane substrates were prepared via cyclodehydration of chiral 1,3-diols. Perhydrolysis with ethereal hydrogen peroxide and catalytic amounts of TMSOTf or Yb(OTf)3, afforded the 1,3-hydroperoxyalkanols in yields of 31–60% (Table 52, entries 1–6). Alkyl peroxides could also be used in place of hydrogen peroxide, affording the target 1,3-peroxyalkanols in similar yields (entries 7–9). It was noted that this methodology was not compatible with secondary oxetanes, even under forcing conditions. This approach was subsequently employed in the asymmetric synthesis of the natural product, plakinic acid A.100
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Lewis acid (eq.) | Yield |
| 1 | Hexadecyl | H | H | TMSOTf (0.4) | 48% |
| 2 | Hexadecyl | H | H | Yb(OTf)3 (0.1) | 60% |
| 3 | 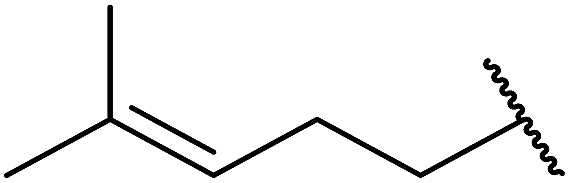 |
H | H | TMSOTf (0.1) | 46% |
| 4 | 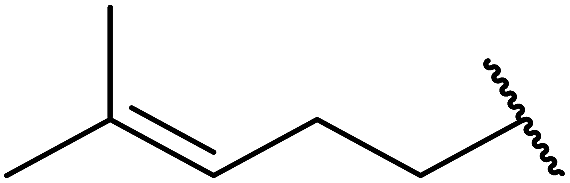 |
H | H | Yb(OTf)3 (0.1) | 48% |
| 5 | 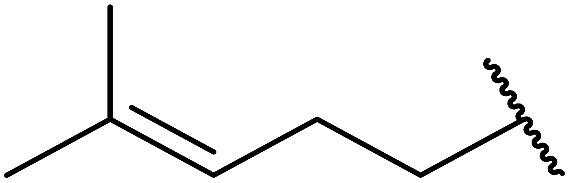 |
Me | H | TMSOTf (0.1) | 31% |
| 6 | 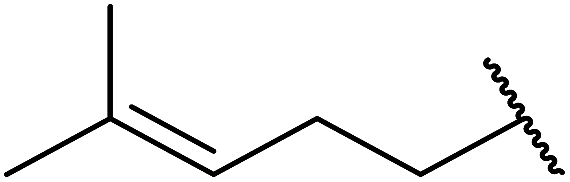 |
Me | H | Yb(OTf)3 (0.1) | 40% |
| 7 | C16H33 | H | THP– | TMSOTf (0.25) | 39% |
| 8 | 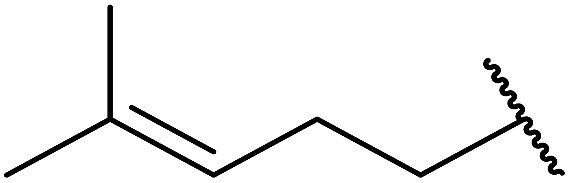 |
H | t-Bu | TMSOTf (0.1) | 54% |
| 9 | 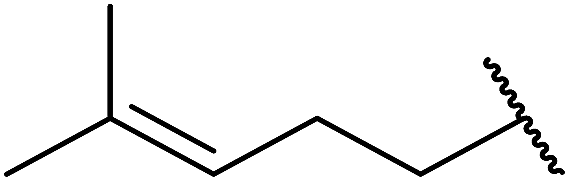 |
Me | t-Bu | TMSOTf (0.1) | 51% |
2.7 Peroxidation of miscellaneous substrates
Tungstic acid was investigated by Jefford et al. as a potential catalyst for the perhydrolysis of ketals with hydrogen peroxide.101 The target hydroperoxides were isolated in good yields along with small amount of ketone by-products (Table 53, entries 1–5). Shorter reaction times limited the formation of unwanted dimers and were associated with higher yields.
|
|||
|---|---|---|---|
| Entry | R1 | R2 | Yield |
| 1 | –(CH2)4– | 83% | |
| 2 | –(CH2)5– | 81% | |
| 3 | –(CH2)11– | 67% | |
| 4 | n-C4H9 | n-C4H9 | 72% |
| 5 | C2H5 | n-C6H13 | 70% |
For their synthetic studies on plakinic acids, Dussault et al. required a robust method for the preparation of enantiomerically enriched 3-peroxyalkanoates.102 A range of silyl ketene acetals (SKAs) were converted to 3-peroxyalkanoates in poor to moderate yields in the presence of a suitable Lewis acid (Table 54, entries 1–3). Contrary to expectations, the level of diastereoselectivity was poor for O-ester SKAs (entries 1–2) and completely absent for S-ester SKAs (entry 3).
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | X | Lewis acid | Yield | d.r. |
| 1 |  |
TBDMS | O | TMSOTf | 18% | 54:46 |
| 2 |  |
TBDMS | O | TiCl4 | 48% | 55:45 |
| 3 | 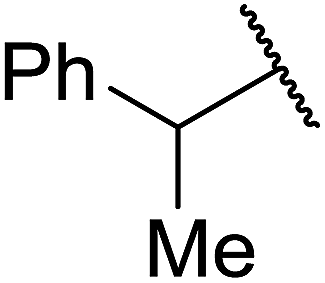 |
TMS | S | TMSOTf | 40% | 50:50 |
Arai et al. have developed a novel bis-(imidazolidine)pyridine ligand (PyBidine) which, when complexed to nickel chloride, catalyses the asymmetric addition of peroxides to isatin-derived N-Boc-imines.103 The highest yields and enantiomeric excesses were obtained with cumene hydroperoxide (CMHP), which was then tested against a library of N-Boc-ketimines in the presence of the PyBidine–NiCl2 complex (Table 55). The desired N,O-acetals were formed in excellent yields and with ee values ranging from 88% to 94% (entries 1–7). Substrates with either electron-donating (entries 2 and 3) or electron-withdrawing (entries 4–7) substituents on the benzene ring reacted readily. The authors propose that the imine lone pair coordinates to the nickel center as a result of the affinity of nickel for nitrogen atoms. This is followed by addition of the peroxide onto the polarized imine group. Finally, the NH functionality of the imidazolidine ring in PyBidine guides the stereoselective attack of the nucleophile via hydrogen bonding.
|
|||
|---|---|---|---|
| Entry | R | Yield | ee |
| 1 | H | 99% | 94% |
| 2 | 5-Me | 99% | 94% |
| 3 | 5-MeO | 99% | 92% |
| 4 | 5-F | 99% | 94% |
| 5 | 5-Cl | 99% | 88% |
| 6 | 5-Br | 99% | 92% |
| 7 | 6-Br | 86% | 91% |
3 Organocatalysed reactions
Organocatalysis is a burgeoning field in organic chemistry owing to the very high stereoselectives and mild conditions associated with these types of reactions. Deng and co-workers reported the first, organocatalytic, enantioselective peroxidation of α,β-unsaturated ketones by the modification of a well-established epoxidation pathway.104 A cinchona-derived catalyst 76 was employed, which not only aids the asymmetric nucleophilic addition of the hydroperoxide to the iminium intermediate, but also influences the partitioning of the peroxyenamine intermediate between epoxidation and peroxidation pathways (Table 56). The β-peroxyketones were isolated in moderate to high yields, with excellent enantioselectivity observed across a range of aliphatic and aromatic unsaturated ketones (entries 1–22). The reaction was compatible with different peroxide sources including TBHP (entries 1–8), cumene hydroperoxide (entries 9–16) and α-methoxy iso-propyl hydroperoxide (entries 17–22). The latter is notable as the resulting alkyl peroxides may be readily converted to hydroperoxides following acidic hydrolysis.
|
||||||
|---|---|---|---|---|---|---|
| Entrya | R1 | R2 | R3 | 74:75 |
Yield | ee 74 |
| a Entries 1–9 were run at 23 °C; entries 10–22 were run at 0 °C. | ||||||
| 1 | Me | CH2Bn | t-Bu | 92:8 |
85% | 91% |
| 2 | Me | Me | t-Bu | 94:6 |
88% | 84% |
| 3 | Me | n-Pr | t-Bu | 94:5 |
91% | 90% |
| 4 | Me | n-Pen | t-Bu | 93:7 |
86% | 93% |
| 5 | Me | (CH2)3OBn | t-Bu | 90:10 |
65% | 91% |
| 6 | Et | Me | t-Bu | 95:5 |
90% | 89% |
| 7 | n-Bu | Me | t-Bu | 94:6 |
89% | 87% |
| 8 | n-Bu | n-Pr | t-Bu | 86:14 |
64% | 94% |
| 9 | Me | CH2Bn | C(Me)2Ph | 86:14 |
74% | 94% |
| 10 | Me | Me | C(Me)2Ph | 77:23 |
70% | 92% |
| 11 | Me | n-Pr | C(Me)2Ph | 88:12 |
75% | 92% |
| 12 | Me | n-Pen | C(Me)2Ph | 87:13 |
77% | 95% |
| 13 | Me | (CH2)3OBn | C(Me)2Ph | 90:10 |
82% | 96% |
| 14 | Et | Me | C(Me)2Ph | 89:11 |
75% | 94% |
| 15 | n-Bu | Me | C(Me)2Ph | 85:15 |
66% | 96% |
| 16 | n-Bu | n-Pr | C(Me)2Ph | 65:35 |
55% | 97% |
| 17 | Me | CH2Bn | C(Me)2OMe | 77:23 |
60% | 92% |
| 18 | Me | n-Pr | C(Me)2OMe | 86:14 |
70% | 95% |
| 19 | Me | n-Pen | C(Me)2OMe | 88:12 |
62% | 95% |
| 20 | Et | Me | C(Me)2OMe | 95:5 |
63% | 95% |
| 21 | Et | n-Hep | C(Me)2OMe | 78:22 |
42% | 94% |
| 22 | n-Bu | Me | C(Me)2OMe | 95:5 |
60% | 94% |
This work was later expanded to encompass α,β-unsaturated aldehydes using a similar catalyst system.105 It was found that the degree of enantioselectivity was dependent on the peroxide source, and that the highest selectivities were recorded with α-methoxydiphenyl hydroperoxide. Accordingly, peroxidation of a range of enals with quinine-derived 76 as the catalyst furnished β-peroxyaldehydes in yields of 54–67% and ee values of 87–91% (Table 57, entries 1–5). Switching to quinidine-derivated catalyst 77, afforded β-peroxyaldehydes with the opposite stereochemical configuration in comparable yields and ee's (entries 6–10).
|
||||
|---|---|---|---|---|
| Entry | R | Catalyst | Yields | ee |
| 1 | n-Hexyl | 76 | 60% | 91% (R) |
| 2 | n-Pentyl | 76 | 62% | 87% (R) |
| 3 | n-Pr | 76 | 67% | 90% (R) |
| 4 | i-Bu | 76 | 57% | 90% (R) |
| 5 | –CH2Bn | 76 | 61% | 90% (R) |
| 6 |  |
77 | 60% | 85% (S) |
| 7 |  |
77 | 58% | 86% (S) |
| 8 |  |
77 | 54% | 87% (S) |
| 9 |  |
77 | 55% | 87% (S) |
| 10 |  |
77 | 58% | 85% (S) |
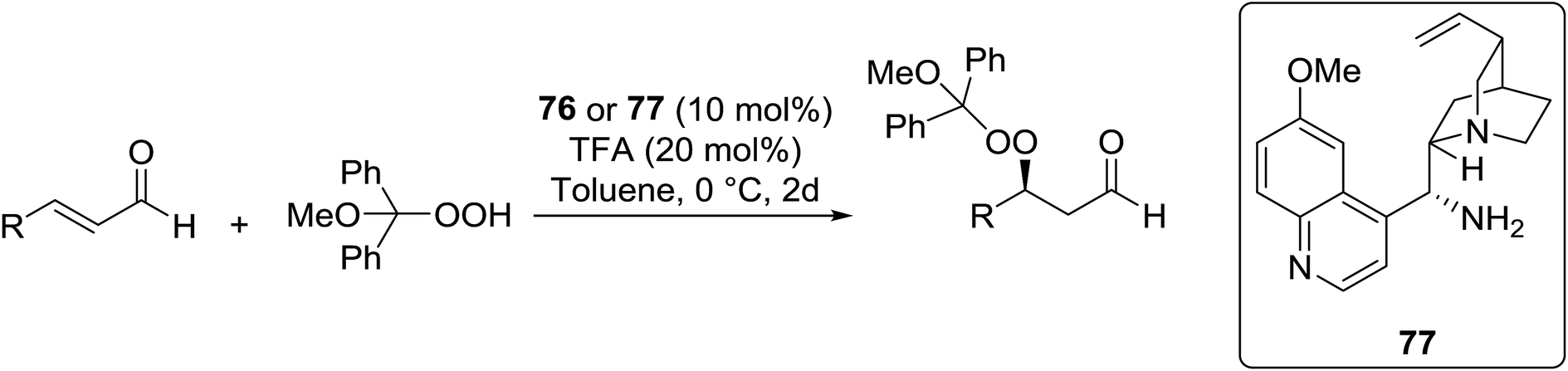
In parallel work, we investigated the cinchona-catalysed, asymmetric peroxidation of α,β-unsaturated aldehydes with TBHP.106 As the intermediate β-peroxyaldehydes were found to be unstable, in situ Pinnick oxidation afforded the corresponding β-peroxycarboxylic acids in high yields and good ee's (Table 58). Formation of the (R)-enantiomer was preferred in all cases, with no discernible relationship between the degree of stereoselectivity and chain length evident (entries 1–9). This methodology was also compatible with cumene hydroperoxide and the resultant β-peroxyaldehydes were reduced to stable β-peroxyalcohols (entries 11–19). The choice of peroxide was found to impact on enantioselectivity, with cumene hydroperoxide resulting in slightly lower ee's than TBHP (entries 1–9 vs. 11–19).
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Method | R3 | Yield | ee |
| 1 | CH3 | CH3 | a | CO2H | 90% | 60% |
| 2 | CH2CH3 | CH3 | a | CO2H | 91% | 63% |
| 3 | (CH2)2CH3 | CH3 | a | CO2H | 87% | 66% |
| 4 | (CH2)3CH3 | CH3 | a | CO2H | 89% | 68% |
| 5 | (CH2)4CH3 | CH3 | a | CO2H | 92% | 65% |
| 6 | (CH2)5CH3 | CH3 | a | CO2H | 86% | 66% |
| 7 | (CH2)6CH3 | CH3 | a | CO2H | 88% | 65% |
| 8 | (CH2)7CH3 | CH3 | a | CO2H | 94% | 64% |
| 9 | (CH2)8CH3 | CH3 | a | CO2H | 94% | 66% |
| 10 | (CH2)2Ph | CH3 | a | CO2H | 77% | 66% |
| 11 | CH3 | Ph | b | CH2OH | 83% | 56% |
| 12 | CH2CH3 | Ph | b | CH2OH | 81% | 64% |
| 13 | (CH2)2CH3 | Ph | b | CH2OH | 77% | 62% |
| 14 | (CH2)3CH3 | Ph | b | CH2OH | 84% | 66% |
| 15 | (CH2)4CH3 | Ph | b | CH2OH | 82% | 59% |
| 16 | (CH2)5CH3 | Ph | b | CH2OH | 76% | 64% |
| 17 | (CH2)6CH3 | Ph | b | CH2OH | 78% | 58% |
| 18 | (CH2)7CH3 | Ph | b | CH2OH | 84% | 65% |
| 19 | (CH2)8CH3 | Ph | b | CH2OH | 80% | 55% |
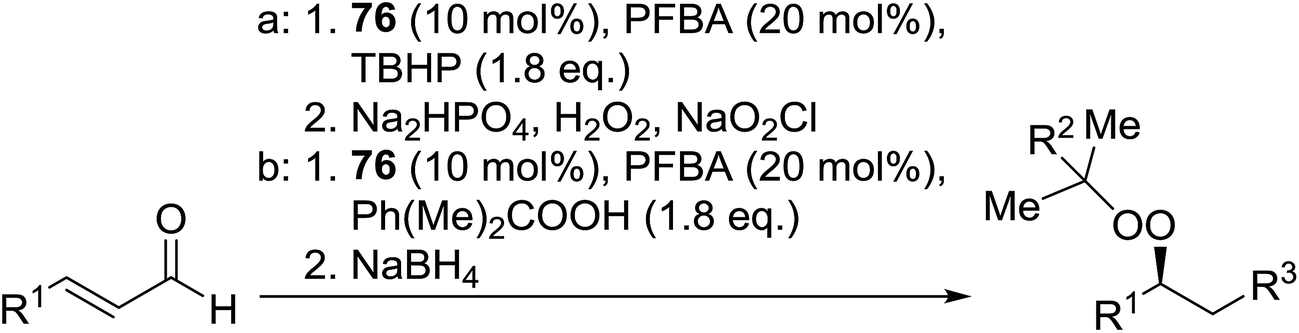
Deng has also reported the enantioselective peroxidation of unsaturated nitroalkenes.107 In this instance, the most efficient catalyst was determined to be 78, a sulphonamide derivative of 76 (Table 59). Further optimisation studies indicated that both yields and enantioselectivity were strongly influenced by the number of equivalents of cumene hydroperoxide and the reaction concentration. Aromatic nitroalkenes bearing either meta- or para-substituted rings were transformed into the corresponding β-peroxy nitroalkanes in good yields and enantioselectivities (entries 2–8). Aliphatic nitroalkenes were also tolerated under these conditions (entries 9–12) as were other peroxides, such as 1-hydroperoxy-1-methoxyethylbenzene, which furnished the target peroxides with high diastereoselectivity (entries 13–17). The synthetic utility of these latter compounds lies in their ability to be transformed into the corresponding hydroperoxides in high yields on treatment with Amberlyst 15.
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Time (h) | Yield | ee or d.r. |
| 1 | Ph | PhC(Me)2 | 80 | 62% | 93% |
| 2 | 4-MeC6H4 | PhC(Me)2 | 120 | 51% | 88% |
| 3 | 3-MeC6H4 | PhC(Me)2 | 96 | 58% | 90% |
| 4 | 4-BrC6H4 | PhC(Me)2 | 96 | 43% | 91% |
| 5 | 3-BrC6H4 | PhC(Me)2 | 120 | 43% | 87% |
| 6 | 4-ClC6H4 | PhC(Me)2 | 96 | 52% | 94% |
| 7 | 4-FC6H4 | PhC(Me)2 | 120 | 63% | 88% |
| 8 | 4-MeOC6H4 | PhC(Me)2 | 120 | 65% | 92% |
| 9 | n-Bu | PhC(Me)2 | 120 | 59% | 83% |
| 10 | n-Heptyl | PhC(Me)2 | 120 | 61% | 90% |
| 11 | –CH2Bn | PhC(Me)2 | 88 | 67% | 84% |
| 12 | i-Bu | PhC(Me)2 | 100 | 55% | 86% |
| 13 | –CH2Bn | PhCMe(OMe) | 144 | 80% | 6.5:1 d.r. |
| 14 | n-Pr | PhCMe(OMe) | 144 | 81% | 6.8:1 d.r. |
| 15 | n-Bu | PhCMe(OMe) | 144 | 67% | 6.2:1 d.r. |
| 16 | n-Heptyl | PhCMe(OMe) | 140 | 74% | 8.3:1 d.r. |
| 17 | i-Bu | PhCMe(OMe) | 148 | 68% | 6.1:1 d.r. |
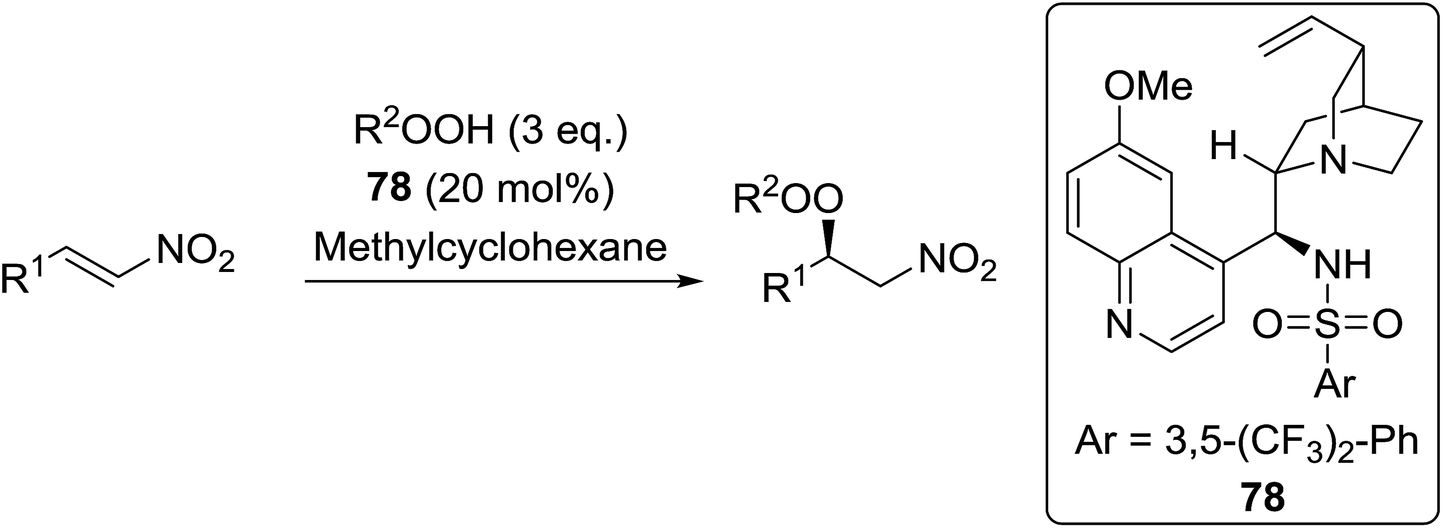
Lattanzi used α,α-diarylprolinol-derived catalyst 79 for the successful peroxidation of aromatic-derived nitroalkenes, generating enantiomerically enriched β-peroxynitroalkanes (Table 60).108 Aromatic β-nitrostyrenes, bearing both electron-donating and electron-withdrawing substituents, afforded the products with high enantioselectivities, albeit in lower yields for the latter group (entries 1–5 & 6–8). Good asymmetric induction was maintained for heteroaryl-substituted substrates but overall yields were negatively impacted (entries 11 and 12). The outcome was more unpredictable with aliphatic substrates with a cyclohexyl nitroalkene affording high enantiomeric excess (entry 13) whereas peroxidation of pentyl nitroalkene furnished a low yielding, racemic mixture (entry 14). It is believed that catalyst 79 serves as a bifunctional promoter working via deprotonation of tert-butyl hydroperoxide to generate the contact ion-pair. The intermolecular hydrogen bonding between the catalyst and the oxygen atom orientates the nitroalkene and favours attack of the peroxy anion on the Si face.
|
|||
|---|---|---|---|
| Entry | R | Yield | ee |
| 1 | Ph | 72% | 84% |
| 2 | 4-MeC6H4 | 59% | 83% |
| 3 | 3,4-(Me)2C6H3 | 70% | 83% |
| 4 | 4-t-BuC6H4 | 83% | 83% |
| 5 | 2-MeC6H4 | 62% | 43% |
| 6 | 4-CF3C6H4 | 60% | 78% |
| 7 | 4-BrC6H4 | 41% | 82% |
| 8 | 4-ClC6H4 | 80% | 84% |
| 9 | 3-MeOC6H4 | 40% | 80% |
| 10 | 2-Naphthyl | 38% | 80% |
| 11 | 2-Furyl | 37% | 81% |
| 12 | 3-Furyl | 45% | 83% |
| 13 | Cyclohexyl | 73% | 72% |
| 14 | n-Pen | 20% | 0% |
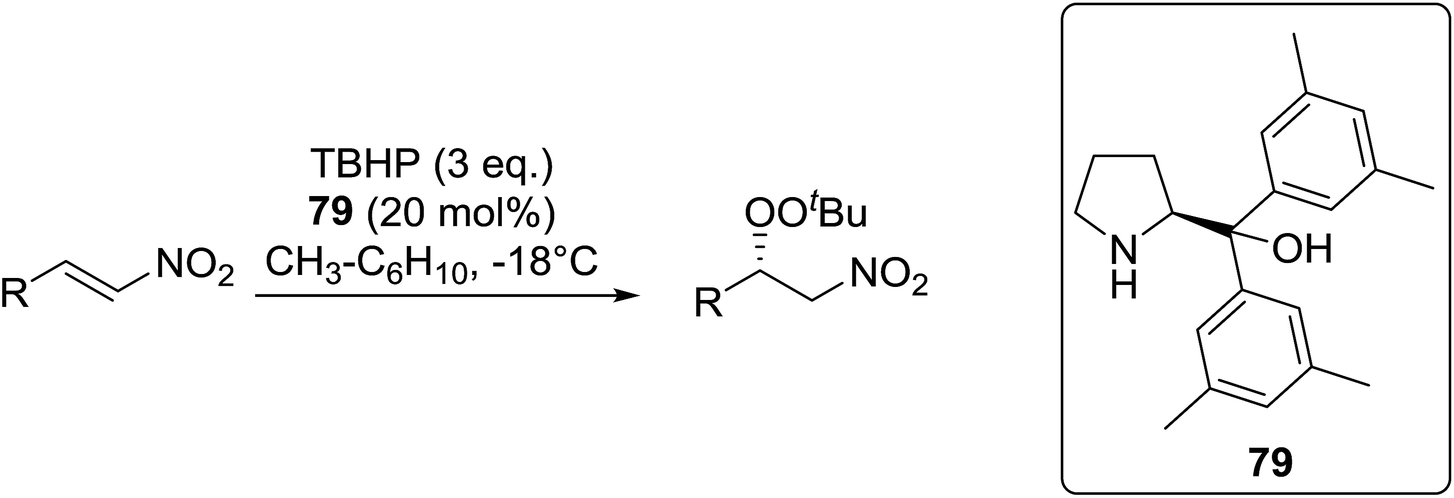
Antilla and co-workers have employed chiral phosphoric acid 80 in the asymmetric peroxidation of acylated imines, forming optically active α-amino peroxides (Table 61).109 It was discovered that the degree of enantioselectivity was dependent on the substitution pattern of the aromatic acyl protecting group, with the introduction of methoxy groups leading to an increase in asymmetric induction (entries 1 vs. 2 vs. 6). With the 3,5-dimethoxybenzoyl protecting group in place, imines with electron-withdrawing or electron-donating groups were subjected to peroxidation with TBHP and consistently high yields and enantioselectivities were recorded (entries 7–15). Peroxidation with cumene hydroperoxide was also feasible under these conditions (entries 16–18). It is believed that the bifunctional nature of catalyst 80 is responsible for the dual activation of both the imine substrate and the peroxide through hydrogen-bonding interactions. This creates a chiral environment which facilitates the highly stereoselective addition of the peroxide to the activated imine.
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Yield | ee |
| 1 | Ph | Ph | t-Bu | 84% | 86% |
| 2 | 4-MeOC6H4 | Ph | t-Bu | 83% | 90% |
| 3 | 2-MeC6H4 | Ph | t-Bu | 92% | 84% |
| 4 | 3-MeC6H4 | Ph | t-Bu | 83% | 88% |
| 5 | 3-MeOC6H4 | Ph | t-Bu | 82% | 97% |
| 6 | 3,5-(MeO)2C6H3 | Ph | t-Bu | 81% | 96% |
| 7 | 3,5-(MeO)2C6H3 | 4-FC6H4 | t-Bu | 95% | 88% |
| 8 | 3,5-(MeO)2C6H3 | 4-ClC6H4 | t-Bu | 98% | 87% |
| 9 | 3,5-(MeO)2C6H3 | 4-BrC6H4 | t-Bu | 98% | 82% |
| 10 | 3,5-(MeO)2C6H3 | 4-MeOC6H4 | t-Bu | 95% | 77% |
| 11 | 3,5-(MeO)2C6H3 | 2-MeOC6H4 | t-Bu | 91% | 75% |
| 12 | 3-MeOC6H4 | 3-MeC6H4 | t-Bu | 91% | 91% |
| 13 | 3-MeOC6H4 | 4-MeOC6H4 | t-Bu | 90% | 86% |
| 14 | 3-MeOC6H4 | 4-ClC6H4 | t-Bu | 89% | 86% |
| 15 | 3-MeOC6H4 | 4-MeC6H4 | t-Bu | 97% | 92% |
| 16 | 3,5-(MeO)2C6H3 | 4-ClC6H4 | C(Me)2Ph | 96% | 91% |
| 17 | 3,5-(MeO)2C6H3 | 4-FC6H4 | C(Me)2Ph | 93% | 90% |
| 18 | 3,5-(MeO)2C6H3 | 4-MeC6H4 | C(Me)2Ph | 98% | 89% |
Chen et al. developed the first, peroxo-asymmetric, allylic alkylation of bulky hydroperoxyalkanes with Morita–Baylis–Hillman carbonates using modified cinchona alkaloids to afford chiral α-methylene-β-hydroxy esters (Table 62).110 Following initial screening studies, it was determined that hydroquinidine 1,4-phthalazinediyl diether [(DHQD)2PHAL] in carbon tetrachloride was the most effective catalyst system. Good to excellent ee's were obtained for both electron poor (entries 2–5) and electron rich (entries 6–9) substrates. Heteroaromatic substrates were also tolerated under these conditions (entries 10 and 11). The reaction proceeds by way of 1,4-addition of the tertiary amine to the activated alkene, resulting in loss of the OBoc group. This is followed by asymmetric allylic addition of the peroxide anion and finally formation of the α-methylene-β-peroxyester.
|
||||
|---|---|---|---|---|
| Entry | R | Time (h) | Yield | ee |
| 1 | Ph | 48 | 79% | 93% |
| 2 | 4-FC6H4 | 58 | 69% | 90% |
| 3 | 4-ClC6H4 | 58 | 71% | 89% |
| 4 | 3-ClC6H4 | 48 | 71% | 83% |
| 5 | 3,4-Cl2C6H3 | 46 | 73% | 88% |
| 6 | 4-MeC6H4 | 50 | 65% | 86% |
| 7 | 3-MeC6H4 | 47 | 67% | 92% |
| 8 | 4-MeOC6H4 | 47 | 73% | 93% |
| 9 | 3,4-(CH2O)2C6H3 | 52 | 50% | 89% |
| 10 | 2-Thienyl | 48 | 53% | 92% |
| 11 | 2-Furyl | 56 | 62% | 91% |
A novel, organocatalytic O–H insertion of α-aryl diazoesters into hydroperoxides to afford α-peroxyesters has been reported by Mattson and Fisher.111 This approach builds on earlier work on the S–H and O–H insertion of aryl diazoesters into thiols and carboxylic acids using urea catalyst 81.112 Initial screening indicated that highest yields were recorded when a twofold excess of the diazoester was employed with TBHP acting as the limiting reagent, while increasing the reaction temperature led to a significant decrease in reaction times. The presence of highly activating substituents were found to afford higher yields (Table 63, entries 1–3) whereas less activating groups returned lower yields (entries 4 and 5). Highly deactivated substrates failed to provide the desired product even at elevated temperatures and with prolonged reaction times (entry 10). Variation of the ester groups (entries 7 and 8) or of the peroxide to cumene hydroperoxide (entry 9) was also tolerated.
|
||||
|---|---|---|---|---|
| Entry | R1 | Ar | R2 | Yield |
| 1 | Me | 4-MeOC6H4 | t-Bu | 71% |
| 2 | Me | 2-MeOC6H4 | t-Bu | 63% |
| 3 | Me | 4-TBSOC6H4 | t-Bu | 85% |
| 4 | Me | 4-MeC6H4 | t-Bu | 32% |
| 5 | Me | 4-AcOC6H4 | t-Bu | 50% |
| 6 | Me | 2-Thienyl | t-Bu | 44% |
| 7 | Et | 4-MeOC6H4 | t-Bu | 73% |
| 8 | t-Bu | 4-MeOC6H4 | t-Bu | 73% |
| 9 | Me | 4-MeOC6H4 | Ph(Me)2C | 66% |
| 10 | Me | 4-CF3C6H4 | t-Bu | — |
Tan and co-workers reported the formation of a hydroperoxide oxindole side product in their investigations of enantioselective α-hydroxylation of oxindoles with chiral pentanidiums (Scheme 15).113 They discovered that the ratio of hydroperoxide to alcohol produced was dependent on the amount of molecular oxygen in the reaction. When an oxygen balloon was used instead of air, the hydroperoxide oxindole was isolated in a high yield (85%) and high enantioselectivity (80% ee). It was also observed that more polar solvents favoured the formation of the hydroperoxide oxindole.
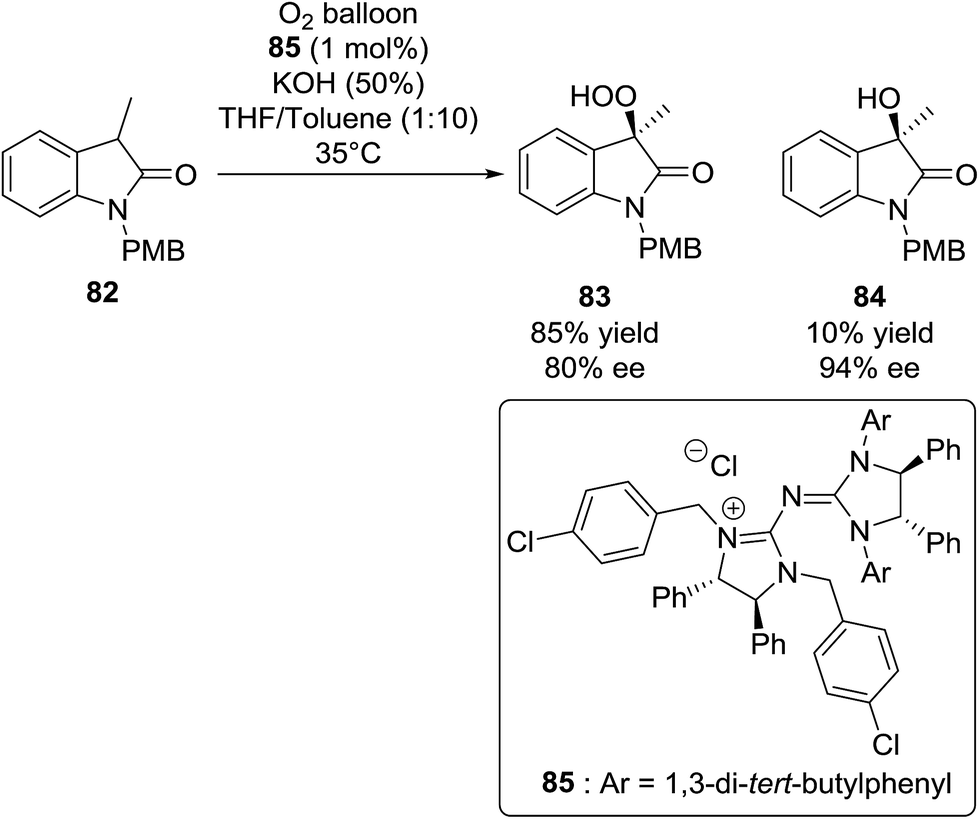 | ||
| Scheme 15 | ||
4 Oxygen- or ozone-mediated reactions
In an effort to synthesise potentially bioactive 1,2,4-trioxanes, Griesbeck and co-workers first prepared a range of β-hydroperoxy alcohols via a solvent-free photooxygenation of chiral allylic alcohols.114 The photooxygenation of a series of allylic alcohols using a polystyrene-divinylbenzene polymer matrix doped with adsorbed porphyrin sensitisers (e.g. tetraphenylporphyrin or TPP) resulted in the formation of a syn or anti diasteromeric mixture of β-hydroperoxy alcohols in good yields (Table 64, entries 1–8). All of the reactions were found to proceed with a preference for the syn-diastereomer.
|
|||
|---|---|---|---|
| Entry | R | Syn–anti d.r. | Yield |
| 1 | Et | 77:23 |
72% |
| 2 | n-Pr | 79:21 |
78% |
| 3 | n-Bu | 79:21 |
78% |
| 4 | i-Bu | 80:20 |
77% |
| 5 | c-Pr | 62:38 |
80% |
| 6 | c-Hex | 88:12 |
65% |
| 7 | CH2CHCH2 |
75:25 |
69% |
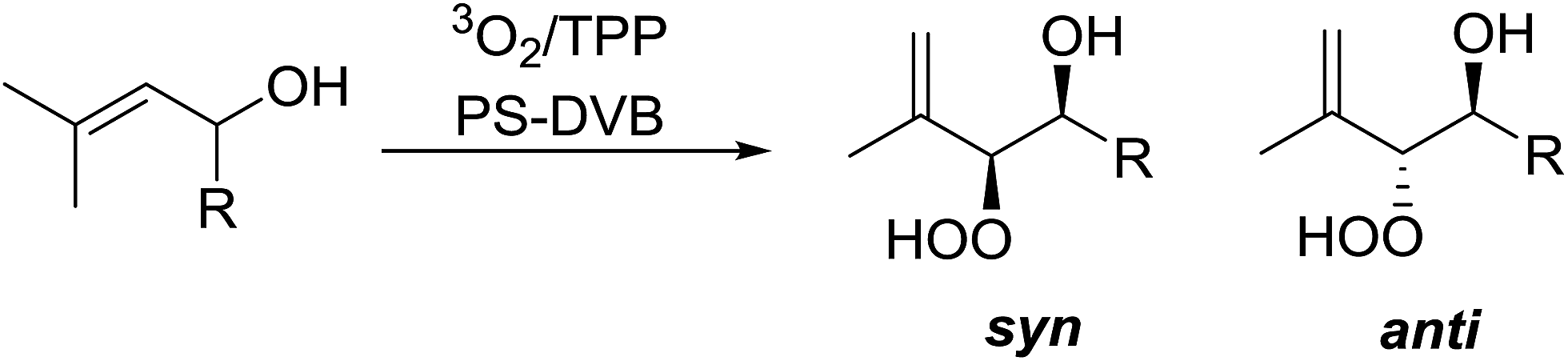
In subsequent work, Griesbeck further expanded upon this approach to encompass the synthesis of perorthoesters and cycloperoxides. This involved a three-step process with the key step comprising the peroxidation of unsaturated aldol adducts by way of a singlet oxygen ene reaction (Scheme 16).115 The photooxygenation proceeded with complete conversion and without any overperoxidation or side products.
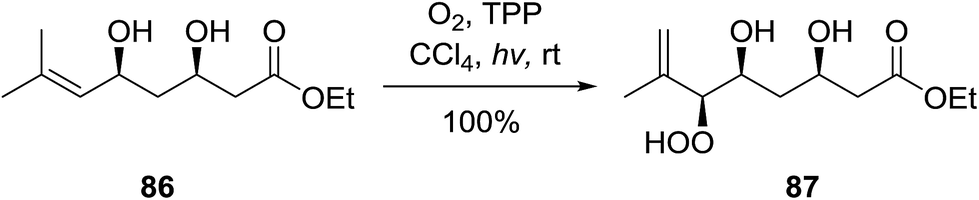 | ||
| Scheme 16 | ||
Gollnick and Knutzen-Mies have conducted an extensive study on the photooxygenation of 2,3-dihydrofurans with singlet oxygen in various solvents.116 They found that the highest ratios of acyclic hydroperoxides were obtained when benzene was used as the reaction medium (Table 65, entries 1–4). These allylic hydroperoxides ultimately decomposed into the corresponding furans with the concomitant loss of hydrogen peroxide. The allylic hydroperoxides were isolated alongside dioxetane side products which also decomposed slowly at ambient temperature to form the corresponding dicarbonyl compounds. In later work, Tung et al. applied this methodology to the synthesis of α,β-unsaturated γ-lactones.117
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Product ratio | |
| 89 | 91 | |||
| 1 | H | H | 32% | 68% |
| 2 | H | CH3 | 52% | 48% |
| 3 | CH3 | CH3 | 82% | 13% |
| 4 | CO2CH3 | CH3 | 4% | 82% |
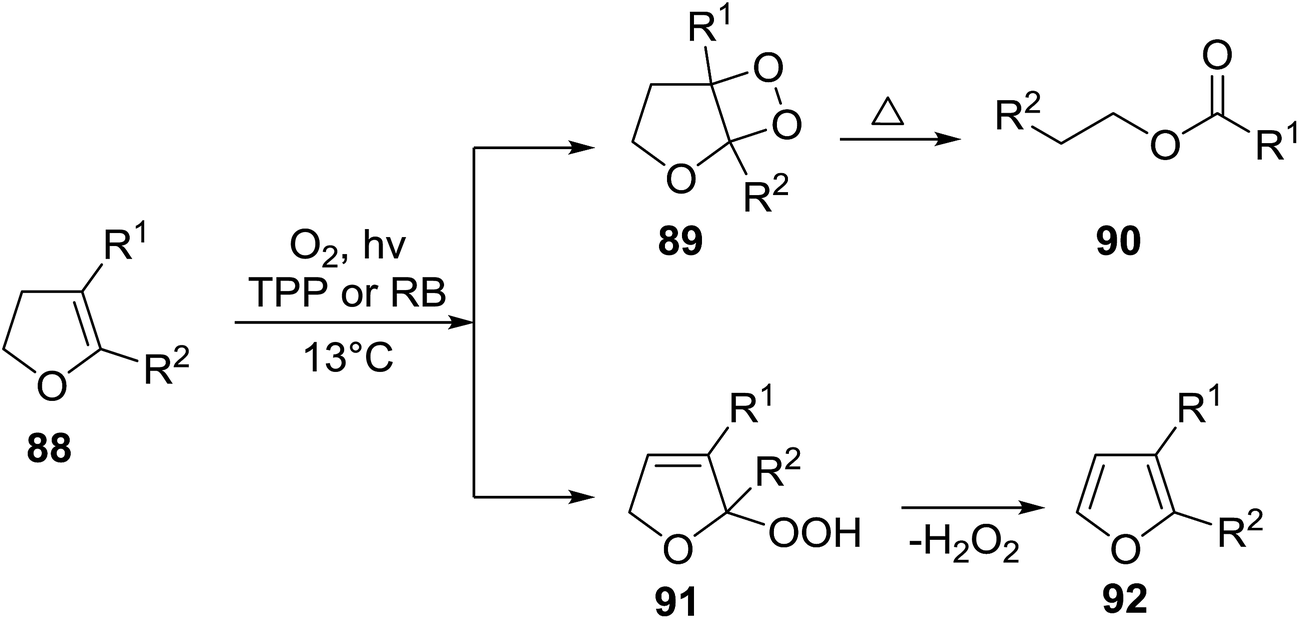
Alberti and Orfanopoulos have conducted an exhaustive study on the choice of solvent and sensitizer on the allylic oxyfunctionalisation of alkenes with singlet oxygen.118 They found that use of aprotic solvents saw a small preference for hydrogen abstraction from the methyl group cis- to the trifluoromethyl substituent (Table 66, entries 1–4). By contrast, in protic solvents, the preference for hydrogen abstraction from this group increases significantly (entries 5–7).
|
|||
|---|---|---|---|
| Entry | Solvent | Sensitiser | 94:95 |
| 1 | CCl4 | TPP | 46:54 |
| 2 | CHCl3 | TPP | 48:52 |
| 3 | Acetone | RB | 49:51 |
| 4 | CH3CN | RB | 49:51 |
| 5 | CF3CH2OH | MB | 40:60 |
| 6 | MeOH | MB | 42:58 |
| 7 | MeOH/D2O (4:1) |
MB | 42:58 |
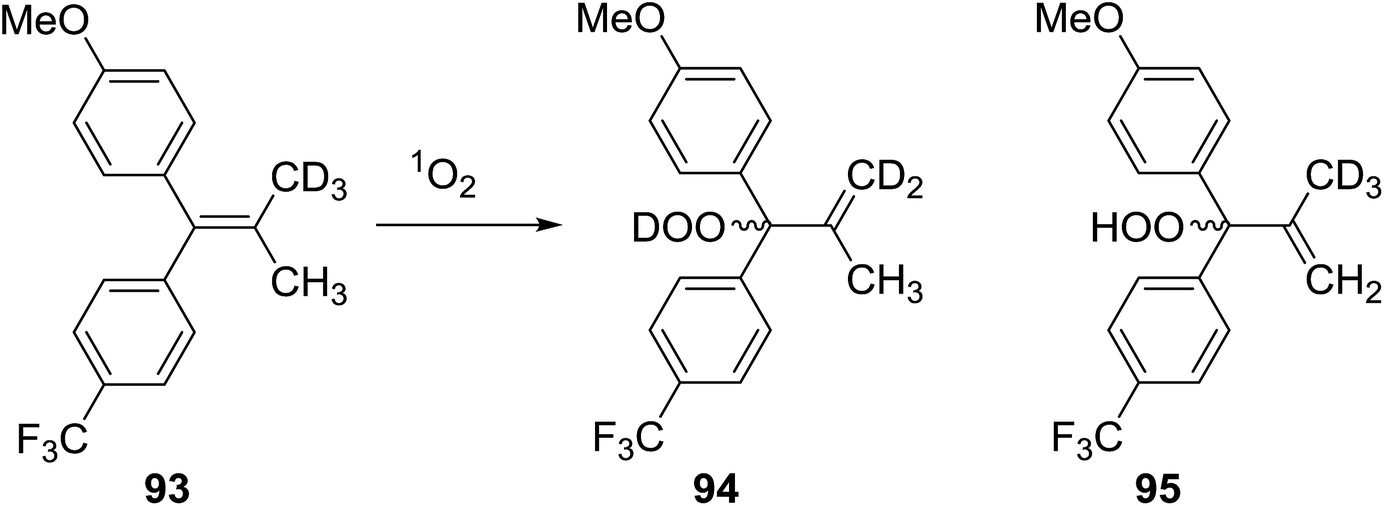
Stratakis et al. have found that the regio- and diastereoselectivity of the ene hydroperoxidation reaction of (R)-(−)-α-phellandrene can be significantly altered on addition of zeolites (Table 67).119 Reaction of singlet oxygen with (R)-(−)-α-phellandrene 96 affords the two diasteromeric endoperoxides 97 and 98 as the major products along with hydroperoxides 99 and 100 (entry 1). When the same substrate is instead subjected to intra-zeolite photo-oxygenation, exomethylene ene adduct 100 is a significant product. The authors suggest that the cation–π interactions within the confined environment of the Na–Y zeolite can affect the profile of the ene pathway to afford 100 as the major ene product. A similar approach has been used by other groups for substrates such as chiral alkenes and isobutenylarenes.120–122
|
|||||
|---|---|---|---|---|---|
| Entry | Conditions | Yield 97 | Yield 98 | Yield 99 | Yield 100 |
| 1 | i-PrOH/Rose Bengal | 30% | 26% | 2% | 1.5% |
| 2 | Thionin/Na–Y | 33% | 15% | 5% | 30% |
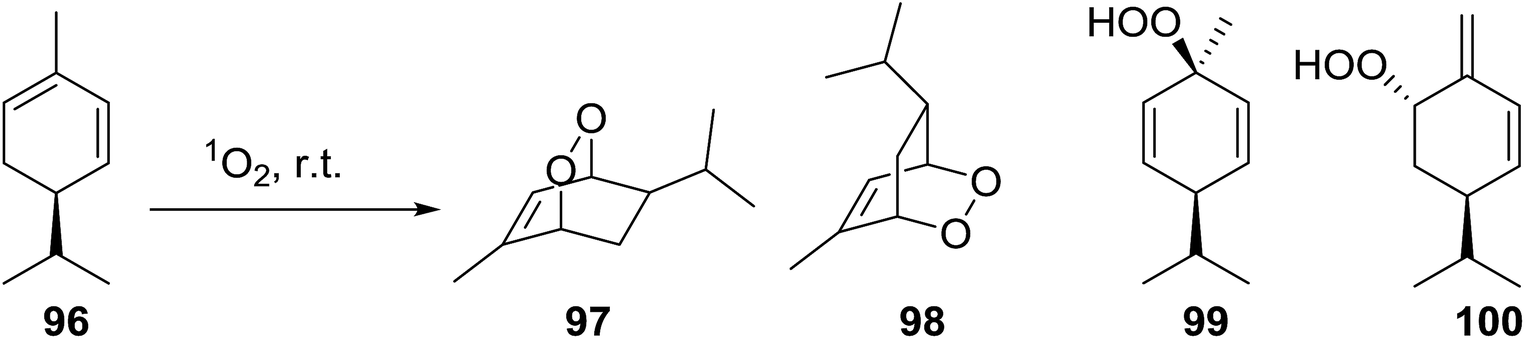
Ramamurthy et al. reported unprecedented selectivity during the singlet oxygen-mediated oxidation of olefins included within dye-exchanged zeolites.123,124 Following irradiation in a Kimax tube containing a zeolite and the alkene in a hexane slurry, the hydroperoxide products were isolated in yields of 65% to 75% (Table 68). The ratio of secondary to tertiary products was strongly influenced by the choice of zeolite (entries 1–5). The authors speculate that the alkene is adsorbed to the surface of the zeolite via a π–cation interaction. Accordingly, if the R-group is bulky, then steric strain will cause internal rotation of the substrate, placing the methylene hydrogens away from the incoming singlet oxygen and no tertiary hydroperoxide will be formed. By contrast, with larger cations such as cesium, the alkene may be forced to adsorb further from the surface, thus reducing the steric strain and allowing for the formation of small amounts of the tertiary hydroperoxide. Similar findings have been made by other groups in this field.125
|
||||
|---|---|---|---|---|
| Entry | R | Condition | Yield 102 | Yield 103 |
| 1 | H | Thionine/CH3CN | 51% | 49% |
| RB/CH3CN | 50% | 50% | ||
| NaY/thionine | 72% | 28% | ||
| 2 | CH3 | Thionine/CH3CN | 40% | 60% |
| NaY/thionine | 100% | — | ||
| 3 | Ph | Thionine/CH3CN | 50% | 50% |
| RB/CH3CN | 54% | 46% | ||
| LiY/thionine | 100% | — | ||
| NaY/thionine | 85% | 15% | ||
| RbY/thionine | 80% | 20% | ||
| CsY/thionine | 66% | 34% | ||
| 4 | 4-Me–C6H4 | RB/CH3CN | 56% | 44% |
| LiY/thionine | 100% | — | ||
| NaY/thionine | 100% | — | ||
| RbY/thionine | 94% | 6% | ||
| CsY/thionine | 92% | 8% | ||
| 5 | 4-MeO–C6H4 | RB/CH3CN | 52% | 48% |
| LiY/thionine | 100% | — | ||
| NaY/thionine | 100% | — | ||
| RbY/thionine | 100% | — | ||
| CsY/thionine | 100% | — | ||
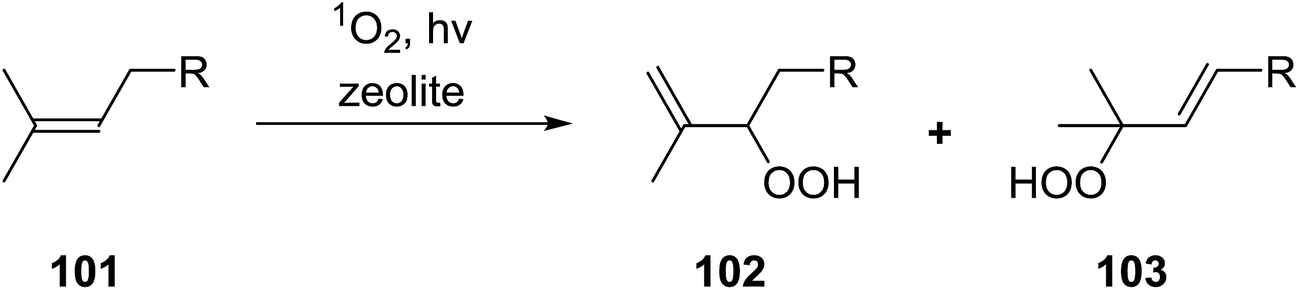
Dussault et al. have demonstrated that allylstannanes and allylsilanes are effective directing groups for controlling the regio- and stereoselectivity of a variety of singlet oxygen addition reactions.126 Allylstannanes were found to be the better directing group as exemplified by the photooxygenation of tributylstannyl enoate (R)-104 (R = Bu) with tetraphenylporphyrin as the sensitizer (Table 69). This oxygenation reaction yielded β-stannyl hydroperoxide (R)-105 with smaller amounts of a 1,2-dioxalane arising from migration of the tri-butyltin group (entries 1 and 2). By contrast, oxygenation of triphenylstannyl enoate (S)-104 (R = Ph) furnished only the H–ene product, possibly reflecting the relative migratory aptitudes of the triphenyl- and tri-butylstannyl groups (entry 3). It is interesting to note that the oxygenation of allylstannanes proceeds preferentially with inversion of chirality (entries 1–3).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | R | Sensitizer | Yield | ee |
| 1 | CH2Cl2 | Bu | TPP | 52% | 80% (R) |
| 2 | CH2Cl2/MeCN | Bu | RB | 52% | 80% (R) |
| 3 | CH2Cl2/MeCN | Ph | RB | 60% | 86% (S) |
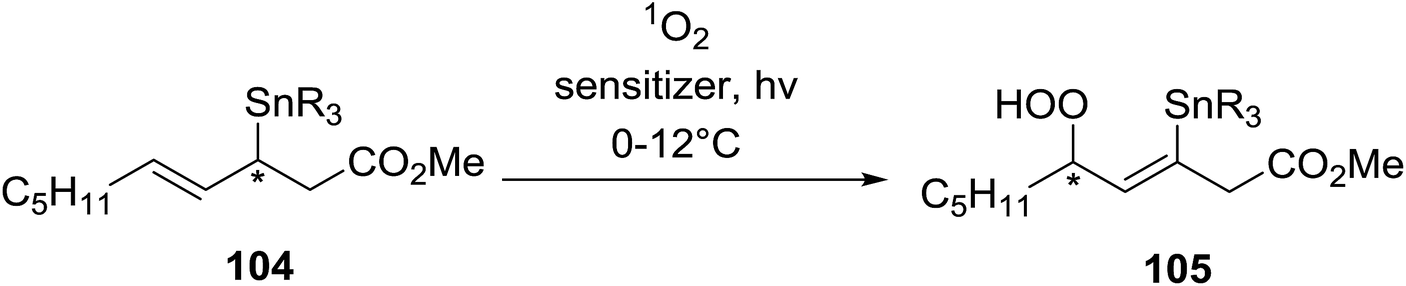
This work was further developed to include functionalised allylsilanes (Table 70). Oxygenation of the E-allylsilanes led to the predominant formation of Z-hydroperoxyvinylsilanes, derived from the H-ene decomposition of the intermediate perepoxide. A reversal in selectivity was observed when Z-allylsilanes were employed (entries 4 and 5) with formation of the E-alkene products being favoured. The reversal in regioselectivity is likely due to the influence of 1,3-allylic strain. A similar methodology has also been successfully employed by others in the synthesis of lophine peroxides.127
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Z-Vinylsilane 107 | E-Alkene 108 |
| 1 | (E)-C4H9 | CH2CO2Me | SiMe3 | 40% | 12% |
| 2 | (E)-C3H7 | CO2Me | SiMe3 | 38% | — |
| 3 | (E)-C3H7 | CH2OH | SiMe3 | 60% | — |
| 4 | (Z)-C3H7 | CO2Et | SiPhMe2 | 15% | 45% |
| 5 | (Z)-C3H7 | CH2OH | SiPhMe2 | 14% | 44% |
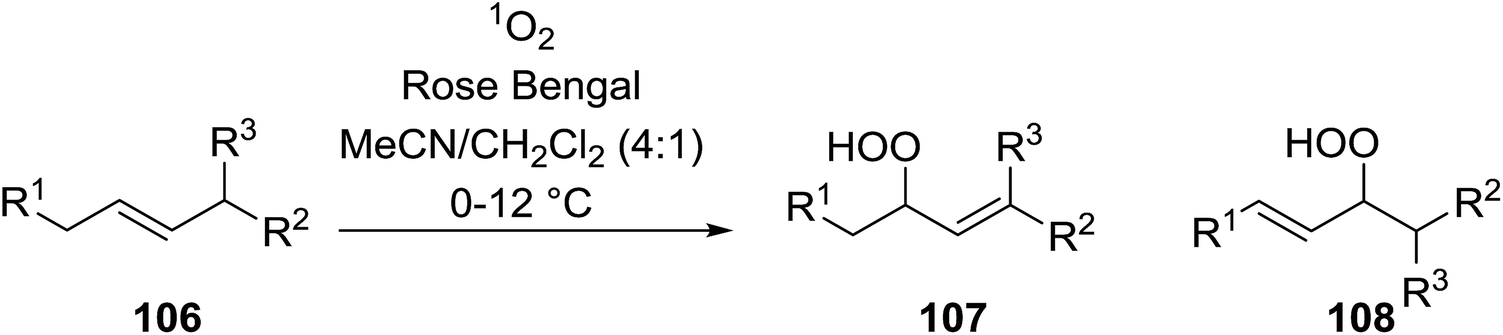
Tomooka et al. has reported the successful peroxidation of γ-silyl allylic alcohols employing ozone as the oxidant.128 Peroxidation of allylic alcohols proceeded smoothly in ethyl acetate affording the target peroxides in good yields following a reductive work-up with sodium borohydride (Table 71, entries 1 and 2). Silyl peroxides were also obtained from silyl ethers indicating that a free hydroxyl group was not essential (entries 3–7). For substrates bearing an α-stereogenic centre, the reaction exhibited high diastereoselectivity with anti-selectivity becoming more pronounced with increasing steric bulk (entries 4–7). In the case of a bulky tert-butyl substitutent, the anti-product was formed exclusively (entry 7). In the absence of a reductive work-up, α-formyl silyl peroxides were formed preferentially.129
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | R4 | Yield (%) | d.r. |
| 1 | Me | Me | H | t-BuMe2 | 73% | |
| 2 | Me | Me | H | t-BuPh2 | 70% | |
| 3 | H | H | TBS | t-BuPh2 | 90% | |
| 4 | Me | H | TMS | i-Pr3 | 67% | 58:42 |
| 5 | Et | H | TMS | i-Pr3 | 78% | 67:33 |
| 6 | c-Hex | H | TMS | i-Pr3 | 84% | 72:28 |
| 7 | t-Bu | H | TMS | i-Pr3 | 71% | >99:1 |
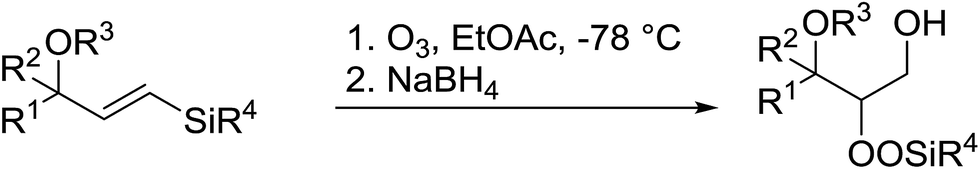
Dussault has demonstrated the application of ozone-mediated peroxidation in the synthesis of peroxyplakoric acids.130 Methanolic ozonolysis of the methylene group in 109 furnished 110 in 88% yield as a 1:1 mixture of hydroperoxyacetals (Scheme 17).
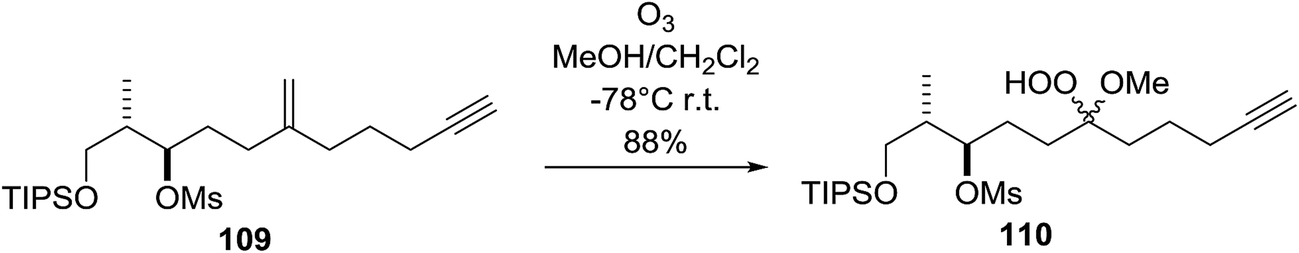 | ||
| Scheme 17 | ||
Andreev observed that the oxidation of pyrrole, furan and thiophene by ozone in a solution of glacial acetic acid returned primarily peroxide-containing compounds although actual yields are not reported (Scheme 18).131 The reaction is believed to proceed by a 1,3-dipolar cycloaddition with the formation of a monoozonide intermediate. The intermediate subsequently forms a diozonide which ring opens to form a hydroperoxy peroxide.
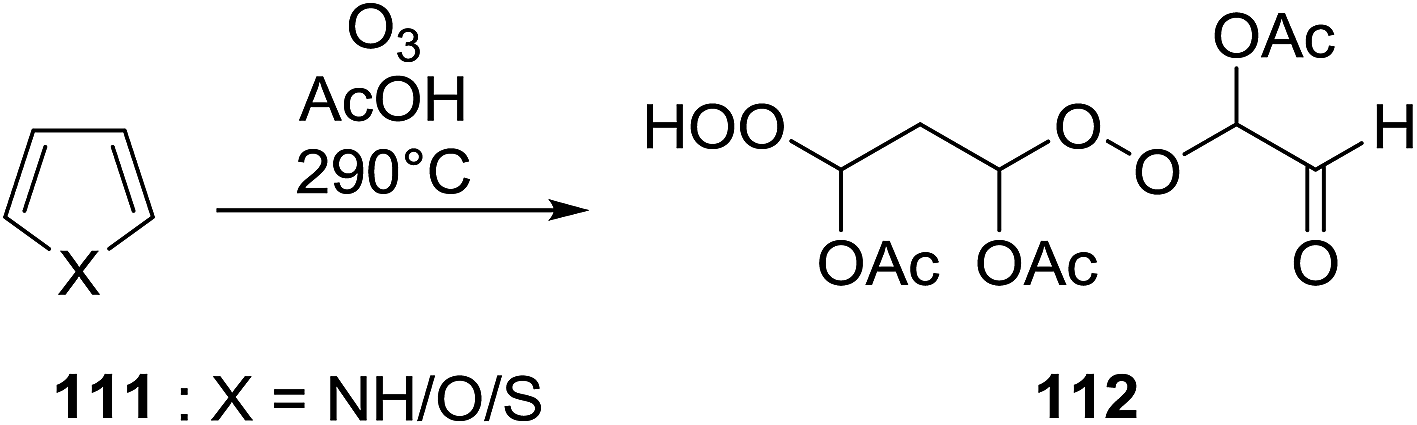 | ||
| Scheme 18 | ||
5 Miscellaneous reactions
Research into C–H activation has attracted the attention of many groups over the past decade.132 Wan et al. have discovered that tetra-butylammonium iodide effectively catalyses the C–H oxidation of aldehydes with TBHP as oxidant.133 Iodide was critical for efficient conversion, as neither tetra-butylammonium bromide nor tetra-butylammonium chloride furnished the target tert-butyl peresters. A wide variety of aromatic aldehydes were tolerated irrespective of the nature of the substituent or the substitution pattern (Table 72, entries 1–18). Both heteroaromatic (entry 18) and aliphatic (entry 19) aldehydes also underwent C–H activation, albeit in more moderate yields for the latter. The authors ascribe a radical mechanism to the transformation as evidenced by the formation of an acylated-adduct on addition of a radical trapping agent (i.e. TEMPO) to a mixture of 2-naphthaldehyde, TBHP and tetra-butylammonium iodide.
|
||
|---|---|---|
| Entry | R | Yield |
| 1 | Ph | 84% |
| 2 | 4-MeC6H4 | 72% |
| 3 | 3-MeC6H4 | 67% |
| 4 | Naphthyl | 88% |
| 5 | 4-MeOC6H4 | 52% |
| 6 | 4-MeSC6H4 | 69% |
| 7 | 4-FC6H4 | 75% |
| 8 | 4-BrC6H4 | 77% |
| 9 | 4-ClC6H4 | 84% |
| 10 | 3-ClC6H4 | 68% |
| 11 | 2-ClC6H4 | 79% |
| 12 | 2-BrC6H4 | 75% |
| 13 | 2-FC6H4 | 75% |
| 14 | 3-PhOC6H4 | 90% |
| 15 | 2-F3CC6H4 | 58% |
| 16 | 4-TsOC6H4 | 67% |
| 17 | 4-MeO2CC6H4 | 54% |
| 18 | Thienyl | 84% |
| 19 | Cyclohexyl | 43% |
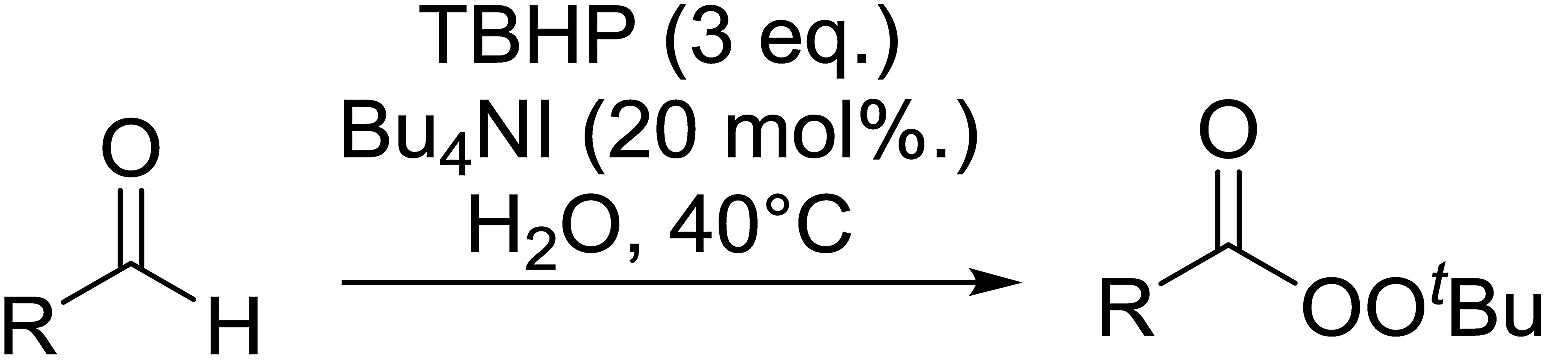
A novel method for the synthesis of peroxides based on the CH activation of malonates, β-ketoesters and cyanoacetic esters has been described by Terent'ev and co-workers.134 Peroxides were prepared in 31–89% yields following treatment with a combination of Bu4NI/TBHP (Table 73). TBHP plays a dual role by acting both as an oxidant and an oxygen source for the C–O coupling. The yields of peroxides prepared from β-ketoesters (entries 9–12) or cyanoacetic esters (entries 13–16) were approximately half those obtained from malonate substrates (entries 1–8). Attempts to peroxidise β-diketones proved unsuccessful.
|
|||
|---|---|---|---|
| Entry | R1 | R2 | Yield |
| 1 | CO2Et | Me | 76% |
| 2 | CO2Et | Et | 73% |
| 3 | CO2Et | n-Bu | 83% |
| 4 | CO2Et | Bn | 85% |
| 5 | CO2Et | CH2–CHCH2 |
83% |
| 6 | CO2Et | Ph | 89% |
| 7 | CO2Et | (CH2)2COMe | 83% |
| 8 | CO2Et | (CH2)2CN | 83% |
| 9 | C(O)Me | Me | 38% |
| 10 | C(O)Me | n-Bu | 47% |
| 11 | C(O)Me | Bn | 35% |
| 12 | C(O)Me | (CH2)2COMe | 33% |
| 13 | CN | n-Bu | 35% |
| 14 | CN | Bn | 35% |
| 15 | CN | (CH2)2CN | 31% |
| 16 | CN | (CH2)2CO2Et | 42% |
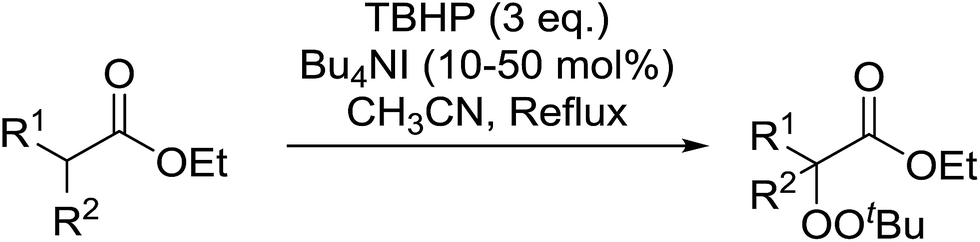
Yu and Shen discovered that tetra-n-butylammonium iodide is an effective catalyst for the C–H peroxidation of amides.135 The reaction requires an excess of TBHP as oxidant and a substoichiometric amount of tetra-n-butylammonium to effect peroxidation adjacent to the nitrogen amide in a variety of amides. Compounds containing electron-rich or electron-poor aroyl groups furnished the target tert-butylperoxyamido acetals in good yields (Table 74, entries 1–4). The transformation of napthalene- and thiophene-based substrates (entries 5 and 6) was similarly successful. Variation of N-benzyl substituents was also investigated, and the methodology was found to be compatible with a variety of aromatic rings bearing electron-donating and electron-withdrawing groups (entries 9–14) as well as pyridyl and vinyl groups (entries 15 & 16). However, attempted peroxidation of N-butylbenzamide proved fruitless. Changes in the reaction temperature or solvent were accompanied by decreased yields. The reaction most likely proceeds by a radical mechanism as evidenced by a significant reduction in yields on addition of TEMPO, a known radical scavenger.
|
|||
|---|---|---|---|
| Entry | R1 | R2 | Yield |
| 1 | 2-MeC6H4 | Ph | 88% |
| 2 | 3-MeC6H4 | Ph | 76% |
| 3 | 4-MeC6H4 | Ph | 73% |
| 4 | 4-ClC6H4 | Ph | 96% |
| 5 | 2-Naphthyl | Ph | 79% |
| 6 | 2-Thienyl | Ph | 87% |
| 7 | Ph–CHCH |
Ph | 80% |
| 8 | i-Pr | Ph | 79% |
| 9 | Ph | 2-MeOC6H4 | 94% |
| 10 | Ph | 3-MeOC6H4 | 76% |
| 11 | Ph | 4-MeOC6H4 | 90% |
| 12 | Ph | 4-MeC6H4 | 90% |
| 13 | Ph | 4-FC6H4 | 66% |
| 14 | Ph | 4-ClC6H4 | 74% |
| 15 | Ph | 2-Pyridyl | 80% |
| 16 | Ph | –CHCH2 |
65% |
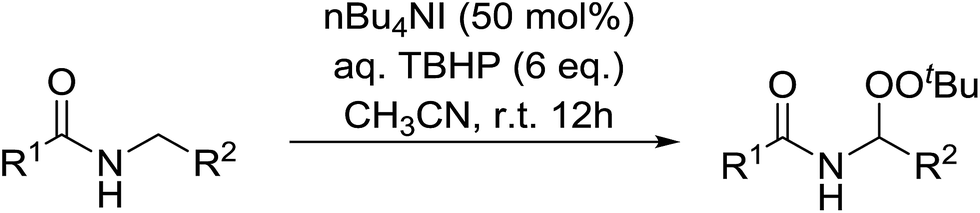
We have previously described the work of Patel et al. on the copper-catalysed cycloalkylation–peroxidation of alkenes (Section 2.2). Recently, this group has successfully developed a metal-free difunctionalisation of 3-acetylcoumarins using cycloalkanes and TBHP.136 A range of cycloalkylated α-peroxy products were formed in moderate to good yields irrespective of type of 3-acyl group (Table 75, entries 1–4) or of the ring substituents present (entries 5–11) in various coumarins. Substituting cyclohexane with cyclopentane furnished the desired product in 46% yield (entry 12). The use of other Csp3–H hydrocarbon solvents in lieu of cyclohexane (e.g. toluene) gave no benzylated α-peroxy product, instead resulting in a mixture of benzaldehyde and benzoic acid.
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | n | Yield |
| 1 | H | Me | 1 | 65% |
| 2 | H | Et | 1 | 65% |
| 3 | H | n-Bu | 1 | 59% |
| 4 | H | Ph | 1 | 60% |
| 5 | 6-Cl | Me | 1 | 64% |
| 6 | 6-Br | Me | 1 | 51% |
| 7 | 6-NO2 | Me | 1 | 66% |
| 8 | 7-MeO | Me | 1 | 44% |
| 9 | 8-MeO | Me | 1 | 65% |
| 10 | 6,8-Dichloro | Me | 1 | 57% |
| 11 | 6,8-Dibromo | Me | 1 | 50% |
| 12 | H | Me | 0 | 46% |
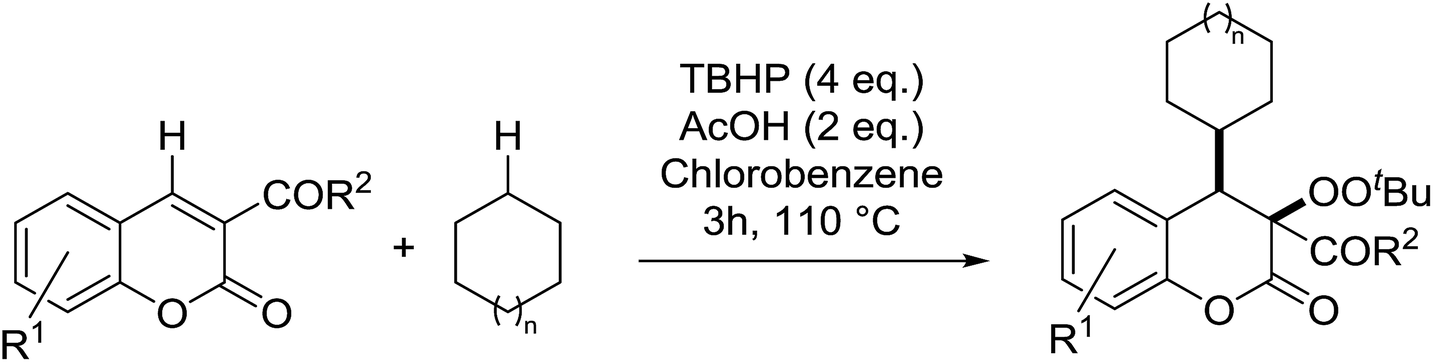
The metal-mediated conversion of carbonyls to gem-dihydrohydroperoxides is a well known process (Section 2.4). Liebscher and co-workers have employed camphorsulfonic acid (CSA) to catalyse the conversion of aldehydes directly to the corresponding gem-dihydroperoxides on treatment with aqueous hydrogen peroxide in a biphasic system with ether.137 The use of a biphasic system is crucial, as the application of ethereal hydrogen peroxide to aldehydes leads instead to perhydrates.138 Both acyclic (Table 76, entries 1–7) and cyclic (entry 6) aliphatic aldehydes afforded the desired products in moderate to good yields. The highest yields were recorded from aromatic aldehydes (entry 7).
|
|||
|---|---|---|---|
| Entry | R | Time (h) | Yield |
| 1 | Me | 16 | 34% |
| 2 | Et | 16 | 42% |
| 3 | n-Pr | 22 | 63% |
| 4 | i-Pr | 21 | 64% |
| 5 | t-Bu | 40 | 28% |
| 6 | n-Pen | 16 | 44% |
| 7 | n-Hep | 18 | 33% |
| 6 | Cy | 16 | 44% |
| 7 | Ph | 16 | 77% |
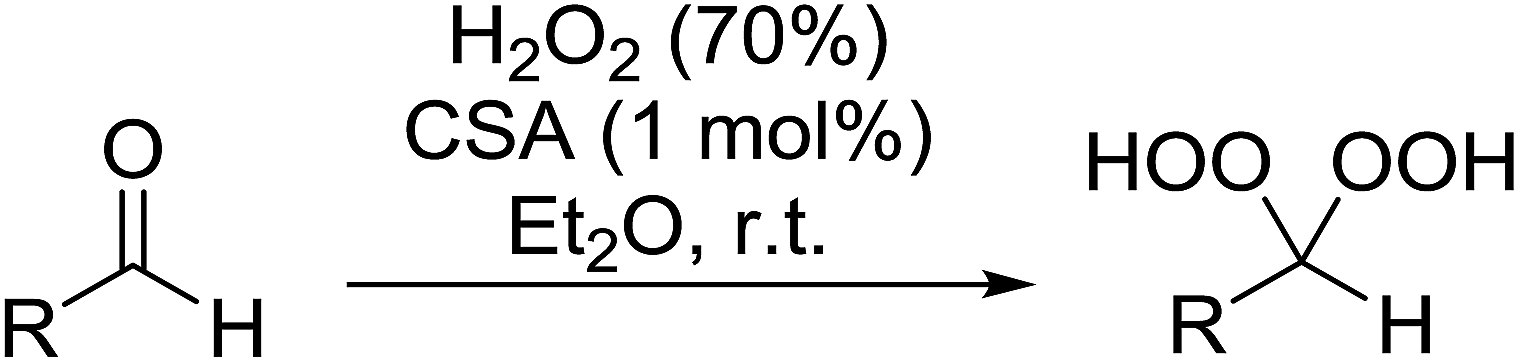
In their search for novel, herbicidal tetraoxanes, Barbosa et al. employed a combination of 30% hydrogen peroxide with concentrated hydrochloric acid as the catalyst to effect the transformation of carbonyls to dihydroperoxides.139 While these conditions were generally compatible with acyclic ketones (Table 77, entries 1–3), reactions involving aldehydes proved more capricious (entries 5 and 6) with no product being isolated from electron rich substrates (entry 7). Cyclic ketones were also tolerated under these conditions (entries 8–11).
|
|||
|---|---|---|---|
| Entry | R1 | R2 | Yield |
| 1 | Me | Me | 62% |
| 2 | Et | Et | 82% |
| 3 | i-Pr | i-Pr | 71% |
| 4 | n-Bu | n-Bu | 55% |
| 5 | Ph | H | 86% |
| 6 | 4-Et–C6H4 | H | 91% |
| 7 | 4-MeO–C6H4 | H | — |
| 8 | Cyclopentanone | 77% | |
| 9 | Cyclohexanone | 75% | |
| 10 | Cycloheptanone | 83% | |
| 11 | Cyclooctanone | 36% | |
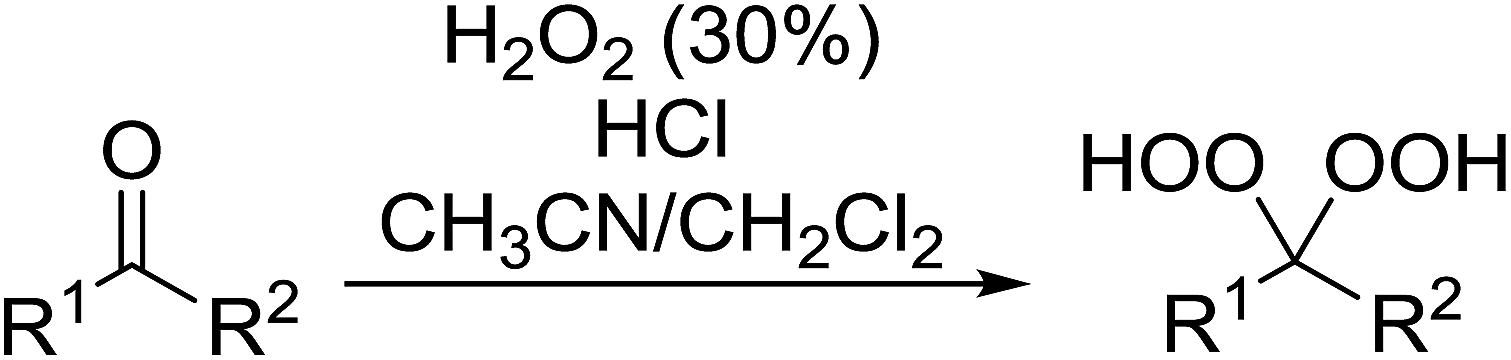
Terent'ev developed a convenient procedure for the preparation of geminal bishydroperoxides by the sulfuric acid-catalysed reaction of ketones with hydrogen peroxide in THF.140 gem-Bishydroperoxides were synthesised from five- to seven-membered cycloalkanones without the need for additional purification in 80–95% yields (Table 78, entries 4–8). Larger ring sizes were accompanied by decreased yields (entries 9 and 10). Acyclic substrates were also transformed under these conditions (entries 1–3).
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Time (h) | H2O2 (eq.) | H2SO4 (eq.) | Yield |
| 1 | CH3 | Iso-butyl | 3 | 10 | 0.5 | 67% |
| 2 | C5H11 | C5H11 | 6 | 10 | 1.0 | 43% |
| 3 | CH3 | C4H9 | 2 | 10 | 0.5 | 72% |
| 4 | Cyclopentanone | 2 | 8 | 0.3 | 80% | |
| 5 | Cyclohexanone | 2 | 8 | 0.3 | 81% | |
| 6 | 4-Methylcyclohexanone | 2 | 4 | 0.3 | 90% | |
| 7 | 4-t-Butylcyclohexanone | 2 | 8 | 0.3 | 95% | |
| 8 | Cycloheptanone | 3 | 10 | 0.4 | 86% | |
| 9 | Cyclooctanone | 3 | 14 | 0.8 | 39% | |
| 10 | Cyclododecanone | 6 | 14 | 0.8 | 12% | |
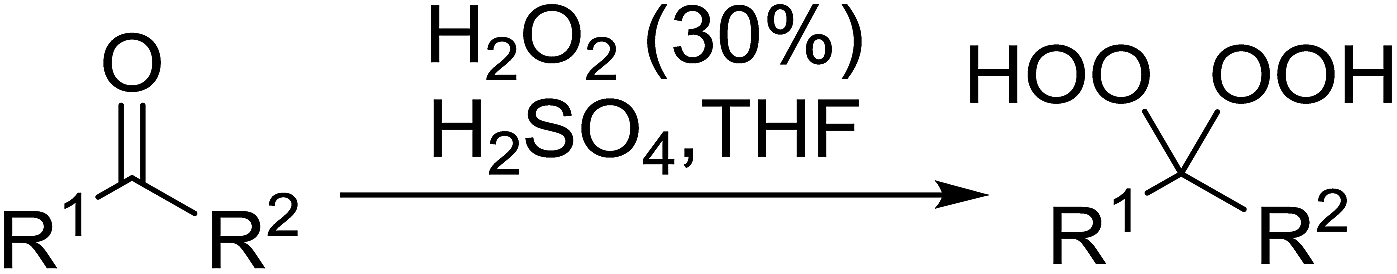
Iskra et al. utilised iodine as a catalyst in the formation of gem-dihydroperoxides from both ketones and aldehydes.141,142 Iodine plays a double role: it both enhances the electrophilic character of the carbonyl atom and also increases the nucleophilic character of hydrogen peroxide. In the absence of iodine, reactions were found to be sluggish or not to proceed at all. Iodine-catalysed hydroperoxidation was effective for acyclic, aliphatic ketones (Table 79, entries 1–4) although lower yields were observed with acyclic, aromatic ketones (entry 4). Hydroperoxidation with cyclic ketones was facile and the corresponding gem-dihydroperoxides were recovered in good to excellent yields (entries 5–8).
|
|||
|---|---|---|---|
| Entry | R1 | R2 | Yield |
| 1 | Et | n-Hep | 91% |
| 2 | Me | n-Hep | 96% |
| 3 | n-Bu | n-Bu | 78% |
| 4 | Ph | Me | 78% |
| 5 | Cyclopentanone | 70% | |
| 6 | Cycloheptanone | 90% | |
| 7 | Cyclodecanone | 60% | |
| 8 | 2-Adamantanone | 92% | |
| 9 | Ph | H | 55% |
| 10 | 4-MeC6H4 | H | 78% |
| 11 | 4-MeOC6H4 | H | 78% |
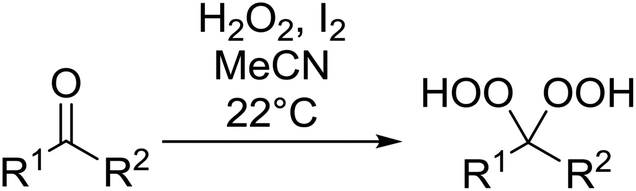
Other peroxide sources, such as TBHP, could be substituted for hydrogen peroxide, allowing for the preparation of various alkylperoxide products (Scheme 19).
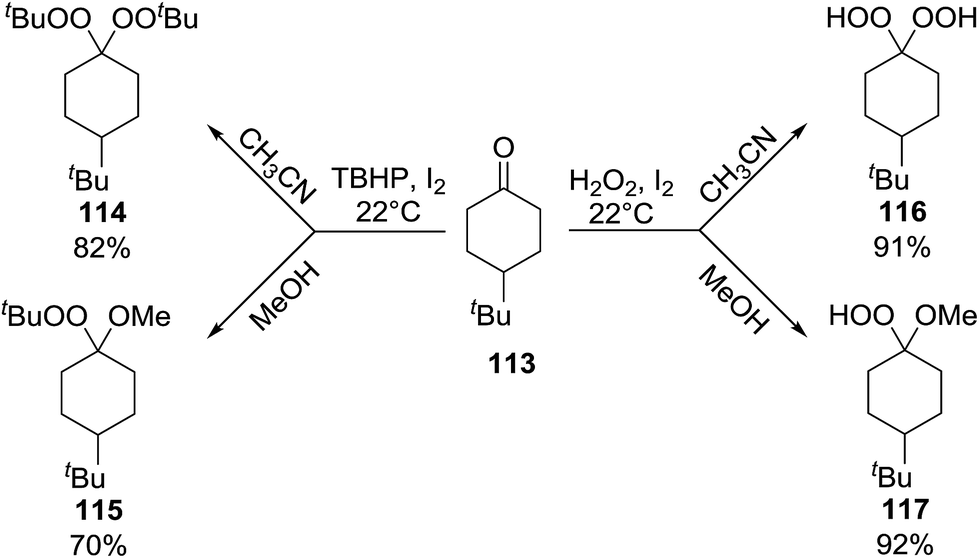 | ||
| Scheme 19 | ||
Building on earlier observations by Itoh et al.,143 Iskra and co-workers reported that cyclic and acyclic ketones could be readily converted to gem-dihydroperoxides in 72–99% yields using aqueous hydrogen peroxide without the need for a catalyst (Table 80, entries 1–12). They discovered the reaction outcome was highly dependent on the concentration of the hydrogen peroxide solution, and that maximum yields were achieved following azeotropic distillation of water from an acetonitrile solution of 30% aqueous hydrogen peroxide and the starting ketone. It is likely that the process of azeotropic evaporation facilitates in situ concentration of 30% hydrogen peroxide to ∼100% hydrogen peroxide and that the elimination of water is accompanied by simultaneous peroxidation of the carbonyl group. Recent computational studies on the catalyst-free preparation of gem-dihydroperoxides offer additional insight into this chemistry.144
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | H2O2 (eq.) | Time (h) | Temp. (°C) | Yield |
| 1 | Me | C7H15 | 8 | 24 | 40 | 82% |
| 2 | n-Bu | n-Bu | 8 | 24 | 40 | 86% |
| 3 |  |
4 | 2 | r.t. | 80% | |
| 4 |  |
4 | 2 | r.t. | 70% | |
| 5 |  |
4 | 5 | r.t. | 89% | |
| 6 | 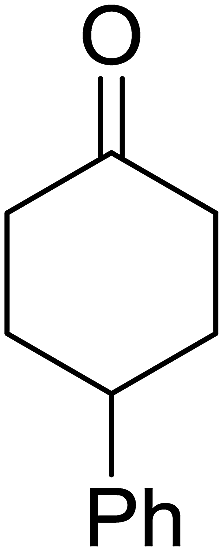 |
8 | 24 | 40 | 99% | |
| 7 | 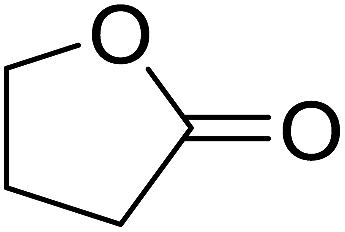 |
4 | 16 | r.t. | 48% | |
| 8 | 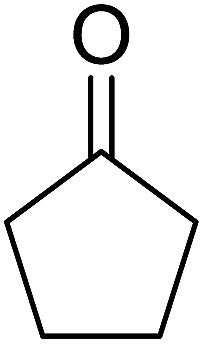 |
4 | 24 | r.t. | 94% | |
| 9 |  |
8 | 24 | 40 | 78% | |
| 10 |  |
8 | 24 | r.t. | 72% | |
| 11 |  |
8 | 24 | 40 | 95% | |
| 12 |  |
8 | 24 | 40 | 94% | |
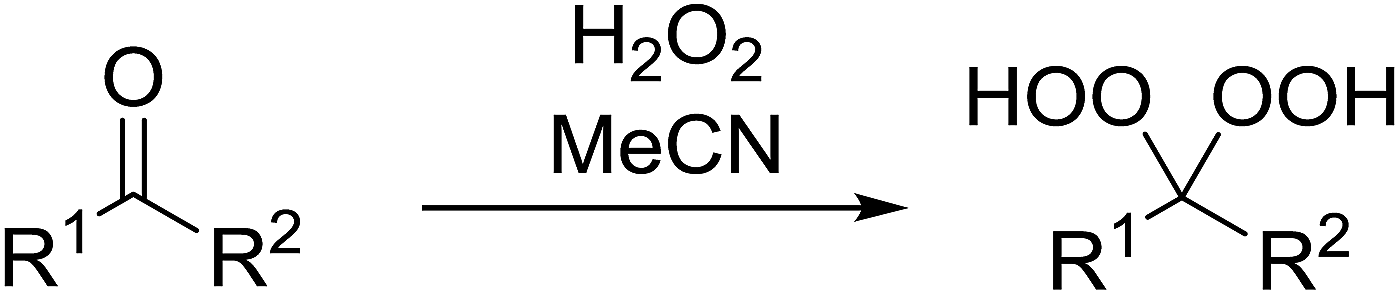
Wu and co-workers have successfully employed urea-hydrogen peroxide (UHP) as a peroxide source in the synthesis of bioactive sprioperoxides.145 It was found that hydroperoxidation of the ketone group was facile by virtue of a γ-hydroxy substituent, as it resulted in the formation of a stable, five-membered hemiketals (Table 81, entries 1–3). Subsequent addition of mild base to the hemiketals effected an intramolecular Michael addition and formation of stable, spiroperoxide products.
|
|||
|---|---|---|---|
| Entry | R | Solvent | Yield |
| 1 | CO2Et | MeOH | 82% |
| 2 | CO2Bn | MeOH | 75% |
| 3 | NO2 | DME | 35% |
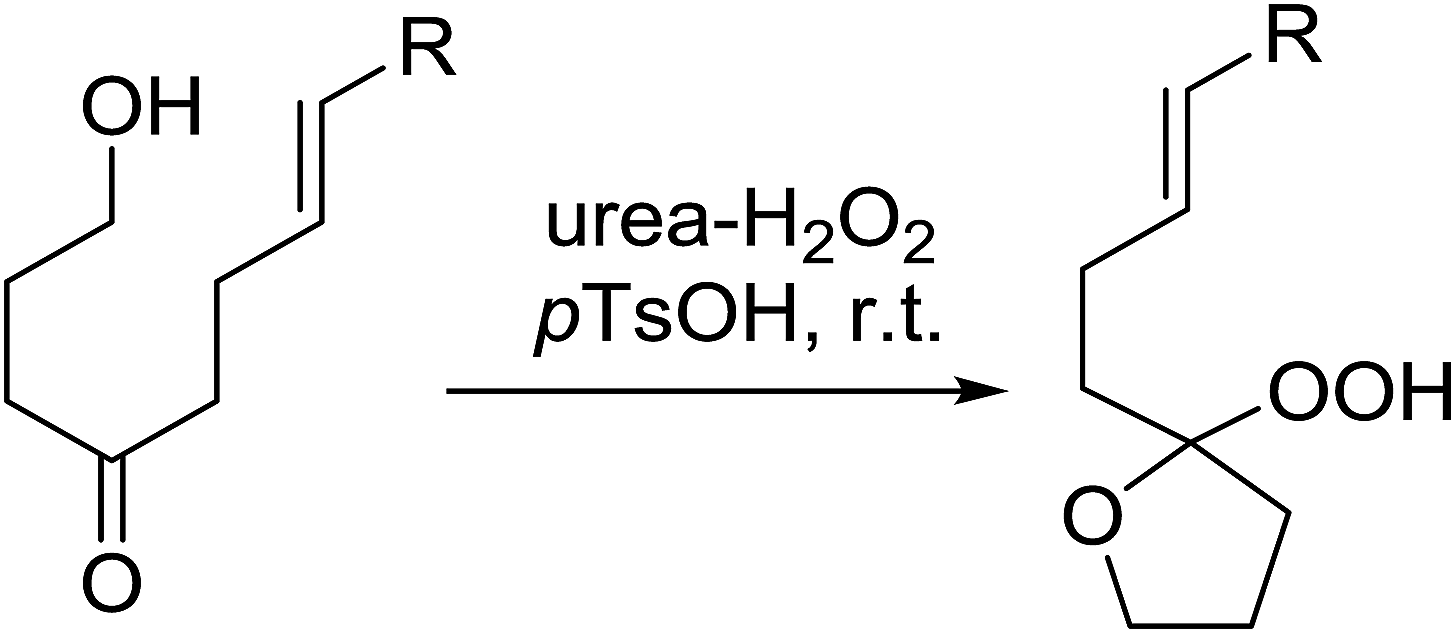
Singh et al. used a combination of ethereal hydrogen peroxide and triflic acid to convert quinoxaline acetals to the corresponding hydroperoxy-hemiacetals.146 The use of mild conditions allowed for the preparation of the target compounds in good yields of 75% and 70% for the phenyl and 1-napthyl substituted derivatives respectively (Scheme 20).
 | ||
| Scheme 20 | ||
Szechner et al. reported the transformation of 2-deoxysugar glycosides to the corresponding glycosyl hydroperoxides (Table 82).147 The oxidation was performed using two different procedures, either hydrogen peroxide in tert-butyl alcohol (Method A) or ethereal hydrogen peroxide (Method B). Mixtures of anomeric hydroperoxides were formed and the ratio of α:β anomers was calculated using the integration of the signals in the 1H-NMR spectra. Overall, oxidation of glycosides of lyxo configuration (entries 1, 3, 5) was more selective than that of their arabino counterparts (entry 2, 4, 6). Additionally, it was observed that Method B gave over 90% α-selectivity in lyxo series and also slightly better selectivity in arabino series.
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | R3 | α:β (Method A) |
α:β (Method B) |
| 1 | C7H15 | OC7H15 | H | 84:16 |
95:5 |
| 2 | C7H15 | H | OC7H15 | 55:45 |
65:35 |
| 3 | C12H25 | OC12H25 | H | 85:15 |
93:7 |
| 4 | C12H25 | H | OC12H25 | 58:42 |
67:33 |
| 5 | C18H31 | OC18H31 | H | 87:13 |
98:2 |
| 6 | C18H31 | H | OC18H31 | 56:44 |
68:32 |
Terent'ev et al. have investigated the catalytic ability of a range of protic and Lewis acids to effect the conversion of acetals to gem-dialkylperoxides. They found fluoroboric acid to be the most active catalyst overall. Several cyclic dimethyl acetals of various ring sizes were converted to the corresponding gem-dialkylperoxides using either aqueous or ethereal solutions of TBHP with increasing ring size leading to a fall in yields (Table 83, entries 1–9).148 For aqueous solutions, the addition of calcium chloride as drying agent led to a noticeable improvement in yields (entry 6 vs. 7, 8 vs. 9). A similar effect was observed with acyclic acetals as starting materials (Table 84, entry 3 vs. 4, 5 vs. 6).
|
||||
|---|---|---|---|---|
| Entry | n | R | Peroxide solution (eq.) | Yield |
| 1 | 1 | H | Aqueous (2.5) | 85% |
| 2 | 1 | n-C8H17 | Aqueous (3.0) + CaCl2 | 33% |
| 3 | 2 | H | Aqueous (2.5) | 87% |
| 4 | 2 | H | Ethereal (4.0) | 92% |
| 5 | 3 | H | Aqueous (3.0) + CaCl2 | 70% |
| 6 | 4 | H | Aqueous (3.0) | 49% |
| 7 | 4 | H | Aqueous (3.0) + CaCl2 | 75% |
| 8 | 8 | H | Aqueous (3.0) | 5% |
| 9 | 8 | H | Aqueous (3.0) + CaCl2 | 69% |
| 10 | 8 | n-C9H19 | Aqueous (3.0) + CaCl2 | 19% |
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | Peroxide solution (eq.) | Yield |
| 1 | n-Oct | H | Aqueous (3.0) | 67% |
| 2 | n-Oct | H | Aqueous + CaCl2 (5.0) | 70% |
| 3 | n-Bu | n-Bu | Aqueous (3.0) | 37% |
| 4 | n-Bu | n-Bu | Aqueous + CaCl2 (5.0) | 62% |
| 5 | i-Bu | Me | Aqueous (3.0) | 21% |
| 6 | i-Bu | Me | Aqueous + CaCl2 (3.0) | 40% |
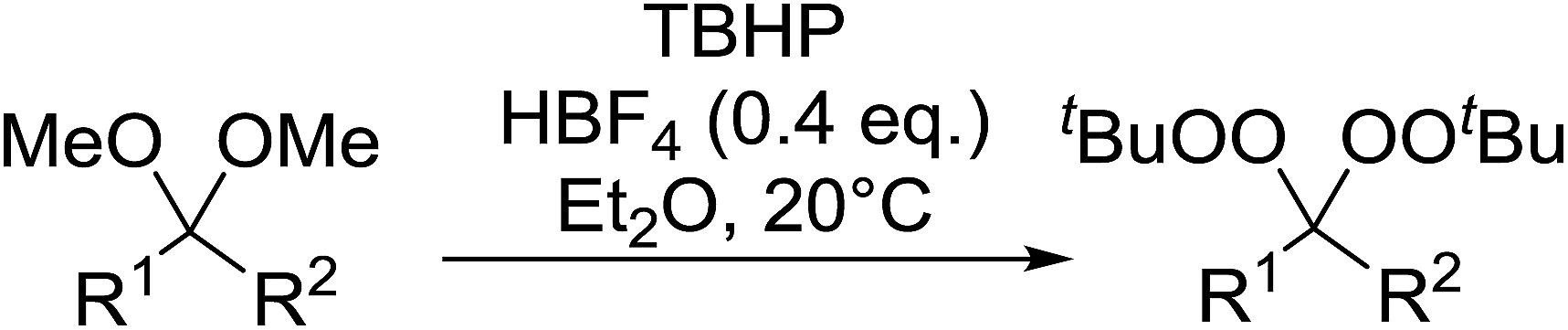
The transformation of enol ethers is also feasible under these conditions. Treatment of 120a with aqueous TBHP in the presence of fluoroboric acid afforded 121a in 85% yield, comparable to that obtained from the corresponding cyclic acetal (Scheme 21). Boron trifluoride-catalysed conversion of enol ether 120b furnished only 16% of 121b, in contrast to a 69% yield from the equivalent dimethyl acetal (Table 83, entry 9).
 | ||
| Scheme 21 | ||
In related work, Terent'ev et al. demonstrated that enol ethers may be transformed into 2-iodoketones using a combination of iodine and hydrogen peroxide.149 These reactions proceed via 2-iodo-1-methoxy hydroperoxide intermediates which are stable and isolable compounds. Accordingly, a range of enol ethers were treated with I2/H2O2 affording a mixture of peroxides and ketones in almost equal yields after six hours (Table 85, entries 1–6). It was found that increasing the number of equivalents of iodine from 1 to 2.5 favoured formation of the 2-iodoketones with only trace amounts of the peroxides remaining.
|
||||
|---|---|---|---|---|
| Entry | n | R | Yield (peroxide) | Yield (ketone) |
| 1 | 1 | H | 33% | 40% |
| 2 | 1 | Me | 38% | 45% |
| 3 | 1 | t-Bu | 35% | 54% |
| 4 | 2 | H | 34% | 41% |
| 5 | 3 | H | 35% | 43% |
| 6 | 7 | H | 41% | 44% |
The same group also studied the peroxidation of bicyclic enol ethers featuring an endocyclic oxygen atom.150 They discovered that treatment of bicyclic enol ethers with I2/H2O2 led to the formation of iodohydroperoxides (Table 86, entries 1 and 2). By contrast, the use of I2/TBHP gave peroxidated oxabicycloalkane products exclusively (entries 3–5). The reaction of monocyclic enol ethers was more unpredictable and furnished multiple products.
|
|||||
|---|---|---|---|---|---|
| Entry | n | R | X | Temp. (°C) | Yield |
| 1 | 1 | H | I | −40 | 82% |
| 2 | 2 | H | I | −40 | 40% |
| 3 | 0 | t-Bu | H | −70 | 89% |
| 4 | 1 | t-Bu | H | −70 | 75% |
| 5 | 2 | t-Bu | H | −70 | 66% |
Hydroperoxy-1′-alkoxyperoxides are versatile intermediates, containing a nucleophilic bishydroperoxide centre as well as a latent electrophilic monoperoxyacetal centre. Terent'ev et al. discovered that the iodine-catalysed reaction of geminal bishydroperoxides with acetals proceeds with the replacement of only one alkoxy group by the peroxide group to give 1-hydroperoxy-1′-alkoxyperoxides in yields of up to 64% with little variation observed with the different substrates (Table 87, entries 1–14).151 The nature of the solvent, however, had a significant impact on the reaction outcome, with diethyl ether providing yields which were over double those obtained in dichloromethane or methanol. It is likely that diethyl ether acts both as a solvent and an activator for iodine in this system. The use of enol ethers in place of acetals afforded the desired products in comparable yields.
|
||||||
|---|---|---|---|---|---|---|
| Entry | n | R1 | R2 | R3 | R4 | Yield |
| 1 | 1 | H | Et | –(CH2)4– | 48% | |
| 2 | 1 | H | Me | –(CH2)5– | 45% | |
| 3 | 1 | H | Me | –(CH2)6– | 64% | |
| 4 | 1 | H | Et | –(CH2)6– | 61% | |
| 5 | 1 | Me | Me | Me | Me | 54% |
| 6 | 1 | Me | Me | Me | C7H15 | 55% |
| 7 | 1 | Me | Et | –(CH2)4– | 59% | |
| 8 | 1 | Me | Me | –(CH2)5– | 54% | |
| 9 | 1 | Me | Me | –(CH2)6– | 60% | |
| 10 | 2 | H | Me | Me | Me | 64% |
| 11 | 2 | H | Me | Me | C7H15 | 63% |
| 12 | 2 | H | Et | –(CH2)4– | 62% | |
| 13 | 7 | H | Me | Me | Me | 51% |
| 14 | 7 | H | Me | –(CH2)5– | 50% | |
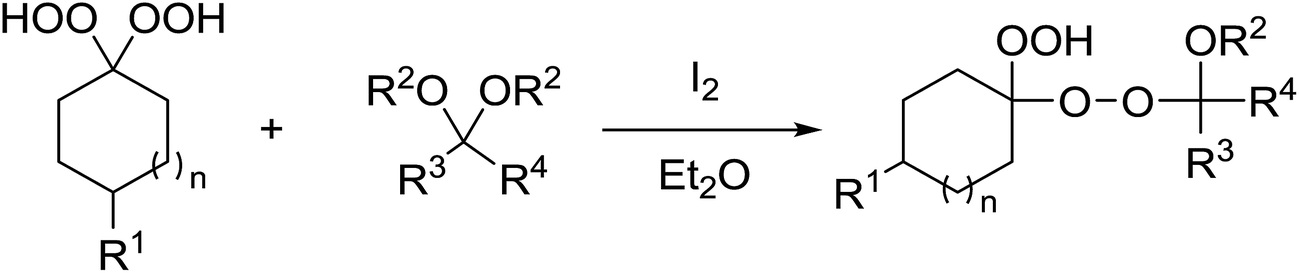
Terent'ev et al. have developed a facile route to vicinal iodoperoxyalkanes by the reaction of alkenes with iodine and various hydroperoxides.152 The best results were obtained with the use of excess iodine, which enables the preparation of the target compounds in yield of up to 70% (Table 88, entries 1–11) along with smaller amounts of the iodohydroxyalkane side products. In addition to hydrogen peroxide and TBHP, 2-hydroperoxytetrahydropyran was also compatible with these conditions. However, the lower reactivity of this particular hydroperoxide necessitated longer reaction times of up to 72 hours (entries 12–15) in comparison to the similar transformation with TBHP (5 hours – entries 1–7) or hydrogen peroxide (3 hours – entries 8–11).
|
||||
|---|---|---|---|---|
| Entry | R1 | R2 | R3 | Yield |
| 1 | n-Bu | H | t-Bu | 57% |
| 2 | n-C5H11 | H | t-Bu | 52% |
| 3 | n-C8H17 | H | t-Bu | 61% |
| 4 | n-C8H16CO2Me | H | t-Bu | 53% |
| 5 | –C3H6– | t-Bu | 68% | |
| 6 | –C4H8– | t-Bu | 61% | |
| 7 | –C6H12– | t-Bu | 67% | |
| 8 | n-Bu | H | H | 62% |
| 9 | –C3H6– | H | 59% | |
| 10 | –C4H8– | H | 61% | |
| 11 | –C6H12– | H | 43% | |
| 12 | n-Bu | H | Tetrahydropyran-2-yl | 31% |
| 13 | –C3H6– | Tetrahydropyran-2-yl | 47% | |
| 14 | –C4H8– | Tetrahydropyran-2-y | 51% | |
| 15 | –C6H12– | Tetrahydropyran-2-yl | 37% | |
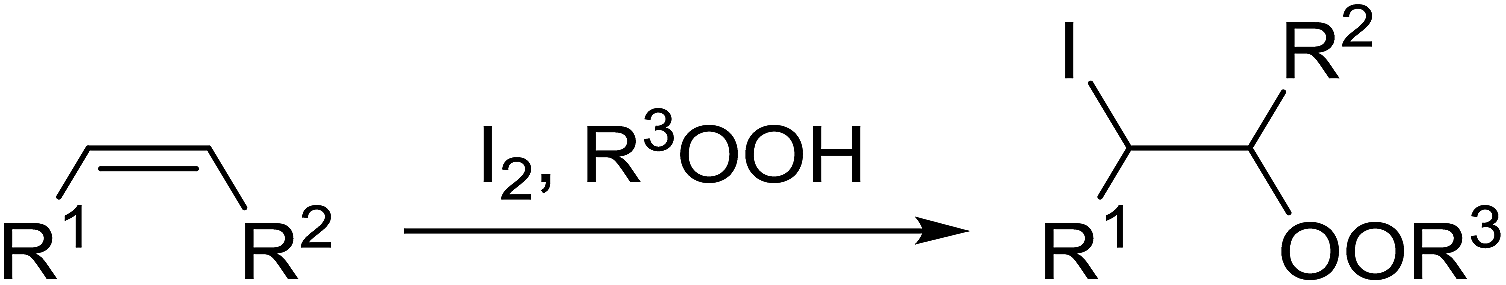
The preparation of peroxides by the displacement of alkyl halides is well established in the literature.153,154 2-Methoxyprop-2-yl hydroperoxide, a reagent developed by Dussault and Sahli,155 allows for the convenient synthesis of hydroperoxides using this approach. Addition of this reagent to an alkyl halide and cesium hydroxide in dimethylformamide led to formation of the corresponding perketal in generally good yields (Table 89, entries 1–3). Subsequent acid hydrolysis afforded the desired hydroperoxides in equally good yields. Displacement of secondary alkyl bromides was more capricious (entry 4) with the combination of potassium tert-butoxide/18-crown-6 providing more reliable results.
|
|||
|---|---|---|---|
| Entry | R | Yield 123 | Yield 124 |
| 1 | CH3(CH2)17 | 76% | 70% |
| 2 | CH3(CH2)13 | 73% | 68% |
| 3 | CH3(CH2)11 | 72% | 65% |
| 4 | 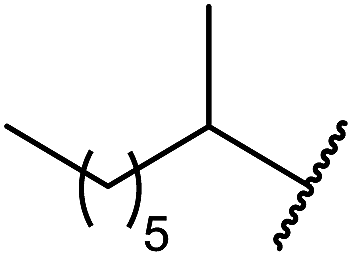 |
25% | 75% |
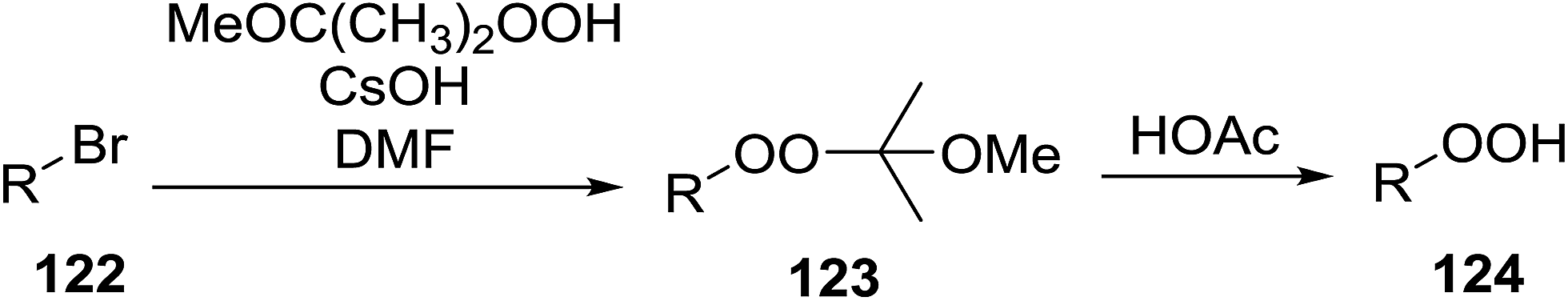
The use of polyethylene glycol (PEG) as a phase transfer catalyst for the preparation of dialkyl peroxides has been pioneered by Baj and Siewniak.156 A triphasic liquid-liquid-liquid system involving cumene hydroperoxide, PEG and an aqueous solution of inorganic base was identified as the optimal setup (Table 90). The reaction proceeded in good yields with various alkyl bromides although the increased steric hindrance associated with branched reagents did result in decreased yields (entries 3 and 5). This methodology also facilitates recovery of the phase transfer catalyst as it forms a separated phase and reuse of the catalyst afforded consistently high yields. The authors have further demonstrated that tetraalkylammonium salts may be used as phase-transfer catalysts in lieu of polyethylene glycol.157
|
||
|---|---|---|
| Entry | R | Yield |
| 1 | Et | 94% |
| 2 | n-Pr | 96% |
| 3 | i-Pr | 72% |
| 4 | n-Bu | 95% |
| 5 | sec-Bu | 68% |
| 6 | n-Pen | 92% |
| 7 | n-Hex | 93% |
| 8 | n-Hep | 80% |
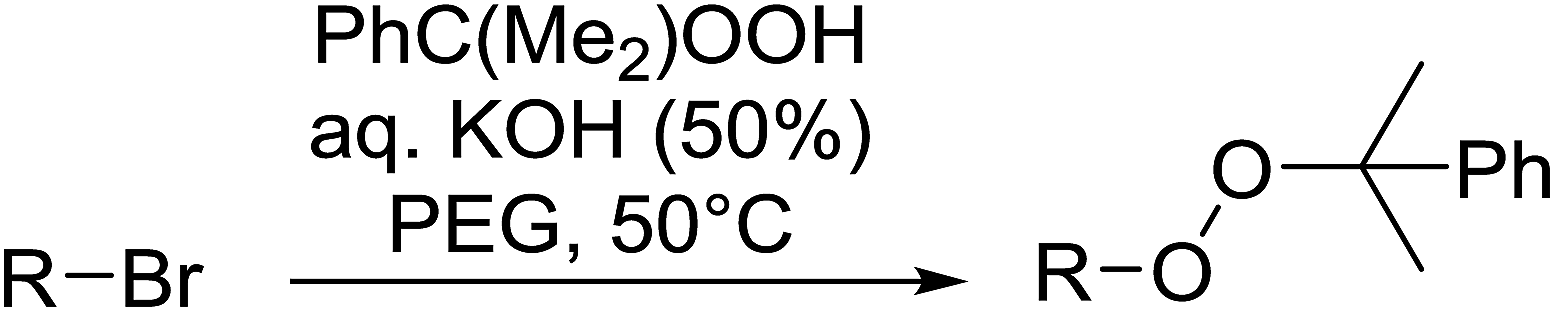
In a similar vein, Foglia and Silbert employed phase transfer catalysis in the preparation of dialkyl peroxides from alkyl bromides.158 To counteract the limited solubility of potassium superoxide in polar solvents and its tendency to decompose in hydroxylic solvents, the authors employed a combination of superoxides and tetraethylammonium bromide (TEAB) in dimethylformamide in the transformation of a range of aliphatic alcohols peroxides to symmetrical diakyl peroxides. Yields were moderate for short chain alkyl bromides but were found to increase with increasing chain length (Table 91, entries 1–8). This methodology was also extended to incorporate secondary alkyl bromides but at the cost of significantly lower yields (entry 9).
|
||
|---|---|---|
| Entry | R | Yield (%) |
| 1 | Bu | 44% |
| 2 | CH2CH(CH2)3 |
54% |
| 3 | n-C6H13 | 51% |
| 4 | n-C8H17 | 66% |
| 5 | n-C10H21 | 71% |
| 6 | n-C12H25 | 85% |
| 7 | n-C14H27 | 76% |
| 8 | n-C16H31 | 87% |
| 9 | CH3(CH2)3(CH3)CH | 23% |
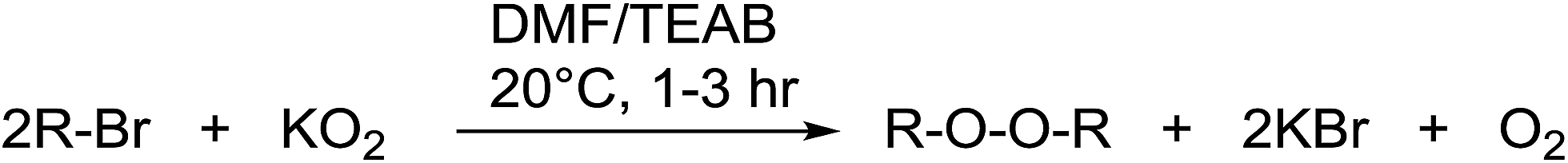
6 Enzymatic catalysis
Synthetically useful examples of peroxidation via enzymatic catalysis are limited in the literature given that many such processes are substrate dependent. Akakabe et al. incubated saturated and unsaturated fatty acids with a crude enzyme from marine green algae, ulva pertusa.159,160 The corresponding (R)-2-hydroperoxy acids were isolated with high enantiopurity (>99% ee) (Table 92, entries 1–6). The reaction was unsuccessful with heptadecanoic acid or stearic acid as starting materials. The authors also found that crude enzymes extracted from either brown or red algae were found to oxidise palmitic acid to the corresponding (R)-2-hydroperoxy acid in >99% ee, suggesting that enzymes capable of α-oxidation are distributed in a variety of organisms.
|
||
|---|---|---|
| Entry | R | ee |
| 1 |  |
>99% |
| 2 |  |
>99% |
| 3 |  |
>99% |
| 4 |  |
>99% |
| 5 |  |
>99% |
| 6 |  |
>99% |
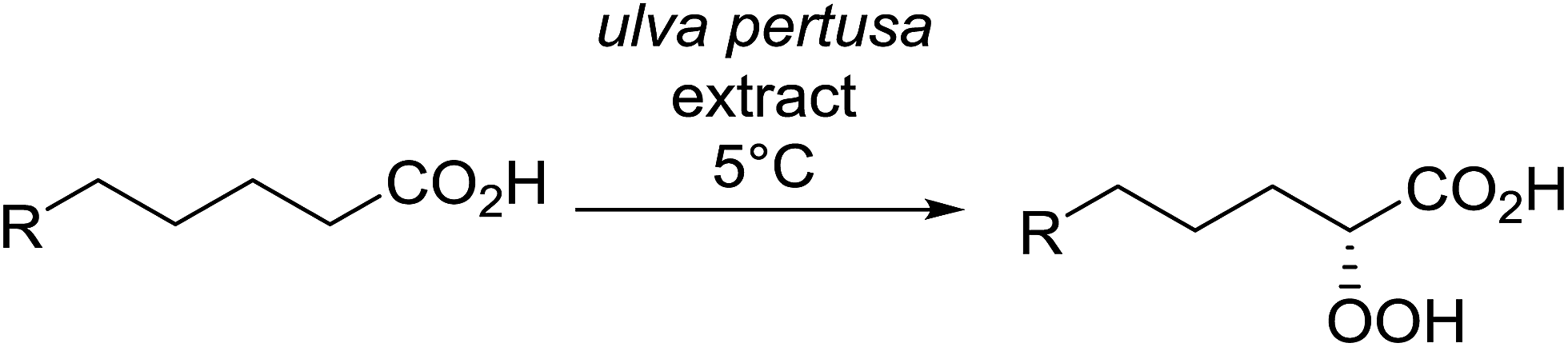
Lipoxygenase enzymes catalyse the highly enantioselective insertion of 3O2 into 1,4-dienes to form lipid peroxides and are important physiological processes which have been widely studied.161 Soybean lipoxygenase mediates the highly enantioselective and regioselective insertion of oxygen into the ω-6 position of polyunsaturated fatty acids to form diene hydroperoxides.162–166 Lipoxygenase enzymes have also been used in the regioselective synthesis of a diacylglyceroposphoncholine hydroperoxide (Scheme 22).167 Enzymatic peroxidation of linoleic acid 125 with soybean lipoxygenase and subsequent treatment with diazomethane afforded methyl ester 126 in 36% overall yield. The enantiomeric excess for this step was not reported.
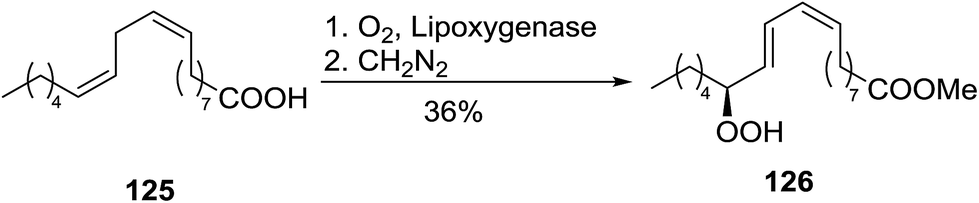 | ||
| Scheme 22 | ||
Manganese lipoxygenase (Mn-LO) successfully catalysed the peroxidation of linoleic acid as reported by Hamberg et al. (Scheme 23).168 Two major product were isolated, namely (11S)-hydroperoxyocatacdecdienoic acid 128 and (13R)-hydroperoxyocatacdecdienoic acid 129 in a ratio of 1:5 – no yields are provided. The reaction proceeds by initial abstraction of a bisallylic pro-S-hydrogen from C11 leading to the formation of a radical. This may be followed by the reversible oxygen insertion to C11 forming the bisallyic hydroperoxide product 128. Alternatively, oxygen attack at the C13 position leads to irreversible formation of 129. Other groups have described similar transformations with lipoxygenase enzymes163,164 including a novel iron mini-lipoxygenase CspLOX2 which catalyses similar reactions.169
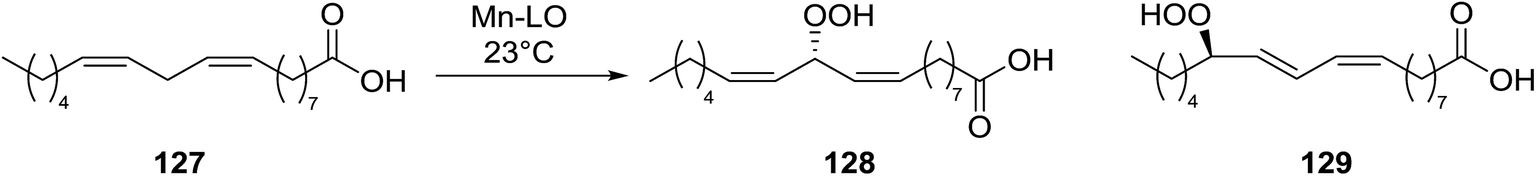 | ||
| Scheme 23 | ||
Horseradish peroxidase was found to be an effective enzyme for the kinetic resolution of hydroperoxides.170 The hydroperoxide structure influenced not only the enantioselectivity but also the enantiomer preference of the enzyme. For aryl hydroperoxides bearing linear alkyl substituents, the (S)-hydroperoxide and (R)-alcohol were the preferred products (Table 93, entries 1–3). By contrast, for similar substrates with branched alkyl substituents, the configuration of the products was reversed (entries 4 and 5). Horseradish peroxidase was found to be less substrate specific than other enzymes, with high enantioselectivities also observed across various substrates including, but not limited to, α-hydroperoxy esters (entries 6 and 7).
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Time (min) | Enantiomeric excess | |
| 131 | 132 | ||||
| 1 | Ph | Me | 5 | >99% (S) | >99% (R) |
| 2 | Ph | Et | 150 | 93% (S) | 95% (R) |
| 3 | Ph | n-Pr | 90 | <5% (S) | <5% (R) |
| 4 | Ph | i-Pr | 180 | 15% (R) | 14% (S) |
| 5 | Ph | i-Bu | 960 | 37% (R) | 48% (S) |
| 6 | Et | CO2Me | 60 | 97% (R) | 97% (S) |
| 7 | i-Pr | CO2Me | 60 | 79% (R) | 64% (S) |
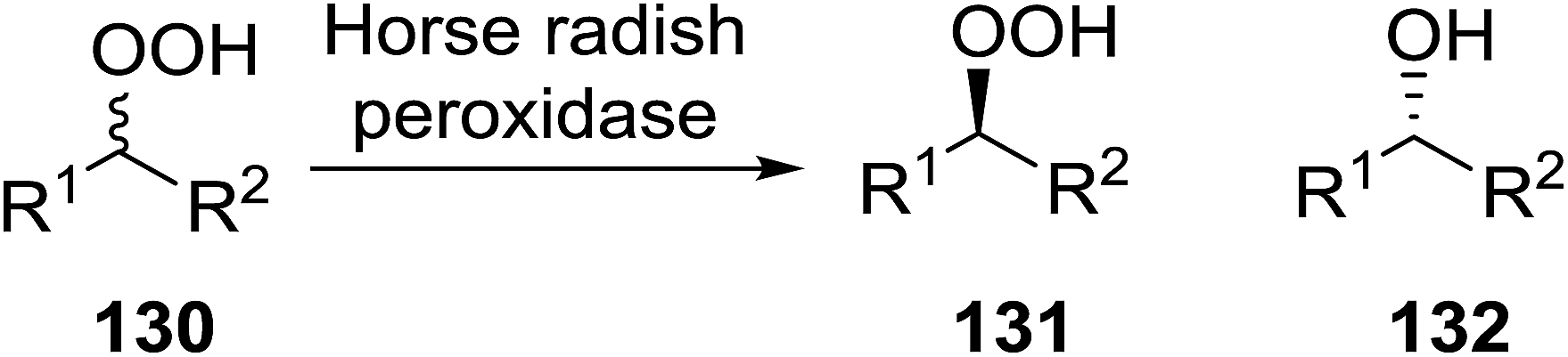
7 Concluding remarks
As outlined in this review, several successful strategies have been developed to access acyclic peroxides. These include the traditional approaches of metal-catalysed reactions, direct oxidation reactions, enzymatic peroxidations and the ever-increasing role of organocatalysis. While peroxides have shown to be stable to a wide-variety of chemical transformations, this area of research is still in its infancy, with its full synthetic potential yet to be realised.Acknowledgements
KOR and HG (IRCGOIPG/2013/113) thank the Irish Research Council for funding. The research leading to these results has received funding from the People Programme (FP7 Marie Sklodowska Curie Actions) under REA grant agreement 272194 (MKG).References
- V. M. Dembitsky, T. A. Gloriozova and V. V. Poroikov, Mini-Rev. Med. Chem., 2007, 571–589 CrossRef CAS.
- V. M. Dembitsky, Eur. J. Med. Chem., 2008, 43, 223–251 CrossRef CAS PubMed.
- K. Zmitek, M. Zupan and J. Iskra, Org. Biomol. Chem., 2007, 5, 3895–3908 CAS.
- G. O. Schenck, Naturwissenschaften, 1948, 35, 28–29 CrossRef CAS.
- G. O. Schenck and K. Ziegler, Naturwissenschaften, 1944, 32, 157 Search PubMed.
- M. S. Kharasch and A. Fono, J. Org. Chem., 1959, 24, 72–78 CrossRef CAS.
- A. O. Terent'ev, D. A. Borisov, V. A. Vil and V. M. Dembitsky, Beilstein J. Org. Chem., 2014, 10, 34–114 CrossRef PubMed.
- S. Isayama and T. Mukaiyama, Chem. Lett., 1989, 18, 573–576 CrossRef.
- M. T. Nojima, T. Kunikawa, S. Masuyama and A. Masuyama, Org. Lett., 2002, 4, 3595–3598 CrossRef.
- S. Isayama and T. Mukaiyama, Chem. Lett., 1989, 18, 1071–1074 CrossRef.
- P. M. O'Neill, S. Hindley, M. D. Pugh, J. Davies, P. G. Bray, B. K. Park, D. S. Kapu, S. A. Ward and P. A. Stocks, Tetrahedron Lett., 2003, 44, 8135–8138 CrossRef.
- T. Tokuyasu, S. Kunikawa, K. J. McCullough, A. Masuyama and M. Nojima, J. Org. Chem., 2004, 70, 251–260 CrossRef PubMed.
- T. Tokuyasu, S. Kunikawa, M. Abe, A. Masuyama, M. Nojima, H.-S. Kim, K. Begum and Y. Wataya, J. Org. Chem., 2003, 68, 7361–7367 CrossRef CAS PubMed.
- J.-M. Wu, S. Kunikawa, T. Tokuyasu, A. Masuyama, M. Nojima, H.-S. Kim and Y. Wataya, Tetrahedron, 2005, 61, 9961–9968 CrossRef CAS.
- P. Ghorai, P. H. Dussault and C. Hu, Org. Lett., 2008, 10, 2401–2404 CrossRef CAS PubMed.
- S. Gemma, E. Gabellieri, S. Sanna Coccone, F. Martí, O. Taglialatela-Scafati, E. Novellino, G. Campiani and S. Butini, J. Org. Chem., 2010, 75, 2333–2340 CrossRef CAS PubMed.
- S. Gemma, S. Kunjir, S. Sanna Coccone, M. Brindisi, V. Moretti, S. Brogi, E. Novellino, N. Basilico, S. Parapini, D. Taramelli, G. Campiani and S. Butini, J. Med. Chem., 2011, 54, 5949–5953 CrossRef CAS PubMed.
- B. Barnych and J.-M. Vatèle, Org. Lett., 2012, 14, 564–567 CrossRef CAS PubMed.
- B. Barnych, B. Fenet and J.-M. Vatèle, Tetrahedron, 2013, 69, 334–340 CrossRef CAS.
- S. Isayama, Bull. Chem. Soc. Jpn., 1990, 63, 1305–1310 CrossRef CAS.
- K. Sugamoto, Y. Matsushita and T. Matsui, J. Chem. Soc., Perkin Trans. 1, 1998, 3989–3998 RSC.
- Y. Matsushita, K. Sugamoto, T. Nakama and T. Matsui, J. Chem. Soc., Chem. Commun., 1995, 31, 567–568 RSC.
- B. Hurlocker, M. R. Miner and K. A. Woerpel, Org. Lett., 2014, 16, 4280–4283 CrossRef CAS PubMed.
- M. S. Kharasch, P. Pauson and W. Nudenberg, J. Org. Chem., 1953, 18, 322–327 CrossRef CAS.
- M. S. Kharasch, A. Fono and W. Nudenberg, J. Org. Chem., 1951, 16, 113–127 CrossRef CAS.
- M. S. Kharasch, W. Nudenberg and G. J. Mantell, J. Org. Chem., 1951, 16, 524–532 CrossRef CAS.
- M. S. Kharasch, A. Fono, W. Nudenberg and B. Bischof, J. Org. Chem., 1952, 17, 207–220 CrossRef CAS.
- M. S. Kharasch, A. Fono and W. Nudenberg, J. Org. Chem., 1950, 15, 748–752 CrossRef CAS.
- M. S. Kharasch, A. Fono and W. Nudenberg, J. Org. Chem., 1950, 15, 763–774 CrossRef CAS.
- M. S. Kharasch, P. Pauson and W. Nudenberg, J. Org. Chem., 1953, 18, 322–327 CrossRef CAS.
- M. S. Kharasch, F. S. Arimoto and W. Nudenberg, J. Org. Chem., 1956, 21, 1556–1565 Search PubMed.
- M. S. Kharasch and A. Fono, J. Org. Chem., 1958, 23, 324–325 Search PubMed.
- M. S. Kharasch and G. Sosnovsky, J. Org. Chem., 1958, 23, 1322–1326 CrossRef CAS.
- M. S. Kharasch and A. Fono, J. Org. Chem., 1959, 24, 72–78 CrossRef CAS.
- M. S. Kharasch, G. Sosnovsky and N. C. Yang, J. Am. Chem. Soc., 1959, 81, 5819–5824 CrossRef CAS.
- M. S. Kharasch, A. Fono, W. Nudenberg and A. C. Phoskus, J. Org. Chem., 1950, 15, 775–781 CrossRef CAS.
- M. S. Kharasch, A. Fono and W. Nudenberg, J. Org. Chem., 1950, 15, 753–762 CrossRef CAS.
- M. B. Meder and L. H. Gade, Eur. J. Inorg. Chem., 2004, 2004, 2716–2722 CrossRef.
- M. Schulz, R. Kluge and F. Gadissa Gelalcha, Tetrahedron: Asymmetry, 1998, 9, 4341–4360 CrossRef CAS.
- A. O. Terent'ev, D. A. Borisov, V. V. Chernyshev and G. I. Nikishin, J. Org. Chem., 2009, 74, 3335–3340 CrossRef PubMed.
- A. O. Terent'ev, D. A. Borisov, I. A. Yaremenko, V. V. Chernyshev and G. I. Nikishin, J. Org. Chem., 2010, 75, 5065–5071 CrossRef PubMed.
- A. O. Terent'ev, D. A. Borisov, V. V. Semenov, V. V. Chernyshev, V. M. Dembitsky and G. I. Nikishin, Synthesis, 2011, 2011, 2091–2100 CrossRef.
- F. Minisci, F. Fontana, S. Araneo and F. Recupero, J. Chem. Soc., Chem. Commun., 1994, 1994, 1823–1824 RSC.
- F. Minisci, F. Fontana, S. Araneo, F. Recupero, S. Banfi and S. Quici, J. Am. Chem. Soc., 1995, 117, 226–232 CrossRef CAS.
- F. Minisci, F. Fontana, S. Araneo, F. Recupero and A. Serri, J. Chem. Soc., Chem. Commun., 1995, 1995, 1399–1400 Search PubMed.
- G. B. Shul'pin, J. Gradinaru and Y. N. Kozlov, Org. Biomol. Chem., 2003, 1, 3611–3617 Search PubMed.
- D. Mandelli, K. C. Chiacchio, Y. N. Kozlov and G. B. Shul'pin, Tetrahedron Lett., 2008, 49, 6693–6697 CrossRef CAS.
- B. M. Choudary, N. Narender and V. Bhuma, Synlett, 1994, 1994, 641–642 CrossRef.
- M. D. Nikalje and A. Sudalai, Tetrahedron, 1999, 55, 5903–5908 CrossRef CAS.
- T. Nishimura, T. Onoue, K. Ohe, J. I. Tateiwa and S. Uemura, Tetrahedron Lett., 1995, 39, 4359–4362 CrossRef.
- B. A. Allal, L. El Firdoussi, S. Allaoud, A. Karim, Y. Castanet and A. Mortreux, J. Mol. Catal. A: Chem., 2003, 200, 177–184 CrossRef CAS.
- J. Q. Yu and E. J. Corey, Org. Lett., 2002, 4, 2727–2730 CrossRef CAS PubMed.
- G. An, W. Zhou, G. Zhang, H. Sun, J. Han and Y. Pan, Org. Lett., 2010, 12, 4482–4485 CrossRef CAS PubMed.
- S. Lee and J. F. Hartwig, J. Org. Chem., 2001, 66, 3402–3415 CrossRef CAS PubMed.
- Y. X. Jia and E. P. Kündig, Angew. Chem., Int. Ed., 2009, 48, 1636–1639 CrossRef CAS PubMed.
- A. O. Terent'ev, M. Y. Sharipov, I. B. Krylov, D. V. Gaidarenko and G. I. Nikishin, Org. Biomol. Chem., 2015, 13, 1439–1445 Search PubMed.
- A. Massa, M. R. Acocella, M. De Rosa, A. Soriente, R. Villano and A. Scettri, Tetrahedron Lett., 2003, 44, 835–837 CrossRef CAS.
- W. Liu, Y. Li, K. Liu and Z. Li, J. Am. Chem. Soc., 2011, 133, 10756–10759 CrossRef CAS PubMed.
- K. Liu, Y. Li, X. Zheng, W. Liu and Z. Li, Tetrahedron, 2012, 68, 10333–10337 CrossRef CAS.
- J.-K. Cheng and T.-P. Loh, J. Am. Chem. Soc., 2015, 137, 42–45 CrossRef CAS PubMed.
- A. Banerjee, S. K. Santra, A. Mishra, N. Khatun and B. K. Patel, Org. Biomol. Chem., 2015, 13, 1307–1312 CAS.
- W.-C. Yang, S.-S. Weng, A. Ramasamy, G. Rajeshwaren, Y.-Y. Liaob and C.-T. Chen, Org. Biomol. Chem., 2015, 13, 2385–2392 CAS.
- M. J. Fuchter, B. M. Hoffman and A. G. Barrett, J. Org. Chem., 2006, 71, 724–729 CrossRef CAS PubMed.
- S. W. Krabbe, D. T. Do and J. S. Johnson, Org. Lett., 2012, 14, 5932–5935 CrossRef CAS PubMed.
- V. P. Mentz, M. Schulz and R. Kluge, Arzneim.-Forsch., 1987, 37, 1229–1232 Search PubMed.
- H. Nishino, M. Rahman and C. Qian, Tetrahedron Lett., 2003, 44, 5225–5228 CrossRef.
- H. Nishino and M. Rahman, Org. Lett., 2003, 5, 2887–2890 CrossRef PubMed.
- H. Nishino and M. A. Haque, Synth. Commun., 2012, 42, 608–619 CrossRef.
- H. Nishino, R. Kumabe, R. Hamada and M. Yakut, Tetrahedron, 2014, 70, 1437–1450 CrossRef CAS.
- X.-F. Xia, S.-L. Zhu, Z. Gu, H. Wang, W. Li, X. Liu and Y.-M. Liang, J. Org. Chem., 2015, 80, 5572–5580 CrossRef CAS PubMed.
- D. Yamamoto, M. Soga, H. Ansai and K. Makino, Org. Chem. Front., 2016, 3, 1420–1424 RSC.
- B. Das, M. Krishnaiah, B. Veeranjaneyulu and B. Ravikanth, Tetrahedron Lett., 2007, 48, 6286–6289 CrossRef CAS.
- K. Khosravi, Cogent Chem., 2015, 1, 1002339–1002348 Search PubMed.
- D. Azarifar, K. Khosravi and F. Soleimanei, Synthesis, 2009, 15, 2553–2556 CrossRef.
- D. Azarifar and K. Khosravi, J. Iran. Chem. Soc., 2011, 8, 1006–1013 CrossRef CAS.
- P. Ghorai and P. H. Dussault, Org. Lett., 2008, 10, 4577–4579 CrossRef CAS PubMed.
- S. Kyasa, B. W. Puffer and P. H. Dussault, J. Org. Chem., 2013, 78, 3452–3456 CrossRef CAS PubMed.
- A. O. Terent'ev, O. B. Krivykh, I. B. Krylov, Y. N. Ogibin and G. I. Nikishin, Russ. J. Gen. Chem., 2010, 80, 1667–1671 CrossRef.
- M. Nojima, Y. Hamada, H. Tokuhara, A. Masuyama, H. Kim, K. Ono, N. Ogura and Y. Wataya, J. Med. Chem., 2002, 45, 1374–1378 CrossRef.
- C.-Y. Tsai, L.-A. Chen and K. Sung, Can. J. Chem., 2012, 90, 321–325 CrossRef CAS.
- N. Murakami, M. Kawanishi, S. Itagaki, T. Horii and M. Kobayashi, Tetrahedron Lett., 2001, 42, 7281–7285 CrossRef CAS.
- S. Pramanik and P. Ghorai, Org. Lett., 2013, 15, 3832–3835 CrossRef CAS PubMed.
- S.-I. Murahashi, T. Naota, N. Miyaguchi and S. Noda, J. Am. Chem. Soc., 1996, 118, 2509–2510 CrossRef CAS.
- S.-I. Murahashi, N. Miyaguchi, S. Noda, T. Naota, A. Fujii, Y. Inubushi and N. Komiya, Eur. J. Org. Chem., 2011, 2011, 5355–5365 CrossRef CAS.
- M. O. Ratnikov, L. E. Farkas, E. C. McLaughlin, G. Chiou, H. Choi, S. H. El-Khalafy and M. P. Doyle, J. Org. Chem., 2011, 76, 2585–2593 CrossRef CAS PubMed.
- M. O. Ratnikov, L. E. Farkas and M. P. Doyle, J. Org. Chem., 2012, 77, 10294–10303 CrossRef CAS PubMed.
- C. M. Marson, A. Khan and R. A. Porter, J. Org. Chem., 2001, 66, 4771–4775 CrossRef CAS PubMed.
- A. Tarlani, A. Riahi, M. Abedini, M. M. Amini and J. Muzart, Appl. Catal., A, 2006, 315, 150–152 CrossRef CAS.
- M. J. Bourgeois, E. Montaudon and B. Maillard, Tetrahedron, 1993, 49, 2477–2484 CrossRef CAS.
- W. H. Richardson and R. S. Smith, J. Org. Chem., 1968, 33, 3882–3885 CrossRef CAS.
- Y. Higuchi, K. Matsuyama and T. Komai, Bull. Chem. Soc. Jpn., 1988, 61, 1821–1823 CrossRef CAS.
- B. Barnych and J.-M. Vatèle, Tetrahedron, 2012, 68, 3717–3724 CrossRef CAS.
- A. Waldemar and R. Agustin, Chem. Commun., 1971, 7, 822–823 Search PubMed.
- Y. Tang, Y. Dong, X. Wang, K. Sriraghavan, J. K. Wood and J. L. Vennerstrom, J. Org. Chem., 2005, 70, 5103–5110 CrossRef CAS PubMed.
- Y. Li, H.-D. Hao and Y. Wu, Org. Lett., 2009, 11, 2691–2694 CrossRef CAS PubMed.
- H.-D. Hao, Y. Li, W.-B. Han and Y. Wu, Org. Lett., 2011, 13, 4212–4215 CrossRef CAS PubMed.
- Y.-H. Liu, Z.-H. Zhang and T.-S. Li, Synthesis, 2008, 2008, 3314–3318 CrossRef.
- P. H. Dussault and C. Xu, Tetrahedron Lett., 2004, 45, 7455–7457 CrossRef CAS.
- P. H. Dussault, T. K. Trullinger and F. Noor-e-Ain, Org. Lett., 2002, 4, 4591–4593 CrossRef CAS PubMed.
- P. Dai, T. K. Trullinger, X. Liu and P. H. Dussault, J. Org. Chem., 2006, 71, 2283–2292 CrossRef CAS PubMed.
- C. W. Jefford, Y. Li, A. Jaber and J. Boukouvalas, Synth. Commun., 1990, 20, 2589–2596 CrossRef CAS.
- P. H. Dussault, T. K. Trullinger and S. Cho-Shultz, Tetrahedron, 2000, 56, 9213–9220 CrossRef CAS.
- T. Arai, K. Tsuchiya and E. Matsumura, Org. Lett., 2015, 17, 2416–2419 CrossRef CAS PubMed.
- X. Lu, Y. Liu, B. Sun, B. Cindric and L. Deng, J. Am. Chem. Soc., 2008, 130, 8134–8135 CrossRef CAS PubMed.
- L. Hu, X. Lu and L. Deng, J. Am. Chem. Soc., 2015, 137, 8400–8403 CrossRef CAS PubMed.
- K. O'Reilly, M. K. Gupta, H. Gandhi, V. P. Kumar, K. S. Eccles, S. E. Lawrence and T. P. O'Sullivan, Curr. Org. Chem., 2016, 20, 2633–2638 CrossRef.
- X. Lu and L. Deng, Org. Lett., 2014, 16, 2358–2361 CrossRef CAS PubMed.
- A. Lattanzi, Adv. Synth. Catal., 2008, 350, 1991–1995 CrossRef.
- W. Zheng, L. Wojtas and J. Antilla, Angew. Chem., Int. Ed., 2010, 49, 6589–6591 CrossRef CAS PubMed.
- X. Feng, Y.-Q. Yuan, H.-L. Cui, K. Jiang and Y.-C. Chen, Org. Biomol. Chem., 2009, 7, 3660–3662 CAS.
- T. J. Fisher and A. E. Mattson, Org. Lett., 2014, 16, 5316–5319 CrossRef CAS PubMed.
- E. D. Couch, T. J. Auvil and A. E. Mattson, Eur. J. Chem., 2014, 20, 8283–8287 CrossRef CAS PubMed.
- Y. Yang, F. Moinodeen, W. Chin, T. Ma, Z. Jiang and C.-H. Tan, Org. Lett., 2012, 14, 4762–4765 CrossRef CAS PubMed.
- A. G. Griesbeck, T. T. El-Idreesy and J. Lex, Tetrahedron, 2006, 62, 10615–10622 CrossRef CAS.
- A. G. Griesbeck and A. Raabe, Synlett, 2009, 2009, 1514–1516 CrossRef.
- K. Gollnick and K. Knutzen-Mies, J. Org. Chem., 1991, 56, 4017–4027 CrossRef CAS.
- C.-H. Tung, Y.-Z. Chen, L.-Z. Wu, M.-L. Peng, D. Zhang and L.-P. Zhang, Tetrahedron, 2006, 62, 10688–10693 CrossRef.
- M. N. Alberti and M. Orfanopoulos, Tetrahedron Lett., 2006, 62, 10660–10675 CrossRef CAS.
- M. Stratakis and N. Sofikiti, J. Chem. Res., 2002, 2002, 374–375 CrossRef.
- M. Stratakis, C. Raptis, N. Sofikiti, C. Tsandarakis, G. Kosmas, I.-P. Zaravinos, D. Kalaitzakis, D. Stavoulakis, C. Baskakis and A. Stathoulopoulou, Tetrahedron, 2006, 62, 10623–10632 CrossRef CAS.
- M. Stratakis, C. Rabalakos, G. Mpourmpakis and G. E. Froudakis, J. Org. Chem., 2003, 68, 2839–2843 CrossRef CAS PubMed.
- M. Stratakis, D. Kalaitzakis, D. Stavroulakis, G. Kosmos and C. Tsangarakis, Org. Lett., 2003, 5, 3471–3474 CrossRef CAS PubMed.
- X. Li and V. Ramamurthy, J. Am. Chem. Soc., 1996, 118, 10666–10667 CrossRef CAS.
- R. J. Robbins and V. Ramamurthy, Chem. Commun., 1997, 33, 1071–1072 RSC.
- C. Tung, Y. Chen, L. Wu and L. Zhang, J. Org. Chem., 2005, 70, 4676–4681 CrossRef PubMed.
- P. H. Dussault, C. T. Eary, R. J. Lee and U. R. Zope, J. Chem. Soc., Perkin Trans. 1, 1999, 2189–2204 RSC.
- M. Kimura, G. Lu, H. Iga, M. Tsunenaga, Z. Zhang and Z. Hu, Tetrahedron Lett., 2007, 48, 3109–3113 CrossRef CAS.
- M. Murakami, K. Sakita, K. Igawa and K. Tomooka, Org. Lett., 2006, 8, 4023–4026 CrossRef CAS PubMed.
- K. Tamooka, M. Murakami and K. Sakita, Synthesis, 2008, 10, 1641–1645 CrossRef.
- C. Xu, C. Schwartz, J. Raible and P. H. Dussault, Tetrahedron, 2009, 65, 9680–9685 CrossRef CAS PubMed.
- P. Y. Andreev, Russ. J. Appl. Chem., 2012, 85, 1395–1398 CrossRef CAS.
- Y. J. Park, J. W. Park and C. H. Jun, Acc. Chem. Res., 2008, 41, 222–234 CrossRef CAS PubMed.
- W. Wei, C. Zhang, Y. Xu and X. Wan, Chem. Commun., 2011, 47, 10827–10829 RSC.
- A. O. Terent'ev, A. T. Zdvizhkov, D. O. Levitsky, F. Fleury, R. A. Pototskiy, A. N. Kulakova and G. I. Nikishin, Tetrahedron, 2015, 71, 8985–8990 CrossRef.
- H. Yu and J. Shen, Org. Lett., 2014, 16, 3204–3207 CrossRef CAS PubMed.
- A. Banerjee, S. K. Santra, N. Khatun, W. Alia and B. K. Patel, Chem. Commun., 2015, 51, 15422–15425 RSC.
- A. Bunge, H.-J. Hamann and J. Liebscher, Tetrahedron Lett., 2009, 50, 524–526 CrossRef CAS.
- A. Rieche, Chem. Ber., 1931, 64, 2328–2335 CrossRef.
- R. C. Cusati, L. C. A. Barbosa, C. R. A. Maltha, A. J. Demuner, A. Oliveros-Bastidasc and A. A. Silva, Pest Manage. Sci., 2014, 71, 1037–1048 CrossRef PubMed.
- A. O. Terent'ev, M. M. Platonov, Y. N. Ogibin and G. I. Nikishin, Synth. Commun., 2007, 37, 1281–1287 CrossRef.
- K. Zmitek, M. Zupan, S. Stavber and J. Iskra, J. Org. Chem., 2007, 72, 6534–6540 CrossRef CAS PubMed.
- K. Zmitek, M. Zupan, S. Stavber and J. Iskra, Org. Lett., 2006, 8, 2491–2494 CrossRef CAS PubMed.
- N. Tada, L. Cui, H. Okubo, H. Miura and A. Itoh, Chem. Commun., 2010, 46, 1772–1774 RSC.
- E. Rozhko, S. Solmi, F. Cavani, A. Albini, P. Righi and D. Ravelli, J. Org. Chem., 2015, 80, 6425–6431 CrossRef CAS PubMed.
- H.-X. Jin, Q. Zhang, H.-S. Kim, Y. Wataya, H.-H. Liu and Y. Wu, Tetrahedron, 2006, 62, 7699–7711 CrossRef CAS.
- N. Chowdhury, M. Gangopadhyay, S. Karthik, N. D. P. Singh, M. Baidya and S. K. Ghosh, J. Photochem. Photobiol., B, 2014, 130, 188–198 CrossRef CAS PubMed.
- B. Szechner, A. Jaromin, S. Parapini, N. Basilico, B. Grzeszczyk, B. Furman and M. Chmielewski, Bioorg. Med. Chem., 2015, 23, 3033–3039 CrossRef CAS PubMed.
- A. O. Terent'ev, A. V. Kutkin, N. A. Troizky, Y. N. Ogibin and G. I. Nikishin, Synthesis, 2005, 2005, 2215–2219 CrossRef.
- A. O. Terent'ev, A. M. Borisov, M. M. Platonov, Z. A. Starikova, V. V. Chernyshev, G. I. Nikishin and N. D. Zelinsky, Synthesis, 2009, 24, 4159–4166 Search PubMed.
- A. O. Terent'ev, A. T. Zdvizhkov, A. N. Kulakova, R. A. Novikov, A. V. Arzumanyana and G. I. Nikishina, RSC Adv., 2014, 4, 7579–7587 RSC.
- A. O. Terent'ev, M. M. Platonov, I. B. Krylov, V. V. Chernyshev and G. I. Nikishin, Org. Biomol. Chem., 2008, 6, 4435–4441 Search PubMed.
- A. O. Terent'ev, I. B. Krylov, D. A. Borisov and G. I. Nikishin, Synthesis, 2007, 19, 2979–2986 CrossRef.
- F. M. Guerra, E. Zubía, M. J. Ortega, F. J. Moreno-Dorado and G. M. Massanet, Tetrahedron, 2010, 66, 157–163 CrossRef CAS.
- L. Cointeaux, J.-F. Berrien and J. Mayrargue, Tetrahedron Lett., 2002, 43, 6275–6277 CrossRef CAS.
- P. Dussault and A. Sahli, J. Org. Chem., 1992, 57, 1009–1012 CrossRef CAS.
- S. Baj and A. Siewniak, Appl. Catal., A, 2007, 321, 175–179 CrossRef CAS.
- S. Baj and A. Siewniak, Appl. Catal., A, 2010, 385, 208–213 CrossRef CAS.
- T. A. Foglia and L. S. Silbert, Synthesis, 1992, 1992, 545–547 CrossRef.
- Y. Akakabe, K. Matsui and T. Kajiwara, Fish. Sci., 2001, 67, 328–332 CrossRef CAS.
- Y. Akakabe, K. Matsui and T. Kajiwara, Tetrahedron Lett., 1999, 40, 1137–1140 CrossRef CAS.
- C. Dufour and M. Loonis, Chem. Phys. Lipids, 2005, 138, 60–68 CrossRef CAS PubMed.
- P. H. Dussault, T. A. Anderson, M. R. Hayden, K. J. Koeller and Q. Jason Niu, Tetrahedron, 1996, 52, 12381–12398 CrossRef CAS.
- G. Coffa, A. N. Imber, B. C. Maguire, G. Laxmikanthan, C. Schneider, B. J. Gaffney and A. R. Brash, J. Biol. Chem., 2005, 280, 38756–38766 CrossRef CAS PubMed.
- B. Gao, W. E. Boeglin and A. R. Brash, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2010, 1801, 58–63 CrossRef CAS PubMed.
- Y. Zheng and A. R. Brash, J. Biol. Chem., 2010, 285, 39876–39887 CrossRef CAS PubMed.
- W. Boeglin, A. Itoh, Y. Zheng, G. Coffa, G. Howe and A. Brash, Lipids, 2008, 43, 979–987 CrossRef CAS PubMed.
- N. Baba, K. Yoneda, S. Tahara, J. Iwasa, T. Kaneko and M. Matsuo, J. Chem. Soc., Chem. Commun., 1990, 1281–1282 RSC.
- M. Hamberg, C. Su and E. Oliw, J. Biol. Chem., 1998, 273, 13080–13088 CrossRef CAS PubMed.
- A. Andreou, C. Göbel, M. Hamberg and I. Feussner, J. Biol. Chem., 2010, 285, 14178–14186 CrossRef CAS PubMed.
- U. Hoch, W. Adam, R. Fell, C. R. Saha-Möller and P. Schreier, J. Mol. Catal. A: Chem., 1997, 117, 321–328 CrossRef CAS.
Footnote |
| † Current address: Department of Chemistry, School of Chemical Sciences, Central University of Haryana, Mahendergarh – 123 031 Haryana, India. |
| This journal is © The Royal Society of Chemistry 2017 |