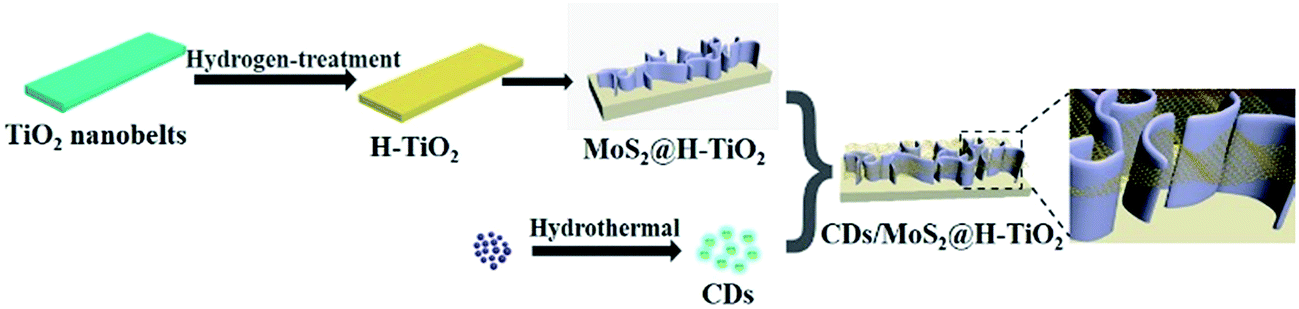
DOI:
10.1039/C6RA28479E
(Paper)
RSC Adv., 2017,
7, 8429-8442
Construction and enhanced photocatalytic activities of a hydrogenated TiO2 nanobelt coated with CDs/MoS2 nanosheets†
Received
20th December 2016
, Accepted 28th December 2016
First published on 25th January 2017
Abstract
A few-layered CDs (carbon dots)/MoS2 nanosheet-coated hydrogenated TiO2 (H-TiO2) nanobelt heterostructures—referred to as CDs/MoS2@H-TiO2—with a flexible three-dimensional (3D) hierarchical configuration were prepared via a facial hydrothermal reaction. Note that the visible photocatalytic activity of H-TiO2 was improved compared with that of pristine rutile TiO2, which can be mainly attributed to the optical absorption and charge carrier trapping of oxygen vacancies and Ti3+ ions in TiO2 nanobelts created by the hydrogenation. The CDs/MoS2@H-TiO2 ternary photocatalysts exhibit excellent UV and visible photocatalytic property. Via optimizing the proportion of each component, the CDs/MoS2@H-TiO2 composite showed the highest photocatalytic degradation activity when the content of the CDs/MoS2 co-catalyst was 5.0 wt% and the content of CDs in this cocatalyst was 25%. Further study revealed that the considerable photodegradation rate under UV irradiation and a large promotion of the photocatalytic activity in both the visible and near-infrared (NIR) region originated from the synergistic effect of oxygen vacancies, interfacial modification, and the vectorial charge-transfer channel design. Our study provides a desired strategy to understand and realize a rationally designed electronic transition between a semiconductor and cocatalysts, which is of great importance for the enhancement of charge separation and obtaining improved photocatalytic performance.
1. Introduction
As part of a high production and usage of drugs in the world, antibiotics are widely applied in the treatment of bacterial infection, but carry serious environmental concerns as they are widely discharged into the aquatic ecosystem via wastewater and waste treatment effluent, animal manure, and soil erosion.1,2 Therefore, the elimination of tetracycline antibiotics from the aquatic ecosystem has awakened global awareness in the need to continuously improve and perfect more efficient methods to deal with their presence in the aquatic ecosystem. Titanium dioxide (TiO2), a representative species of semiconductor photocatalysts that can convert solar energy into chemical energy, has been extensively researched for environmental purification applications and in water splitting for hydrogen generation.3–6 Although TiO2 has a strong oxidation strength and high chemical stability, making it suitable as a photocatalyst, its primary drawback is its wide band gap of 3.23 eV.7–11 Its wide band gap means it is only active in the UV light region of the solar spectrum, which accounts for only 5% of the total incident solar energy. Therefore, conspicuous efforts have been made to extend the response spectrum of TiO2 towards the visible spectral region.12–15 This can be achieved by sensitizing it with narrow-band-gap semiconductors or via forming new donor states below the conduction band of TiO2, e.g. through the incorporation of metal or non-metal elements into its crystal matrix.16–18 An alternative method for ameliorating the optical absorption and photocatalytic properties of TiO2 is to prepare disordered nanophase TiO2 through the hydrogenation of TiO2 nanocrystals at 20 bar H2.19 Hydrogen treatment is a more desired strategy as introducing oxygen vacancies or Ti3+ incorporation into TiO2 can dramatically narrow the band gap to ≈1.54 eV and thus enhance the visible light absorption, which consequently has attracted great interest in photocatalysis.20,21 Mao's group reported that the optical absorption of hydrogenated TiO2 nanocrystals shifted from ultraviolet light to the near infrared (NIR), accompanied by the appearance of oxygen vacancies and Ti3+ (Ti3+/oxygen vacancies).19 The introduced Ti3+/oxygen vacancies are believed to act as electronic charge carriers for suppressing the recombination of photogenerated electrons and holes (e–h pairs), thereby improving the photocatalytic activity of TiO2.22 Therefore, hydrogen treatment is a desired strategy that can significantly enhance the photocatalytic property of TiO2 nanostructures.
Considering the superiority of the two-dimensional (2D) nanolayered materials as a composition in photocatalysts, it is expected that coupling 2D layer cocatalysts with a visible light-responsive H-TiO2 semiconductor may create a new type of photocatalyst with an efficient separation rate of e–h pairs, which would thus improve the photodegradation activity.23,24 Accordingly, research on the nanohybrids, named “van der Waals heterostructures”, has aroused great interest. These materials are typically composed of MoS2 nanoplates and others nanomaterials, formed by a layer-by-layer self-assembly.25,26 Generally, this type of nanohybrid can exhibit superior photocatalytic activity by efficiently utilizing the intergrated merits of these components. Hinnemann's group concluded, through density function calculations of the free energy, that the S atoms on the exposed edges of MoS2 are highly active, greatly improving the MoS2/G-CdS photocatalytic H2 evolution activity.27 Zhou et al. indicated that the matched energy band of the TiO2@MoS2 heterojunction favours the charge transfer and suppresses the photoelectron–hole recombination between MoS2 and TiO2, which induces the significant photocatalytic hydrogen production and photocatalytic degradation activity of organic materials.4 Xiang et al. reported that the MoS2–graphene nanocomposite, when used as an efficient co-catalyst over TiO2 nanoparticles, can help realize highly efficient hydrogen production without the need for a noble metal co-catalyst.6 Despite the great efforts made so far, the study of photocatalysts based on van der Waals heterojunctions is still in its infancy. From a practical point of view, further work is needed to enhance their photoabsorption performance, enabling them to harvest a wide spectrum of sunlight, from ultraviolet to near-infrared wavelengths, and to achieve efficient solar energy conversion. Monodispersed carbon dots (CDs) retain the graphite structure with a size below 10 nm and have natural merits over other counterparts. First, a zero dimensional (0D) uniform distribution of CDs and less flocculation would be propitious to the interspaces and pathways for ions transfer over a multidimensional carbon material.28 Second, CDs are thin enough so that there is no negative effect on the photoabsorption of the semiconductor.29–31 Most importantly, CDs possess a broad cross-section of up-conversion emission, which means they can realize a better utilization of NIR light in the solar spectrum.12,29 For example, CQDs/Ag/Ag2O (where CQDs = carbon quantum dots), CQDs/hydrogenated-gamma-TaON and a CQDs/C3N4 composite successfully revealed photocatalytic activity and a photo-response ability over the photodecomposition of organic compounds, including for hydrogen evolution from water splitting.29,30,32 This has given us useful enlightenment: similarly, the feasibility of applying a solution-based carbon precursor inspires the idea of forming a thin carbon coating layer on 3D nanostructures surface to absorb more sunlight and to enhance the vector transfer of photogenerated electrons.
Herein, we report the fabrication of a nano-sized layer-structured CDs/MoS2 nanosheet-coated H-TiO2 nanobelts with a 3D hierarchical configuration through a facile hydrothermal method, and furthermore, we report investigations into its tetracycline (TC) degradation activities under simulated solar irradiation. The CDs/MoS2@H-TiO2 composite photocatalysts showed the highest photocatalytic degradation activity for the sample containing 5% CDs/MoS2 (CM) hybrid co-catalyst, consisting of 80% MoS2 and 20% CDs. To be an effective photocatalyst the following strategies are needed: (1) a high surface area of 3D architectures consisting of H-TiO2 nanobelts and a MoS2 nanosheet as this will enhance the contact between the charge carriers and surface species; (2) the development of ultrathin MoS2 nanosheets encapsulating the H-TiO2 nanobelts for efficient and stable photogenerated charge separation; and (3) the formation of van der Waals heterostructures in a 3D CDs/MoS2@H-TiO2 system, which would result in the further migration of electrons from MoS2 to the CDs layer due to the electrical conductivity of the MoS2 nanosheet.
2. Experimental
2.1 Materials
Titania P25, sodium hydroxide (NaOH), citric acid, ethylenediamine, hydrochloric acid (HCl), sulfuric acid (H2SO4), sodium molybdate (Na2MoO4·2H2O), and L-cysteine were purchased from Sinopharm Chemical Reagent Beijing Co., Ltd.
2.2 Synthesis of hydrogenated TiO2 (H-TiO2) nanobelts
TiO2 nanobelts were synthesized by a hydrothermal process. Typically, we mixed the P25 powder (0.1 g) with an aqueous solution of NaOH (20 mL, 10 mol L−1), followed by a hydrothermal treatment at 180 °C in a 25 mL Teflon-lined autoclave for 48 h. Then, we eluted it with deionized water, followed by filtration and drying. The obtained sample was immersed in hydrochloric acid aqueous solution (0.1 mol L−1) for 48 h and then the sample was mixed with 20 mL H2SO4 aqueous solution (0.02 mol L−1) and then transferred into a reaction caldron and heated at 80 °C for 2 h. Finally, the TiO2 nanobelts were obtained via annealing at 600 °C for 2 h. After that, the TiO2 nanobelts were heated to 600 °C at a ramping rate of 5 °C min−1 and maintained for 5 h under a flow of hydrogen with a flow rate of 50 sccm.
2.3 Synthesis of the MoS2@H-TiO2 nanobelts
Few-layers MoS2@H-TiO2 heterostructures (5 wt% of MoS2) were prepared by a facile hydrothermal method. The formation process of the MoS2@H-TiO2 heterostructures (5 wt% of MoS2) can be described as follows. Typically, 45 mg L-cysteine and 30 mg sodium molybdate (Na2MoO4·2H2O) were dissolved in 20 mL deionized water to form a transparent solution, and then 20 mg of prepared H-TiO2 nanobelts were added into the solution. Next, the homogeneous solution was transferred into a Teflon-lined stainless steel autoclave and held at 200 °C for 24 h. A black product, MoS2@H-TiO2 heterostructures, was collected after centrifugation and was dried at 50 °C for 12 h. For comparison, pure MoS2 nanosheets powders were synthesized under the same conditions as in the hydrothermal process but without the presence of H-TiO2 nanobelts.
2.4 Synthesis of the CDs/MoS2@H-TiO2
The multistep preparation process of CDs/MoS2@H-TiO2 is displayed in Scheme 1. The CDs were prepared according to the literature, followed by the rotation-evaporation method.33
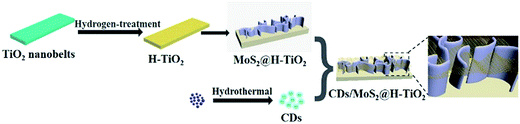 |
| | Scheme 1 Synthesis process of the CDs/MoS2@H-TiO2 nanocomposites. | |
First, 9.8 mg of the prepared MoS2@H-TiO2 heterostructures (WTi/WMo = 90/8) was sonicated thoroughly in 20 mL DS stock solution (0.1 mg mL−1). After ultrasonication for 30 min, the mixture was transferred into a 50 mL Teflon-lined autoclave and kept at 140 °C for 24 h. To investigate the effect of the CDs/MoS2 content on the photocatalytic degradation activity of H-TiO2, different amounts of CDs (according to the mass ratio of MoS2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CDs 1:1, 2:1, 4:1, 6:1 and 8:1, corresponding to a mass ratio of MoS2 in composite of 1%, 3%, 5%, 7%, 10% and 15%) was added into the starting materials solution. The as-prepared CDs/MoS2@H-TiO2 could be obtained after washing and drying at 60 °C overnight.
:CDs 1:1, 2:1, 4:1, 6:1 and 8:1, corresponding to a mass ratio of MoS2 in composite of 1%, 3%, 5%, 7%, 10% and 15%) was added into the starting materials solution. The as-prepared CDs/MoS2@H-TiO2 could be obtained after washing and drying at 60 °C overnight.
2.5 Characterization
The morphologies of the as-obtained samples were observed using FESEM (S-4800 field emission scanning electron microscope) and TEM (Tenai G2 F30 S-Twin, FEI). XRD patterns were recorded by a powder X-ray diffraction method using a D/Max-IIIC instrument with Cu Kα radiation (λ = 1.54178 Å). The Brunauer–Emmett–Teller specific surface areas (SBET) and porosity of the samples were measured at −196 °C on a TriStar II 3020 Micrometrics apparatus. Raman spectra were recorded on an Ihr550 Raman microscope with 532 nm solid laser as an excitation source. FT-IR spectroscopy was recorded on a Bruker Vertex 70 spectrometer. UV-Vis absorption spectra were obtained using a UV-Vis spectrophotometer (UV-3600, Shimadzu, Japan). XPS spectra were recorded on a Thermo ESCALAB 250X (America) electron spectrometer. Time-resolved photoluminescence spectroscopy (TRPS) spectra were obtained on a Model FES 920 system with an excitation wavelength of 337 nm and a detection wavelength of 469 nm. The PL spectra were obtained on a Horiba JobinYvon Luminescence spectrometer.
2.6 Photocatalytic experiments
The photodegradation experiments with a tetracycline (TC) solution (10 mg L−1) were carried out under UV, visible, and NIR light irradiation. In a typical photocatalytic experiment, 50 mg of as-synthesized samples was dispersed in 100 mL pollutants aqueous solution (10 mg L−1). Then, the suspension was stirred in the dark for 40 min to ensure the adsorption–desorption equilibrium prior. A 250 W xenon lamp was used as the visible light source with filter glasses (λ < 420 nm). A 350 W mercury lamp was used as the UV source for photocatalysis. A 250 W infrared lamp was used as the NIR light source. At given irradiation time intervals, the residual pollution concentration was detected after centrifugation by the UV-Vis spectrophotometer at a specific wavelength. The concentration changes of TC were monitored by measuring the UV-Vis absorption of the suspensions at 357 nm using a TU-1810 UV-Vis spectrophotometer.
2.7 Photoelectrochemical measurements
The photoelectric current (PC) response measurements were performed using a CHI 660B electrochemical workstation with a Pt plate and a Ag/AgCl electrode as the counter electrode and reference electrode, respectively. A 0.5 M Na2SO4 aqueous solution was added to the electrolytic tank as the electrolyte. Electrochemical impedance spectroscopy (EIS) was performed in the frequency range of 10−2 to 106 Hz, with the initial potential (0 V) in 0.5 M KCl solution containing 5.0 mM K3 [Fe(CN)6]/K4 [Fe(CN)6] under open circuit potential conditions. The working electrode was prepared on fluoride tin oxide (FTO) conductor glass. Typically, the sample powder (0.1 g), absolute ethanol (3 mL), polyvinylpyrrolidone (0.03 g) and oleic acid (30 μL) were mixed and ultrasonicated to obtain a slurry. The slurry was spread onto the surface of an FTO plate dried at 80 °C for 2 h to improve the adhesion. A 300 W Xenon lamp with a UV cut-off filter (λ > 420 nm) was used as the light source.
3. Result and discussion
3.1 Characterization of the as-prepared photocatalysts
The TiO2 nanobelts were prepared by a hydrothermal reaction of P25 in concentrated NaOH solution. Fig. S1 (ESI†) showed the TiO2 nanobelts with a wide of 50–300 nm, a thickness of ≈20 nm and a length of several micrometres. After further hydrogenation treatment, the size and morphology remained the same, as seen in Fig. 1a. However, the colour of the TiO2 powder changes from white to black. A typical SEM image of the CDs/MoS2@H-TiO2 heterostuctures is shown in Fig. 1b and c (with 5 and 10 wt% CDs/MoS2, respectively). The thin CDs/MoS2 nanosheets are uniformly in situ grown on the surface of the H-TiO2 nanobelts and are interpenetrated with each other, forming 3D nanosheet networks. The pure MoS2 nanosheets were also obtained by the same preparation process without the presence of H-TiO2 nanobelts, self-assembled to form MoS2 microsphere (Fig. 1d). However, no aggregation could be found in the SEM image of the CDs/MoS2@H-TiO2, which can be attributed to the acid etching process providing nucleation sites for the nucleation and growth of the MoS2 nanosheets. Therefore, by simply increasing the precursor concentration, the density of the MoS2 nanosheets in CDs/MoS2@H-TiO2 can be increased significantly.
 |
| | Fig. 1 SEM images of: (a) pure H-TiO2 nanobelts, (b and c) CDs/MoS2@H-TiO2 nanocomposite (5 wt% and 10 wt% of CDs/MoS2), and (d) pure MoS2 microspheres assembled from MoS2 microspheres. | |
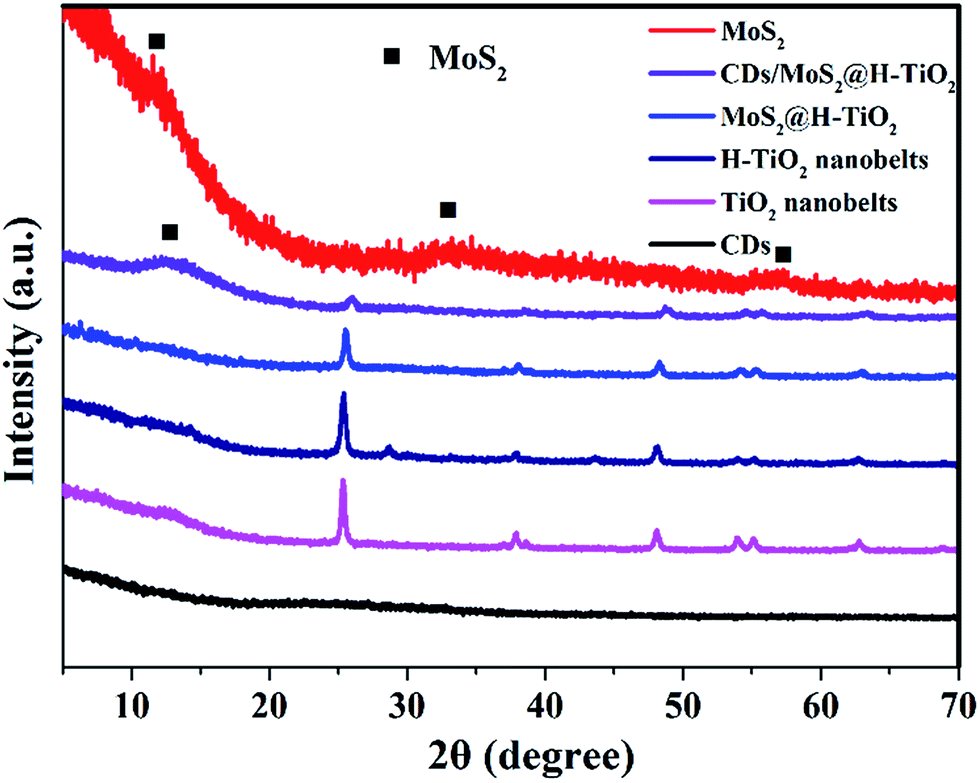
The CDs/MoS2@H-TiO2 composite was characterized by powder X-ray diffraction (XRD), as shown in Fig. 2. The TiO2 nanobelts and H-TiO2 nanobelts exhibited similar diffraction peaks. Strong XRD diffraction peaks at 2θ = 25.56°, 37.84°, 48.15°, 53.79°, 55.06° and 62.69° can be clearly observed, and are consistent with the (110), (101), (111), (211) and (301) crystal faces of anatase phase TiO2 indexed to the JCPDS card 21-1272 with a space group of P42/mnm (a = b = 4.584 Å and c = 2.953 Å). This indicated that both TiO2 nanobelts and H-TiO2 nanobelts displayed highly crystalline anatase phases, without other phases after hydrogenation, such as a rutile or brookite phase. Furthermore, the intensity of the peak increased after hydrogenation and the average crystal size of the H-TiO2 nanobelts and TiO2 nanobelts was 22.2 and 26.8 nm, respectively, as deduced from the peak broadening of the X-ray diffraction pattern using the Scherrer equation. In addition, a slight peak at 28.32 nm was observed. This peak can be attributed to the lattice oxygen on the surface of TiO2 nanobelts interacting dramatically with the hydrogen atoms when the electrons of the H atoms are transferred to the Ti4+ of TiO2, which result in Ti3+ defects being formed.20 However, little signal attributable to the MoS2 or CDs diffraction peaks can be observed in the complex samples, which may due to the low content.
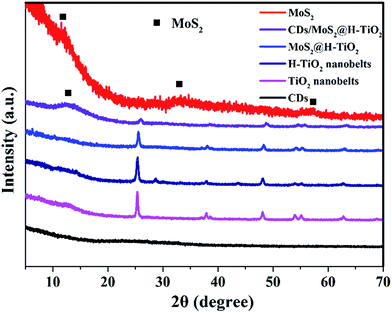 |
| | Fig. 2 XRD pattern of the as-prepared CDs, TiO2, H-TiO2, MoS2@H-TiO2, CDs/MoS2@H-TiO2, and MoS2. | |
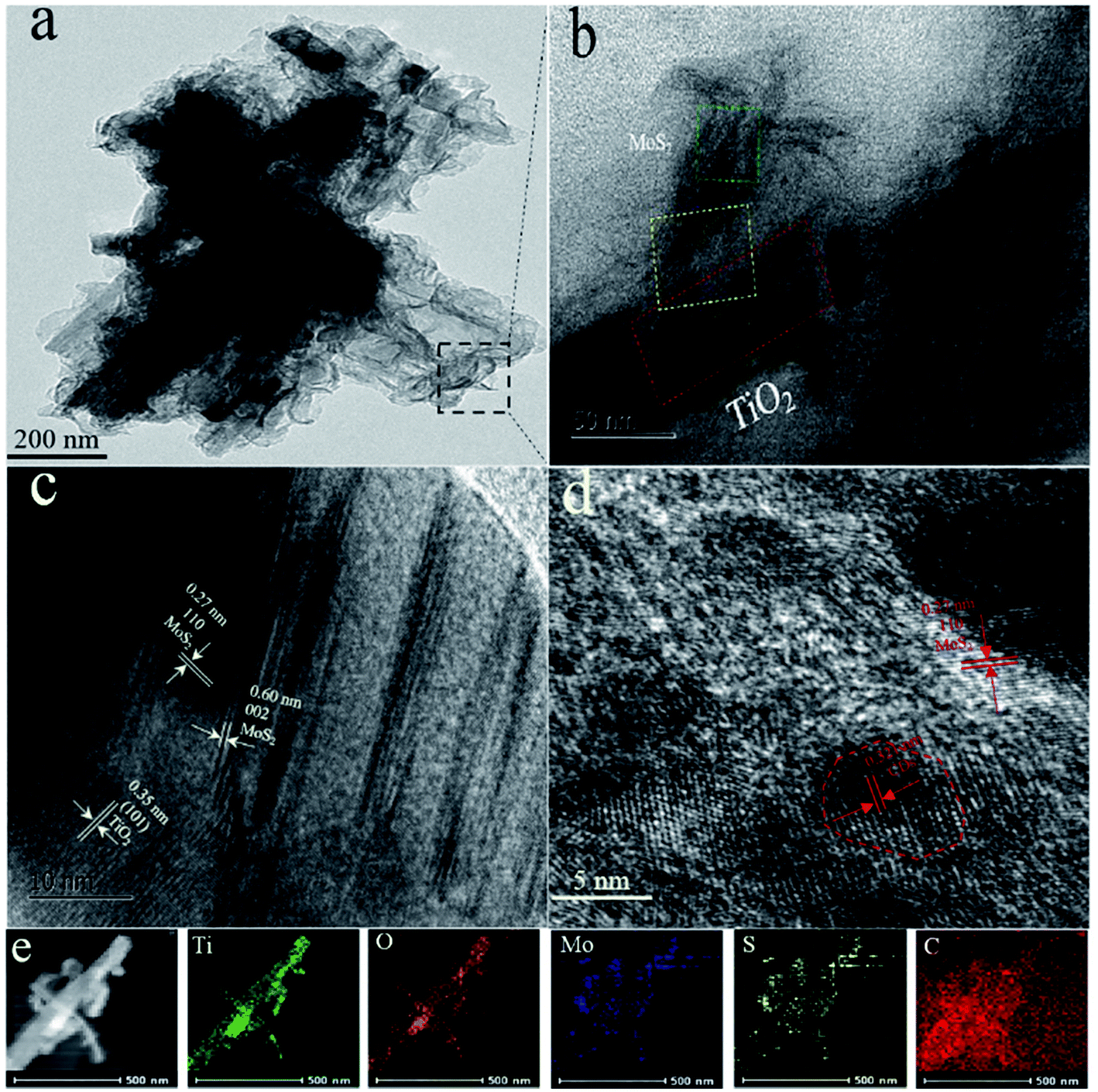
Fig. 3a and b show the TEM images of CDs/MoS2@H-TiO2 heterostructure, where the few-layer MoS2 nanosheets cover the surface of the H-TiO2 nanobelts and their thickness is about 5 nm. Fig. 3c shows three type of lattice fringes: the lattice fringes with a lattice spacing of 0.35 nm correspond to the (101) facet of H-TiO2, while another set of fringe spacings around 0.27 nm can be found, which agree well with the (002) lattice spacing of the hexagonal phase MoS2. From the HRTEM images in Fig. 3d, the lattice fringes of the MoS2 nanosheets can be clearly observed, suggesting the well-defined crystal structure. The fringes with a lattice spacing of 0.27 nm correspond to the (100) plane of MoS2. The (002) plane of MoS2 with a lattice spacing of 0.60 nm is also observed, which comprises only 5 layers of MoS2 nanosheets. The energy dispersive X-ray spectroscopy (EDS) elemental mapping from CDs/MoS2@H-TiO2 also confirmed the core (H-TiO2) and the shell (CDs/MoS2 nanosheets) hierarchical structures (Fig. 3e).
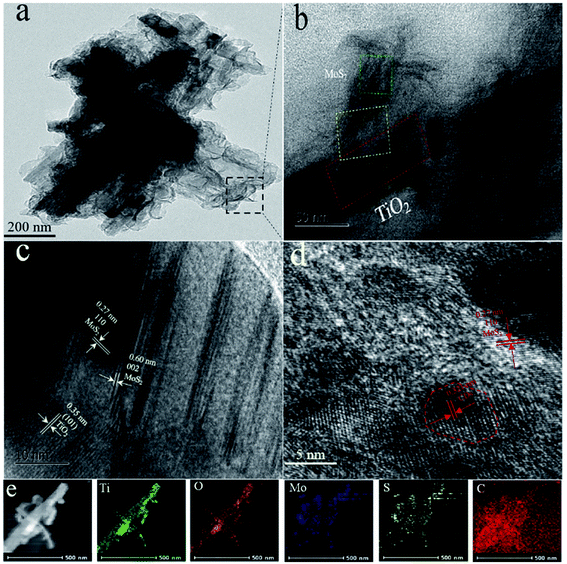 |
| | Fig. 3 (a and b) Low-magnification TEM images of CD/MoS2@H-TiO2 (5 wt% of cocatalysts); (c and d) HRTEM images of the CD/MoS2@H-TiO2, and (e) EDS mapping results from CD/MoS2@H-TiO2 (5 wt% of cocatalysts). | |
Fig. 4 shows the N2 adsorption–desorption isotherms of the as-prepared pure H-TiO2 and CDs/MoS2@H-TiO2 samples. It can be seen that the CDs/MoS2 shell has an effect on the specific surface area of the samples. The Brunauer–Emmett–Teller (BET) specific surface area obtained for CDs/MoS2@H-TiO2 was 27.557 m2 g−1, a value higher than the corresponding value, namely 13.618 m2 g−1, estimated for H-TiO2 nanobelts. The nanocomposites surface area increase, which indicates that the nanosheets do not restack in the composites and instead form a 3D hierarchical porous structure, as evidenced by SEM.24 This kind of 3D hierarchical structure is supposed to be beneficial for solution infiltration and photogenerated mobility, consequently improving the photocatalytic activity.
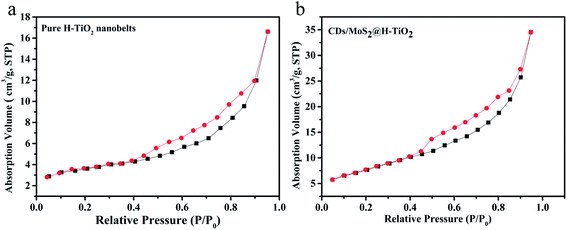 |
| | Fig. 4 BET absorption–desorption isotherms obtained of: (a) pure H-TiO2 nanobelts and (b) CDs/MoS2@H-TiO2 composites, in which the mass ratio of CDs to MoS2 is 1:4 and the amount of CDs/MoS2 is 5.0 wt%. | |
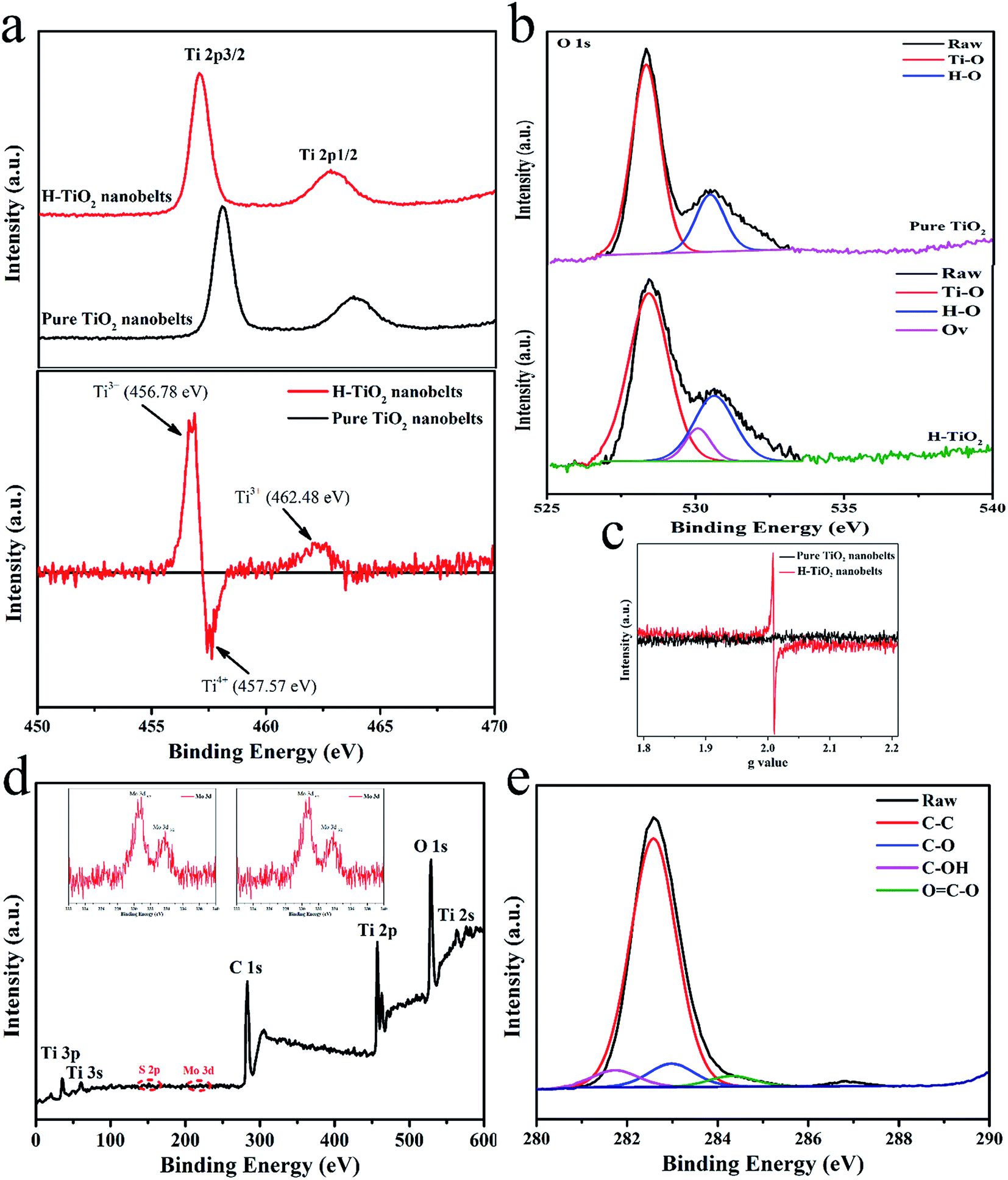
High-resolution XPS was employed to further investigate the surface binding properties of the as-synthesized samples. As illustrated in Fig. 5a (top), the Ti 2p peaks of the TiO2 nanobelts are located at 460.78 and 466.03 eV, respectively, while a radically different characteristic can be found in the Ti 2p XPS spectrum of H-TiO2, which may be reasonably attributed to the formation of high density defects.20 To observe these differences, a different Ti 2p XPS spectrum was obtained by subtracting the normalized spectra value of TiO2 with that of the H-TiO2 sample (“H-TiO2” minus “TiO2”). As illustrated in Fig. 4a (bottom), the fitting peaks at 456.78 and 462.48 eV are ascribed to Ti 2p3/2 and Ti 2p1/2 and should originate from Ti3+ of the H-TiO2 nanobelts. Similarly, the fitted result of the O 1s XPS spectrum in Fig. 4b suggests that obvious oxygen vacancies are introduced into H-TiO2 via hydrogenation treatment (Fig. 3b). Furthermore, in terms of EPR spectroscopy (Fig. 3c), no obvious signal could be observed from the TiO2 nanobelts, while the H-TiO2 samples developed a noteworthy peak at g = 2.0010. It was thought that the above signal peak occurred due to the existence of oxygen vacancies in the H-TiO2 nanobelts.34 These results manifest that Ti3+ was successfully doped into H-TiO2 under the reduction atmosphere via taking the electrons from the hydrogen atoms and transferring them to Ti4+. The XPS spectrum (Fig. 4d) showed peaks for Ti, O, Mo, S and C, with a Mo/S atomic ratio of ∼1:2, which is in good agreement with the nominal atomic composition of Mo2. The binding energies of the Mo 3d5/2 and Mo 3d3/2 peaks at 230.8 and 233.6 eV were observed (Fig. 4d inset), respectively, indicating that the Mo element was present in the Mo4+ chemical state. In addition, the XPS spectrum of C 1s (Fig. 3e) proved the existence of CDs in the CDs/MoS2@H-TiO2 sample and that it efficiently combines with the MoS2 to form bonds.
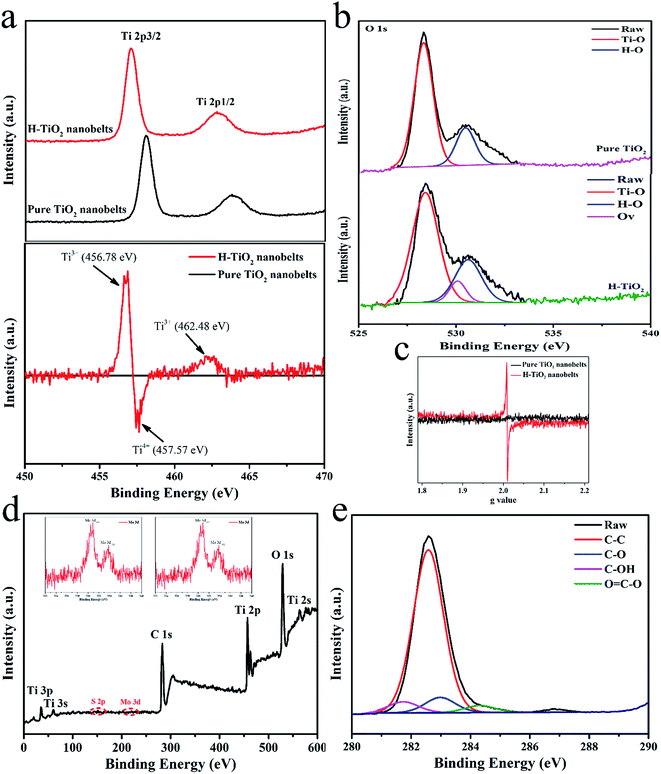 |
| | Fig. 5 (a) Original XPS spectra of Ti 2p for pure TiO2 (top) and H-TiO2 together with their different spectra (“H-TiO2” minus “pure TiO2”) (bottom); (b) high-resolution XPS spectra of O 1s; (c) low-temperature EPR for pure TiO2 and H-TiO2; (d) XPS spectrum of CDs/MoS2@H-TiO2 composite and (e) XPS spectrum of C 1s. | |
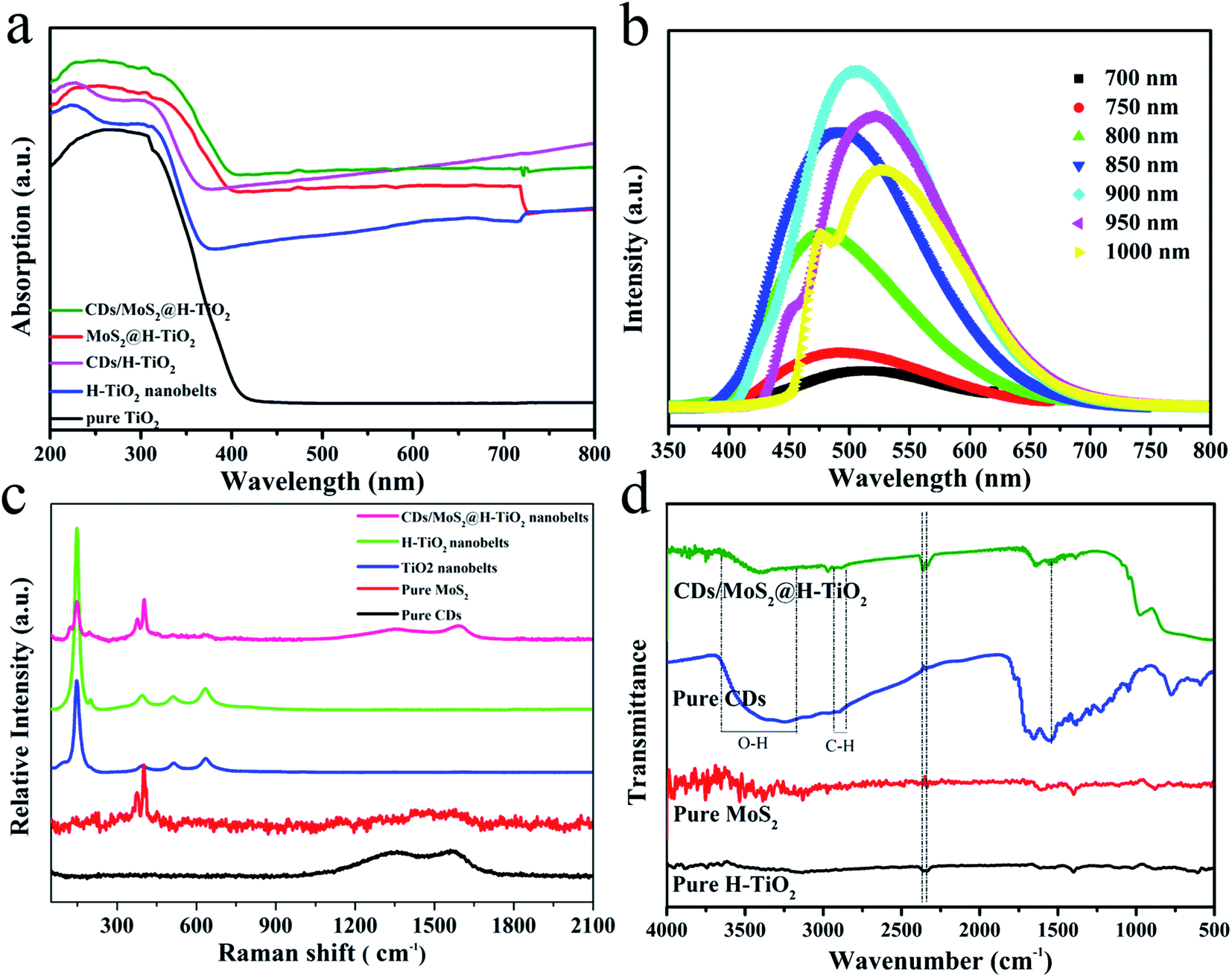
The UV-Vis absorption spectrum of the as-synthesized samples is shown in Fig. 6a. For the pure TiO2 nanobelts, an apparent absorption in the UV region, which was coincident with the bandgap of TiO2 (e.g. 3.2 eV, corresponding to λ = 387 nm). A remarkable characteristic of the UV-Vis absorption spectra of the H-TiO2 samples is the appearance of a broad and almost continuous absorption band in the visible region, which can be attributed to the low-energy photon of trapped electrons in the localized states of oxygen vacancies associated with Ti3+ just below the lowest conduction band.20 Adding MoS2, CDs or CDs/MoS2 as cocatalysts, their light absorption was enhanced, without exception. This phenomenon may originate from the deep colour and the strong quantum confinement effect of the CDs/MoS2 thin nanosheets.27 The optical band gaps of pure MoS2 and the CDs/MoS2 nanohybrid were calculated by UV-Vis spectra. As shown in Fig. S2,† the band gap of the synthesized MoS2 and CDs/MoS2 were estimated to be 1.0 eV, which is narrower than that of the graphene/MoS2 values reported in the previous literature (1.1–1.3 eV). This might be due to the exceptional advantages of CDs, such as high optical absorptivity. In addition, the up-converted photoluminescence spectra of the CDs under the excitation of (N)IR light were obtained (Fig. 6b). The results show that the emissions peaks are strands in the visible light wavelength of 350–600 nm. That is to say, the H-TiO2 nanobelts can realize the indirect utilization of NIR by using the fluorescence effect of the CDs, so as to enhance the optical absorption response range.
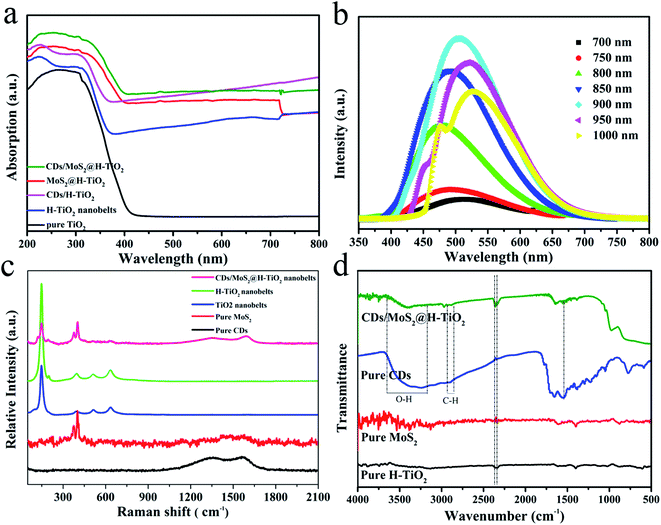 |
| | Fig. 6 (a) UV-Vis absorption spectra; (b) up-converted photoluminescence spectra of CDs; (c) Raman spectra and (d) FT-IR spectra of the prepared samples. | |
To investigate in-depth the atomic structure of the CDs/MoS2@H-TiO2 sample, Raman and FT-IR spectroscopy were conducted. As shown in Fig. 6c, the Raman spectra for the CDs/MoS2@H-TiO2 composite shows several characteristic bands at 148, 399, 518 and 639 cm−1, corresponding to the Eg(1), B1g(1), A1g + B1g(2) and Eg(2) modes of TiO2, respectively.6 Interestingly, two peaks at about 1343 cm−1 and 1586 cm−1 were also observed, which were associated with disordered sp3 carbon (D band) and conjugated sp2 (G-band) of the carbon atoms.35 Also, the observed D and G bands of the composite were slightly shifted compared with pure CDs, which provides further evidence for the presence of a chemical bond between the CDs and composites in the CDs/MoS2@H-TiO2 samples. The FT-IR spectrum was used to analyze the change in functional groups during the synthesis process. From Fig. 6d, the stretching vibrations of OH at 3430 cm−1 and C–H at 2923 cm−1 and 1380 cm−1 can be observed. The absorption bands at 3238 cm−1 and 3389 cm−1 are ascribed to the O–Ti–O stretching mode and S–Mo–S bridging stretching modes, which imply the MoS2 composites can efficiently combine with the H-TiO2 substrate to form bonding. Based on the above description and discussion, we can conclude that the synergetic catalytic effect of the MoS2 nanoplate and CDs layer by interfacial modulation plays an important role in the enhancement of photocatalytic activity.
3.2 Charge-transfer properties
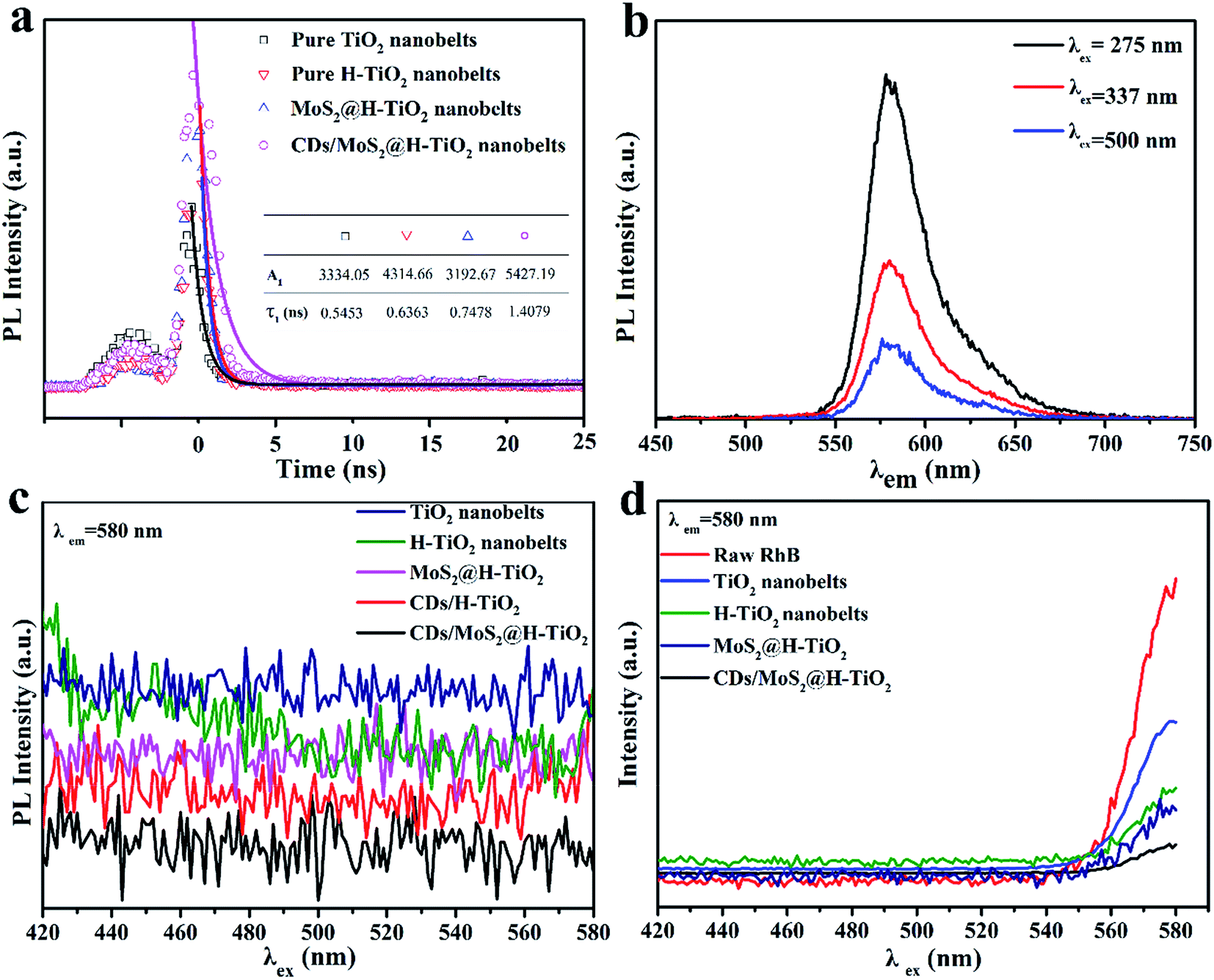
As part of the in-depth research into the electron-assisted charge dynamics of CDs/MoS2@H-TiO2, we also measured the time-resolved and steady-state photoluminescence spectra. As shown in Fig. 7a, the resulting fluorescence was analyzed by fitting the time-resolved fluorescence decay curves with the following multi-exponential regress model (excitation at λ = 337 nm, for the excitation of H-TiO2):| | |
I(t) = A1exp(−t/τ1) + A2exp(−t/τ2)
| (1) |
| | |
τave = (A1τ1 + A2τ2)/(A1 + A2)
| (2) |
where τ1 and τ2 are the decay times and A1 and A2 represent the relative weights of the components at t = 0.29 In comparison with the TiO2 nanobelts, the lifetime of H-TiO2 was significantly prolonged over the lifetime of the charge carriers, which indicates that the oxygen atoms on the surface of TiO2 nanoparticles can easily be removed and an oxygen-deficient TiO2−x is generated during the hydrogenation process.20 The lifetime of the as-prepared MoS2@H-TiO2 nanobelts and CDs/MoS2@H-TiO2 nanobelts were 0.7478 and 1.4079 nm, respectively. Notably, the CDs/MoS2@H-TiO2 nanobelts yielded the longest decay time as compared with the H-TiO2 nanobelts and MoS2@H-TiO2 nanobelts, indicating an accelerated charge transfer induced via the coating of the CDs/MoS2 layer. Fig. 7b shows the PL emission of 1 μM RhB, where the emission peak is located at 584 nm, which is a constant independent of the excitation wavelength. The PL emission of the prepared samples in aqueous solution can be seen in Fig. 7c, where no obvious radiation peaks were observed in any of the samples. Fig. 7d displays the steady-state PL quenching spectra of the as-synthesized samples with suspended rhodamine B aqueous solution (RhB as an indicator dye used to monitor the interfacial charge transfer) with an excitation wavelength of 337 nm. The PL emission of RhB can be quenched by H-TiO2, since the lowest unoccupied molecular orbital (LUMO) level of RhB (−1.1 V vs. NHE) is higher than the CB of H-TiO2 (−0.29 V vs. NHE).36,37 The H-TiO2 dispersed in the RhB solution displayed a stronger PL emission than pure RhB at the emission wavelengths in the range 405–480 nm (Fig. 5d). This phenomenon should be ascribed to the overlapping emission of H-TiO2 and RhB. However, a weaker PL emission peak of RhB in the presence of H-TiO2 was noticed for excitation wavelengths longer than 480 nm, which suggests the significance of intramolecular charge transfer. The MoS2@H-TiO2 exhibited a remarkable quenching in the PL emission of RhB, considering the PL emission of MoS2@H-TiO2, indicating that the photoexcited electrons from RhB were transferred to the CB of H-TiO2 as well as to the MoS2 nanosheets. In particular, a remarkable depression in the PL intensity of RhB was also noted in the CDs/MoS2@H-TiO2 system when the excitation wavelength was shorter than 480 nm. The depression was due to the introduction of CDs, which facilitated the further transfer of electronic from the CB of MoS2 to the CDs layer. As a result, a fast electron transfer via H-TiO2 nanobelts → MoS2 nanosheet → CDs layer was proved. The accelerated charge transfer is bound to reduce the recombination of photogenerated electrons–holes and enhance the photocatalytic activity.
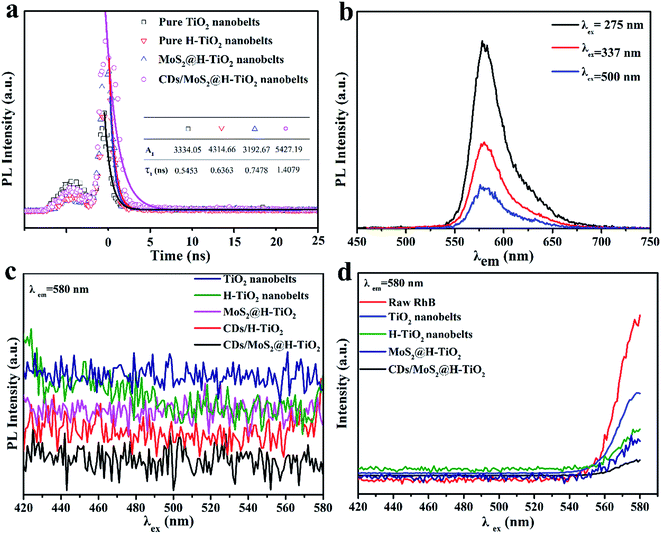 |
| | Fig. 7 (a) Time-resolved fluorescence decay curves of TiO2, H-TiO2, MoS2@H-TiO2 and CDs/MoS2@H-TiO2; (b) steady-state PL spectra of 1 μM RhB, where the excitation wavelengths were 275, 337 and 500 nm; (c) PL emission of an aqueous solution in the presence of each sample; and (d) PL emission of an aqueous solution of 1 μM RhB in the presence of each sample. | |
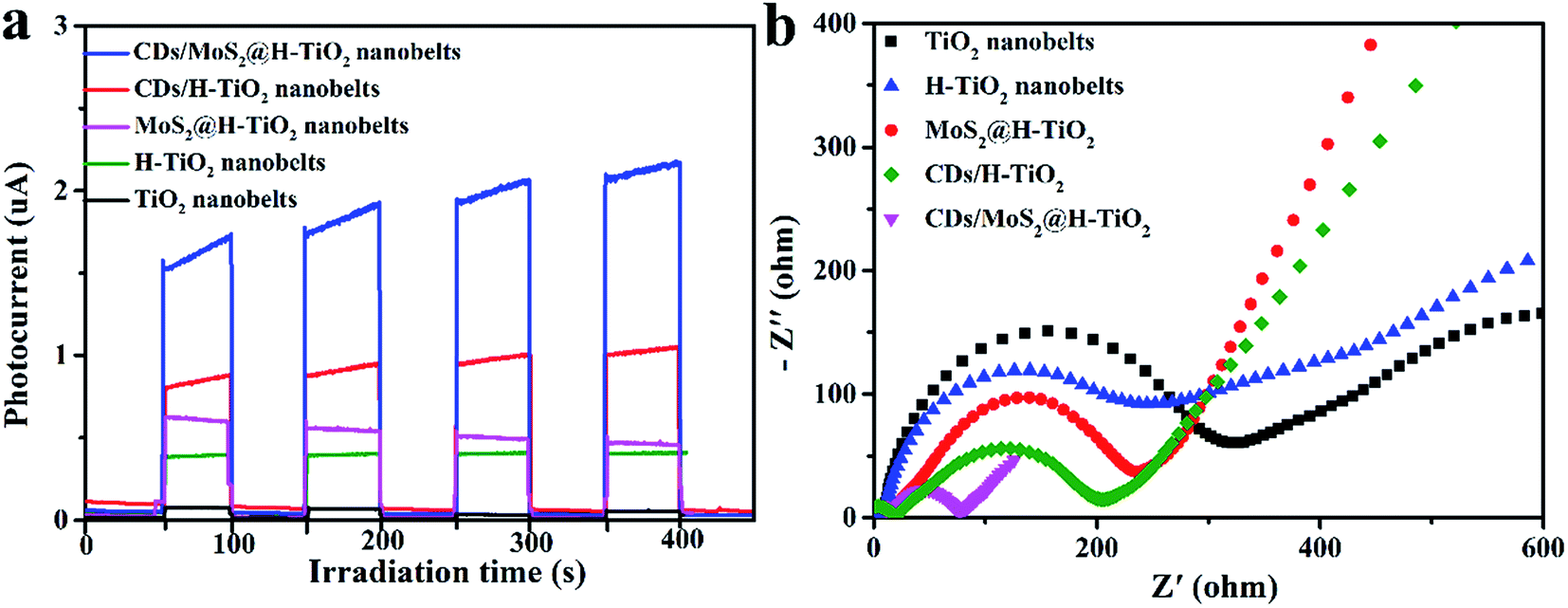
To provide additional evidence for the kinetics of charge transfer, the transient photocurrent responses of as-prepared composites coated on the FTO substrate were measured for several cycles under visible light irradiation. Fig. 8a shows that the photocurrent increased rapidly under visible light irradiation. Meanwhile, after several periods of intermittent switching, the photocurrent still remains steady and repeatable. Compared to pure TiO2 nanobelts, the H-TiO2 nanobelts displayed a higher photocurrent intensity. Previous studies have reported that surface O vacancies promote the separation of photogenerated carriers in H-TiO2. After adding MoS2 or CDs cocatalysts, the photocurrent densities were significantly enhanced, indicating that the recombination of electron–hole pairs is delayed. As expected, the CDs/MoS2@H-TiO2 composites exhibited the highest photocurrent response, which demonstrates that constructing CDs/MoS2@H-TiO2 can significantly improve the photogenerated electron–hole pair's separation. Here, the H-TiO2 nanobelts not only provide a confined substrate for the in situ growth of few-layer MoS2 nanoplates, but also form interconnected 3D conductive networks for efficiently transferring photogenerated electrons through the harness charge flow of the H-TiO2 nanobelts → MoS2 nanoplates → CDs layer. Additionally, electrochemical impedance spectroscopy (EIS) was employed to investigate the charge-transfer resistance of the system in the battery enclosure. As shown in Fig. 8b, the Nyquist plot indicates the charge-transfer rate occurring at the contact interface between the working electrode and the electrolyte solution. It should be noted that the H-TiO2 electrode is much smaller in comparison to that of the TiO2 electrode. In other words, hydrogenation treatment of the oxygen atoms on the surface of the TiO2 nanobelts greatly promotes charge transfer across the electrode/electrolyte interface. Correspondingly, the CDs/MoS2@H-TiO2 sample shows the smallest arc radius of Nyquist curve, which shows that MoS2 may serve as an electron transfer medium in the vectorial electron transfer from H-TiO2 → MoS2 →CDs, thus promoting interfacial charge separation and migration, also efficiently reducing the excitron quenching and energy dissipation. In short, the PL measurements and electrochemical data demonstrate that the CDs/MoS2 nanohybrids in CDs/MoS2@H-TiO2 could generate a notable positive synergistic effect that significantly improves the separation of photogenerated electrons and holes.
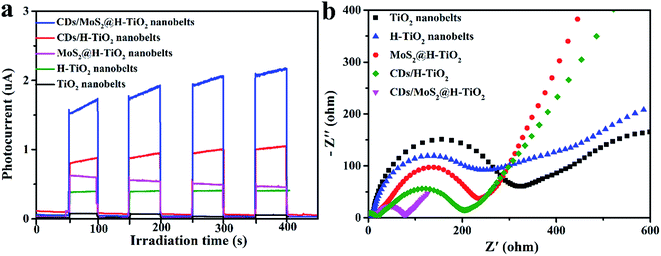 |
| | Fig. 8 Transient photocurrent response (a) and EIS (b) for TiO2, H-TiO2, MoS2@H-TiO2, CDs/TiO2 and CDs/MoS2@H-TiO2 samples. | |
3.3 Photocatalytic activity and photostability
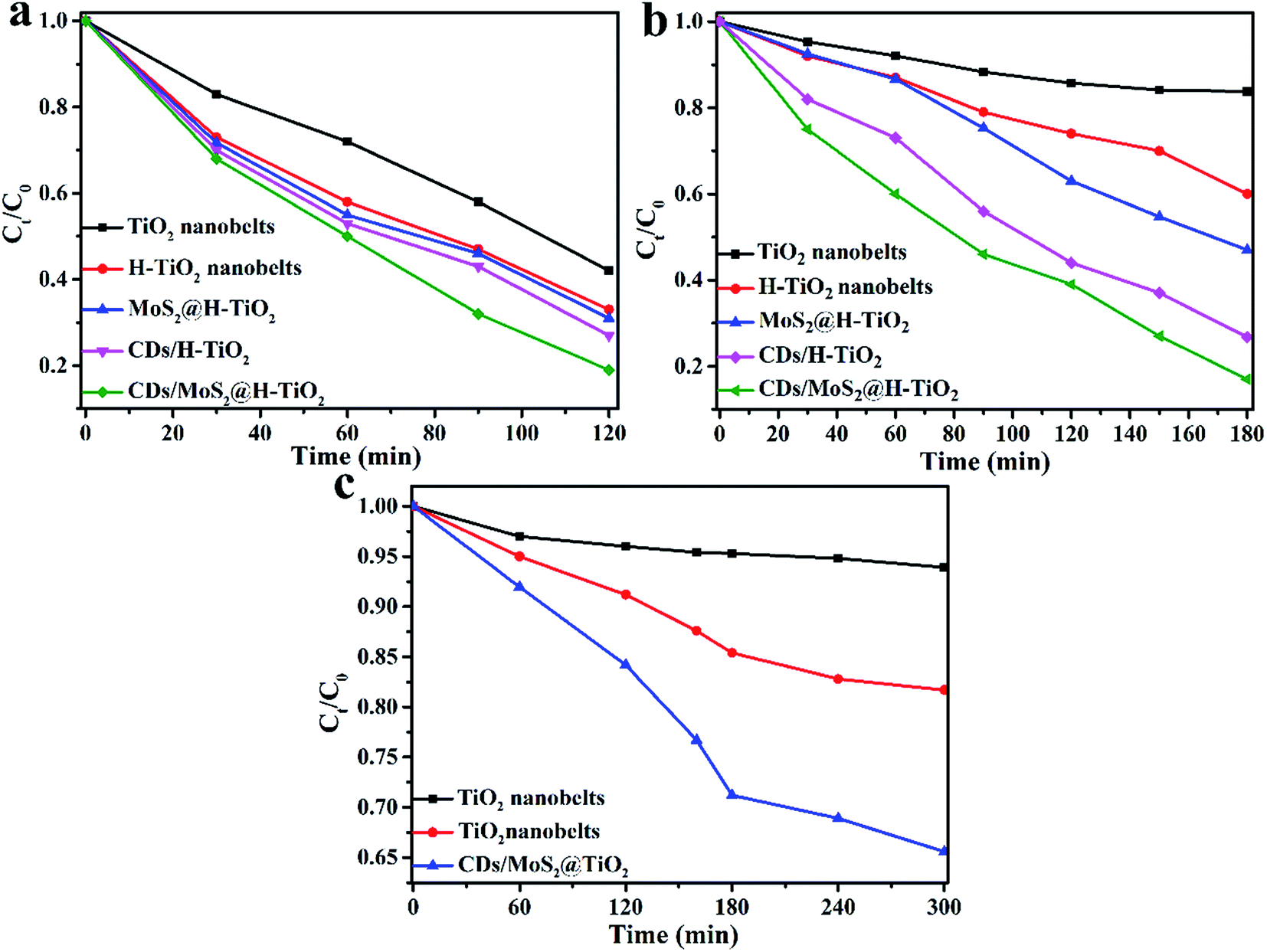
The photocatalytic degradation activities of the aforementioned samples was carried out and the results plotted and compared in Fig. 9. Fig. 9a shows the degradation rate of TC of the photocatalysts with different weight amounts of CDs/MoS2 cocatalysts. It can be seen that the CDs/MoS2 composites show that the photolysis of the TC molecule is very slowly, which suggests that CDs/MoS2 plays the role of only a co-catalyst. Pure TiO2 nanobelts show negligible activities because of the absence of optical absorption under visible light irradiation. However, the H-TiO2 nanobelts exhibit obvious photocatalytic degradation activities due to the introduction of an oxygen vacancy and Ti3+ in the H-TiO2 nanobelts. Furthermore, the CDs/MoS2@H-TiO2 composites exhibit remarkable photocatalytic activities due to the recombination delay of electron–hole pairs by the targeted electron transfer. In particular, the sample with 5.0 wt% content of CDs/MoS2 nanohybrids showed the highest photocatalytic degradation activity towards TC (81.6%) within 3 h under visible light irradiation. The activities showed a gradual downward trend with the increase in the content of CDs/MoS2 nanohybrids, which might be because excessive CDs/MoS2 can hinder the production of active radicals by preventing the electron–hole pairs reacting with the adsorbed oxidants/reducers. Everyone knows the co-catalyst activities of MoS2 originate from the unsaturated active S atoms on its exposed edges, while the saturated S atoms at the basal plane have no activity. Happily, the introduction of CDs can ameliorate this disadvantage due to its superior electron mobility. Obviously, as shown in Fig. 9b, the co-catalytic activity of CDs/MoS2 hybrids is much better than that of pure MoS2 nanosheets. The remarkably improved photocatalytic activity could be reasonably attributed to the positive synergistic effect of the CD layer and the few-layer MoS2 nanosheets on the H-TiO2. After the above experiments, we demonstrated that CDs/MoS2 could uniformly grow on H-TiO2 when the mass ratios of CDs/MoS2 to H-TiO2 was 5:95, and this kind of composite displayed a perfect synergetic effect between the CDs and MoS2, further facilitating the electronic separation. To optimize the effects of the content of CDs/MoS2 on the photodegradation activities of H-TiO2, a series of CDs/MoS2@H-TiO2 composites with different mass ratios of CDs to MoS2 in which the amount of the CDs/MoS2 co-catalyst was 5% were investigated. From Fig. 9b, it can be seen that all the ternary CDs/MoS2@H-TiO2 composites show higher photocatalytic performance as compared with the binary MoS2@MoS2 composites. For the ternary CDs/MoS2@H-TiO2 nanocomposites, CDs/MoS2 with a mass ratio of 1:4 of CDs:MoS2 exhibited the highest photocatalytic activity.
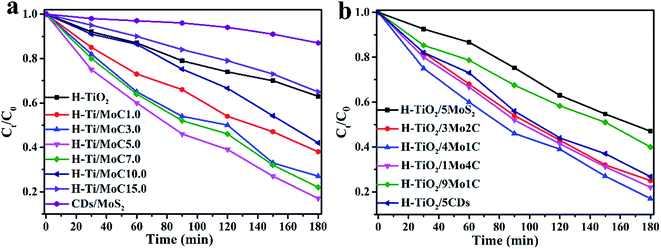 |
| | Fig. 9 Photocatalytic degradation of TC with the as-prepared samples. (a) Comparison of the photocatalytic degradation rate of different samples with different weights of cocatalysts; (b) photocatalytic degradation rate of different samples with different weight contents of CDs/MoS2 in which the amount of co-catalyst is 5.0 wt%. | |
To further demonstrate that the photocatalytic activity can be improved in all directions, the photocatalytic activity of CDs/MoS2@H-TiO2 was evaluated for the degradation of TC under UV, visible and NIR light irradiation (Fig. 10a–c). As shown in Fig. 10a, the photodegradation efficiency of TC over the CDs/MoS2@H-TiO2 ternary photocatalyst was more than 80% within 120 min under UV light irradiation. The photocatalytic activity of the CDs/MoS2@H-TiO2 sample was superior to the others samples. This can be attributed to the oxygen vacancies and Ti3+ created by the hydrogenation. The Ti3+ species can serve as hole scavengers. Also, oxygen vacancies on the surface of H-TiO2 can act as O2 binding sites and electron scavengers, thus resisting the electron–hole pair recombination and remarkably ameliorating the photocatalytic activity of the H-TiO2 nanobelts. Additionally, the visible photodegradation performance of H-TiO2 was better than that of the TiO2 nanobelts as well (Fig. 10b). This is because H-TiO2 has intense absorption in the visible spectrum, so the solar spectrum response range has been expanded from 250 nm to about 800 nm. This guess also coincides with the above-mentioned DR tests (Fig. 6a). Under NIR light irradiation, we can see that the TC degradation degree over CDs/MoS2@H-TiO2 is about 33% after 5 h. This result demonstrates that the up-converted PL effect of the CDs can play a key role in the ameliorated NIR photocatalytic performance. The NIR light is harvested by CDs, converted to visible light, and subsequently excites H-TiO2 to complete the NIR photocatalysis process for the hybrid nanobelts.
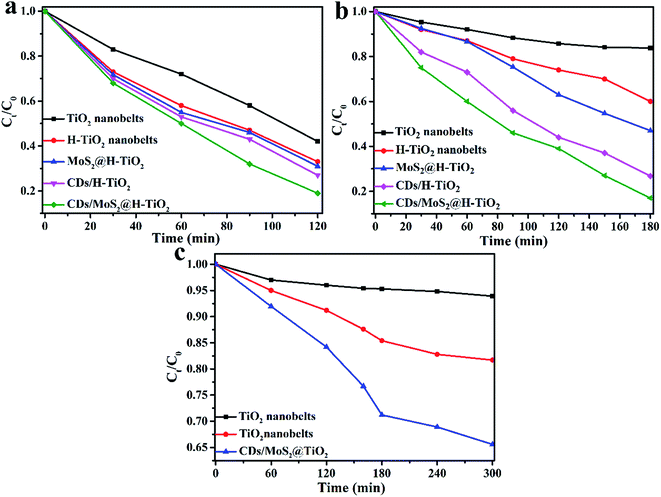 |
| | Fig. 10 Photocatalytic degradation of TC in the presence of different as-prepared photocatalysts under: (a) UV, (b) visible, and (c) NIR light irradiation. | |
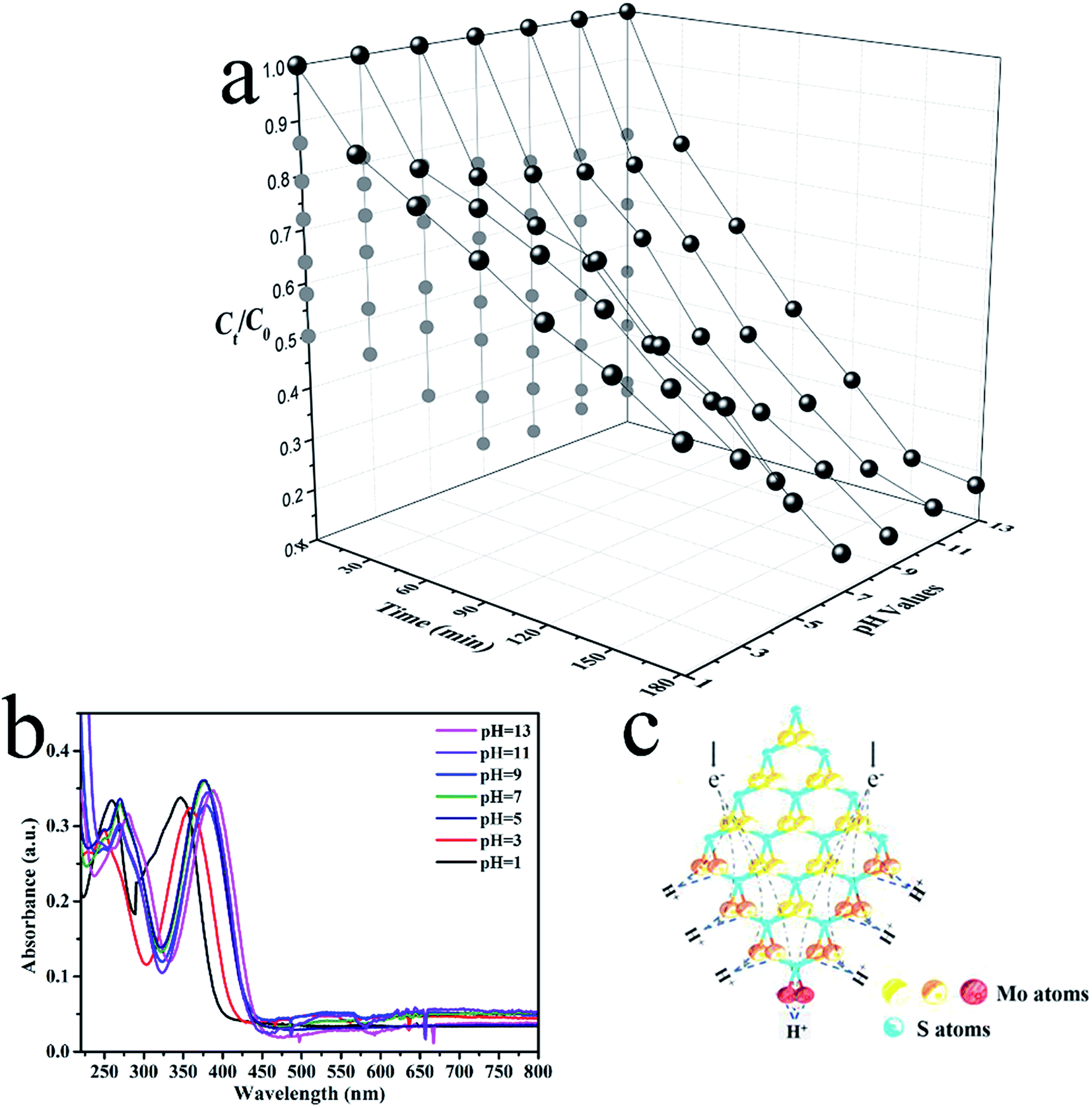
Prior studies suggested that the photodegradation activity of TC was affected by the temperature and pH.38,39 The pH level features most among these contributing factors. Experiments on the photodegradation of TC were carried out using CDs/MoS2@H-TiO2 at pH levels between 1 and 13 to assess the effect of pH (Fig. 11a). The UV-Vis absorption spectra of TC solution under various pH conditions are shown in Fig. 11b, where it can be clearly observed that the main absorption peaks of TC-HCl are slightly red-shifted at pH below 7. Besides, the intensity of the peaks gradually decrease with time, and no additional peaks appears (Fig. S3, ESI†), indicating that the photocatalytic reaction induced the degradation of TC and not a molecular structural change. Fig. 11a shows that the CDs/MoS2@H-TiO2 have a remarkable photocatalytic activity at all pH levels, but for the degradation of TC, the pH value should be as high as possible. This phenomenon could be ascribed to the presence of an unsaturated active S atom in MoS2. Hence, a diagrammatic sketch of the microstructure of MoS2 and its electron reaction mechanisms in the photocatalytic process under an acidic environment are shown in Fig. 11c. As illustrate in Fig. 11c, different kinds of S atoms in the MoS2 microstructure lead to varied catalytic activities. The unsaturated S atoms signalled by a red colour have a strong attraction to H+ ions in the solution, which is higher than from the saturated atoms marked by an orange colour. Due to the abundance of H+ ions in the acidic solutions, the unsaturated active S atoms expose the edges of MoS2 more easily to aid capturing H+ ions than in alkaline solutions, which are easily reduced by consuming a large amount of electrons. Thereby, considering the number of electrons loss in the acidic solutions, more ·O2− will be generated in the alkaline solutions. In summary, the experiments demonstrated that the CDs/MoS2@H-TiO2 system possesses excellent alkaline resistance and can be efficiently used for pollutant purification under alkaline conditions.
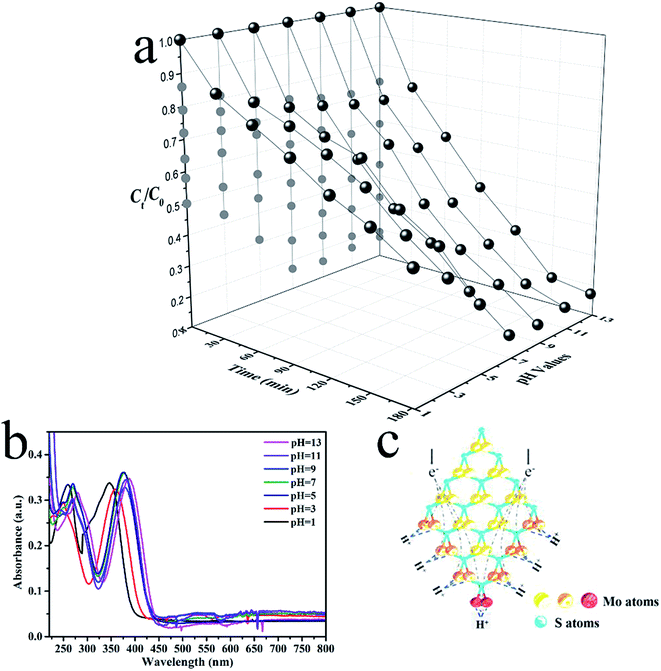 |
| | Fig. 11 (a) Photocatalytic degradation of TC at various pH conditions in the presence of CDs/MoS2@H-TiO2 with visible light irradiation; (b) UV-Vis absorption spectra of TC under various pH conditions; (c) schematic of the microstructure of MoS2. | |
The CDs/MoS2@H-TiO2 catalyst is robust, as was shown when performing repeat runs for the photocatalytic degradation of TC (pH = 7) under visible light irradiation, as shown in Fig. 12a. After five successive cycles, CDs/MoS2@H-TiO2 degraded around 82% of the TC within 180 min, which is similar to the activity for the first cycle. Furthermore, the XRD and Raman patterns of before and after 5 cycles of reactions are provided in Fig. 11b and c. Notably, ever after 5 successive cycles, the as-synthesized sample showed no obvious differences, indicating its high stability and great prospect for application.
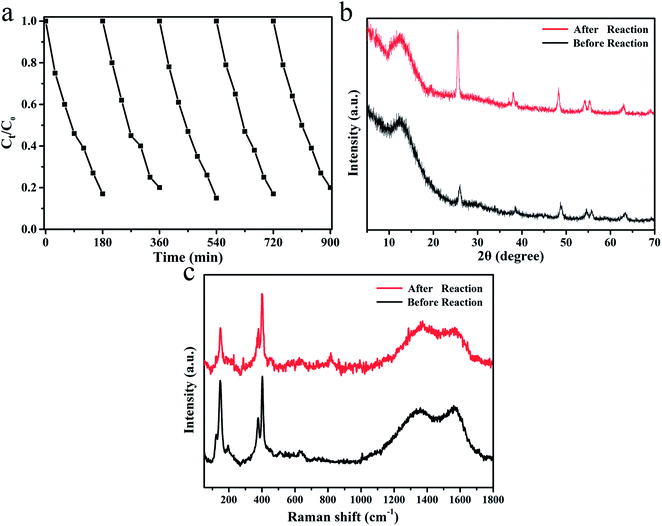 |
| | Fig. 12 (a) Five cycling experiments of CDs/Mos2@H-TiO2 for TC degradation; (b) XRD pattern (b) and Raman spectrum (c) of CDs/MoS2@H-TiO2 before and after 5 cycles of photocatalytic experiments. | |
3.4 Possible mechanism of the photocatalytic reaction process
Based on the above results and discussion, a tentative mechanism proposed for the high photocatalytic activity of the CDs/MoS2@H-TiO2 composite is illustrated in Fig. 13. Under simulated sunlight irradiation, H-TiO2 could absorb visible light. Simultaneously, the CDs absorb NIR light and then emit visible light (300–600 nm) as a result of up-conversion, which can in turn excite H-TiO2 to generate electron–hole pairs, which is a result of the fact that H-TiO2 nanobelts have a more negative potential of conduction band (CB) and valence band (VB) than that of MoS2 nanosheet.6 Therefore, the photogenerated electrons in the H-TiO2 surface can be easily transferred to the surface of the MoS2 by the built-in potential in the heterojunction, leaving holes on the surface. Meanwhile, the holes left in the VB of the TiO2 nanosheets and those transferred from the VB of TiO2 can be consumed by the pollutants.4 Some of the electrons approaching the edge of MoS2 directly react with O2 to produce ·O2− under the co-catalytic activities of unsaturated active S atoms, which can accept electrons and act as active sites for TC degradation. Other unreacted electrons on the MoS2 basal planes can be transferred to the CDs energy level and then can react with the target pollutant.29,39–43 In view of this, the mechanism of an efficient muti-step electrons transfer process was proposed: (1) when the CDs/MoS2@H-TiO2 nanocomposite is suspended in aqueous captured solar illumination, H-TiO2 simultaneously generates photoelectrons and holes; (2) the photogenerated electrons can be easily transferred to the MoS2 layer from the conduction band (CB) of H-TiO2; (3) the formation of a heterojunction between MoS2 and the CDs bring about the further transfer of photoelectrons from MoS2 to CDs. And above all, the relative band alignment of H-TiO2, MoS2, and CDs makes it feasible for migration of the photoexcited electrons through the vectorial electron transfer of H-TiO2 → MoS2 nanosheets → CDs layer. This process greatly inhibits the recombination of electron–hole pairs. Eventually, the accumulated electrons on the surface CD layer can capture and reduce the oxygen to form the reactive ·O2−, which is a powerful oxidizing species responsible for degradation of the target pollutant. Meanwhile, the holes on the surface of H-TiO2 can also react with H2O to generate ·OH. Therefore, the synergetic catalytic effect through the interfacial modulation and design of the charge-transfer channels by use of the integration of MoS2 nanosheets and the CD layer plays a crucial role in the photocatalytic degradation.
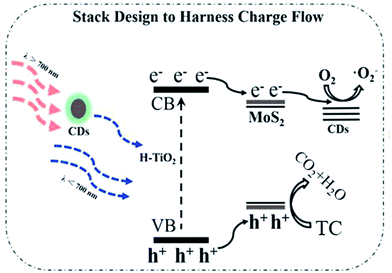 |
| | Fig. 13 Schematic of the TC photodegradation mechanism for the CDs/MoS2@H-TiO2 samples. | |
4. Conclusion
In summary, a novel and stable carbon dots/MoS2 few-layer nanosheet-coated hydrogenated TiO2 nanobelt heterostructured catalyst was successfully synthesized by a facile process. Through optimizing the proportion of each component proportion, the CDs/MoS2@H-TiO2 catalysts showed the highest photodegradation activity when the content of the CDs/MoS2 co-catalyst is 5.0 wt% and the mass ratio of MoS2 to CDs was 4:1. The enhancement of the photocatalytic activity was mainly attributed to the mutual-benefit cooperation from the following three aspects: (1) the synergistic effect effectively suppresses the recombination of photogenerated electrons and holes; (2) the cooperative contribution of nano-sized CDs/MoS2 few-layer sheets have more active edges and provide an excellent up-converted PL effect; (3) the CDs/MoS2@H-TiO2 3D hierarchical structure provides a favourable electron transfer pathway, H-TiO2 → MoS2 → CDs, for the efficient separation of electron–hole pairs. This kind of ternary CDs/MoS2@H-TiO2 composite achieved a remarkable photocatalytic activity and demonstrated remarkable electron transport capabilities. Therefore, the findings from this research not only involved a systematic study of electron transfer, but also can be easily extended to the preparation of other CDs/MoS2-based 3D catalysts for a broad range of applications, such as solar cells, fuel cells, etc.
Acknowledgements
We would like to acknowledge the Innovation Program Foundation of JiangSu Province (SJLX15-0502), National Natural Science Foundation of China (21546006) and Six Tanlents Peaks Project in Jiangsu Province (XNY-009) and the Foundation Research Project of JiangSu Province (the Natural Science Fund BK20150536).
References
- M. Yan, Y. Q. Hua, F. F. Zhu, W. Gu, J. H. Jiang, H. Q. Shen and W. D. Shi, Appl. Catal., B, 2017, 202, 518–527 CrossRef CAS.
- N. Zhang, M. Q. Yang, S. Q. Liu, Y. G. Sun and Y. J. Xu, Chem. Rev., 2015, 115, 10307–10377 CrossRef CAS PubMed.
- C. T. Dinh, H. Yen, F. Kleitz and T. O. Do, Angew. Chem., Int. Ed., 2014, 53, 6618–6623 CrossRef CAS PubMed.
- W. J. Zhou, Z. Y. Yin, Y. P. Du, X. Huang, Z. Y. Zeng, Z. X. Fan, H. Liu, J. Y. Wang and H. Zhang, Small, 2013, 9, 140–147 CrossRef CAS PubMed.
- S. J. Zhuo, M. W. Shao and S. T. Lee, ACS Nano, 2012, 6, 1059–1064 CrossRef CAS PubMed.
- Q. J. Xiang, J. G. Yu and M. Jaroniec, J. Am. Chem. Soc., 2012, 134, 6575–6578 CrossRef CAS PubMed.
- Z. Zheng, B. B. Huang, X. D. Meng, J. P. Wang, S. Y. Wang, Z. Z. Lou, Z. Y. Wang, X. Y. Qin, X. Y. Zhang and Y. Dai, Chem. Commun., 2013, 49, 868–870 RSC.
- J. Chen, W. X. Song, H. S. Hou, Y. Zhang, M. J. Jing, X. N. Jia and X. B. Ji, Adv. Funct. Mater., 2015, 25, 6793–6801 CrossRef CAS.
- Y. H. Lin, H. T. Hsueh, C. W. Chang and H. Chu, Appl. Catal., B, 2016, 199, 1–10 CrossRef CAS.
- S. N. Phattalung, S. Limpijumnong and J. J. Yu, Appl. Catal., B, 2017, 200, 1–9 CrossRef.
- H. Zhang, L. H. Guo, D. B. Wang, L. X. Zhao and B. Wan, ACS Appl. Mater. Interfaces, 2015, 7, 1816–1823 CAS.
- J. Tian, Y. H. Leng, Z. H. Zhao, Y. Xia, Y. H. Sang, P. Hao, J. Zhan, M. C. Li and H. Liu, Nano Energy, 2015, 11, 419–427 CrossRef CAS.
- Z. Bian, J. Zhu, S. Wang, Y. Cao, X. Qian and H. Li, J. Phys. Chem. C, 2008, 112, 6258–6262 CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- J. Wang, P. Zhang, X. Li, J. Zhu and H. Li, Appl. Catal., B, 2013, 134, 198–204 CrossRef.
- S. M. El-Sheikh, G. Zhang, H. M. El-Hosainy, A. A. Ismail, K. E. O'Shea, P. Falaras, A. G. Kontos and D. D. Dionysiou, J. Hazard. Mater., 2014, 280, 723–733 CrossRef CAS PubMed.
- Y. H. Lin, S. H. Chou and H. Chu, J. Nanopart. Res., 2014, 16, 1–12 CAS.
- Z. Wang, X. Liu, W. Li, H. Wang and H. Li, Ceram. Int., 2014, 40, 8863–8867 CrossRef CAS.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- J. Zheng, L. Liu, G. B. Ji, Q. F. Yang, L. R. Zheng and J. Zhang, ACS Appl. Mater. Interfaces, 2016, 8, 20074–20081 CAS.
- T. Xia, C. Zhang, N. A. Oyler and X. Chen, Adv. Mater., 2013, 25, 6905–6910 CrossRef CAS PubMed.
- Y. H. Hu, Angew. Chem., Int. Ed., 2012, 51, 12410–12412 CrossRef CAS PubMed.
- A. K. Geim and I. V. Grigorieva, Van Der Waals Heterostructures, Nature, 2013, 499, 419–425 CrossRef CAS PubMed.
- J. J. Duan, S. Chen, M. Jaroniec and S. Z. Qiao, ACS Nano, 2015, 9, 931–940 CrossRef CAS PubMed.
- B. R. Sathe, X. Zou and T. Asefa, Catal. Sci. Technol., 2014, 4, 2023–2030 Search PubMed.
- H. Wang, Z. Lu, D. Kong, J. Sun, T. M. Hymel and Y. Cui, ACS Nano, 2014, 8, 4940–4947 CrossRef CAS PubMed.
- K. Chang, Z. W. Mei, T. Wang, Q. Kang, S. Ouyang and J. H. Ye, ACS Nano, 2014, 8, 7078–7087 CrossRef CAS PubMed.
- J. S. Wei, H. Ding, P. Zhang, Y. F. Song, J. Chen, Y. G. Wang and H. M. Xiong, Small, 2016, 12, 5927–5934 CrossRef CAS PubMed.
- J. B. Chen, H. N. Che, K. Huang, C. B. Liu and W. D. Shi, Appl. Catal., B, 2016, 192, 134–144 CrossRef CAS.
- J. Liu, Y. Liu, N. Y. Liu, Y. Z. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhou and Z. H. Kang, Science, 2015, 347, 970–974 CrossRef CAS PubMed.
- H. C. Zhang, H. Huang, H. Ming, H. T. Li, L. L. Zhang, Y. Liu and Z. H. Kang, J. Mater. Chem., 2012, 22, 10501–10506 RSC.
- J. G. Hou, H. J. Cheng and Y. Chao, Nano Energy, 2015, 18, 143–153 CrossRef CAS.
- S. J. Zhou, Q. N. Meng, L. Wang, J. H. Zhang, Y. B. Song, H. Jin, K. Zhang, H. C. Sun, H. Y. Wang and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 3953–3957 CrossRef PubMed.
- L. M. Xu, L. F. Ming and F. Chen, ChemCatChem, 2015, 7, 1797–1800 CrossRef CAS.
- J. Di, J. X. Xia, Y. P. Ge, H. P. Li, H. Y. Ji, H. Xu, Q. Zhang, H. M. Li and M. N. Li, Appl. Catal., B, 2015, 168–169, 51–61 CrossRef CAS.
- S. J. Yu, Y. H. Kim, S. Y. Lee, H. D. Song and J. H. Yi, Angew. Chem., Int. Ed., 2014, 53, 11203–11207 CrossRef CAS PubMed.
- Y. H. Sang, Z. H. Zhao, M. W. Zhao, P. Hao, Y. H. Leng and H. Liu, Adv. Mater., 2014, 27, 363–369 CrossRef PubMed.
- F. Deng, K. Li, X. B. Luo, S. L. Luo, G. S. Zeng, S. L. Wu and C. T. Au, Sci. Adv. Mater., 2014, 6, 363–369 Search PubMed.
- J. G. Hong, H. J. Cheng, O. Takeda and H. M. Zhu, Angew. Chem., Int. Ed., 2015, 54, 8480–8484 CrossRef PubMed.
- J. Di, J. X. Xia, M. Ji, B. Wang, S. Yin, Q. Zhang, Z. G. Chen and H. M. Li, ACS Appl. Mater. Interfaces, 2015, 7, 20111–20123 CAS.
- J. Liu, H. C. Zhang, D. Tang, X. Zhang, L. K. Yan, Y. Z. Han, H. Huang, Y. Liu and Z. H. Kang, ChemCatChem, 2014, 1–9 Search PubMed.
- H. Zhang, L. X. Zhao, F. L. Geng, L. H. Guo, B. Wan and Y. Yang, Appl. Catal., B, 2016, 180, 656–662 CrossRef CAS.
- J. Di, J. X. Xia, M. X. Ji, H. P. Li, H. Xu, H. M. Li and R. Chen, Nanoscale, 2015, 7, 11433–11443 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28479e |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b and
HongJun Dong
*b and
HongJun Dong