
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
β-Lactoglobulin–chlorogenic acid conjugate-based nanoparticles for delivery of (−)-epigallocatechin-3-gallate
Yuting Fana,
Yuzhu Zhangb,
Wallace Yokoyamab and
Jiang Yi *a
*a
aDepartment of Food Science and Engineering, College of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen, Guangdong 518060, China. E-mail: yijiangjnu@gmail.com; Fax: +86-755-26536141; Tel: +86-755-26557377
bWestern Regional Research Center, ARS, USDA, Albany, California 94710, USA
First published on 18th April 2017
Abstract
β-Lactoglobulin (BLG)–chlorogenic acid (CA) conjugates were generated by a free radical induced grafting method. BLG–CA conjugates showed better antioxidant activities than BLG. The antioxidant activity increased with the increase of CA substitution. The particle sizes of (−)-epigallocatechin-3-gallate (EGCG)-loaded nanoparticles prepared by the anti-solvent method were 110.3, 107.4, and 105.8 nm for BLG, BLG–CA (low), and BLG–CA (high), respectively. The encapsulation efficiencies of EGCG in BLG, BLG–CA conjugate (low), and BLG–CA conjugate (high) nanoparticles were 72.9%, 71.8%, and 73.5%, respectively. The chemical stabilities of EGCG in both BLG–CA nanoparticles were significantly higher than in BLG nanoparticles. BLG–CA conjugate (high) showed better EGCG retention than BLG–CA conjugate (low) in simulated gastrointestinal digestion fluid. Little EGCG was released in both BLG nanoparticles and BLG–CA nanoparticles under simulated gastric digestion. The release of EGCG in BLG–CA nanoparticles was less than that in BLG nanoparticles, indicating that CA conjugating protected BLG from the digestive enzymes.
Introduction
(−)-Epigallocatechin-3-gallate (EGCG) is the most active catechin of tea polyphenols. It accounts for 50–80% of the total catechin present in green tea.1,2 Potential health-promoting properties of EGCG include strong antioxidant activity, cancer chemoprevention, radioprotection, cardiovascular health improvement, anti-obesity ability.3–5 EGCG was reported to be approximately 10 times as effective as β-carotene, and L-ascorbate in alkyl peroxyl radical scavenging.6 It was more effective than TBHQ in preventing fish meat from oxidizing (p < 0.05).3 EGCG was also reported to have anticancer effects through suppressing angiogenesis by blocking the induction of vascular endothelial growth factor (VEGF) in human colon carcinoma cells.7However, EGCG's bioactivity is limited by its low oral bioavailability, and poor stability. EGCG is highly soluble in water. It can be easily oxidized in aqueous phase. Thus, it is an unstable bioactive compound, especially under neutral and alkaline conditions. In a phosphate buffer (PB, pH 7.4), more than 80% of EGCG was quickly degraded in only 3 h.8 The increase of either pH, oxygen concentration or storage temperature accelerated its degradation. The bioabsorption of EGCG was less than 1.1% after oral administration.9,10 Recent studies showed that the absorption of EGCG was enhanced when delivered in nanoparticles. The result was mainly attributed to the stabilization of EGCG in nanoparticles rather than the effect of nanoparticles on intestinal transport or on efflux proteins.9
EGCG can be protected against chemical degradation or oxidation by nano-encapsulation with proteins mainly through hydrophilic and hydrophobic interactions.11–13 Even though the chemical stability of EGCG in protein nanoparticles can be significantly improved, more than 30% of EGCG was oxidized during only 10 days of storage at 4 °C in the dark.13 Predictably, EGCG oxidation will be accelerated when stored at room temperature or higher temperature or exposed to light. Furthermore, the early release during in vitro digestion due to proteolysis also facilitated the oxidation or degradation and resulted in poor bioavailability.
β-Lactoglobulin (BLG), a ligand-binding protein, was extensively studied. BLG is the most abundant whey protein.14 It was widely used for encapsulating bioactive molecules to enhance their bioavailability.15,16 Among all tea polyphenols, EGCG exhibited the highest affinity for BLG.17 Recent studies showed that thermal treatment (75–85 °C) of BLG led to the increase of its affinity for EGCG by 3.5-fold due to the exposure of hydrophobic amino acid residues. Compared with native BLG, encapsulation with thermal treatment BLG resulted in improved of chemical stability of EGCG and better protection of its antioxidant activity.18–20 This may be mainly due to the increased thiol group, more exposed hydrophobic group of protein, and more EGCG bound to protein after heat treatment.21 The functional properties (heat-induced insolubility, emulsifying activity, gelling property, and calcium phosphate solubilizing ability) of BLG can also be remarkably improved with glycation and phosphorylation.14,22 Our previous study showed that BLG is resistant to pepsin hydrolysis partly due to its compact structure with almost 50% of β-sheet.23 Whereas it can be digested by trypsin. Pepsin and trypsin are the main proteases in human digestive system. Pepsin, acted in the stomach, is specialized in hydrolyzing peptide bonds between hydrophobic and preferably aromatic amino acids (Phe, Trp, and Tyr). Whereas trypsin, produced in the small intestine, mainly cleaves proteins on the carboxyl side of the amino acids (Lys or Arg).24,25 Recently, it was reported that protein–polyphenols conjugates with potential antioxidant activity can be obtained through free radical induced grafting method.26–28
Nevertheless, the potential of these protein–phenolic conjugates in the development of nanoparticles delivery systems for labile hydrophilic molecules has not been well investigated. Study in our group found that compared to BLG, BLG–catechin conjugate exhibited better protection against β-carotene oxidation in nanoemulsion.28
In this study, conjugation between BLG and chlorogenic acid (CA) was optimized with various mass ratio of BLG![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CA. The obtained BLG–CA conjugate was characterized with SDS-PAGE, far UV CD, and ATR-FTIR. EGCG-loaded BLG and BLG–CA nanoparticles were prepared with an anti-solvent method and the storage stability was compared. In vitro release profile and digestive stability of EGCG was also investigated under simulated gastrointestinal tract. The goal of this paper was to study the potential of BLG–CA conjugates used as nano-delivery matrices for protecting and delivering of bioactive nutraceuticals.
:CA. The obtained BLG–CA conjugate was characterized with SDS-PAGE, far UV CD, and ATR-FTIR. EGCG-loaded BLG and BLG–CA nanoparticles were prepared with an anti-solvent method and the storage stability was compared. In vitro release profile and digestive stability of EGCG was also investigated under simulated gastrointestinal tract. The goal of this paper was to study the potential of BLG–CA conjugates used as nano-delivery matrices for protecting and delivering of bioactive nutraceuticals.
Materials and methods
Bovine beta-lactoglobulin (≥90%, L0130), chlorogenic acid, L-ascorbic acid, hydrogen peroxide (30%, w/w), DPPH, AAPH, fluorescein, and Trolox, were obtained from Sigma-Aldrich (St. Louis, MO). EGCG (purity > 95% by HPLC) was purchased from Chuangsai Co., Ltd. (Shanghai, China). All other analytical grade chemicals and reagents were obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China).Preparation of BLG–CA conjugates
BLG–CA conjugates were produced with a free radical method as previously reported26 with slight modifications. In brief, 0.5 g BLG was fully dissolved in 50 mL distilled water, then, 0.5 mL of 10.0 M H2O2 and 0.25 g L-ascorbic acid (1.4 mmol) were added gradually, while the mixture was stirred at 25 °C under atmospheric air. After 2 h, different amount of CA was added to aliquots of the mixture (at the mass ratios of CA to BLG: 0.1:1, 0.25:1, 0.5:1, 0.75:1, and 1:1, respectively). After 24 h, the free unreacted CA in reaction solution was removed by dialysis against ultrapure water 10 times the volume of the reaction solution. The dialysis bags had a 3 kDa molecular weight cutoffs for proteins. The dialysis was carried out for 48 h at 4 °C with 10 water changes. RP-HPLC results showed that no free CA or free L-ascorbic acid could be detected in the dialyzed conjugate solutions. The remaining BLG–CA conjugates solution was lyophilized (Labconco, MO) and stored in a freezer for further use.
Evaluation of phenolic groups by Folin–Ciocalteu reagent
The CA amount in BLG–CA conjugates was analyzed with previous protocols with minor modification.29 In brief, 0.5 mL of BLG–CA conjugates was mixed with 1 mL of Folin–Ciocalteu reagent for 5 min in the dark, then 2 mL of 20% sodium carbonate (Na2CO3) was added. The mixture was vortexed and incubated 1 h at room temperature. The absorbance was recorded with Nanodrop 2000c (Thermos-Scientific, U.S.) at a wavelength of 747 nm. Gallic acid was used as a standard. The conjugating amounts of BLG–CA conjugates were expressed as milligrams of gallic acid equivalent per gram of conjugates.Characterization of BLG–CA conjugates
For analyzing the secondary structure of BLG and BLG–CA conjugate, the IR spectra (1700 to 1600 cm−1) were further subjected to Fourier self-deconvolution (FSD) using the OMNIC software (Thermo Scientific, West Palm Beach, FL, USA). The FSD spectra were then curve fitted assuming a Gaussian band profile. The FTIR deconvolution spectra were curve-fitted by Gaussian–Lorentzian function with PeakFit software (Version 4.12, SeaSolve Software Inc., Framingham, MA). Peak assignment was carried out on the FSD spectra following literatures on native BLG, and the content of each secondary structure was expressed as the percentage of area of the corresponding peaks.34
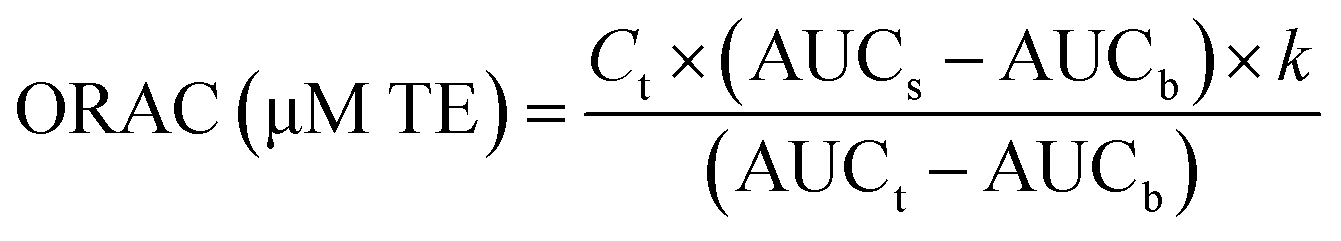 | (1) |
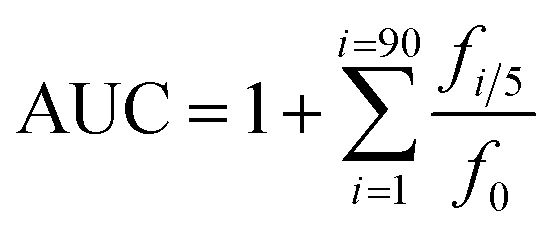 | (2) |
:20 (v/v). After desolvation, glutaraldehyde was added and sample was incubated with stirring for 2 h. Acetone was removed with an evaporator and the same volume of water was added. EGCG stock solution were added dropwise and mixed for 1 h (molar ratio of EGCG/BLG was 4:1). The obtained samples were freeze-dried and put in a refrigerator for further use.The encapsulation efficiency (EE) and loading amount (LA) were calculated with the following equations:
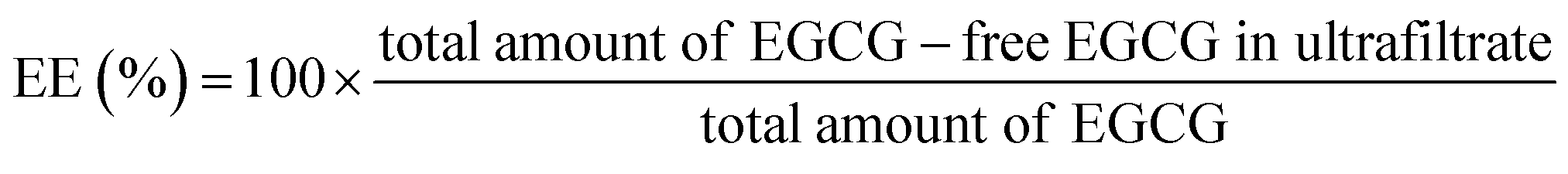 | (3) |
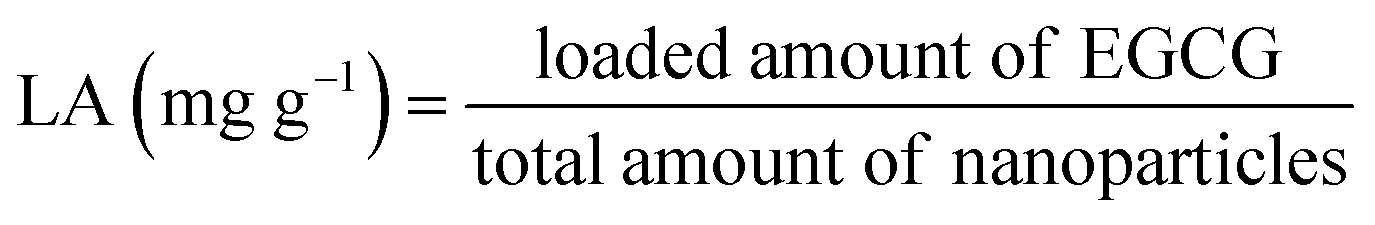 | (4) |
An Agilent 1100 HPLC system equipped with a DAD UV-vis absorption detector (Agilent, Santa Clara, CA) and a Nova-Pak C18 3.9 × 150 mm column (Waters) was used. The aqueous solution of 30% methanol containing 0.1% acetic acid was used as eluent with a flow-rate of 1.0 mL min−1 and the injection volume was 10 μL. The detection temperature was kept at 25 °C and the detection wavelength was 280 nm.38
:1 (w/w).39 SGF was 0.1 M HCl (pH 1.5) containing 0.1% pepsin. SIF is mainly composed of 50 mM PB (pH 6.5) with 0.2% pancreatin and 0.25% bile salts. For gastric digestion, the dialysis bags containing SGF were put in acid release medium I (0.1 M HCl, pH 1.5). For intestinal stage, the dialysis bags containing SIF were put in release medium II (50 mM PB, pH 6.5). Aliquots of dissolution medium (0.5 mL) were withdrawn at certain time intervals, and the concentration of EGCG was determined by HPLC as described above. The same volume of buffer (0.5 mL) was fed back to the release media. The percent cumulative amount of EGCG released from the nanocomplexes was calculated as a function of time.Results and discussion
CA conjugation optimization
The hydroxyl radicals, generated by the reaction between L-ascorbic acid and H2O2, can react with the side chain of sensitive amino residues (tryptophan, lysine, and cysteine) of a protein, producing radical species on its structure. The radical species react with phenolic acid producing protein–phenolic acid covalently grafted conjugates.26 The more phenolic acid conjugated, the higher the antioxidant activity of the protein–phenolic acid conjugate. BLG–CA conjugates with various mass ratios of CA to BLG (0.1:1, 0.25:1, 0.5:1, 0.75:1, and 1:1) were prepared and the conjugation degree was estimated to obtain the optimal BLG–CA conjugates. As shown in Fig. 1, lowest amount of CA was bound to BLG (31.3 mg g−1) at mass ratio of 0.1:1 (CA:BLG). The conjugation degree increased with the increase of the CA:BLG mass ratio between 0.1:1 to 0.5:1. Whereas, CA substitution reached a plateau when further increase the mass ratio. The conjugation amounts were 102.1, 106.8, and 107.7 mg g−1, corresponding to the mass ratio of CA to BLG of 0.5:1, 0.75:1, and 1:1, respectively. No significant differences were observed for these three mass ratios. However, the efficiency decreased with the increase of mass ratio between CA and BLG. The substitution efficiency decreased from 32.4% to 12.1% with the increase of CA:BLG mass ratio from 0.1:1 to 1:1.
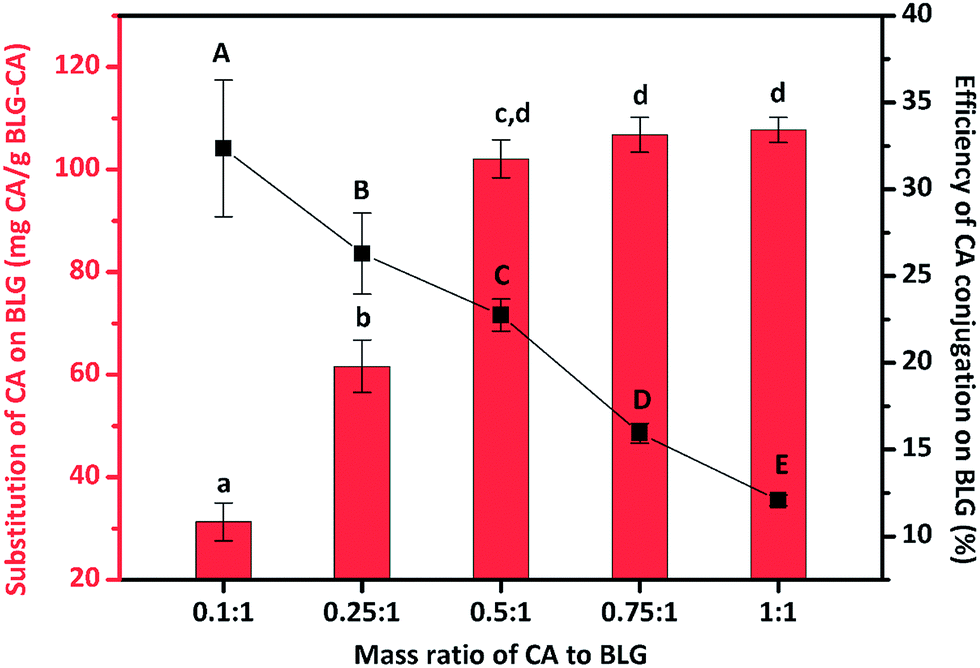 | ||
| Fig. 1 Impact of the various mass ratios of chlorogenic acid (CA) to β-lactoglobulin (BLG) on the substitution and conjugation efficiency of CA on BLG. Values with different letters (a–d; A–E) are significantly different (p ≤ 0.05). | ||
Characterization of BLG–CA conjugates
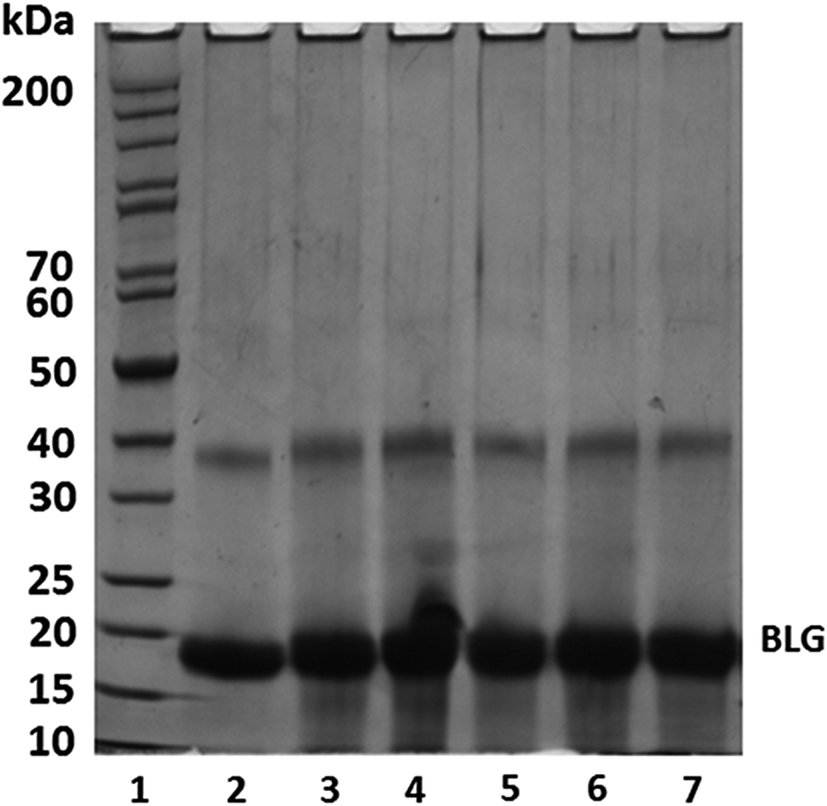 | ||
| Fig. 2 SDS-PAGE results of BLG and BLG–CA conjugates under reducing conditions. Lanes: 1, protein markers; 2, BLG (control); 3–7, BLG–CA conjugates synthesized at different mass ratio of CA:BLG (0.1:1, 0.25:1, 0.50:1, 0.75:1, and 1:1, respectively). | ||
The molecular weight of BLG increased after CA conjugation at CA:BLG of 0.1:1, indicating CA was successfully conjugated onto BLG with the free radical method. There is an increase in the molecular weight of BLG with the increase of the weight ratio of CA:BLG from 0.1:1 to 0.5:1, indicating more CA was covalently bound to BLG. Similar results were also reported by Rawel et al.40 The increase of the molecular weight of BLG dimer was also found when the weight ratio of CA:BLG increased. This was consistent with the phenolic acid determination results. However, further increasing of mass ratio resulted in no appreciable increase of BLG's molecular weight. Similar results were also reported in grafting gallic acid onto chitosan.41 It is likely that all available binding sites on BLG were saturated with CA at the CA:BLG mass ratio of 0.5:1.
It is important to note that little CA non-covalently bound to BLG through hydrophobic interactions and hydrogen bonding may exist,21 possibly leading to the increase of bound CA and the increase of antioxidant activity of BLG–CA conjugate.
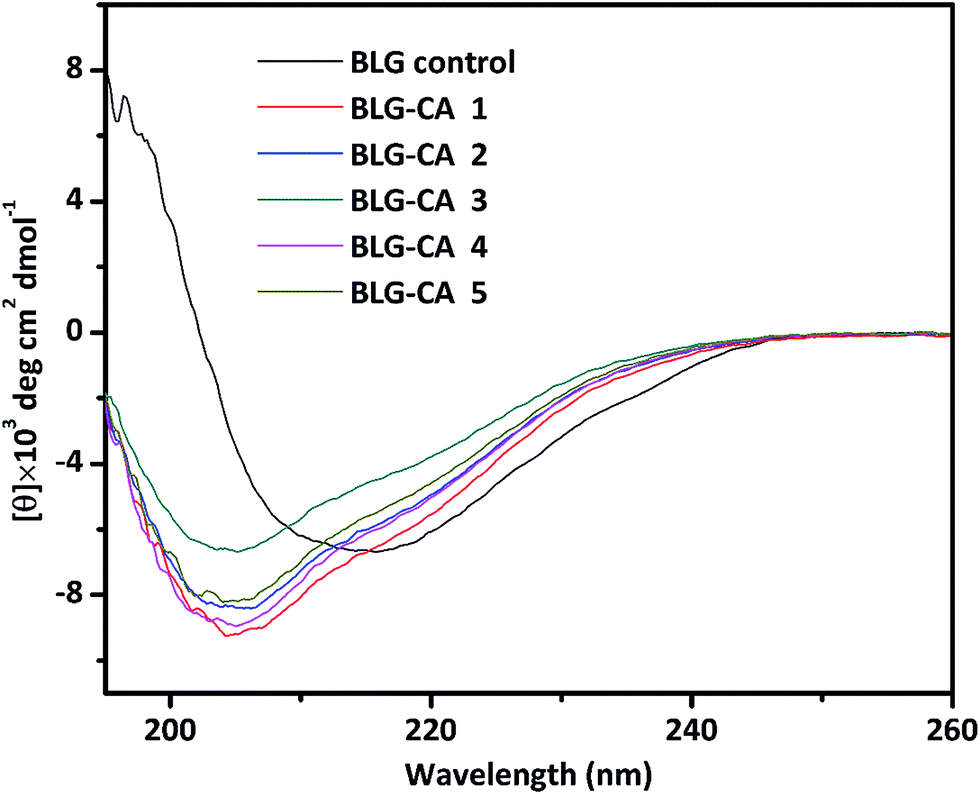 | ||
| Fig. 3 Far-UV CD spectra (195–260 nm) of BLG control, and BLG–CA conjugates obtained at different mass ratio of CA:BLG (0.1:1, 0.25:1, 0.50:1, 0.75:1, and 1:1, respectively). | ||
:BLG mass ratios (0.1:1 to 1:1) from far UV CDa
| Samples | α-Helix (%) | β-Sheet (%) | Turns (%) | Unordered (%) |
|---|---|---|---|---|
| a The secondary structure composition was calculated from far-UV CD spectra data using DichroWeb online (http://dichroweb.cryst.bbk.ac.uk/html/home.shtml). | ||||
| BLG | 18.1 | 38.3 | 18.7 | 25.2 |
| BLG–CA conjugate 1 | 11.9 | 27.8 | 20.6 | 37.6 |
| BLG–CA conjugate 2 | 10.7 | 30.2 | 19.7 | 35.7 |
| BLG–CA conjugate 3 | 7.6 | 24.8 | 21.9 | 41.7 |
| BLG–CA conjugate 4 | 9.7 | 29.7 | 21.4 | 36.3 |
| BLG–CA conjugate 5 | 11.2 | 30.2 | 19.2 | 38.4 |
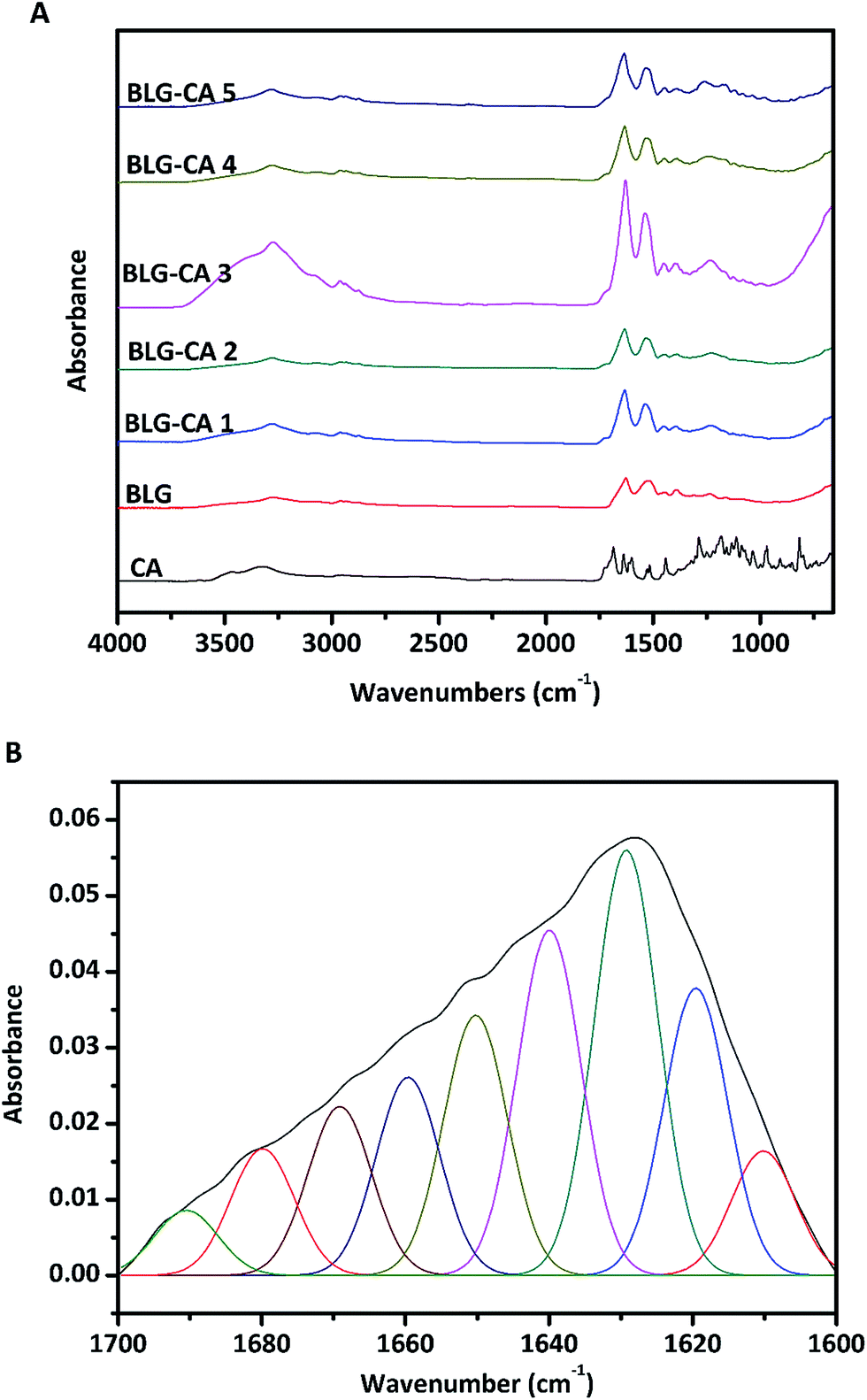 | ||
| Fig. 4 ATR-FTIR spectra (A) of BLG control, and BLG–CA conjugates obtained at different mass ratio of CA:BLG (0.1:1, 0.25:1, 0.50:1, 0.75:1, and 1:1, respectively). Deconvolution of the spectra (B) depicted detailed secondary structure profiles in the amide I band region of BLG (1700–1600 cm−1). | ||
:BLG mass ratios (0.1:1 to 1:1) from ATR-FTIRa
| Samples | α-Helix (%) | β-Sheet (%) | Turns (%) | Unordered (%) |
|---|---|---|---|---|
| a The secondary structure composition was calculated from ATR-FTIR curve-fitting individual components. | ||||
| BLG | 12.0 | 57.3 | 16.0 | 14.7 |
| BLG–CA conjugate 1 | 9.1 | 49.3 | 23.4 | 18.1 |
| BLG–CA conjugate 2 | 8.7 | 47.2 | 22.1 | 22.0 |
| BLG–CA conjugate 3 | 8.4 | 47.3 | 26.3 | 18.0 |
| BLG–CA conjugate 4 | 7.9 | 50.6 | 21.7 | 19.9 |
| BLG–CA conjugate 5 | 8.3 | 46.1 | 23.0 | 22.5 |
Antioxidant activity of BLG–CA conjugate
The DPPH radical scavenging abilities of BLG, BLG–CA (low), BLG–CA (high), and CA were shown in Fig. 5A. DPPH radical scavenging ability increased with the increase of concentrations for all samples (BLG, BLG–CA (low), BLG–CA (high), and CA). BLG was reported to have anti-oxidative activity mainly due to cysteine-121 group.45 However, BLG showed the least DPPH radical scavenging ability in this study. The value was only 9.8% at the highest concentration (1 mg mL−1). After CA conjugation, the DPPH radicals scavenging ability was significantly improved. At 0.1 mg mL−1, the value was 22.3% and 30.3% for BLG–CA (low) and BLG–CA (high), respectively. BLG–CA (high) has better DPPH radical scavenging ability than BLG–CA (low). CA exhibited greatest ability in scavenging DPPH radicals for its strong hydrogen-donating ability. The value was 92.5%, which was remarkably higher than both BLG–CA conjugates at the same concentration. DPPH radical scavenging abilities increased with the increase of the concentration of both BLG–CA conjugates. At 0.6 mg mL−1, the values were 91.8%, 92.6% and 93.0% for BLG–CA (low), BLG–CA (high) and CA, respectively. No significant differences were observed for either BLG–CA conjugates or CA when concentration is higher than 0.6 mg mL−1.
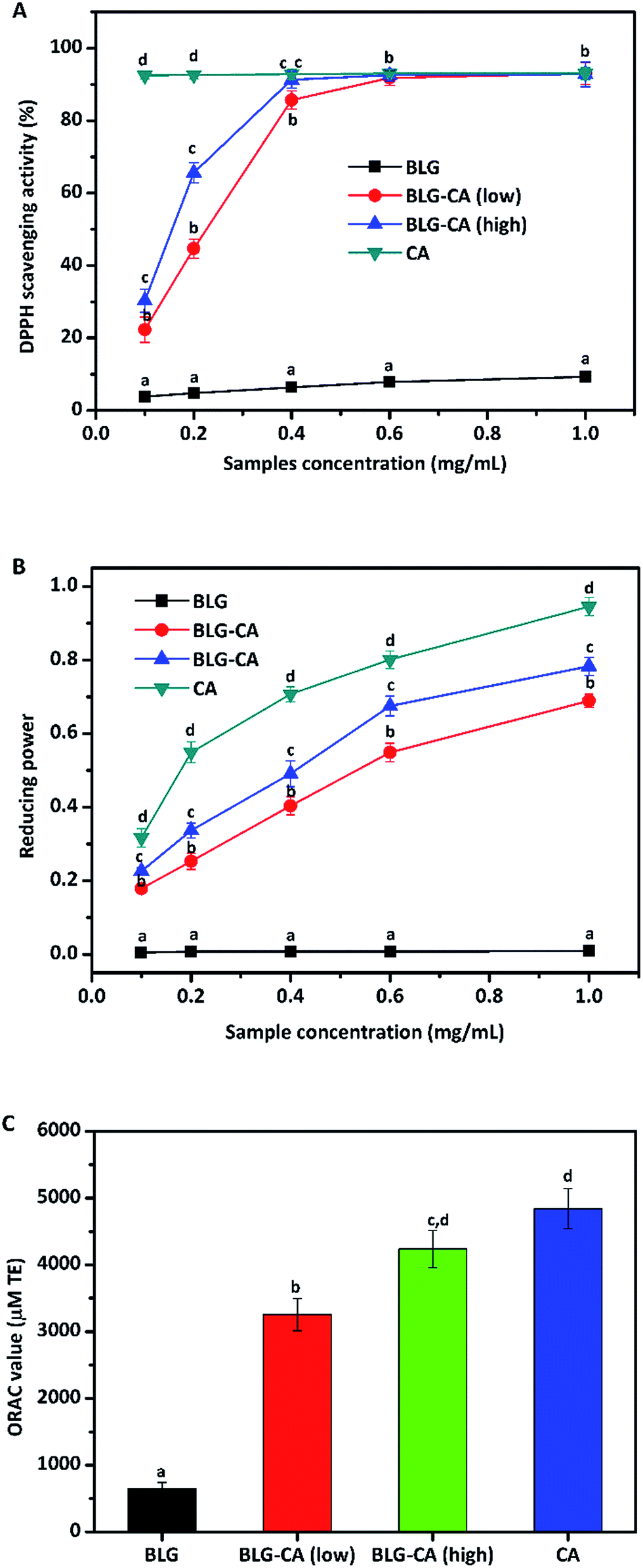 | ||
| Fig. 5 Antioxidant activity (DPPH scavenging ability, oxygen-radical antioxidant capacity (ORAC), and reducing power) of BLG, BLG–CA conjugate, and CA. Values at the same concentration with different letters (a–d) are significantly different (p ≤ 0.05). | ||
The above results (DPPH radical scavenging ability, reducing power, and ORAC value) obviously suggested that BLG–CA conjugate can be used as an effective stabilizer for nutraceuticals protection, encapsulation, and delivery.
| Sample | Mean particle diameter (nm) | Zeta-potential (mV) | EE (%) | LA (%) |
|---|---|---|---|---|
| a Significant differences within a column are denoted with letters (a) (P < 0.05). | ||||
| BLG | 110.3 ± 3.2 (a) | −44.3 ± 1.6 (a) | 72.9 ± 1.3% (a) | 7.3 ± 0.1% (a) |
| BLG–CA conjugate (low) | 107.4 ± 2.0 (a) | −45.6 ± 2.1 (a) | 71.8 ± 1.4% (a) | 7.2 ± 0.2% (a) |
| BLG–CA conjugate (high) | 105.8 ± 1.7 (a) | −47.7 ± 2.3 (a) | 73.5 ± 1.5% (a) | 7.4 ± 0.2% (a) |
TEM results showed that both BLG–CA (low) and BLG–CA (high) nanoparticles were spherical, homogeneous, and uniform (Fig. 6). The mean particle sizes were less than 50 nm, far smaller than the results obtained with DLS. Similar results was also reported.10 This is mainly attributed to that DLS data were obtained in solution where nanoparticles were fully hydrated, while TEM images were got in a dry state on carbon-coated copper grid.
 | ||
| Fig. 6 Transmission electron microscopy (TEM) images of EGCG-loaded nanoparticles. EGCG encapsulated with BLG–CA (low) nanoparticles (A), and EGCG encapsulated with BLG–CA (high) nanoparticles (B). The mass ratio of CA to BLG was 1:2. The scale bar represents 50 nm in both (A) and (B). | ||
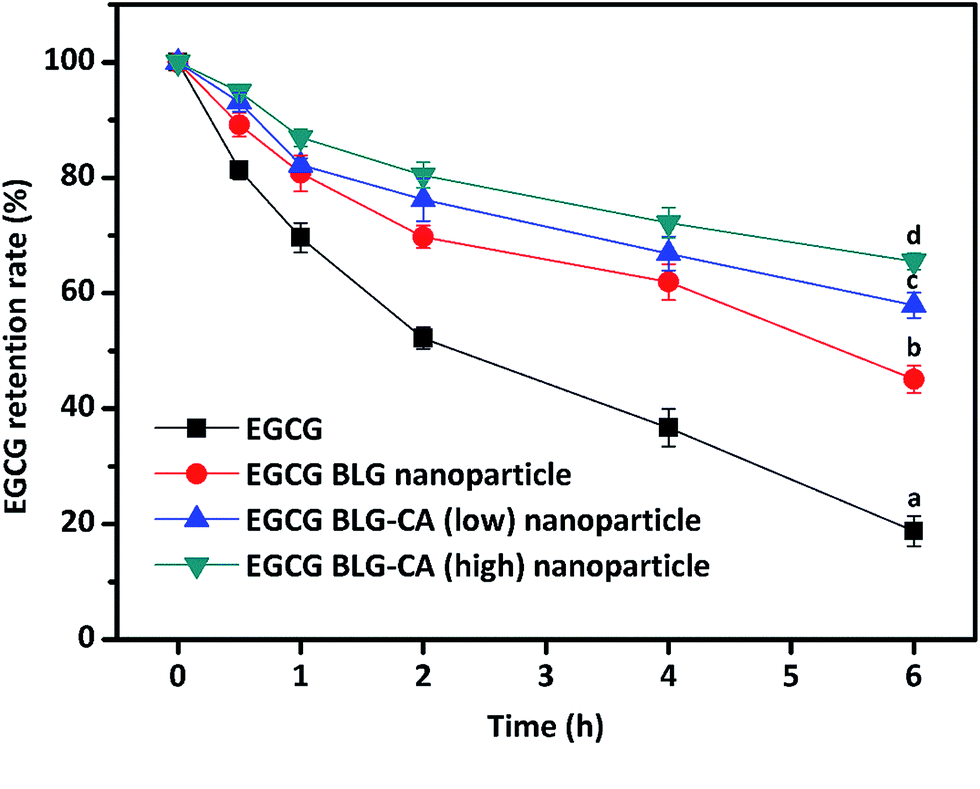 | ||
| Fig. 7 Chemical stabilities of free EGCG and EGCG in nanoparticles during storage. The chemical stabilities of free EGCG (black square), EGCG-loaded BLG nanoparticles (red circle), BLG–CA (low) (blue triangle), and BLG–CA (high) nanoparticles (green inverse triangle) at pH 7.0 after 6 h at room temperature are shown. Values with different letters (a–d) at the same time intervals are significantly different (p ≤ 0.05). Data are expressed as mean ± STD. | ||
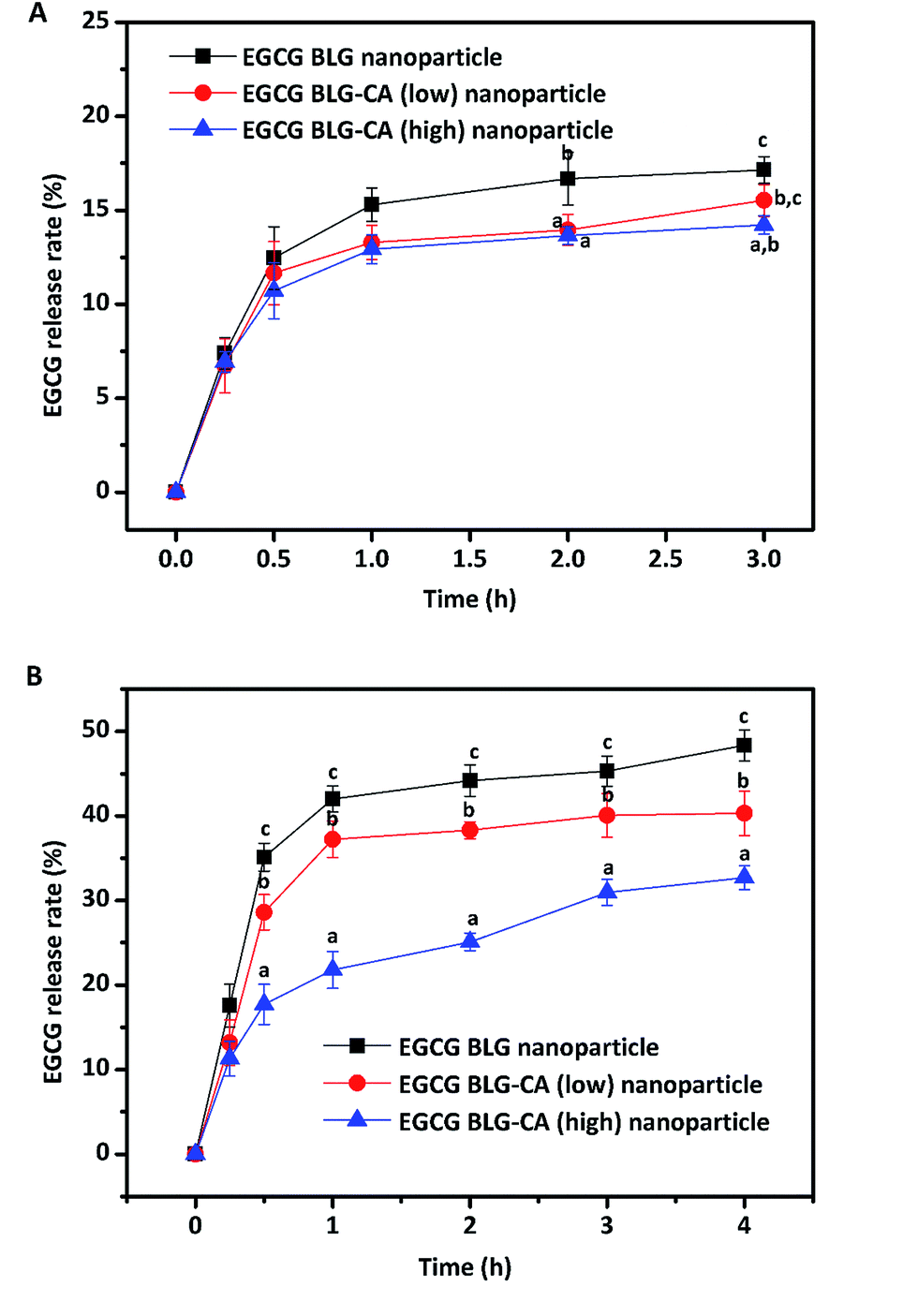 | ||
| Fig. 8 In vitro release of EGCG in nanoparticles digested with pepsin and pancreatin at simulated gastrointestinal juice. Values with different letters (a–c) at the same time intervals are significantly different (p ≤ 0.05). Data are expressed as mean ± STD. | ||
At the intestinal stage, all three nanoparticles showed a rapid release of EGCG at the beginning and a sustained release of EGCG during the whole digestion process. After the first 30 min of digestion, 35.1, 28.6, 17.7% of EGCG were released in BLG, BLG–CA (low), and BLG–CA (high) nanoparticles, respectively. The release of EGCG was relatively slow from BLG–CA conjugates nanoparticle than from BLG nanoparticle (p < 0.05). After 4 h of digestion with pancreatin, the extents of EGCG release were 48.3%, 40.3%, 32.7% for BLG, BLG–CA conjugate (low), and BLG–CA conjugate (high) nanoparticles, respectively. The results indicated that covalently conjugated CA inhibited the activity of the digestive enzyme, which led to the reduction of the release rate and extent of EGCG. Furthermore, the release of EGCG was further inhibited with the increase of CA conjugation degree.
In conclusion, BLG–CA conjugate was prepared by the free radicals method and characterized with SDS-PAGE, far UV-CD, and ATR-FTIR. SDS-PAGE results clearly suggested that CA was successfully covalently bound to BLG. α-Helix and β-sheet structure of BLG decreased with a corresponding increase of turns and unordered structure after CA conjugation. The mean particle diameters of EGCG-loaded BLG or BLG–CA (low) and (high) nanoparticles were all approximately between 105 and 110 nm. BLG–CA showed better protection than BLG against EGCG degradation or oxidation. Simulated gastric digestion of EGCG-loaded nanoparticles showed limited release of EGCG, indicating they could potentially be used as vehicles for protection of EGCG in the stomach, and for its sustained release in the intestine. The release of EGCG from BLG–CA nanoparticles was slower and less than that from BLG nanoparticles. BLG–CA (high) showed higher inhibition of EGCG release than BLG–CA (low), suggesting that CA exhibited inhibition for digestive enzymes in intestinal stage. The results demonstrated the potential benefits of using BLG–CA conjugate as nano-carriers for nutraceuticals.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 31601512) and Young Scholars' Scientific Research Startup Funding from Shenzhen University (No. 2016010).References
- N. Khan, F. Afaq, M. Saleem, N. Ahmad and H. Mukhtar, Cancer Res., 2006, 66, 2500–2505 CrossRef CAS PubMed
.
- D. G. Nagle, D. Ferreira and Y.-D. Zhou, Phytochemistry, 2006, 67, 1849–1855 CrossRef CAS PubMed
- Y. He and F. Shahidi, J. Agric. Food Chem., 1997, 45, 4262–4266 CrossRef CAS
- Y.-H. Kao, H.-H. Chang, M.-J. Lee and C.-L. Chen, Mol. Nutr. Food Res., 2006, 50, 188–210 CAS
- B. N. Singh, S. Shankar and R. K. Srivastava, Biochem. Pharmacol., 2011, 82, 1807–1821 CrossRef CAS PubMed
- M. Nakao, S. Takio and K. Ono, Phytochemistry, 1998, 49, 2379–2382 CrossRef CAS PubMed
- Y. D. Jung, M. S. Kim, B. A. Shin, K. O. Chay, B. W. Ahn, W. Liu, C. D. Bucana, G. E. Gallick and L. M. Ellis, Br. J. Cancer, 2001, 84, 844–850 CrossRef CAS PubMed
- Y. Lun Su, L. K. Leung, Y. Huang and Z.-Y. Chen, Food Chem., 2003, 83, 189–195 CrossRef
- A. Dube, J. A. Nicolazzo and I. Larson, Eur. J. Pharm. Sci., 2010, 41, 219–225 CrossRef CAS PubMed
- S. Kumar, R. Meena and P. Rajamani, J. Agric. Food Chem., 2016, 64, 6024–6034 CrossRef CAS PubMed
- M. C. Bohin, J.-P. Vincken, H. T. W. M. van der Hijden and H. Gruppen, J. Agric. Food Chem., 2012, 60, 4136–4143 CrossRef CAS PubMed
- P. Lestringant, A. Guri, İ. Gülseren, P. Relkin and M. Corredig, J. Agric. Food Chem., 2014, 62, 8357–8364 CrossRef CAS PubMed
- J. Xue, C. Tan, X. Zhang, B. Feng and S. Xia, J. Agric. Food Chem., 2014, 62, 4677–4684 CrossRef CAS PubMed
- H. Enomoto, C.-P. Li, K. Morizane, H. R. Ibrahim, Y. Sugimoto, S. Ohki, H. Ohtomo and T. Aoki, J. Agric. Food Chem., 2007, 55, 2392–2398 CrossRef CAS PubMed
- L. Liang and M. Subirade, J. Phys. Chem. B, 2010, 114, 6707–6712 CrossRef CAS PubMed
- Z. Teng, R. Xu and Q. Wang, RSC Adv., 2015, 5, 35138–35154 RSC
- C. D. Kanakis, I. Hasni, P. Bourassa, P. A. Tarantilis, M. G. Polissiou and H.-A. Tajmir-Riahi, Food Chem., 2011, 127, 1046–1055 CrossRef CAS PubMed
- B. Li, W. Du, J. Jin and Q. Du, J. Agric. Food Chem., 2012, 60, 3477–3484 CrossRef CAS PubMed
- A. Shpigelman, Y. Cohen and Y. D. Livney, Food Hydrocolloids, 2012, 29, 57–67 CrossRef CAS
- A. Shpigelman, G. Israeli and Y. D. Livney, Food Hydrocolloids, 2010, 24, 735–743 CrossRef CAS
- C. Yin, L. Yang, H. Zhao and C.-P. Li, Food Res. Int., 2014, 64, 855–863 CrossRef CAS
- C. P. Li, H. Enomoto, S. Ohki, H. Ohtomo and T. Aoki, J. Dairy Sci., 2005, 88, 4137–4145 CrossRef CAS PubMed
- J. Yi, T. I. Lam, W. Yokoyama, L. W. Cheng and F. Zhong, J. Agric. Food Chem., 2014, 62, 8900–8907 CrossRef CAS PubMed
- B. M. Dunn, in Current Protocols in Protein Science, John Wiley & Sons, Inc., 2001, ps2103s25 Search PubMed
- L. Polgár, Cellular and Molecular Life Sciences CMLS, 2005, vol. 62, pp. 2161–2172 Search PubMed
- U. G. Spizzirri, F. Iemma, F. Puoci, G. Cirillo, M. Curcio, O. I. Parisi and N. Picci, Biomacromolecules, 2009, 10, 1923–1930 CrossRef CAS PubMed
- J. Yi, Y. Fan, Y. Zhang and L. Zhao, Food Chem., 2016, 205, 73–80 CrossRef CAS PubMed
- J. Yi, Y. Zhang, R. Liang, F. Zhong and J. Ma, J. Agric. Food Chem., 2015, 63, 297–303 CrossRef CAS PubMed
- B. Hu, F. Ma, Y. Yang, M. Xie, C. Zhang, Y. Xu and X. Zeng, J. Agric. Food Chem., 2016, 64, 3422–3429 CrossRef CAS PubMed
- J. Yi, T. I. Lam, W. Yokoyama, L. W. Cheng and F. Zhong, Food Hydrocolloids, 2015, 43, 31–40 CrossRef CAS
- N. Dyballa and S. Metzger, J. Visualized Exp., 2009, 30, e1431 Search PubMed
- J. Yi, T. I. Lam, W. Yokoyama, L. W. Cheng and F. Zhong, J. Agric. Food Chem., 2014, 62, 8900–8907 CrossRef CAS PubMed
- K. Brew, F. J. Castellino, T. C. Vanaman and R. L. Hill, J. Biol. Chem., 1970, 245, 4570–4582 CAS
- A. S. Eissa, C. Puhl, J. F. Kadla and S. A. Khan, Biomacromolecules, 2006, 7, 1707–1713 CrossRef CAS PubMed
- A. Zulueta, M. J. Esteve and A. Frígola, Food Chem., 2009, 114, 310–316 CrossRef CAS
- S. Ko and S. Gunasekaran, J. Microencapsulation, 2006, 23, 887–898 CrossRef CAS PubMed
- J. Yi, T. I. Lam, W. Yokoyama, L. W. Cheng and F. Zhong, J. Agric. Food Chem., 2014, 62, 1096–1104 CrossRef CAS PubMed
- M. Ding, H. Yang and S. Xiao, J. Chromatogr. A, 1999, 849, 637–640 CrossRef CAS PubMed
- J. Maldonado-Valderrama, A. P. Gunning, P. J. Wilde and V. J. Morris, Soft Matter, 2010, 6, 4908–4915 RSC
- H. M. Rawel, S. Rohn, H.-P. Kruse and J. Kroll, Food Chem., 2002, 78, 443–455 CrossRef CAS
- B. Hu, Y. Wang, M. Xie, G. Hu, F. Ma and X. Zeng, J. Funct. Foods, 2015, 15, 593–603 CrossRef CAS
- G. Panick, R. Malessa and R. Winter, Biochemistry, 1999, 38, 6512–6519 CrossRef CAS PubMed
- L. Bekale, P. Chanphai, S. Sanyakamdhorn, D. Agudelo and H. A. Tajmir-Riahi, RSC Adv., 2014, 4, 31084–31093 RSC
- H. R. Costantino, K. Griebenow, P. Mishra, R. Langer and A. M. Klibanov, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1995, 1253, 69–74 CrossRef
- H. C. Liu, W. L. Chen and S. J. T. Mao, J. Dairy Sci., 2007, 90, 547–555 CrossRef CAS PubMed
- Z. Li and L. Gu, J. Agric. Food Chem., 2014, 62, 1301–1309 CrossRef CAS PubMed
- R. Gonçalves, N. Mateus and V. de Freitas, J. Agric. Food Chem., 2010, 58, 11924–11931 CrossRef PubMed
- Q. You, F. Chen, X. Wang, Y. Jiang and S. Lin, LWT–Food Sci. Technol., 2012, 46, 164–168 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2017 |