
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hybrid preparation of terbium(III)-doped mesoporous silica particles with calcium phosphates†
T. Kataokaa,
K. Shiba *b,
L. Y. Wanga,
S. Yamadaa and
M. Tagayaac
*b,
L. Y. Wanga,
S. Yamadaa and
M. Tagayaac
aDepartment of Materials Science and Technology, Nagaoka University of Technology, 1603-1 Kamitomioka, Nagaoka, Niigata 940-2188, Japan
bWorld Premier International Research Center Initiative (WPI), International Center for Materials Nanoarchitectonics (MANA), National Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: SHIBA.Kota@nims.go.jp; Tel: +81-29-860-4603
cTop Runner Incubation Center for Academica-Industry Fusion, Nagaoka University of Technology, 1603-1 Kamitomioka, Nagaoka, Niigata 940-2188, Japan
First published on 31st March 2017
Abstract
The preparation of terbium(III) (Tb3+)-doped mesoporous silica (Tb:MPS) particles and subsequent hybrid formation with calcium phosphate (CP) were proposed in this study. The surfactant/Tb:MPS particles with an initial doping concentration of Tb3+ ions to (Si + Tb) of 2 mol% were synthesized by a sol–gel reaction based on the cooperative organization of tetraethoxysilane, terbium(III) chloride and cetyltrimethylammonium bromide. Subsequently, the Tb:MPS particles were hybridized with CP through precipitation to form CP/Tb:MPS, where the formation was demonstrated by XRF and EDS analyses. The Tb:MPS showed a mesoporous structure which is typically seen in silica-surfactant mesostructured materials and the mesopores were preserved after precipitating the CP with crystalline phases. PL spectra were measured before and after the precipitation and some spectral changes were recognized. The PL spectral shapes due to the transitions of Tb3+ ions indicated that the Tb3+ ions were located inside the silica framework to interact with the siliceous O atoms. After the CP precipitation, the hybrid particles demonstrated higher quantum efficiency as well as longer PL life time, indicating the improved PL properties based on the CP–Tb3+-silica hybrid states. The interfacial manipulation of the present CP–Tb3+-silica hybrid allowed for unique material design in terms of its morphology and properties. This finding will be utilized for the encapsulation techniques for porous silica including with lanthanide ions and can be applicable for optical devices.
Introduction
Mesoporous materials have been prepared by the cooperative organization of surfactants and inorganic species; especially, the synthesis, characterization and applications of mesoporous silicas (MPSs) have been widely investigated.1,2 The MPSs prepared by supramolecular templating methods are known to possess attractive features such as well-defined and controllable pore sizes, large surface areas, and reactive surfaces.3,4 The preparation of multifunctional MPSs is also a topic of interest toward a wider variety of applications. Thus, synthetic pathways for MPSs with various compositions have been explored for decades.5–10Among various approaches, the incorporation of heteroatoms into the silica frameworks of MPSs has received much attention. The framework structures can provide attractive features as a host organization by the charge-compensation of non-network oxygen species.11 The lanthanide ion-doped MPSs demonstrated an improved thermal stability.12 The doping of cerium ions was accomplished for catalytic applications.13,14 Europium (III) ion (Eu3+) is known as an optical probe for the dopant site environment due to the luminescence properties.11,15 We have reported the synthesis of Eu3+-containing MPSs which exhibited the efficient luminescence by the Eu3+ ions coordinated in the non-bridging oxygen atoms.16 Terbium(III) ion (Tb3+) doped MPSs have also been synthesized and the presence of Tb3+ ions in the O–Si–O frameworks were investigated in detail.17 As a result, the obtained materials showed large specific surface areas. However, the efficient synthesis and luminescence of the Tb3+-doped MPSs by controlling the interactions between the doped ions and silica frameworks have not been reported. Thus, the effective control of Tb3+ ions at the near-surfaces in the silica frameworks is worth investigating.
Trivalent rare earth ions doped calcium phosphates (CPs) have been found to exhibit favorable photoluminescence properties.18–21 The CPs can display luminescence under visible excitation if it is doped with lanthanide ions, in which Eu3+ and Tb3+ are the most strongly emitting elements.22 Their luminescence is characterized by narrow emission bandwidths, high photochemical stability and long luminescent lifetime.23 Accordingly, Eu3+-doped24,25 and Tb3+-doped26,27 CPs have been studied. For the Tb3+-doped CP, the near-surface calcium ions can be partially replaced by a small amount of Tb3+. Interestingly, the presence of an extremely low concentration of the doped Tb3+ on the surfaces greatly improved the luminescence, while the main physicochemical properties and bioactivity of the CP compounds were maintained.26 Therefore, the Tb3+ ions at the near-surfaces in MPS would easily interact with CP compounds to form the novel hybrids and the complexed Tb3+ ions at the interfaces should show the unique luminescent properties.
In this study, we introduce the synthesis and properties of the hybrid composed of the Tb3+ doped luminescent MPS particles and CP (CP/Tb:MPS) that induces the unique luminescence behaviors. The synthetic procedure of the CP/Tb:MPS hybrid particles is schematically shown in Scheme 1 in comparison with the Tb3+-doped MPS (Tb:MPS) particles. By a sol–gel reaction based on the cooperative organization of tetraethoxysilane, terbium(III) chloride and cationic surfactant (cetyltrimethylammonium bromide: CTAB), CTAB/Tb:MPS particles were synthesized, and CP was precipitated on the particle surfaces. Then, the CTAB/Tb:MPS particles were calcined to resultantly obtain the mesoporous hybrid (CP/Tb:MPS). The unique interfacial interactions between CP–Tb3+-silica at the hybrid interfaces were examined to explain the enhanced luminescence properties.
 | ||
| Scheme 1 Illustration for the preparation of the CTAB/Tb:MPS hybrid particles with CP. A possible Tb3+ ion state in the surface silica framework is also proposed. | ||
Experimental
Preparation of Tb:MPS and CP/Tb:MPS
Tb3+-doped MPSs were synthesized according to our previous report.17 All the reagents used here were special grades and were used as received without further purification. A 1.0 g (2.75 mmol) of cetyltrimethylammonium bromide (CTAB: Wako Co., Ltd.), 225 g of ultrapure water and 3.5 mL of 2.0 M sodium hydroxide (NaOH: Wako Co., Ltd.) solution were mixed and then stirred at 80 °C for 1 h. A 5.515 mL (24.7 mmol) of tetraethoxysilane (TEOS: Tokyo Kasei Industries Co., Ltd.) and 15 mL of an Tb3+ aqueous solution containing 180 mg of TbCl3·6H2O (Wako Co., Ltd.) at the initial molar concentration of Tb to (Si + Tb) of 2.0 mol% were admixtured, added and stirred at 80 °C for 4 h. The resultant turbid solution was filtered. The separated solid was washed with ultrapure water and ethanol and then dried at 60 °C for 12 h. The sample is named as CTAB/Tb:MPS, which was calcined in a furnace under air at 550 °C for 6 h and the resultant sample is named as Tb:MPS, which was used as the reference for the following CP hybrids.K2HPO4 (2.13 g) was dissolved into deionized water (62.5 mL), and the CTAB/Tb:MPS was added and dispersed into the phosphate solution. The pH value of the solution was adjusted to 9 using tetramethylammonium hydroxide (TMAOH: Wako Co., Ltd.). 62.5 mL of deionized water containing CaCl2·2H2O (3.67 g) was added to the CTAB/Tb:MPS phosphate solution and vigorously stirred at the room temperature 3 h. Then, the solution was centrifuged to sediment solid product and washed with ultrapure water and ethanol. The washed product was dried at 60 °C and the sample is named as CTAB/CP/Tb:MPS. Then, CTAB/CP/Tb:MPS was calcined in a furnace under air at 550 °C and the resultant sample is named as CP/Tb:MPS.
Characterization
The elemental compositions were characterized by an X-ray fluorescence analysis (XRF: ZSX Primus II, Rigaku, Japan). The XRF analysis was performed using a sample pellet which was made by pressurizing sample powder without diluting it. The fundamental parameter method by the software (EZ scan program, Rigaku) was carried out for the semi-quantitative analysis. All the measurements and data analysis were conducted with an optional software (EZ scan program, Rigaku, Japan). Infrared spectra were recorded on a Fourier transform infrared spectrometer (FT-IR: JASCO Co., Ltd., FT/IR-4600ST). The FT-IR spectra were measured with a KBr pellet containing a small amount of sample powder. The weight ratio between sample powder and KBr was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. All the spectra were recorded after subtracting a background spectrum of pristine KBr. The measurement range, sampling time and resolution were 4000–500 cm−1, 100 and 2.0 cm−1, respectively. X-ray diffraction (XRD) patterns were recorded with a powder X-ray diffractometer (Smart Lab, Rigaku, Japan). XRD patterns were measured under the following conditions: X-ray source of CuKα line (λ: 0.15418 nm), voltage/current of 40 kV/30 mA, scan speed of 5.0° min−1 and sampling width of 0.01°. The morphologies and elemental mapping were observed using a field emission scanning electron microscopy (FE-SEM: SU8230, Hitachi High-Technologies Co., Ltd.) equipped with an energy dispersive spectroscopy (EDS: X-MaxN large area silicon drift detector, Oxford Instruments). Nitrogen adsorption and desorption isotherms were measured at 77 K on a BELSORP-mini II instrument (Microtrac/BEL Co., Ltd.). Prior to the measurement, the samples were degassed under vacuum at 393 K for 4 h. The surface area was evaluated by the Brunauer–Emmett–Teller (BET) method.28
:10. All the spectra were recorded after subtracting a background spectrum of pristine KBr. The measurement range, sampling time and resolution were 4000–500 cm−1, 100 and 2.0 cm−1, respectively. X-ray diffraction (XRD) patterns were recorded with a powder X-ray diffractometer (Smart Lab, Rigaku, Japan). XRD patterns were measured under the following conditions: X-ray source of CuKα line (λ: 0.15418 nm), voltage/current of 40 kV/30 mA, scan speed of 5.0° min−1 and sampling width of 0.01°. The morphologies and elemental mapping were observed using a field emission scanning electron microscopy (FE-SEM: SU8230, Hitachi High-Technologies Co., Ltd.) equipped with an energy dispersive spectroscopy (EDS: X-MaxN large area silicon drift detector, Oxford Instruments). Nitrogen adsorption and desorption isotherms were measured at 77 K on a BELSORP-mini II instrument (Microtrac/BEL Co., Ltd.). Prior to the measurement, the samples were degassed under vacuum at 393 K for 4 h. The surface area was evaluated by the Brunauer–Emmett–Teller (BET) method.28
Photoluminescence properties were evaluated by a photoluminescence spectroscopy and microscopy. The excitation and luminescence spectra were recorded on a FP-8500 spectrophotometer (JASCO Co., Ltd.) with the monitored luminescence at 544 nm and excitation wavelength at 369 nm from Xe lamp under room temperature (atmosphere: air, excitation-slit/detection-slit: 2 nm/2 nm, measure time: 0.1 s, step width: 1.0 nm, sample weight: 150 mg, shape: pellet), and the detection was used by photomultiplier tube. The fluorescent microscope images were obtained using a luminescence microscope (OLYMPUS Co., Ltd., CKX41N-FL, excitation wavelength: 360–400 nm, exposure time: 100 ms, sensitivity: ISO 400) through the emission source (OLYMPUS Co., Ltd., U-RFLT50).
The internal quantum efficiency (ηint) was measured using an integrating sphere (internal diameter: 60 mm). The excitation and luminescence spectra were also recorded by the above spectrophotometer with each monitored luminescence maxima and excitation wavelength. The incident, scattering, and luminescence light intensity spectra were measured, and their integrated peak intensities were calculated and abbreviated I0, I1 and I2, respectively. Thus, ηint was calculated by the eqn (1).
| ηint = I2/(I0 − I1) × 100 | (1) |
The decay kinetics was also recorded by the above spectrophotometer with the same excitation and luminescence wavelength, which can be explained with an exponential function. The excitation was measured on a wavelength at 369 nm from a Xe flash lamp (150 W), and the decay of the luminescence intensity was detected on a wavelength at 544 nm. The data plot time was 0.05 ms using a successive rotation chopper. The measured decay was recorded by a numerical fitting to an exponential function based on the eqn (2).
| I(t) = I(0)exp(−t/τ) | (2) |
Results and discussion
As shown in Table 1, the elemental concentrations, i.e. Ca/P and Tb/(Si + Tb), were determined and calculated by the XRF results. Although most of the carbons were removed during the precipitation and washing process, some carbon was still remained even after the calcination. It would be due to the residual CTAB. The resulting Tb/(Tb + Si) concentrations were almost the same between the samples. The resulting Tb/(Tb + Si) concentration was 1.8–2.0 times larger than those in the initial solution composition, suggesting the decrease in the Si content of the particles by the Tb added synthetic process. The Ca and P elements were seen only in the case of the CP hybrid and the Ca/P ratio was not changed after the calcination.| C (mol%) | O (mol%) | Si (mol%) | P (mol%) | K (mol%) | Ca (mol%) | Tb (mol%) | Ca/P | Tb/(Tb + Si) (mol%) | |
|---|---|---|---|---|---|---|---|---|---|
| CTAB/Tb:MPS | 32.9 | 46.2 | 19.5 | 0.0 | 0.0 | 0.0 | 1.4 | — | 4.3 |
| CP/CTAB/Tb:MPS | 2.6 | 63.6 | 1.5 | 13.3 | 1.1 | 17.0 | 0.9 | 1.28 | 3.7 |
| CP/Tb:MPS | 2.5 | 63.8 | 2.0 | 13.1 | 1.2 | 16.5 | 0.9 | 1.26 | 4.8 |
| Tb:MPS | 14.4 | 59.3 | 24.6 | 0.0 | 0.0 | 0.0 | 1.1 | — | 4.1 |
In the FT-IR spectra (see Fig. S1 in ESI†), the absorption bands originated from the Si–O–Si asymmetric and symmetric stretching were observed at around 1070 and 1225 cm−1 and these spectral shapes were almost same irrespective of the doping. The characteristic bands seen at around 1480, 2854 and 2925 cm−1 can be assigned to the C–H stretching and bending of the CTAB that apparently decreased after the calcination. The present hybrid states would suppress the complete removal of the carbon compounds, while the carbon compounds were completely removed in the case of Tb:MPS. Based on the previous reports on the FT-IR spectroscopic studies,29,30 in the CP/CTAB/Tb:MPS and CP/Tb:MPS, the several absorption bands appeared at around 1090–1030, 600 and 560 cm−1 are attributed to P–O stretching of phosphate groups, suggesting that the CP formation on Tb:MPS was verified.
Fig. 1 shows the XRD patterns of the particles at lower and higher 2θ regions. In the CTAB/Tb:MPS and Tb:MPS particles, the diffractions indexed as 100, 110 and 200 reflections of a hexagonal structure were observed (Fig. 1(a)). With the calcination of CTAB/Tb:MPS, the shapes of each diffraction pattern were preserved while slight shift to higher angles was recognized, indicating the pore contraction by the condensation among residual silanol groups of the silica framework. The Tb–O bond length was expected to be longer than that of Si–O, resulting in the enlargement of the unit cell in the presence of Tb3+ as compared to the bare mesoporous silicas. This was also demonstrated by the metal incorporation into the MCM-41 framework.32 With the CP formation, the d100 – spacing value increased and the diffraction peak shape was broadened. The CP nucleation and crystal growth at the mesopore surfaces would induce the pore dilatation.
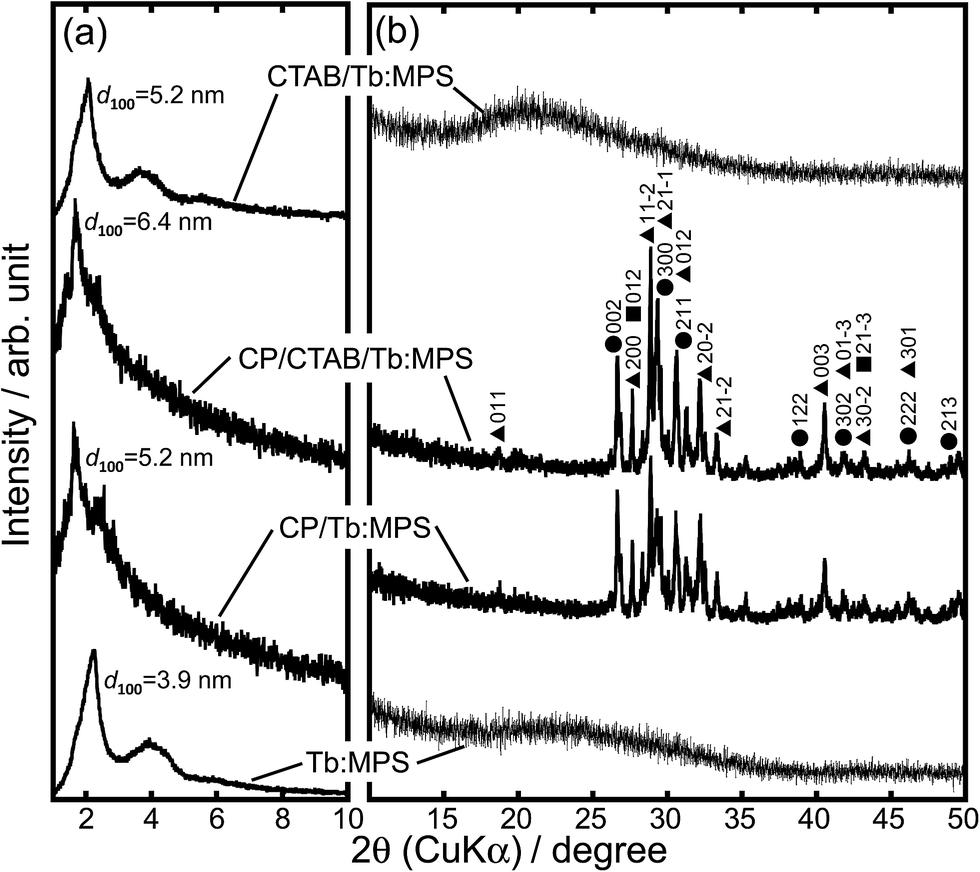 | ||
| Fig. 1 XRD patterns of the CTAB/Tb:MPS, CP/CTAB/Tb:MPS, CP/Tb:MPS and Tb:MPS particles at (a) lower and (b) higher 2θ regions (●: hydroxyapatite (ICDD 00-009-0432), ▲: monetite (ICDD 01-071-1759); ■: calcium pyrophosphate (ICDD 01-075-2756)). | ||
In the higher-angle regions (Fig. 1(b)), the CTAB/Tb:MPS and Tb:MPS do not display any detectable diffraction that can be assigned to terbium oxide, for example. Thus, Tb3+ ions should be incorporated into the amorphous silica networks. It was found that the Tb3+-doping into the silica frameworks can expand the mesopores and then rearrange the Si–O–Si networks without any crystalline impurities (e.g., terbium oxides and silicates), which would be based on the interactions with Tb3+ ions. With the CP formation, some CP crystalline phases (hydroxyapatite (Ca10(PO4)6(OH)2, ICDD 00-009-0432), monetite (CaHPO4, ICDD 01-071-1759), calcium pyrophosphate (Ca2P2O7·4H2O, ICDD 01-075-2756)) were observed, allowing the presence of significant amounts of crystalline CP phases. Considering only single phase formation of hydroxyapatite without CTAB/Tb:MPS particles, the cationic surfactant CTAB, Tb3+ ions and mesopore surfaces would affect the crystalline CP phases. Here, it should also be mentioned that both Ca2+ and Tb3+ ions can provide a similar coordination environment and hardness, ionic radii, and geometric characteristics. Thus, the Tb3+ ion in the silica phase would partially be incorporated into the CP phases at the precipitation process. Therefore, the interfacial reactions between the CTAB/Tb:MPS and CP occurred in the precipitation and calcination processes. To simply demonstrate the properties of CP/Tb:MPS by comparing to those of Tb:MPS, the samples after the calcination will be discussed hereafter.
Fig. 2(a) shows the N2 adsorption and desorption isotherms of the CP/Tb:MPS and Tb:MPS particles. The shape of the isotherms for CP/Tb:MPS is categorized into type III according to the IUPAC classification, whereas that for Tb:MPS is categorized into typical type IV, suggesting the mesopore formation. As shown in Table 2, the BET surface area (SBET) decreased with the CP formation. The Barrett–Joyner–Halenda (BJH) pore size distributions33 indicated the contraction of the pore diameter with the formation, although the distribution width was broadening. Thus, the mesopore entrances on the particle surfaces were encapsulated by the nucleated CP and CP crystals, which result in the decrease in the SBET. The broadening of the X-ray diffraction pattern and pore distribution suggested the presence of less ordered structure, but the mesopore structures were still retained, being also supported by the previous reports.17,31
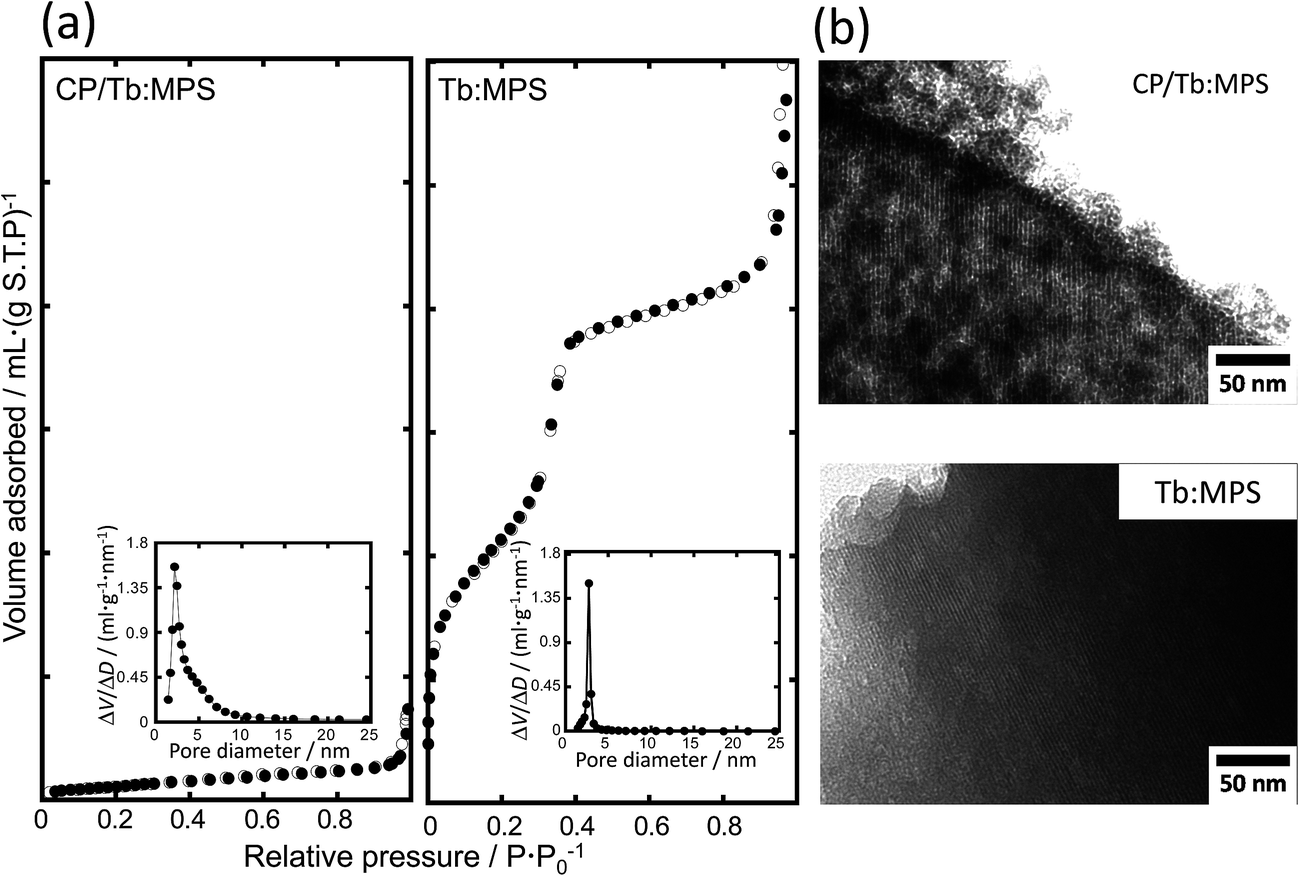 | ||
| Fig. 2 (a) Nitrogen adsorption (●) and desorption (○) isotherms and (inset) corresponding pore size distributions of the CP/Tb:MPS and Tb:MPS particles, and (b) their TEM images of the mesopores. | ||
| SBET (m2 g−1) | rBJH (nm) | hint (%) | t (ms) | |
|---|---|---|---|---|
| CP/Tb:MPS | 48 | 2.2 | 4.4 | 5.0 |
| Tb:MPS | 894 | 2.7 | 2.8 | 1.6 |
In order to investigate the hybrid nanostructures, we observed the TEM images shown in Fig. 2(b). All the TEM images clearly exhibited the mesopore arrangements and the pore diameters were approximately estimated to be 1.5–4.0 nm from the images. In CP/Tb:MPS, the precipitated CP crystallites were significantly covered on the Tb:MPS surfaces without closing the mesopores, while the silica framework exhibited the disordering of the mesopore structures (i.e., worm-like pores). Thus, these results are in agreement with those obtained by the XRD and N2 adsorption/desorption measurements.
In the FE-SEM images (Fig. 3(a) and (c)), the CP/Tb:MPS exhibited irregular shapes with the smaller particles, whereas the Tb:MPS had the spherical shapes, indicating that the CP precipitation on the silica frameworks induced the morphological changes with collapsing the shapes. In the EDS mapping images (Fig. 3(b) and (d)), the CP/Tb:MPS particle shapes corresponded with the Ca, P, Si and O mapping areas and the Si intensity was relatively week, indicating the CP precipitation on the silica surfaces. In contrast, no detectable Ca and P elements was observed in the case of the Tb:MPS particles. Therefore, the Tb:MPS particles were successfully hybridized with the CP phases.
 | ||
| Fig. 3 (a and c) FE-SEM and (b and d) FE-SEM/EDS mapping (element: Ca, P, Si and O) images of (a and b) CP/Tb:MPS and (c and d) Tb:MPS particles. | ||
Fig. 4(a) and (b) show the excitation and photoluminescence spectra. These spectral shapes are almost same irrespective of the CP formation. Thus, no reflection of the electronic transitions of Tb3+ indicates the same local O atom environments in the silica host matrix. The excitation spectra exhibited the peaks associated with 4f–4f transitions of Tb3+ ion (Fig. 4(a)). The features are mainly assigned to the excitations based on the 5LJ (J = 10, 9, 7) levels from the ground state of Tb3+ ion.34,35 The luminescence intensity under the excitation at 369 nm by the 4f–4f transition from 5L10 level to the ground state is relatively stronger in the observed peaks.
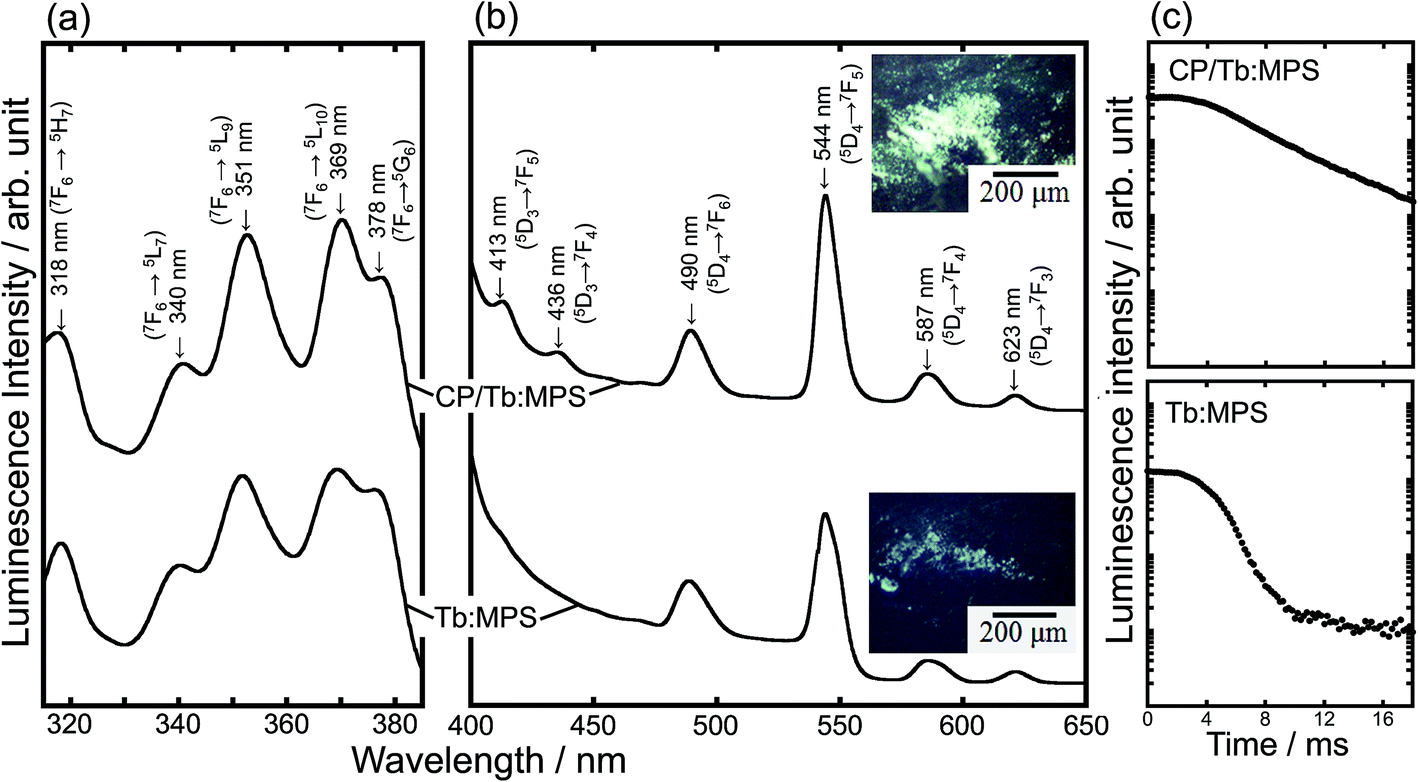 | ||
| Fig. 4 (a) Excitation and (b) photoluminescence spectra of the CP/Tb:MPS and Tb:MPS particles and their (inset) fluorescent images, and (c) fluorescence intensity decay plots. | ||
The luminescence spectra showed the narrow peaks assigned to the f–f transitions of the Tb3+ ions (Fig. 4(b)). The luminescence transitions are assigned to those from 5D4 → 7FJ (J = 6, 5, 4, 3)36 as usual for Tb3+ luminescence, being centered at 490, 544, 587 and 623 nm, respectively. The green luminescence is seen in the microscope image (Fig. 4 (inset)), and the color is also observed in the other Tb3+-doped systems (e.g., Tb3+-doped SiO2–PVA37). By the CP hybrid, the luminescence peaks due to 5D3 → 7FJ (J = 4, 5) were enhanced (Fig. 4(b)) and the apparent luminescence intensities increased (Fig. 4 (inset)). In fact, as shown in Table 2, the ηint significantly increased with the CP formation, although the residual carbon compounds of CTAB would affect the luminescence properties.
To understand the luminescence relaxation, the decay kinetics of the excited states was investigated as shown in Fig. 4(c) and Table 2. The decay kinetics was fitted with an exponential function. The CP precipitation extended τ, even though only the silica framework exhibited the longer τ of the Tb3+ as compared with the previous report.34 The τ clearly reveals the dispersion of the Tb3+ ions into the silica frameworks and the encapsulation of CP induce the preferred interfacial interactions between the CP–Tb3+-silica to exhibit the longer τ, suggesting the interfacial design for the unique luminescence properties. At the interfaces between CP and Tb:MPS, the preferred CP–Tb3+-silica interactions would be present. Then, the near-surface Tb3+ ions in the silica phases were partially incorporated into the CP phases. Such an incorporation would change the luminescence properties.
The Eu3+ ion states in the silica frameworks have been investigated in our previous report.11 The Eu3+ ions were located inside the structure to electrostatically interact with the siliceous O atoms. Under the sol–gel process, the Eu3+ ions would be homogeneously dissolved to resultantly prevent aggregation among the Eu3+ ions in the MPS formation. A homogeneous sol containing CTAB, silica precursor and Tb3+ was used in the synthetic process. Although the mesopores with the large surface area effectively adsorb water molecules, the Tb3+ doped inside the framework would little interact with the water molecules. Therefore, it is suggested that the Tb3+ ions homogeneously diffused inside the non-crystalline silica frameworks, and would be stabilized by the charge-compensation of the non-network oxygen species (e.g., Si–O−). The near-surface Tb3+ ions easily interact with the hydroxyl groups of the adsorbed water molecules or silanol groups. The presence of high energy stretching vibrations of the hydroxyl groups causes the luminescence quenching by a non-radiative decay mechanism. In this study, the interfacial design through the encapsulation by CP induced the unique luminescence properties. This finding provides a merit in the use of the interfacial design between the CP–Tb3+-silica hybrid states for the activity of lanthanide ions. Here, the ionic radius of Tb3+ (1.04 Å) is larger than that of Si4+ (0.41 Å) and same as Ca2+ (1.14 Å). In the present case, the near-surface Tb3+ ions in the silica phases were partially incorporated into the CP phases. Such an incorporation could be explained by the sector zoning model in minerals.38,39 Therefore, the silica frameworks and subsequent encapsulation of CP induced the preferred interfacial interactions between the CP–Tb3+-silica for the unique luminesce properties. After achieving the efficient luminescent properties by the further investigation, the hybrids will be utilized as bio-imaging materials based on the biocompatibility of CP.
Conclusions
The preparation of Tb3+-doped MPS particles and subsequent hybrid formation with CP were investigated. The CP was precipitated on the CTAB/Tb:MPS particles which were synthesized by a sol–gel reaction of tetraethoxysilane in the presence of terbium(III) chloride and CTAB. The CTAB containing hybrid was converted to mesoporous solid by the calcination and the mesopores were preserved even after precipitating the CP with crystalline phases. The formation of the present CP/Tb:MPS hybrid particles were confirmed by XRF and EDS analyses. PL spectra were also measured before and after the CP precipitation and some spectral changes were recognized. The PL spectral shapes due to the transitions of Tb3+ ions indicated that the Tb3+ ions were located inside the silica frameworks, interacting with the siliceous O atoms. After the CP precipitation, the hybrid particles demonstrated higher quantum efficiency as well as longer PL life time, suggesting the improved PL properties based on the CP–Tb3+-silica hybrid states. This finding will be utilized for the encapsulation techniques of the porous silica including lanthanide ions and can be applicable to optical devices.Acknowledgements
This study was supported by a grant from the Japan Society for the Promotion of Science (JSPS) KAKENHI (Grant-in-Aid for Young Scientists (A), Grant No. 26709052).Notes and references
- T. Yanagisawa, T. Shimizu, K. Kuroda and C. Kato, Bull. Chem. Soc. Jpn., 1990, 63, 988–992 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- R. I. Nooney, D. Thirunavukkarasu, Y. Chen, R. Josephs and A. E. Ostafin, Chem. Mater., 2002, 14, 4721–4728 CrossRef CAS.
- M. Ogawa, K. Kato and N. Shimura, Bull. Chem. Soc. Jpn., 2009, 82, 121–125 CrossRef CAS.
- I. I. Slowing, B. G. Trewyn and V. S. Y. J. Lin, J. Am. Chem. Soc., 2007, 129, 8845–8849 CrossRef CAS PubMed.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T.-W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. J. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834–10843 CrossRef CAS.
- M. Tagaya, T. Ikoma, T. Yoshioka and J. Tanaka, Chem. Commun., 2011, 47, 8430–8433 RSC.
- M. Tagaya, S. Motozuka, T. Kobayashi, T. Ikoma and J. Tanaka, J. Mater. Chem., 2012, 22, 18741–18743 RSC.
- M. Tagaya, N. Hanagata, T. Ikoma, T. Kobayashi, K. Shiba, T. Yoshioka and J. Tanaka, Key Eng. Mater., 2013, 529–530, 630–635 CAS.
- M. Tagaya, T. Ikoma, Z. Xu and J. Tanaka, Inorg. Chem., 2014, 53, 6817–6827 CrossRef CAS PubMed.
- M. Tagaya, T. Ikoma, T. Yoshioka and J. Tanaka, J. Colloid Interface Sci., 2011, 363, 456–464 CrossRef CAS PubMed.
- A. S. Araujo and M. Jaroniec, J. Colloid Interface Sci., 1999, 218, 462–467 CrossRef CAS PubMed.
- A. S. Araujo and M. Jaroniec, Thermochim. Acta, 2000, 345, 173–177 CrossRef CAS.
- V. C. Costa, M. J. Lochhead and K. L. Bray, Chem. Mater., 1996, 8, 783–790 CrossRef CAS.
- Q. Xu, L. Li, B. Li, J. Yu and R. Xu, Microporous Mesoporous Mater., 2000, 38, 351–358 CrossRef CAS.
- M. Tagaya, T. Ikoma, T. Yoshioka, F. Minami and J. Tanaka, Mater. Lett., 2011, 65, 2287–2290 CrossRef CAS.
- T. Kataoka, L. Y. Wang, K. Kobayashi, M. Nishikawa and M. Tagaya, Jpn. J. Appl. Phys., 2016, 55, 105503 CrossRef.
- L. D. Deloach, S. A. Payne, W. L. Kway, J. B. Tassano, S. N. Dixit and W. F. Krupke, J. Lumin., 1994, 62, 85–94 CrossRef CAS.
- J. P. M. van Vliet and G. Blasse, Mater. Res. Bull., 1990, 25, 391–394 CrossRef CAS.
- M. Tachihante, D. Zambon, A. Arbus, M. Zahir, A. Sadel and J. C. Cousseins, Mater. Res. Bull., 1993, 28, 605–613 CrossRef CAS.
- R. Ternane, M. Trabelsi-Ayedia, N. Kbir-Ariguibb and B. Piriouc, J. Lumin., 1999, 81, 165–170 CrossRef CAS.
- W. D. Horrocks Jr and D. R. Sudnick, Acc. Chem. Res., 1981, 14, 384–392 CrossRef.
- S. P. Mondejar, A. Kovtun and M. Epple, J. Mater. Chem., 2007, 17, 4153–4159 RSC.
- A. Doat, M. Fanjul, F. Pellé, E. Hollande and A. Lebugle, Biomaterials, 2003, 24, 3365–3371 CrossRef CAS PubMed.
- T. Kataoka, K. Shiba and M. Tagaya, Colloid Interface Sci. Commun., 2016, 13, 1–5 CrossRef CAS.
- L. Li, Y. Liu, J. Tao, M. Zhang, H. Pan, X. Xu and R. Tang, J. Phys. Chem. C, 2008, 112, 12219–12224 CAS.
- Y. Han, X. Wang, S. Li and X. Ma, J. Sol-Gel Sci. Technol., 2009, 49, 125–129 CrossRef CAS.
- S. Brunauer, P. H. Emmett and E. Teller, J. Am. Chem. Soc., 1938, 60, 309–319 CrossRef CAS.
- B. O. Fowler, Inorg. Chem., 1974, 13, 194–207 CrossRef CAS.
- K. C. Blakeslee and R. A. Condrate, J. Am. Ceram. Soc., 1971, 54, 559–563 CrossRef CAS.
- Y. Li and B. Yan, J. Solid State Chem., 2008, 181, 1032–1039 CrossRef CAS.
- V. Parvulescu and B. L. Su, Catal. Today, 2001, 69, 315–322 CrossRef CAS.
- E. P. Barrett, L. G. Joyner and P. H. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 CrossRef CAS.
- B. Szpikowska-Sroka, N. Pawlik, A. S. Swinarew and W. A. Pisarski, Ceram. Int., 2015, 41, 1167–11679 CrossRef.
- H. Yang and Y. Kim, J. Lumin., 2008, 128, 1570–1576 CrossRef CAS.
- H. Weng and B. A. Yan, Sens. Actuators, B, 2016, 228, 702–708 CrossRef CAS.
- H. Ikeda, T. Murata and S. Fujino, Opt. Mater., 2014, 36, 1119–1122 CrossRef CAS.
- Y. Nakamura, Am. Mineral., 1973, 58, 986–990 CAS.
- E. Dowty, Am. Mineral., 1976, 61, 460–469 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR spectra of the CTAB/Tb:MPS, CP/CTAB/Tb:MPS, CP/Tb:MPS and Tb:MPS. See DOI: 10.1039/c6ra28457d |
| This journal is © The Royal Society of Chemistry 2017 |