
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural, electronic and catalytic performances of single-atom Fe stabilized by divacancy-nitrogen-doped graphene†
Zhiyong
Liu
a,
Tingwei
He
a,
Kaikai
Liu
a,
Weiguang
Chen
b and
Yanan
Tang
 *ab
*ab
aCollege of Physics and Materials Science, Henan Normal University, Xinxiang, Henan 453007, China. E-mail: yntang2010@163.com
bQuantum Materials Research Center, College of Physics and Electronic Engineering, Zhengzhou Normal University, Zhengzhou 450044, China
First published on 23rd January 2017
Abstract
Inspired by the experimental discovery of the configuration of a one central transition metal and four surrounding N atom doped graphene sheet (M–GN4), we systemically study the geometry and electronic and catalytic properties of a single-atom Fe embedded GN4 sheet (Fe–GN4) using first-principles calculations. It is found that the neighboring N atoms in graphene strongly stabilize a single Fe atom and make the doped Fe atom more positively charged, which helps to regulate the stability of reactive gases. Besides, the adsorption of gas molecules can modulate the electronic structure and magnetic property of the Fe–GN4 system. Moreover, the catalytic reactions of CO oxidation on the Fe–GN4 substrate are comparably investigated in terms of the Langmuir–Hinshelwood (LH) and Eley–Rideal (ER) mechanism. The results show that the LH reaction as the starting state is energetically more favorable than the ER reaction, since the catalytic process has much smaller energy barriers (0.13 eV) and then promotes the CO oxidation reaction. Therefore, the stable configuration of Fe–GN4 would be a highly efficient catalyst for CO oxidation, which provides a clue for designing atomic-scale catalysts in energy-related devices.
1. Introduction
The catalytic oxidation of carbon monoxide (CO) as a vital prototype reaction plays a very important role in solving the serious environmental problems caused by automobile exhaust and industrial processes,1–3 and can be used to evaluate the reactive activity, selectivity and durability of catalysts in surface chemistry.4–6 Following the previous works on many noble metals surfaces are studied for catalyzing CO oxidation,7–9 such as Pt,2,10,11 Rh,12 Au4,13–15 and Pd.3 Recently, novel low dimensional nanoparticles16–19 and even single atoms20 have been suggested for improving the catalytic reactivity of CO oxidation. Single-atom catalyst loading on various metal oxide substrates (FeOx,21,22 ZnO,23 CeO2,24,25 MnO2 (ref. 26) and MoS2 (ref. 27)) demonstrates the excellent catalytic property toward CO oxidation. These results illustrated that the CO oxidation reaction takes place mainly on the surface of doped metal oxide, and the adsorption of CO can react with surface oxygen to form CO2. In addition, the large surface-to-volume ratio of graphene as a promising substrate exhibits outstanding physical and chemical properties.28–31 The experimental results confirmed that Pt catalysts were well-dispersed on graphene nanosheets32 or graphene oxide,33 and can reduce both CO adsorption and the activation barriers for CO oxidation, which is of great significance in energy-related applications.34–39 However, the metal adatoms easily aggregate into large clusters and then affect the efficiency of the catalyst.40 Therefore, it is a major challenge to explore appropriate substrates that can enhance the stability and chemical reactivity of catalysts.To solve this problem, chemical doping has been confirmed to be an effective approach to tailor the properties of graphene;41–48 especially, the substitutional dopants in graphene can control the size and degree of catalyst dispersion49–53 and result in excellent catalytic activity.54–57 Although the single-atom Au-,58 Fe-,59,60 Cu-,61 Pt-,62 Pd-63 and Al-64 embedded graphene substrates have high catalytic activity for CO oxidation, it is still highly desirable to develop the realization of a single-atom catalyst which relies on the controllable generation of vacancies in graphene sheets. Recently, works have proposed that active sites would consist of a metal atom coordinated by four pyridinic N atoms and are incorporated into graphitic sheets (M–GN4),65–67 which can modify the electronic and magnetic properties of graphene68 and show good activity for the oxygen reduction reaction (ORR).69,70
The computational studies revealed that the ORR activity of Fe–GN4 moieties is comparable with that of Pt-based catalysts,71,72 which stimulates the ongoing debate on whether the Fe–GN4 sheets can be efficient catalysts for CO oxidation. This issue is unresolved, largely due to the lack of knowledge on the origin of the reaction mechanism toward CO oxidation on the metal–N4 moiety doped graphene. Recently, Zhang et al.73 investigated the catalytic activity for CO oxidation on Co atoms incorporated with pyridinic nitrogen graphene (Co–GN3). Yet, there is a lack of systematic analysis of the sensing performance of the M–GN4 surface toward adsorbed gases and their interaction in catalytic reactions.
The previous works studied the catalytic reaction of CO oxidation on different non-noble metal (NNM, Ni, Co and Al) atom anchored graphene systems.74,75 Herein, we chose Fe as the catalyst, because it is inexpensive and abundant in the earth. Moreover, the metal Fe coupled with N-doped graphene plays a vital role in enhancing ORR activity by improving the content of the Fe–N active sites.72 It is natural to ask whether the Fe–GN4 sheet can be used to achieve new functionalities. In this work, the adsorption stability and interaction of reactive gases (CO and O2) on the Fe–GN4 surface are investigated. The sequential CO oxidation reactions on Fe–GN4 are comparably analyzed through considering both the Langmuir–Hinshelwood (LH) and Eley–Rideal (ER) mechanisms.
2. Computational model and methods
Spin-polarized density functional theory (DFT) calculations are carried out using the Vienna ab initio simulation package (VASP)76,77 with the projector augmented wave (PAW) pseudo-potentials.78 Exchange–correlation functions are described with the generalized gradient approximation (GGA) in the form of the Perdew, Burke, and Ernzerhof (PBE) functional.79 The kinetic energy cutoff for the plane-wave basis set was chosen to be 450 eV. The C 2s22p2, Fe 3d64s2, N 2s22p3 and O 2s22p4 states are treated as valence electrons. An armchair edge of graphene ribbon with a 5 × 7 supercell is adopted and the vacuum layer is set to 20 Å to avoid the interaction among mirror images. The calculated lattice constant of the graphene sheet is 2.47 Å, which approximates well to the experimental value of 2.46 Å.80 The Brillouin zone (BZ) integration is sampled using a 3 × 3 × 1 Γ-centered Monkhorst–Pack (MP) grid and a Γ-centered MP grid of 15 × 15 × 1 is used for the final density of states (DOS) calculations.Bader charge analysis81 was used to evaluate the atomic charges and electron transfer in the reactions. The adsorption energies and site preferences for each type of gas molecule are tested on the Fe–graphene surfaces. The climbing image nudged elastic band method (CI-NEB)82–84 was employed to investigate the saddle points and minimum energy path (MEP) for the formation (or dissociation) of reaction gases on graphene substrates. The geometric optimization and the search for the transition states (TS) are tested by means of frequency calculations, while those with one imaginary frequency correspond to the metastable states. A number of intermediate images are constructed along the reaction pathways between the initial state (IS) or TS and the final state (FS), and the spring force between adjacent images was set to be 5.0 eV Å−1. The images are optimized until the forces on each atom are less than 0.02 eV Å−1. The energy barriers (Ebar) of each chemical reaction are calculated by the energy difference between the IS and TS.
The adsorption energy (Eads) is calculated using the formula Eads = EA + EB − EAB, where EA, EB and EAB are the total energies of the optimized adsorbates in the molecules or atoms (A: Fe, O2 O, CO and CO2), the clean graphene substrates (B: GN4 or Fe–GN4) and the adsorbate-substrate systems, respectively. With this definition, a higher Eads value means a stronger adsorption.
3. Results and discussion
3.1. Geometric stability and electronic properties of the Fe–GN4 sheet
Based on the previous calculations,85,86 the formation configuration of one central transition metal and four surrounding N atoms is energetically favorable for three reasons: (1) creation of carbon divacancies, (2) N atom substitution of unsaturated carbons and (3) the metal atom incorporation into the center of the N atoms, as depicted in Fig. 1(a). It was reported that the Fe–GN4 structure is energetically more favorable than the formation of Fe–GN3, Fe–GN2, Mn–GN4 and Co–GN4 configurations.65 In order to ensure the most stable configuration of the Fe atom on the GN4 surface, we have performed a scan in energy of the adsorbed Fe atom at the possible adsorption sites, which include the centers of the substitution N atoms and the neighboring hexagonal carbon ring (H, the hollow site), as well as the bridge (B) sites of the C–C bonds in the same carbon rings. The calculated results show that the Fe atom anchors at the H (or B) of the a and b sites, where the adatom would be drawn back to the vacancy site of the GN4 sheet, as shown in Fig. 1(a). For the other sites (c, d, e and f) further away from the vacancy site, the Fe adatom would be anchored at the neighboring H site with Eads of about 1.53 eV. These results indicate that the Fe adatom is more likely to be anchored at the vacancy site of the GN4 surface and forms the more stable configuration of the Fe–GN4 sheet. The calculated adsorption energy, bond length and transferred electrons of the Fe–GN4 structure are shown in Table 1; the Fe, N and C atoms are all in the same plane in the optimized structure.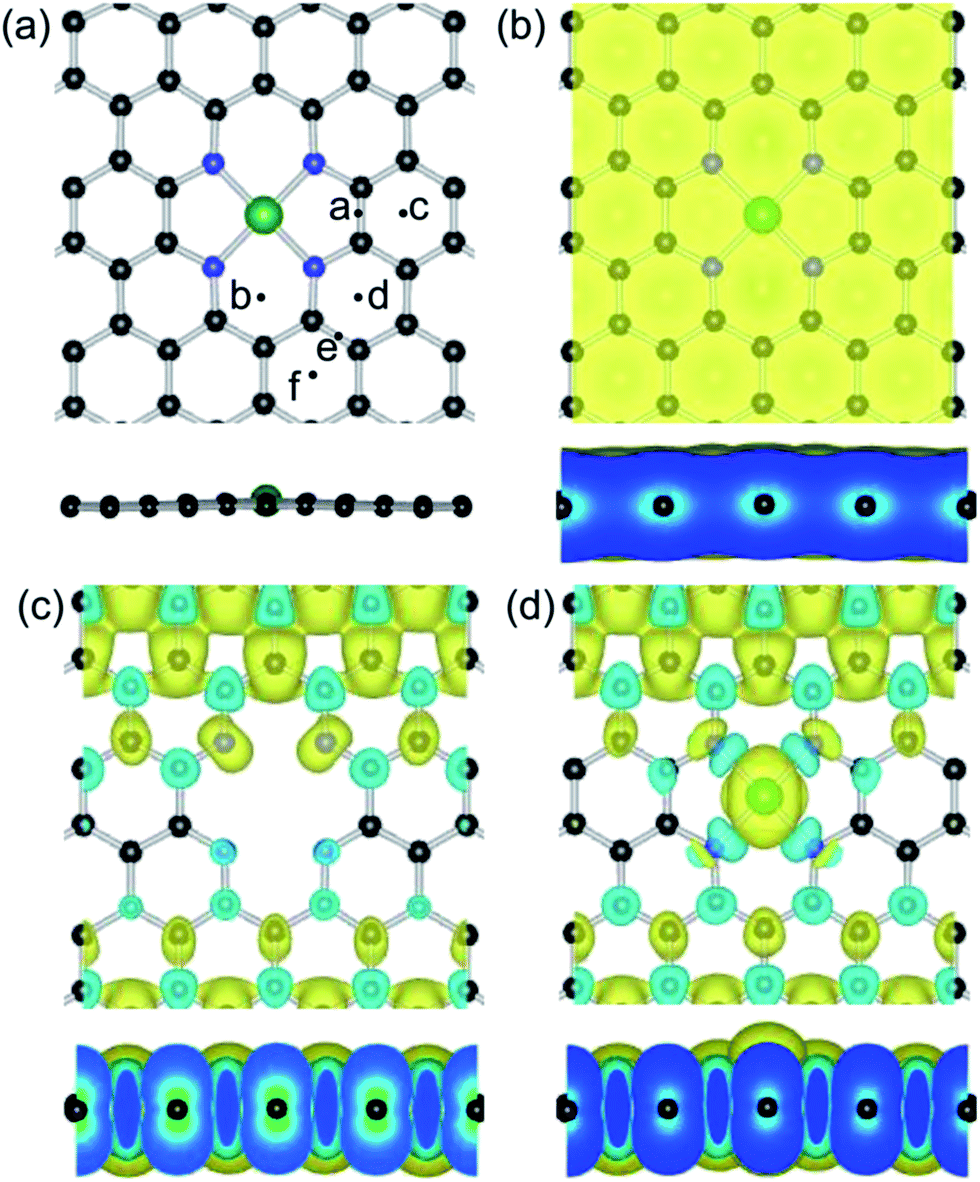 | ||
| Fig. 1 Top and side views of the (a) geometric structure and (b) charge distribution for the Fe–GN4 sheet, as well as the spin charge density plots for the (c) GN4 and (d) Fe–GN4 systems. Black, blue and green balls represent the C, N and Fe atoms, respectively. | ||
| Systems | E ads (eV) | d 1 (Å) | Δq (e) | d 2 (Å) | |
|---|---|---|---|---|---|
| GN4 | Fe | 7.77 | 1.87 | −1.36 | 0.06 |
| Fe–GN4 | O2 | 0.95 | 1.39 | +0.81 | 1.85 |
| CO | 1.19 | 1.17 | +0.32 | 1.71 | |
| O | 4.56 | 1.91 | +0.72 | 1.65 | |
| CO2 | 0.12 | 1.16 | +0.18 | 1.65 | |
Based on the Bader charge analysis,81 the atomic Fe exhibits positive charge due to the transferring electrons (1.36e) that move from the Fe atom to the neighboring N atoms and form the covalent feature between the Fe and N atoms, while the four N atoms are negatively charged to about 0.66e. Compared with the Fe adatom on the pristine graphene surface (1.19 eV), the doped Fe atom at the GN4 sheet (Fe–GN4) has a much larger Eads (7.77 eV), which is larger than that of the cohesive energy of the Fe element of 4.94 eV per atom, and the monovacancy and divacancy sites of the graphene sheet (7.28 and 6.47 eV, respectively). Thus, the formation of the Fe–GN4 configuration is energetically favorable and stable enough in the chemical reaction. Besides, the bond distance between the Fe atom and the neighboring N atoms is 1.87 Å, which is in agreement with the reported results.65,85 As shown in Fig. 1(b), the distributions of charge density occur at interfaces between the Fe atom and neighboring N atoms, where the corresponding contour lines in the plots are drawn at intervals of 0.01 e Å−3. The large value of the electron distribution occurs at the Fe–N bonds, illustrating that the transferred electrons (>1.0e) can enhance the stability of the Fe atom. In order to confirm the stable configuration of the Fe–GN4 sheet, first principles molecular dynamics calculations were performed. Herein, a time step of 1.0 fs was used and the temperature was controlled by velocity scaling at each step. In a period of 2000 fs (2 ps), with calculations at temperatures of 700 K, 1000 K and 1300 K, the embedded Fe atom and the neighboring N atoms in the plane have slight distortion, as shown in Fig. S1.† The configuration of Fe–GN4 is even still stable in a period of 2 ps at 1300 K with some distortion over time, which is confirmed by the change in height of the Fe dopant (from 0.07 to 0.21 Å) and the change in distance of the Fe–N bonds (from 1.79 to 1.97 Å). This result indicates that the GN4 configuration could efficiently stabilize the single-atom catalyst, and the Fe–GN4 sheet as an active site possesses high thermal stability at a certain temperature.
Secondly, we investigate the spin charge density (SCD) for the GN4 and Fe–GN4 systems as depicted in Fig. 1(c) and (d), where the corresponding contour lines in the plots are drawn at intervals of 0.001 e Å−3. It is found that more electrons dominantly accumulate in the vicinity of the Fe–N bonds, and fewer electrons are located on neighboring carbon atoms. It is clearly shown that the doped Fe atom induces the greater spin electron redistributions of the GN4 system, resulting in the magnetic moment of the system increasing from 10.0 µB (GN4) to 12.0 µB (Fe–GN4). In order to analyze the origin of the high stability of the Fe–GN4 sheet, more insight into the corresponding electronic structure of the system is shown in Fig. 2. It is found that the total DOS (TDOS) and partial DOS (PDOS) plots of the Fe–GN4 system have been obviously altered as compared with those of the bare GN4 sheet. A peak at the Fermi level (EF) vanished in the GN4 system, and there is a small band gap at the EF in the Fe–GN4 system, meaning that the semimetal GN4 system converts into a semiconducting material. Besides, the broadened Fe 3d states strongly overlap with the TDOS of the system around the EF, suggesting that the strong hybridization between the Fe atom and the GN4 system can enhance their interaction. The asymmetry of the spin channels in the GN4 and Fe–GN4 sheets indicates that these systems exhibit magnetic character. Thus, the doped Fe atom can regulate the electronic structure and magnetic properties of the GN4 system.
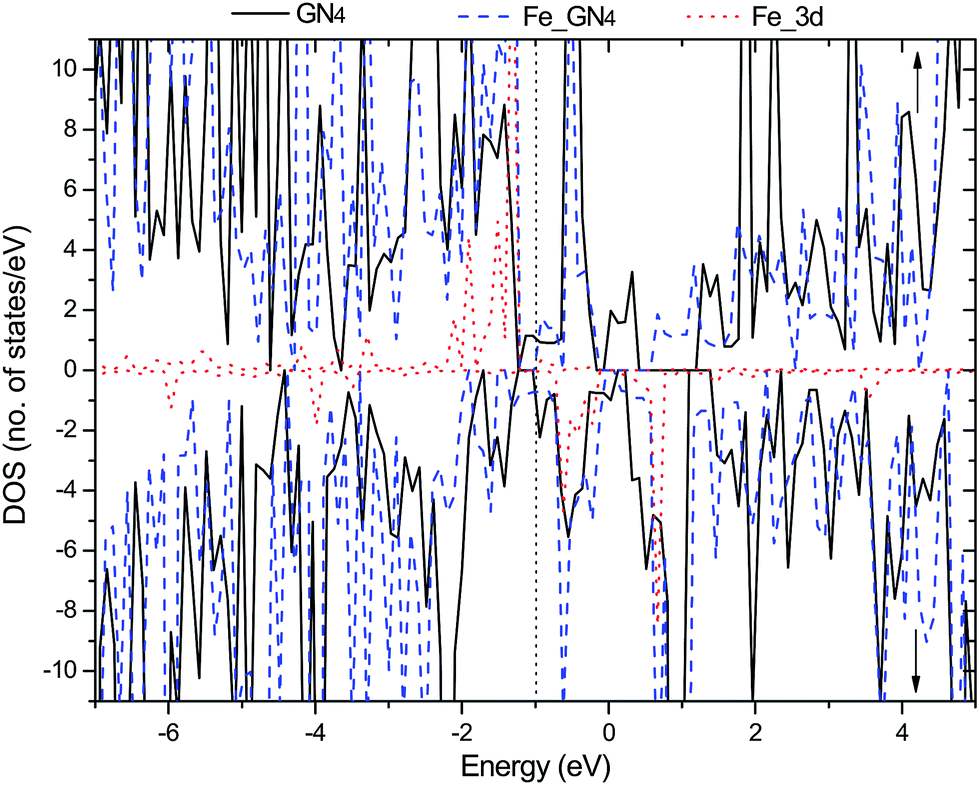 | ||
| Fig. 2 Spin-resolved total DOS (TDOS) and partial DOS (PDOS) (spin-up labeled with ↑ and spin-down labeled with ↓) for the Fe–GN4 system. The black solid, blue dashed and red dotted curves represent the TDOS of GN4 and Fe–GN4 and the PDOS of the Fe 3d states, respectively. The vertical dotted line denotes the Fermi level. | ||
3.2. Adsorption properties of CO and O2 molecules
Based on the optimized configuration of the Fe–GN4 substrate, we firstly investigate the stable configurations of various adsorbed species (O2, CO, O and CO2). For the most stable adsorption configurations, the corresponding adsorption energies and structural parameters are listed in Table 1. It is found that the adsorption of CO on the Fe–GN4 surface is more stable than that of the O2 molecule. As shown in Fig. 3(a), the O2 prefers to form two bonds with the Fe atom and the O–O bond is parallel to the Fe–GN4 surface (1.85 Å), and the corresponding Eads is 0.95 eV. As shown in Fig. 3(b), the end-on configuration of CO is nearly vertical on the Fe–GN4 surface with the distance between Fe and CO being 1.71 Å; it has a larger Eads (1.19 eV) than the adsorbed O2. Compared with the CO adsorption (0.32e), more electrons (0.81e) are transferred from the Fe–GN4 to the adsorbed O2, which subsequently leads to elongation of the O–O bond (1.39 Å). Hence, the elongation of the O–O bond is connected to the number of transferred electrons from the Fe–GN4 substrate. The more charge that is transferred from Fe–GN4 to O2, the more elongated the O–O bonds become.62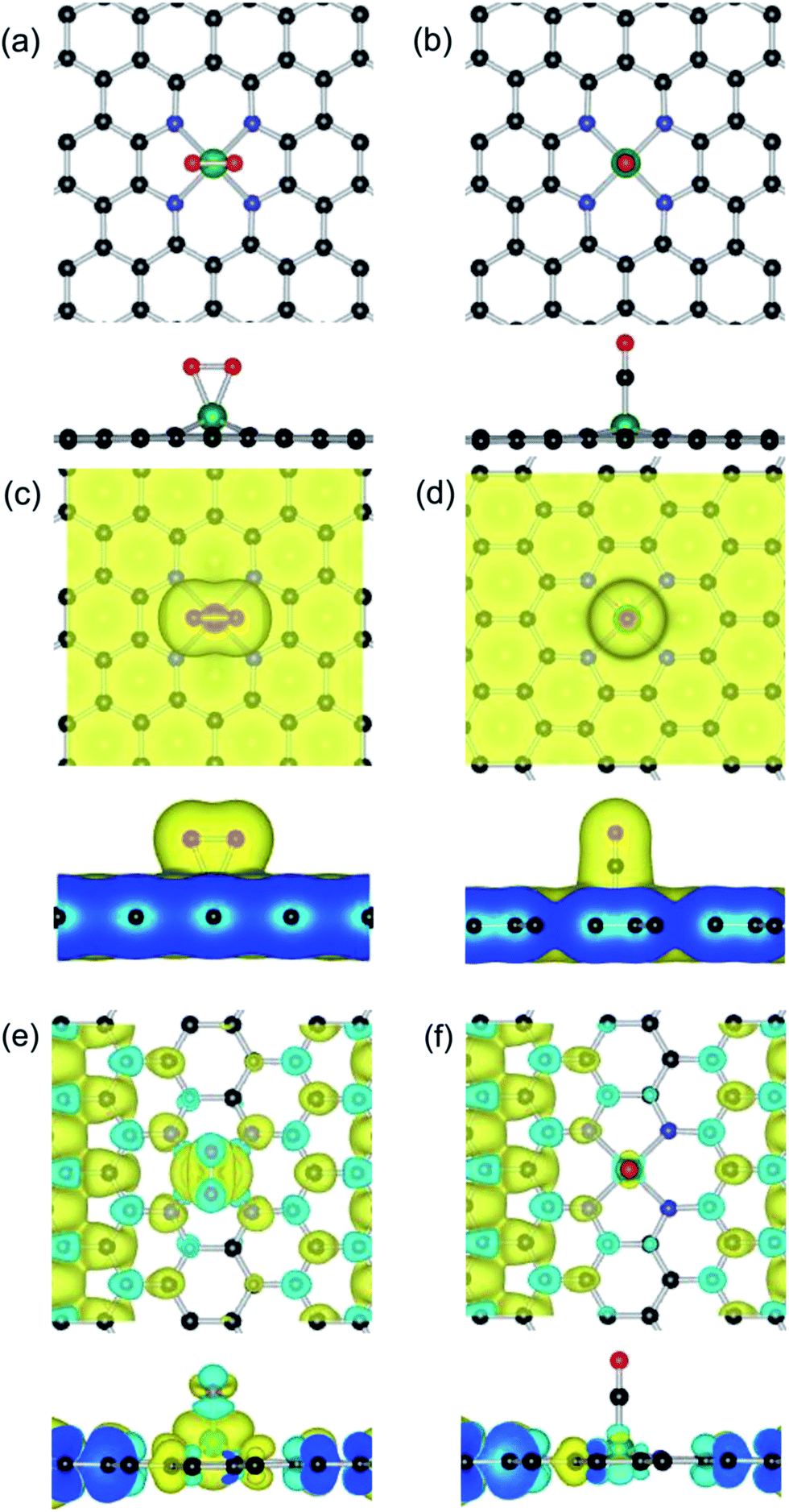 | ||
| Fig. 3 Top and side views of the (a and b) geometric structures, (c and d) charge distribution and (e and f) spin charge density plots for O2 and CO adsorbed on the Fe–GN4 surface. Black, blue, red and green balls represent the C, N, O and Fe atoms, respectively. | ||
As shown in Fig. 3(c) and (d), we investigate the valence charge density between reactive gases and the Fe–GN4 sheet. It is found that the more the electrons dominantly accumulate in the vicinity of the CO–Fe and O2–Fe interfaces, the fewer electrons are located on the graphene substrates. Bader charge analysis shows that the doped Fe atom provides transferred electrons of about 1.38 and 1.61e in the two systems, which are partly transferred to the adsorbed gases (0.32e for CO and 0.81e for O2) and the rest are transferred to the GN4 substrates, illustrating that the doped metal atoms can enhance the interaction between reactants and substrates. In addition, we further investigate the spin electron redistribution for gas molecules on the Fe–GN4 substrate, as shown in Fig. 3(e) and (f). It is found that the adsorbed O2 and CO induce the spin charge redistribution of the GN4 surfaces. The more the electrons dominantly accumulate in the vicinity of the O2–Fe interfaces, the much fewer electrons are located at the CO–Fe interfaces and the GN4 surface. Compared to the adsorbed CO (10.0 µB), the adsorption of O2 on the Fe–GN4 system exhibits a more pronounced spin charge distribution and thus has a better magnetic property (12.0 µB). Hence, the positive charge of the Fe dopant can regulate the adsorption behaviors for gas molecules and the adsorbed gases can control the magnetic property of the Fe–GN4 system. Although the adsorbed CO has a relatively large Eads on the Fe–GN4 surface, the small energy difference of the adsorption gases indicates a relatively smaller repulsion between the reactive gases, which may facilitate the interaction between CO and O2.57
Electronic structure, which fundamentally determines the physical and chemical properties of a system, is directly related to the kind of reactant gases on the substrate. For the adsorbed O2 and CO on the Fe–GN4 systems, the corresponding DOS plots are investigated, which are shown in Fig. 4. As shown in Fig. 4(a), the broadened PDOS of the Fe 3d states strongly hybridize with the O2 2π*, 5σ and 1π orbitals around the EF. The reduced peaks below the EF indicate that the transferred electrons (0.81e) are closer to the O2 and occupy the 2π* and 5σ states of O2, resulting in the elongation of the O–O bond to 1.39 Å. Besides, the increased unpaired electrons between the adsorbed O2 and Fe atom induce the magnetic moment of the whole system (12.0 µB), so the spin-up and spin-down channels of the system become asymmetric, which is similar to Au-58 and Cu-graphene.61 The DOS plot for the adsorbed CO on the Fe–GN4 system is shown in Fig. 4(b); the hybridization between the PDOS of the Fe 3d states and CO-2π*, 5σ states is observed near the EF. Compared to the adsorbed O2, fewer electrons are transferred from the Fe atom to the CO molecule (0.32e). According to the asymmetry of the spin channels, the adsorbed CO on the Fe–GN4 system exhibits the magnetic property (10 µB). Therefore, the electronic and magnetic properties of the Fe–GN4 sheet can be regulated by choosing the kinds of reactive gases, which may have a bearing on important applications in electronic and spintronic devices.
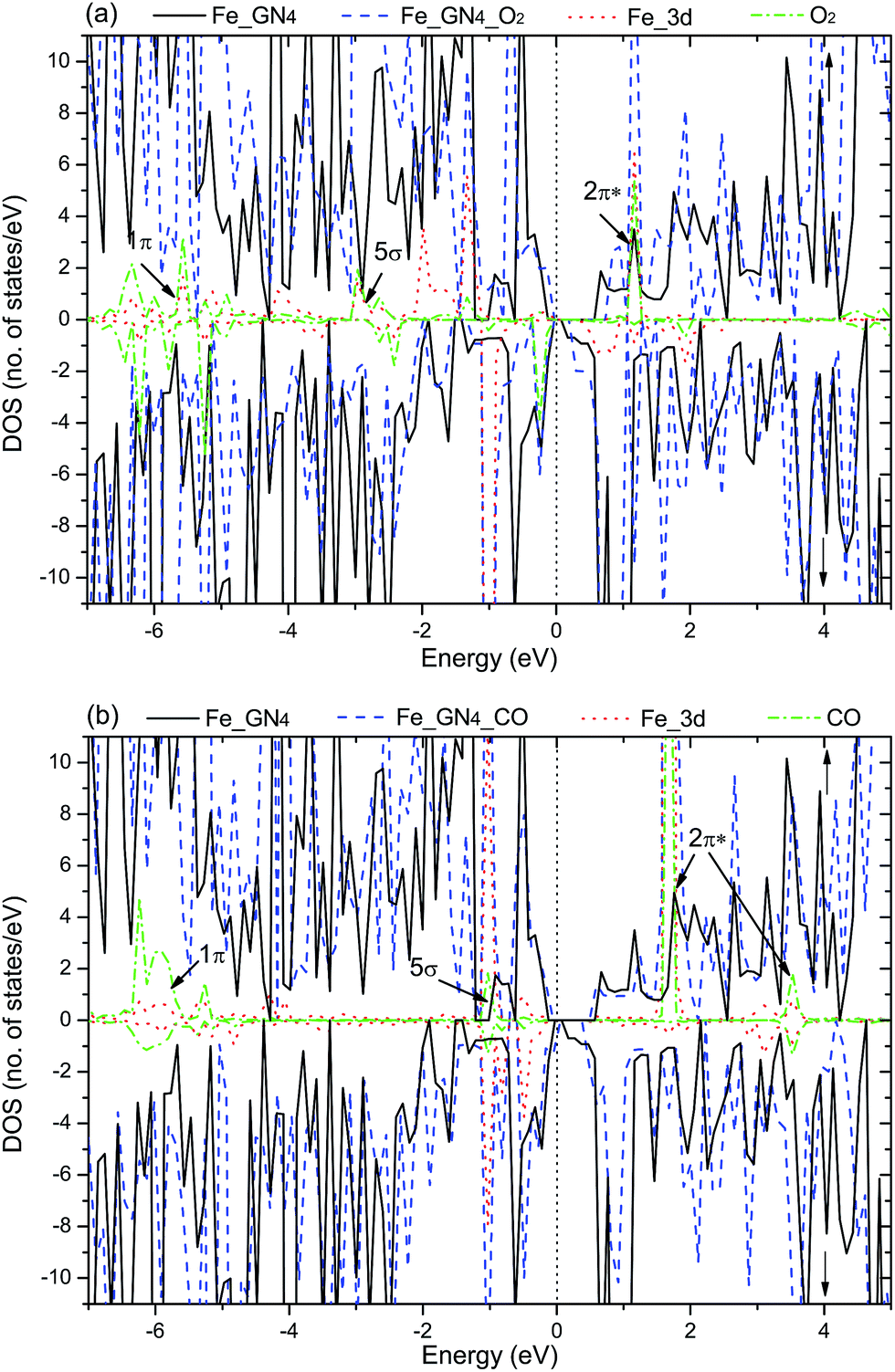 | ||
| Fig. 4 Spin-resolved TDOS, local DOS (LDOS), PDOS (spin-up labeled with ↑ and spin-down labeled with ↓) for (a) O2 and (b) CO adsorbed on the Fe–GN4 sheet. The black solid and blue dashed curves represent the TDOS of the Fe–GN4 system without and with, respectively, O2 or CO adsorption. The red dotted and green dash dotted curves represent the PDOS of Fe 3d states with O2 or CO adsorption and the LDOS of adsorbed O2 (or CO), respectively. The vertical dotted line denotes the Fermi level. | ||
3.3. Reaction mechanism of CO oxidation
Two types of reaction mechanism for CO oxidation have been established, namely, the LH and ER mechanisms.87–89 The LH mechanism occurs with the coadsorption of CO and O2 molecules before reaction, followed by formation and dissociation of a peroxo-type OOCO intermediate state (MS). Finally, the formation and desorption of a CO2 molecule occurs, leaving an adsorbed O atom (Oads) at the catalyst. The ER mechanism features the CO molecule directly reacting with the preadsorbed O2 and the subsequent dissociation of the carbonate-like state CO3 (MS). In this study, both reaction mechanisms are comparatively investigated here to identify the optimal reaction pathway.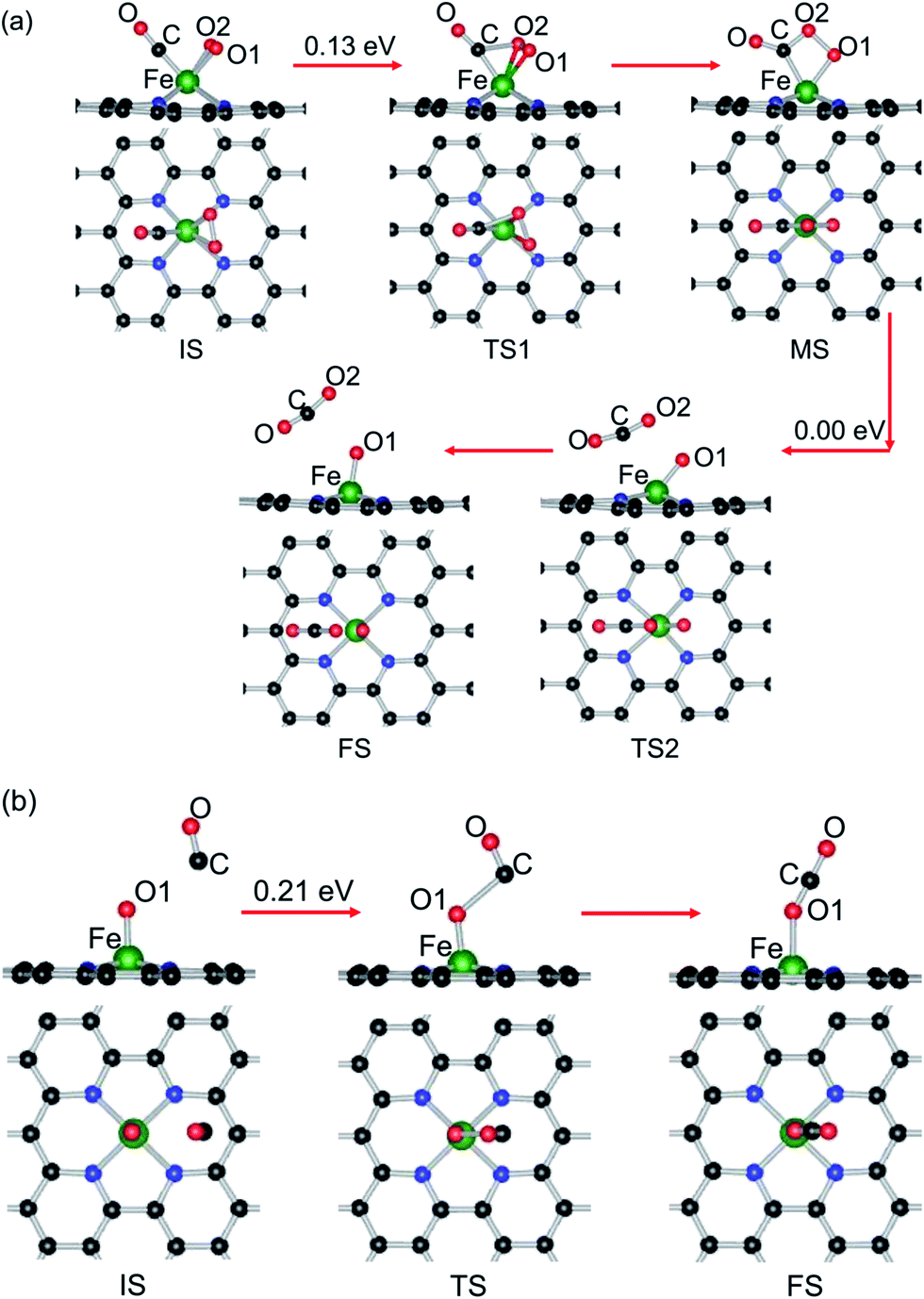 | ||
| Fig. 5 The minimum energy profiles and the configurations of different states for CO oxidation on Fe–GN4, (a) CO + O2 by the LH mechanism and (b) CO + Oads by the ER mechanism. Black, blue, red and green balls represent the C, N, O and Fe atoms, respectively. | ||
| (a) | |||||
|---|---|---|---|---|---|
| Distance (Å) | IS | TS1 | MS | TS2 | FS |
| d C–O | 1.16 | 1.17 | 1.21 | 1.17 | 1.08 |
| d C–Fe | 1.77 | 1.83 | 1.98 | 2.75 | 3.70 |
| d C–O2 | 2.29 | 1.82 | 1.33 | 1.18 | 1.20 |
| d O1–Fe | 1.93 | 1.97 | 1.93 | 1.69 | 1.65 |
| d O2–Fe | 1.90 | 1.99 | 2.56 | 2.96 | 4.17 |
| d O1–O2 | 1.37 | 1.42 | 1.47 | 2.14 | 2.69 |
| (b) | |||
|---|---|---|---|
| Distance (Å) | IS | TS | FS |
| d O1–Fe | 1.65 | 1.66 | 1.93 |
| d O1–C | 2.85 | 2.05 | 1.16 |
| d C–O | 1.15 | 1.16 | 1.18 |
In the IS, the distance between O2 and CO is about 2.29 Å, and the Fe–CO and Fe–O2 distances are 1.77 and 1.90 Å, respectively. To react, the O2 molecule turns around with one of the oxygen atoms breaking away from the Fe atom and approaching the carbon atom of CO to generate a new C–O bond, as shown in Fig. 5(a). Passing over the TS1, the OOCO complex (MS) is formed and the corresponding energy barrier (Ebar1) along the MEP is estimated to be 0.13 eV. In this reaction process, the bond length of O2 (dO1–O2) is gradually elongated from 1.37 to 1.47 Å. The reaction continuously proceeds from the MS to FS through the TS2 without any energy barrier, where the O1–O2 and Fe–C bonds are broken and a CO2 molecule is generated, leaving an atomic O1 adsorbed at the Fe atom, which is expected to be active and can be used for the CO oxidation reaction. Once formed, the physisorbed CO2 will desorb easily from the reactive site. Then, we further check the oxidation process of a second CO reacting with the atomic O1 to produce CO2 through the ER reaction, the corresponding structural parameters and energy barrier of which are depicted in Table 2(b) and Fig. 5(b).
The configuration with a CO molecule more than 2.85 Å away from the preadsorbed Oads (Eads, 4.56 eV) on the Fe atom is chosen as the IS. The adsorption configuration of CO2 at Fe–GN4 is viewed as the FS. In the reaction process, the C atom of CO approaches the adsorbed O1 and the CO–O1 distance is 2.05 Å, developing a new C–O1 bond and attracting the adsorbed O1 at the Fe atom (TS). The calculated results show that the ER reaction has a relatively larger energy barrier (0.21 eV) than that of the first step (TS1, 0.13 eV) through the LH reaction. It is noted that the Eads of CO2 on Fe–GN4 is only 0.12 eV and it is easily desorbed from the reactive site at room temperature.
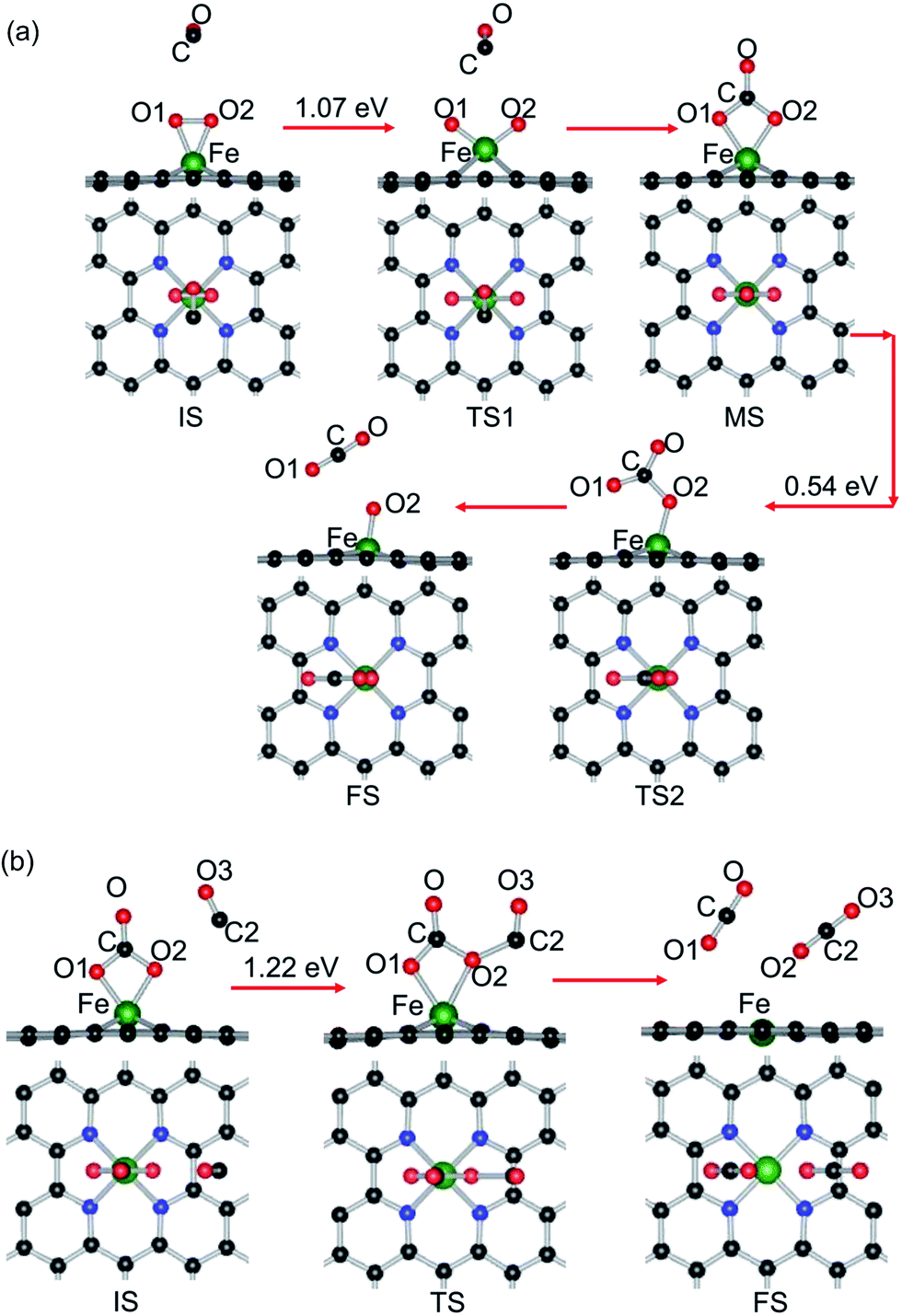 | ||
| Fig. 6 The minimum energy profiles and the configurations of different states for CO oxidation on Fe–GN4, (a) CO + O2 and (b) CO3 + CO reactions by the ER mechanism. Black, blue, red and green balls represent the C, N, O and Fe atoms, respectively. | ||
| (a) | |||||
|---|---|---|---|---|---|
| Distance (Å) | IS | TS1 | MS | TS2 | FS |
| d C–O1 | 3.57 | 3.34 | 1.34 | 1.25 | 1.18 |
| d C–O2 | 3.57 | 3.34 | 1.34 | 1.44 | 2.58 |
| d C–O | 1.14 | 1.14 | 1.22 | 1.24 | 1.17 |
| d O1–Fe | 1.85 | 1.63 | 1.93 | 2.95 | 3.90 |
| d O2–Fe | 1.85 | 1.63 | 1.93 | 1.80 | 1.65 |
| d O1–O2 | 1.40 | 2.60 | 2.14 | 2.28 | 2.91 |
| (b) | |||
|---|---|---|---|
| Distance (Å) | IS | TS | FS |
| d C–O2 | 1.34 | 1.44 | 2.97 |
| d C2–O2 | 3.05 | 1.66 | 1.18 |
| d O1–Fe | 1.93 | 2.05 | 3.80 |
| d O2–Fe | 1.94 | 2.18 | 3.28 |
| d O1–O2 | 2.15 | 2.14 | 3.35 |
| d C–C2 | 3.70 | 2.75 | 3.65 |
For the reactive gases, although the Eads of CO is slightly larger than that of O2, the small energy difference may reduce the repulsion between the reactants and promote the catalytic reaction for CO oxidation, which is in agreement with that on Au–graphene.58 Compared with the catalytic processes through the ER mechanism, the first step through the LH mechanism has a much smaller energy barrier (CO + O2 (b) OOCO, 0.13 eV), while the second CO oxidation step (OOCO → CO2 + O) is without any energy barrier, illustrating that the energy barrier for CO oxidation mainly comes from the formation of the OOCO complex, so the coadsorption of CO and O2 as the starting state is energetically favorable and then the reaction proceeds with the second CO oxidation by the ER reaction. In addition, if the ER reaction is taken as the starting reaction step, the formation of the CO3 complex needs to overcome a large energy barrier. In contrast to the commonly used noble metal catalysts, such as Pt1,2 and Au92 (>1.0 eV), the CO oxidation reactions through the LH mechanism have a lower energy barrier on the Fe–GN4 surface (0.13 eV); meanwhile, the energy barrier is also lower than that of the similar reaction on Fe- (0.58 eV),59 Cu- (0.54 eV),61 Pd- (0.54 eV),63 Pt- (0.59 eV),62 Co- (0.42 eV),74 Al- (0.32 eV)64 and Au-embedded graphene (0.31 eV)58 and Co–N3–graphene (0.86 eV),73 as well as Pd- (0.66 eV)93 and Co-embedded (0.55 eV)94 hexagonal boron nitride nanosheets. Hence, the formation of the Fe–GN4 sheet exhibits a much higher catalytic activity for CO oxidation.
In light of the aforementioned discussion, it is concluded that the adsorption of gas molecules can effectively regulate the electronic structure and magnetic property of the Fe–GN4 system. Besides, the sequential reactions of CO oxidation on the Fe–GN4 surface include the LH and ER reactions. For the LH reaction, the formation of the OOCO complex (CO + O2 → OOCO, 0.13 eV) has a small energy barrier. Then, the second CO oxidation (CO + O → CO2, 0.21 eV) has a slightly larger energy barrier, which can be viewed as the rate limiting step in the catalytic reactions. It is worth noting that the two-step CO oxidation reactions on Fe–GN4 have low enough energy barriers, so the CO oxidation reactions on the Fe–GN4 surface are more likely to proceed rapidly in practical reactions. Therefore, the calculated results show that the high quality Fe–GN4 sheet could be experimentally achieved,71,72 and the small energy barriers for the CO oxidation reaction indicate that the Fe–GN4 configuration has low cost and high activity.
4. Conclusions
We carried out a comprehensive study on the structural stability and CO oxidation reactions on a single-atom Fe incorporated GN4 sheet (Fe–GN4) by using the first-principles method. Firstly, it is found that the single-atom Fe can be stably embedded into the center of four N atoms and the small energy difference between reactant gases can facilitate the catalytic reaction towards CO oxidation. Secondly, the Fe dopant in the GN4 sheet introduces electronic doping and can effectively regulate the electronic structure and magnetic property of the graphene system. In the catalytic reactions, the LH mechanism as a starting state is more preferable than the ER mechanism, since the formation of the intermediate product (OOCO) has a smaller energy barrier (0.13 eV). In comparison, the larger reaction barriers of CO3 formation and decomposition (1.02, 0.54 and 1.22 eV) reactions indicate that the CO oxidation through the ER mechanism is not preferable. As a result, the complete CO oxidation reactions on the Fe–GN4 surface include a two-step process: the LH as a starting step (CO + O2 → OOCO → CO2 + Oads, 0.13 eV) followed by the ER reaction (CO + Oads → CO2, 0.21 eV). This result demonstrates the high catalytic properties of the Fe–GN4 sheet for CO oxidation, which provides a new opportunity for their applications in heterogeneous catalysis.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. U1404109 and 11504334), the Application Foundation and Advanced Technology Research Program of Henan Province (Grant No. 152300410167), the Natural Science Foundation of Henan Province (Grant No. 162300410325), Science Funds for Young Scholar of Henan Normal University (No. 5101029279082) and the Doctoral Research Fund of Henan Normal University (No. 5101029170256).References
- A. Alavi, P. Hu, T. Deutsch, P. L. Silvestrelli and J. Hutter, Phys. Rev. Lett., 1998, 80, 3650–3653 CrossRef CAS.
- M. Ackermann, T. Pedersen, B. Hendriksen, O. Robach, S. Bobaru, I. Popa, C. Quiros, H. Kim, B. Hammer, S. Ferrer and J. W. M. Frenken, Phys. Rev. Lett., 2005, 95, 255505 CrossRef CAS PubMed.
- C. Zhang and P. Hu, J. Am. Chem. Soc., 2001, 123, 1166–1172 CrossRef CAS PubMed.
- A. A. Herzing, C. J. Kiely, A. F. Carley, P. Landon and G. J. Hutchings, Science, 2008, 321, 1331–1335 CrossRef CAS PubMed.
- S. H. Oh and G. B. Hoflund, J. Catal., 2007, 245, 35–44 CrossRef CAS.
- X. Xie, Y. Li, Z. Q. Liu, M. Haruta and W. Shen, Nature, 2009, 458, 746–749 CrossRef CAS PubMed.
- Z. P. Liu, X. Q. Gong, J. Kohanoff, C. Sanchez and P. Hu, Phys. Rev. Lett., 2003, 91, 266102 CrossRef PubMed.
- X. Q. Gong, Z. P. Liu, R. Raval and P. Hu, J. Am. Chem. Soc., 2004, 126, 8–9 CrossRef CAS PubMed.
- L. Molina and B. Hammer, Phys. Rev. Lett., 2003, 90, 206102 CrossRef CAS PubMed.
- I. Nakai, H. Kondoh, K. Amemiya, M. Nagasaka, A. Nambu, T. Shimada and T. Ohta, J. Chem. Phys., 2004, 121, 5035 CrossRef CAS PubMed.
- M. Nagasaka, H. Kondoh, I. Nakai and T. Ohta, J. Chem. Phys., 2007, 126, 044704 CrossRef PubMed.
- M. Chen, Y. Cai, Z. Yan, K. Gath, S. Axnanda and D. W. Goodman, Surf. Sci., 2007, 601, 5326–5331 CrossRef CAS.
- X. Liu, A. Wang, X. Wang, C. Y. Mou and T. Zhang, Chem. Commun., 2008, 27, 3187–3189 RSC.
- S. N. Rashkeev, A. R. Lupini, S. H. Overbury, S. J. Pennycook and S. T. Pantelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 035438 CrossRef.
- C. Chang, C. Cheng and C. Wei, J. Chem. Phys., 2008, 128, 124710 CrossRef CAS PubMed.
- S. Dobrin, Phys. Chem. Chem. Phys., 2012, 14, 12122–12129 RSC.
- B. H. Morrow, D. E. Resasco, A. Striolo and M. B. Nardelli, J. Phys. Chem. C, 2011, 115, 5637–5647 CAS.
- W. Liu, Y. Zhao, R. Zhang, Y. Li, E. J. Lavernia and Q. Jiang, ChemPhysChem, 2009, 10, 3295–3302 CrossRef CAS PubMed.
- C. Liu, Y. Tan, S. Lin, H. Li, X. Wu, L. Li, Y. Pei and X. C. Zeng, J. Am. Chem. Soc., 2013, 135, 2583–2595 CrossRef CAS PubMed.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- F. Li, Y. Li, X. C. Zeng and Z. Chen, ACS Catal., 2015, 5, 544–552 CrossRef CAS.
- D. Ma, Q. Wang, T. Li, Z. Tang, G. Yang, C. He and Z. Lu, J. Mater. Chem. C, 2015, 3, 9964–9972 RSC.
- V. Shapovalov and H. Metiu, J. Catal., 2007, 245, 205–214 CrossRef CAS.
- W. C. Ding, X. K. Gu, H. Y. Su and W. X. Li, J. Phys. Chem. C, 2014, 118, 12216–12223 CAS.
- J. Gao, C. Jia, L. Zhang, H. Wang, Y. Yang, S.-F. Hung, Y.-Y. Hsu and B. Liu, J. Catal., 2016, 341, 82–90 CrossRef CAS.
- D. Ma, Y. Tang, G. Yang, J. Zeng, C. He and Z. Lu, Appl. Surf. Sci., 2015, 328, 71–77 CrossRef CAS.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385–388 CrossRef CAS PubMed.
- K. Novoselov, A. Geim, S. Morozov, D. Jiang, M. Katsnelson, I. Grigorieva, S. Dubonos and A. Firsov, Nature, 2005, 438, 197–200 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902–907 CrossRef CAS PubMed.
- A. Geim and K. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- E. Yoo, T. Okada, T. Akita, M. Kohyama, I. Honma and J. Nakamura, J. Power Sources, 2011, 196, 110–115 CrossRef CAS.
- S. Sharma, A. Ganguly, P. Papakonstantinou, X. Miao, M. Li, J. Hutchison, M. Delichatsios and S. Ukleja, J. Phys. Chem. C, 2010, 423–427 Search PubMed.
- Y. Tang, Z. Yang and X. Dai, J. Nanopart. Res., 2012, 14, 844 CrossRef.
- Y. Tang, Z. Lu, W. Chen, W. Li and X. Dai, Phys. Chem. Chem. Phys., 2015, 17, 11598–11608 RSC.
- E. Yoo, T. Okata, T. Akita, M. Kohyama, J. Nakamura and I. Honma, Nano Lett., 2009, 9, 2255–2259 CrossRef CAS PubMed.
- R. Siburian and J. Nakamura, J. Phys. Chem. C, 2012, 116, 22947–22953 CAS.
- G. Chen, S. J. Li, Y. Su, V. Wang, H. Mizuseki and Y. Kawazoe, J. Phys. Chem. C, 2011, 115, 20168–20174 CAS.
- B. Seger and P. Kamat, J. Phys. Chem. C, 2009, 113, 7990–7995 CAS.
- N. Cuong, A. Sugiyama, A. Fujiwara, T. Mitani and D. Chi, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 235417 CrossRef.
- Q. Tang, Z. Zhou and Z. Chen, Nanoscale, 2013, 5, 4541–4583 RSC.
- A. Kasry, M. A. Kuroda, G. J. Martyna, G. S. Tulevski and A. A. Bol, ACS Nano, 2010, 4, 3839–3844 CrossRef CAS PubMed.
- M. N. Groves, C. Malardier-Jugroot and M. Jugroot, J. Phys. Chem. C, 2012, 116, 10548–10556 CAS.
- T. W. Chen, J. Y. Xu, Z. H. Sheng, K. Wang, F. B. Wang, T. M. Liang and X. H. Xia, Electrochem. Commun., 2012, 16, 30–33 CrossRef CAS.
- A. Ambrosi, S. Y. Chee, B. Khezri, R. D. Webster, Z. Sofer and M. Pumera, Angew. Chem., Int. Ed., 2012, 51, 500–503 CrossRef CAS PubMed.
- J. A. Rodriguez-Manzo and F. Banhart, Nano Lett., 2009, 9, 2285–2289 CrossRef CAS PubMed.
- Y. J. Gan, L. T. Sun and F. Banhart, Small, 2008, 4, 587–591 CrossRef CAS PubMed.
- H. Wang, Q. Wang, Y. Cheng, K. Li, Y. Yao, Q. Zhang, C. Dong, P. Wang, U. Schwingenschlögl, W. Yang and X. X. Zhang, Nano Lett., 2012, 12, 141–144 CrossRef CAS PubMed.
- M. N. Groves, C. Malardier-Jugroot and M. Jugroot, J. Phys. Chem. C, 2012, 116, 10548–10556 CAS.
- F. Li, J. Zhao and Z. Chen, J. Phys. Chem. C, 2012, 116, 2507–2514 CAS.
- A. Krasheninnikov, P. Lehtinen, A. Foster, P. Pyykkö and R. Nieminen, Phys. Rev. Lett., 2009, 102, 126807 CrossRef CAS PubMed.
- O. Cretu, A. V. Krasheninnikov, J. A. Rodríguez-Manzo, L. Sun, R. M. Nieminen and F. Banhart, Phys. Rev. Lett., 2010, 105, 196102 CrossRef PubMed.
- J. Rodriguez-Manzo, O. Cretu and F. Banhart, ACS Nano, 2010, 4, 3422–3428 CrossRef CAS PubMed.
- M. N. Groves, A. S. W. Chan, C. Malardier-Jugroot and M. Jugroot, Chem. Phys. Lett., 2009, 481, 214–219 CrossRef CAS.
- H. M. Jeong, J. W. Lee, W. H. Shin, Y. J. Choi, H. J. Shin, J. K. Kang and J. W. Choi, Nano Lett., 2011, 11, 2472–2477 CrossRef CAS PubMed.
- X. Zhang, Z. Lu, G. Xu, T. Wang, D. Ma, Z. Yang and L. Yang, Phys. Chem. Chem. Phys., 2015, 17, 20006–20013 RSC.
- Y. Tang, Z. Yang, X. Dai, D. Ma and Z. Fu, J. Phys. Chem. C, 2013, 117, 5258–5268 CAS.
- Y. Lu, M. Zhou, C. Zhang and Y. Feng, J. Phys. Chem. C, 2009, 113, 20156–20160 CAS.
- Y. Li, Z. Zhou, G. Yu, W. Chen and Z. Chen, J. Phys. Chem. C, 2010, 114, 6250–6254 CAS.
- Y. Tang, J. Zhou, Z. Shen, W. Chen, C. Li and X. Dai, RSC Adv., 2016, 6, 93985–93996 RSC.
- E. H. Song, Z. Wen and Q. Jiang, J. Phys. Chem. C, 2011, 115, 3678–3683 CAS.
- Y. Tang, Z. Yang and X. Dai, Phys. Chem. Chem. Phys., 2012, 14, 16566–16572 RSC.
- T.-T. Jia, C.-H. Lu, Y.-F. Zhang and W.-K. Chen, J. Nanopart. Res., 2014, 16, 1–11 Search PubMed.
- Q. G. Jiang, Z. M. Ao, S. Li and Z. Wen, RSC Adv., 2014, 4, 20290–20296 RSC.
- W. Orellana, J. Phys. Chem. C, 2013, 117, 9812–9818 CAS.
- F. Calle-Vallejo, J. I. Martínez and J. Rossmeisl, Phys. Chem. Chem. Phys., 2011, 13, 15639–15643 RSC.
- S. Kattel, P. Atanassov and B. Kiefer, Phys. Chem. Chem. Phys., 2012, 15, 148–153 RSC.
- S. Kattel, P. Atanassov and B. Kiefer, J. Phys. Chem. C, 2012, 116, 8161–8166 CAS.
- C. W. B. Bezerra, Z. Lei, K. Lee, H. Liu, A. L. B. Marques, E. P. Marques, H. Wang and J. Zhang, Electrochim. Acta, 2008, 53, 4937–4951 CrossRef CAS.
- Z. Lu, G. Xu, C. He, T. Wang, L. Yang, Z. Yang and D. Ma, Carbon, 2015, 84, 500–508 CrossRef CAS.
- D. Deng, X. Chen, Y. Liang, X. Wu, Q. Liu, Y. Liu, H. Yang, H. Tian, Y. Hu and P. Du, Sci. Adv., 2015, 1, 1–9 Search PubMed.
- K. Liu, S. Kattel, V. Mao and G. Wang, J. Phys. Chem. C, 2016, 120, 1586–1596 CAS.
- X. Zhang, Z. Lu and Z. Yang, J. Mol. Catal. A: Chem., 2016, 417, 28–35 CrossRef CAS.
- Y. Tang, D. Ma, W. Chen and X. Dai, Sens. Actuators, B, 2015, 211, 227–234 CrossRef CAS.
- Y. Tang, X. Dai, Z. Yang, Z. Liu, L. Pan, D. Ma and Z. Lu, Carbon, 2014, 71, 139–149 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- J. Carlsson and M. Scheffler, Phys. Rev. Lett., 2006, 96, 46806 CrossRef PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- G. Henkelman, B. Uberuaga and H. Jónsson, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- G. Henkelman and H. Jónsson, J. Chem. Phys., 2000, 113, 9978–9985 CrossRef CAS.
- T. Zhu, J. Li and S. Yip, Phys. Rev. Lett., 2004, 93, 25503 CrossRef PubMed.
- W. I. Choi, S. H. Jhi, K. Kim and Y. H. Kim, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 085441 CrossRef.
- Y. Feng, F. Li, Z. Hu, X. Luo, L. Zhang, X. F. Zhou, H. T. Wang, J. J. Xu and E. G. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 4506 Search PubMed.
- L. M. Molina and B. Hammer, J. Catal., 2005, 233, 399–404 CrossRef CAS.
- W. An, Y. Pei and X. C. Zeng, Nano Lett., 2008, 8, 195–202 CrossRef CAS PubMed.
- M. Zhou, A. Zhang, Z. Dai, C. Zhang and Y. Feng, J. Chem. Phys., 2010, 132, 194704 CrossRef PubMed.
- Y. Tang, L. Pan, W. Chen, C. Li, Z. Shen and X. Dai, Appl. Phys. A, 2015, 119, 475–485 CrossRef CAS.
- Y. Tang, X. Dai, Z. Yang, L. Pan, W. Chen, D. Ma and Z. Lu, Phys. Chem. Chem. Phys., 2014, 16, 7887–7895 RSC.
- N. Lopez and J. K. Nørskov, J. Am. Chem. Soc., 2002, 124, 11262–11263 CrossRef CAS PubMed.
- Z. Lu, P. Lv, J. Xue, H. Wang, Y. Wang, Y. Huang, C. He, D. Ma and Z. Yang, RSC Adv., 2015, 5, 84381–84388 RSC.
- Z. Lu, P. Lv, Y. Liang, D. Ma, Y. Zhang, W. Zhang, X. Yang and Z. Yang, Phys. Chem. Chem. Phys., 2016, 18, 21865–21870 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28387j |
| This journal is © The Royal Society of Chemistry 2017 |