DOI:
10.1039/C6RA28297K
(Paper)
RSC Adv., 2017,
7, 12486-12495
Synthesis and catalytic activity of Cu–Cr–O–TiO2 composites for the thermal decomposition of ammonium per-chlorate: enhanced decomposition rate of fuel for solid rocket motors
Received
16th December 2016
, Accepted 2nd February 2017
First published on 21st February 2017
Abstract
This study presents the sol–gel synthesis of Cu–Cr–O·nTiO2 particles calcined at different temperatures and their catalytic effects on thermal decompostion of AP. The study focuses on the impact of crystallite size, shape and concentration of TiO2 in the catalyst composition on the thermal decomposition behaviour of ammonium per-chlorate (AP). During synthesis, the molar ratio of Cu/Cr was kept to 0.7 and TiO2 nanoparticles were added into Cu–Cr–O–citric acid solution at different molar ratios to form three different compositions of the catalyst Cu–Cr–O·nTiO2 (n = 0.5, 0.7 and 0.9 mol%). The effect of temperature on the thermal, structural and spectroscopic properties of the different Cu–Cr–O compositions was also studied by calcining them at two different temperatures, 300 and 1050 °C. Post synthesis characterizations of the prepared catalysts were carried out by using XRD, FT-IR, SEM, EDAX and TEM (with SAED pattern) techniques. The desired qualification of Cu–Cr–O–citric acid (the precursor of the catalyst) and the final compositions of the catalysts were carried out by using thermogravimetric and differential thermal analysis (TG-DTA) techniques. The efficiency of the synthesized catalysts was evaluated on thermal decomposition behaviour of AP using TG-DTA techniques. The Cu–Cr–O·nTiO2 composition with the molar ratio of n = 0.7 was found to be the most efficient catalyst for decomposition of AP; it was much better than other laboratory prepared samples (n = 0.5 and 0.9) as well as the industrial catalyst (i.e. activated copper chromite (ACR); Cu–Cr–O). Further experimental work showed that addition of 10 wt% Cu–Cr–O·0.7TiO2 into AP significantly lowered the AP decomposition temperature to 306 °C from 385 °C and was accompanied by a very sharp exothermic peak indicating a single stage decomposition. The excellent finding of the study was also verified by heat of reaction (i.e. calibrated delta H) values. This study finds potential application due to the remarkable enhancement in the thermal decomposition rate of the AP used as oxidizer in propellant of solid rocket motors (SRMs) and space vehicles (SVs) at lower decomposition temperature. The fast decomposition rate of oxidizer at lower decomposition temperature enhances the efficiency of fuel which ultimately will enhance the efficiency of SRMs and SVs.
1. Introduction
Solid composite propellants are the vital fuel system used in modern rocketry. Solid rocket motors (SRMs) and space vehicles (SVs) derive energy through solid composite propellants.1 The key ingredients of these solid propellants are hydroxyl terminated polybutadiene (HTPB), aluminium (aluminum) powder (Al) and ammonium per-chlorate (AP). The role of HTPB is to provide the structural integrity to the propellant, Al acts as the metallic fuel and AP is the most popular oxidizer used worldwide in the propellant. The burn rate of a conventional propellant is governed by the combustion behavior of the solid composite propellant and is a function of the thermal decomposition of AP.2–7 Transition metals and metal oxides are used as catalysts to enhance the rate of thermal decomposition of AP, eventually modifying the burn rate. The oxides of iron, manganese, copper, nickel and copper chromites are widely used as the burn rate modifiers in the solid composite propellant.8 Activated copper chromite (ACR), with chemical formula CuCr2O4 (Cu–Cr–O), is the industrial catalyst used extensively in solid composite propellants. Solid composite propellants with high burn rate and thereby reduced ignition delay and operational time are needed for modern space vehicles.
Liu et al. (2004) carried out a comprehensive study to investigate the effect of Ni, Cu, Al, and NiCu on the thermal decomposition of AP. The study reported the shift of the high temperature decomposition (HTD) peak of AP by 130.2, 112.9, 51.8, 35.1 and 140.4 °C, respectively.9 Song et al. (2010) studied the thermal decomposition of AP in the presence of synthesized α-Fe2O3 with different particle sizes and morphologies. In this study, the absence of a low temperature decomposition (LTD) peak and the shift of the HTD peak to 344 °C from 454 °C was observed.10 Patil et al. (2008) synthesized nano sized particles of nano sized CuO and CuCr2O4 and studied their individual effects on thermal decomposition of AP. They reported the shift of the HTD peak to 360 °C for nano CuO and 348 °C for nano CuCr2O4 from 467 °C with high heat release.11 Xu et al. (2008) and Zhang et al. (2011) synthesized sphere and rod like α-Fe2O3 particles and observed that α-Fe2O3 shows better catalytic activity.12,13 These individal groups have lowered the HTD of thermal decomposition of AP with remarkable heat release. Chandru et al. (2012) synthesized β-MnO2 and found that mesoporous β-MnO2 is an excellent catalyst, lowering the HTD from 426.3 to 301 °C.14 The effect of the shape and size of CuO and CoFe2O4 on the thermal decomposition of AP was also studied and a significant impact was observed on the decomposition pattern of AP.15–21 In previous research, copper chromite (CuO·CuCr2O4),22–24 ACR and titanium oxide (TiO2)25,26 have been the most utilized metal oxide catalysts which have considerably decreased the onset temperature of AP decomposition when used individually as additives.
The lack of available information on the individual use of copper chromites and TiO2 leads to a tremendous gap requiring further study. In spite of the extensive individual use of copper chromites and TiO2 in AP, the physics and chemistry of their catalytic roles in the AP decomposition are not fully known. The aim of the present investigation is to achieve the highest burn rate of fuel at the minimum possible temperature. In the present work we investigate the simultaneous effects of copper chromites and TiO2 catalysts on the thermal decomposition of AP. Several methods such as co-precipitation, solid state synthesis, the ceramic method, the solution combustion method, the hydrothermal method and sol–gel methods have previously been reported for preparation of the catalysts. Due to the better control of homogeneity and the ease of the procedure, we have adopted the sol–gel method, as reported in the literature.27 Thus, after the successful preparation of different concentrations of TiO2-doped copper chromite compositions, i.e. Cu–Cr–O·nTiO2 (n = 0.5, 0.7 and 0.9), calcined at two different temperatures, 300 and 1050 °C, we introduced them into AP to enhance its burn rate. The results are compared with the effect of the industrial catalyst (ACR) on the thermal decomposition of AP. The pronounced effect of TiO2-doped copper chromite modification of AP on the characteristics of thermal decomposition of AP enhanced the burn rate considerably.
2. Experimental
2.1 Materials and methods
All the starting reagents were of analytical grade and high purity (>99.9%). AP (NH4ClO4) with an average particle size of 300 μm was purchased from Pandian Chemicals Cuddalore, India. The average particle size was reduced to 45 μm using an air classifying mill. Copper nitrate tri-hydrate (CuNO3·3H2O) was purchased from Titan Biotech Ltd., India. Chromium nitrate nonahydrate (Cr(NO3)3·9H2O) of laboratory grade was purchased from Central Drug House, India. Anhydrous citric acid (C6H8O7) was purchased from Fisher Scientific, India. Ethanol (C2H5OH) was purchased from Merck, Germany, and TiO2 (P-25) of nano-metric size was purchased from Evonik Industries, Germany. The ready-made industrial catalyst (i.e. activated copper chromite (ACR); CuCr2O4) was purchased from BLI Kerala, India.
2.2 Synthesis of nano-composites by the sol–gel method
In order to prepare Cu–Cr–Ti mixed oxides via the sol–gel technique, the mother solution of Cu–Cr–citric acid was first prepared. For the preparation of mother solution, 0.021 moles of CuNO3·3H2O and 0.03 moles of Cr(NO3)3·9H2O were added into 20 ml of ethanol. The mixture was stirred until it formed a homogenous solution. Citric acid (0.042 moles) was added into the obtained homogeneous mixture with further stirring. The mother solution thus obtained was divided into three equal parts to prepare three different solutions by adding 0.86, 1.172, and 1.516 g of TiO2 nanoparticles respectively. A further 5 ml of ethanol was added into each solution and stirring was carried out for 4 h to give transparent suspension-cum-solutions. The solutions thus obtained were kept for aging at room temperature (30 °C) for 48 h. After this initial aging, each suspension was again mixed thoroughly to ensure the homogeneity of the suspended particles and then heated with continuous stirring at 80 °C until a paste-like material was obtained. The pastes thus formed were dried for 3 h in an oven maintained at a temperature of 130 °C and the obtained materials were individually crushed to fine particles. Each of the three powders thus obtained was divided into two equal parts for calcination at two different temperatures. The first part of each sample was calcined individually at 300 °C (below the crystallization temperature of Cu–Cr–O) in a N2 atmosphere for 3 h; the second part was calcined individually at 1050 °C (above the crystallization temperature of Cu–Cr–O) in the same atmosphere for 3 h. The crystallization temperature of Cu–Cr–O is 700 °C.
2.3 Characterization of the catalysts
The phase transformation of the catalyst precursor was confirmed by thermogravimetric analysis (TGA) using a PerkinElmer Thermal Analyzer (Model no. SPA-6000). Crystal structures of the powders were investigated by X-ray diffraction (XRD) using an X-ray diffractometer (Rigaku Miniflex-II, Desktop X-ray D) with Cu-Kα radiation (λ = 1.5418 Å) and a Ni filter operated at 30 kV and 15 mA. The scanning speed and scanning range were kept at 3° min−1 and 20–80° respectively. The surface morphologies and particle sizes were monitored by scanning electron microscopy (SEM) (FEI Quanta 200F). Elemental analysis of the synthesized powders was carried out with the energy dispersive X-ray analysis method (EDAX) inbuilt in the SEM. Chemical bonding and molecular interaction analysis was investigated using Fourier transform infrared (FT-IR) spectra recorded in the wavenumber range from 4000 to 400 cm−1 using a PerkinElmer 577 FT-IR. The catalytic activities of the catalysts were confirmed by thermogravimetric analysis (TGA) and differential thermogravimetric analysis (DTA) in a N2 atmosphere using LABSYSTM Setaram Instrumentation (serial no. 1-3347-1 DTA/TGA/DSC) from room temperature (30 ± 2 °C) to 600 °C with a heating rate of 10 °C min−1. Transmission electron microscopy (TEM) (TECNAI 20 G2) was employed for viewing the interior morphologies/structures of the samples.
3. Results and discussion
3.1 Thermogravimetric analysis
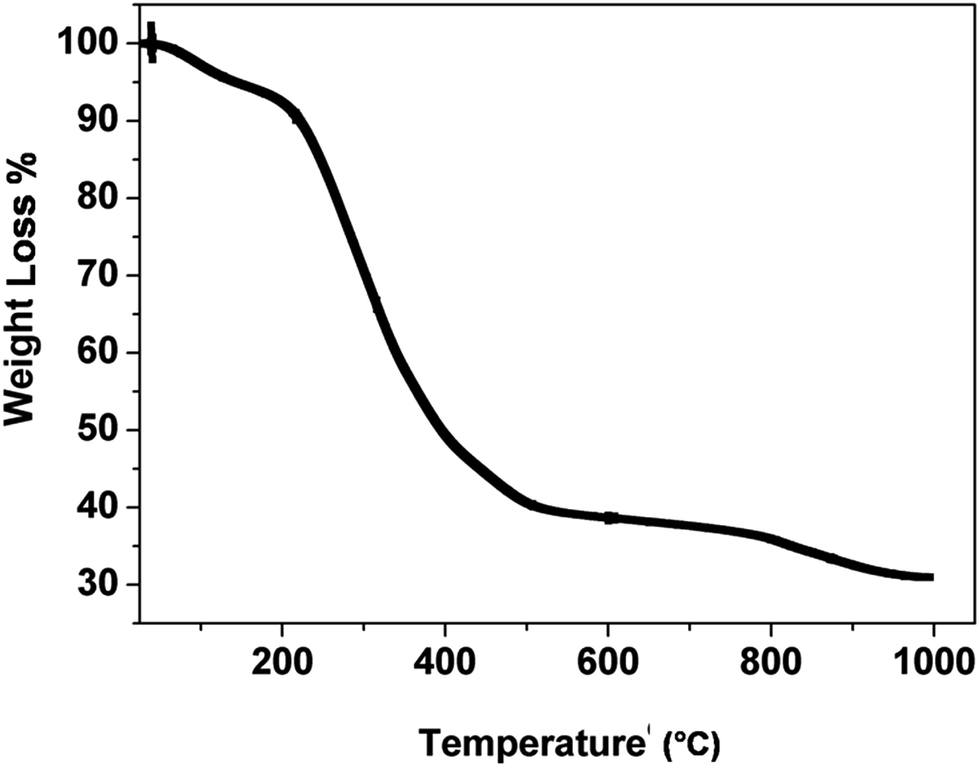
Fig. 1 shows the TGA curve of the catalyst precursor (Cu–Cr–O–citric acid). The curve consists of three regions of weight loss within the temperature range of 30–1000 °C.
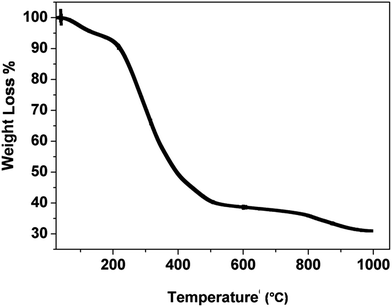 |
| | Fig. 1 TGA curve for the as synthesized precursor of the catalyst, Cu–Cr–O–citric acid. | |
The first region (30–180 °C) shows a weight loss of 6.6% due to removal of surface moisture. The second region (180–580 °C) shows the major weight loss of 54.2% attributed to removal of volatile organics from the decomposition of contained groups. The partial decomposition of nitrate bonds and the formation of intermediate complexes of copper, chromium, TiO2 and nitrates accounts for the weight loss of 7% in the third segment (580–980 °C). Above the temperature of 980 °C, the curve looks almost parallel to the temperature axis which shows that no noticeable weight loss occurs.
3.2 X-ray diffraction analysis
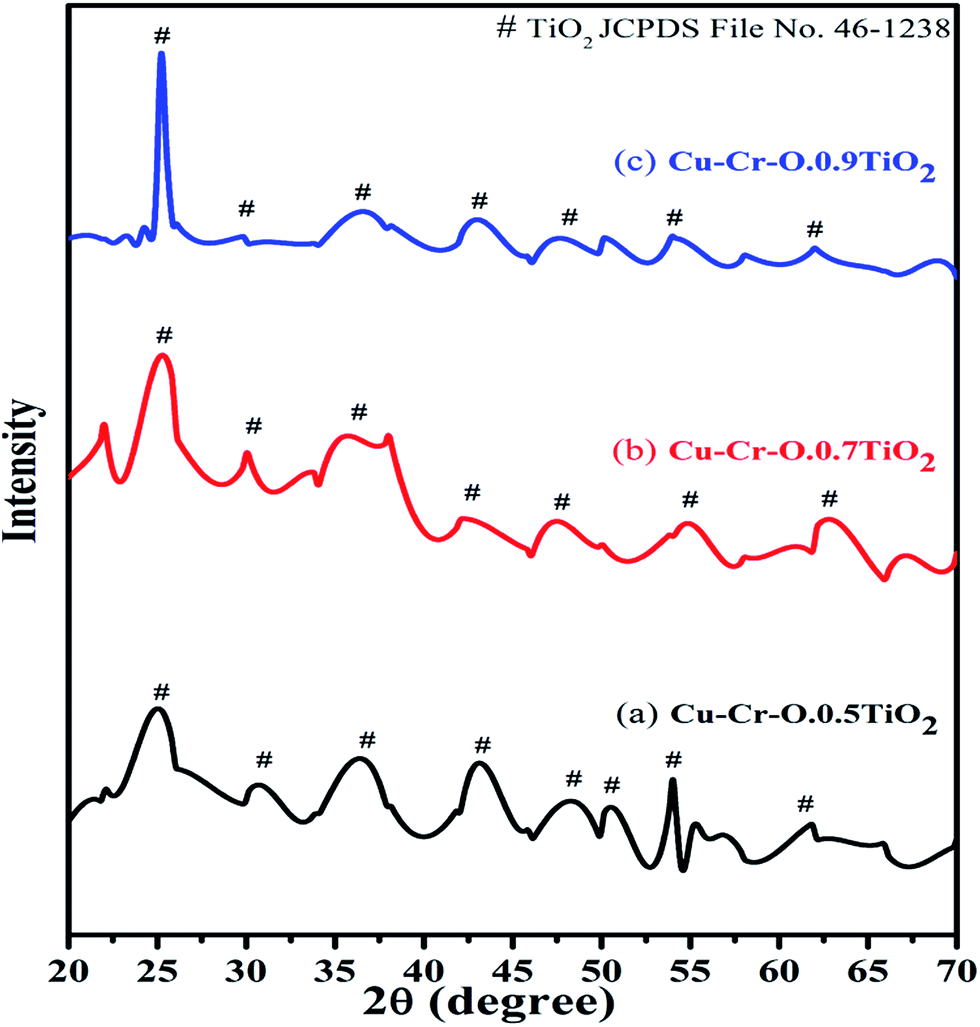
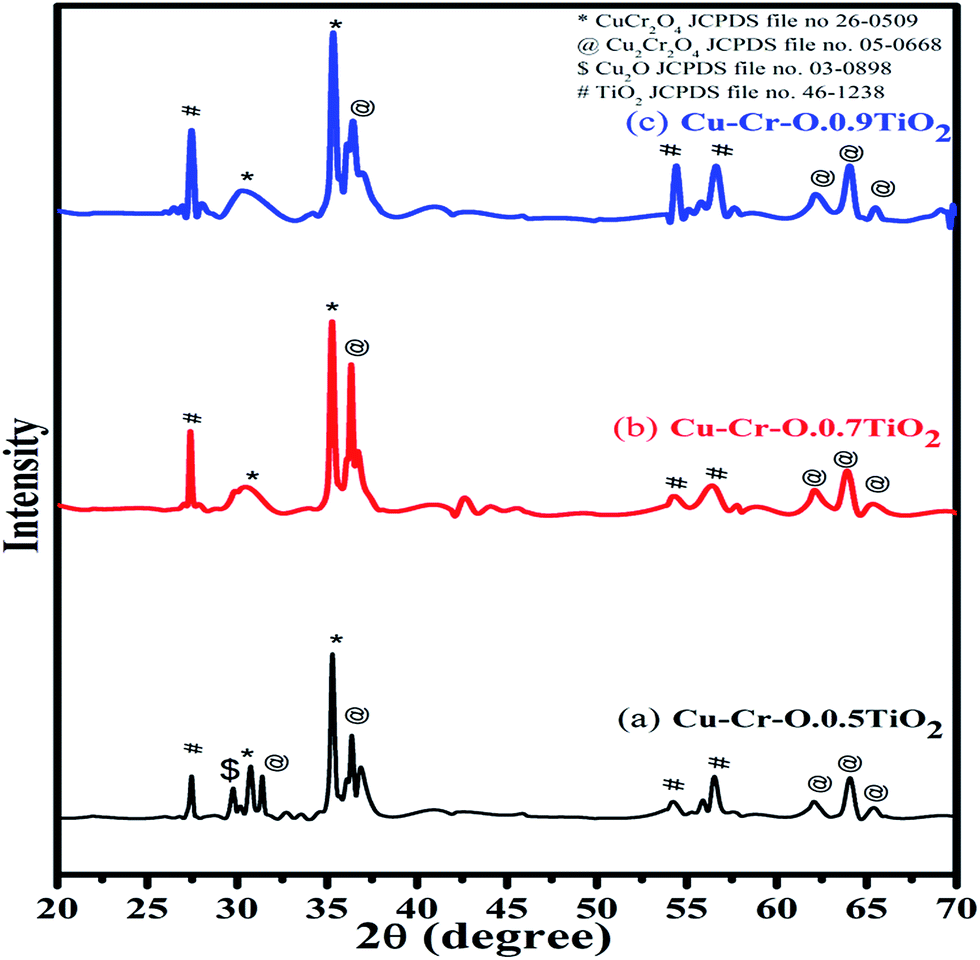
Fig. 2 shows the typical XRD patterns of the (a) Cu–Cr–O·0.5TiO2, (b) Cu–Cr–O·0.7TiO2 and (c) Cu–Cr–O·0.9TiO2 catalysts calcined at 300 °C for 3 h. XRD patterns show diffraction peaks corresponding only to TiO2 (JCPDS file no. 46–1238). This may happen due to the crystallization of TiO2 in a mixed state at around 300 °C, at which temperature the rest of the compounds such as CuCr2O4, Cu2Cr2O4, Cu2O are not found to be crystallized and hence show no diffraction peaks. As the calcination temperature is increased up to 1050 °C, the transformation of the composite materials (CuCr2O4, Cu2Cr2O4, Cu2O) from amorphous to crystalline phases takes place due to the well-known crystallization temperature of Cu–Cr–O at 700 °C. Fig. 3(a–c) shows XRD profiles of the same compositions after calcination at 1050 °C for 3 h. The diffraction patterns show the peaks due to CuCr2O4, Cu2Cr2O4 and Cu2O along with TiO2. The prominent diffraction peaks of CuCr2O4 (JCPDS file no. 26-0509) appear at the diffraction angles (2θ) of 30.76° and 35.31°. The diffraction peaks of Cu2Cr2O4 appear at the diffraction angles of 31.4°, 36.38°, 62.06°, 64.08° and 65.40° (JCPDS file no. 05-0668).
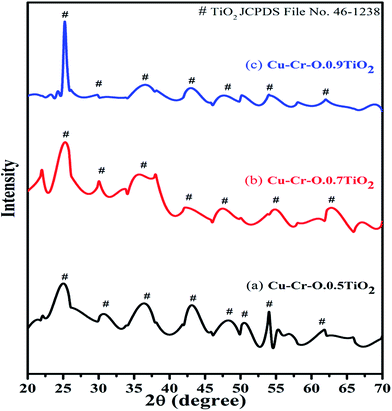 |
| | Fig. 2 XRD patterns of (a) Cu–Cr–O·0.5TiO2, (b) Cu–Cr–O·0.7TiO2 and (c) Cu–Cr–O·0.9TiO2 catalysts calcined at 300 °C. | |
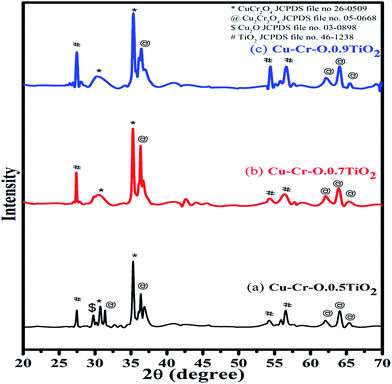 |
| | Fig. 3 XRD patterns of (a) Cu–Cr–O·0.5TiO2, (b) Cu–Cr–O·0.7TiO2 and (c) Cu–Cr–O·0.9TiO2 catalysts calcined at 1050 °C. | |
The presence of Cu2O is noticed due to its characteristic peak at 29.78° (JCPDS file no. 03-0898). Only three diffraction peaks of TiO2, at 27.48°, 54.24° and 56.54°, are observed for the compositions calcined at 1050 °C.
3.3 Fourier transform infrared analysis
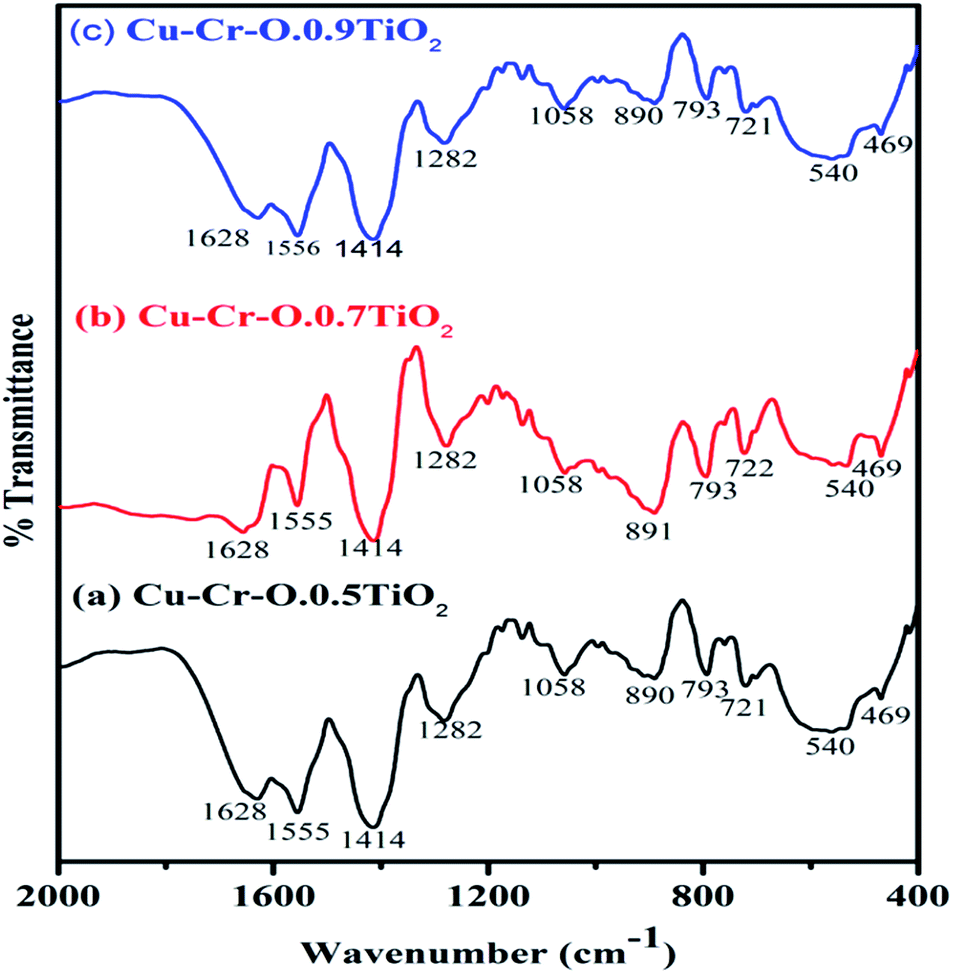
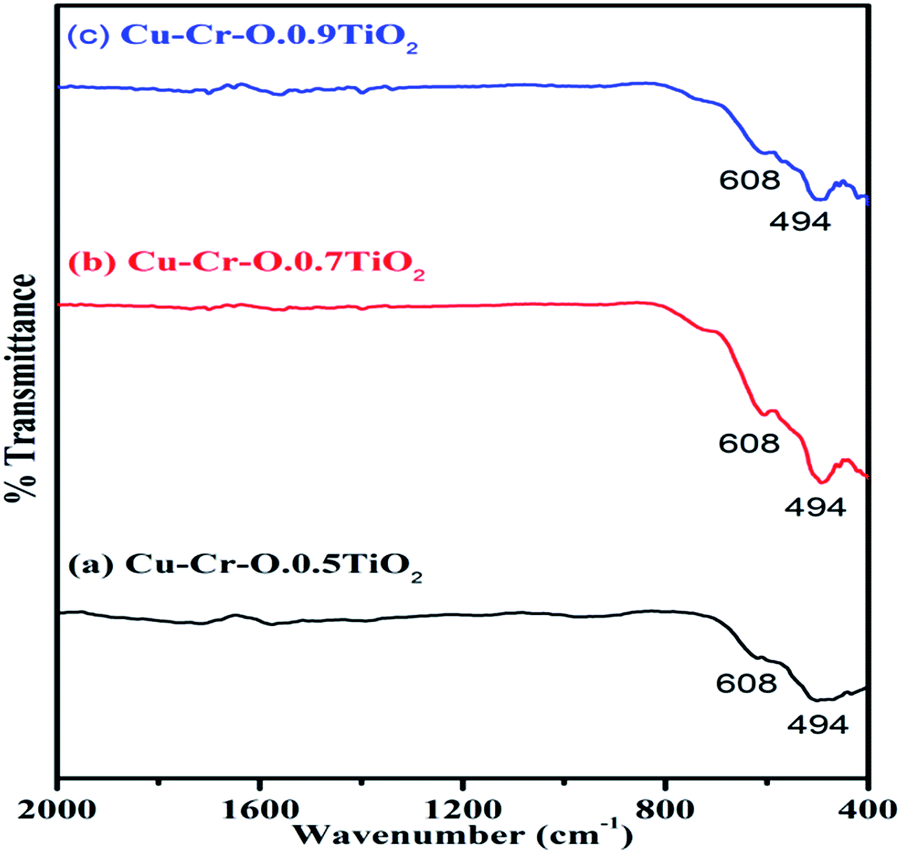
Fig. 4(a–c) shows the typical FT-IR spectra of Cu–Cr–O·0.5TiO2, Cu–Cr–O·0.7TiO2 and Cu–Cr–O·0.9TiO2 calcined at 300 °C for 3 h. Fig. 5(a–c) shows the FT-IR spectra of similar compositions calcined at 1050 °C for the same duration. The respective spectra of similar compositions calcined at 300 °C (see Fig. 4) are quite different from the spectra of the samples calcined at 1050 °C (see Fig. 5). It is interesting to note that most of the absorption bands (8 out of 10) disappear dramatically at the higher temperature. Such dramatic vanishing of absorption bands occurred due to the effect of calcination temperature on the spectroscopic properties of these compositions. The crystallization temperature of Cu–Cr–O is 700 °C, below which the compositions are in an amorphous state showing similar IR almost patterns for 300 °C. No spectroscopic changes are observed due to the varying concentrations of TiO2 on calcination either at 300 °C or at 1050 °C. One of the strong shoulders observed at wavenumber 1628 cm−1 is due to the ν2 bending mode of H–O–H in H2O.28,29 The position of this band may vary by ±10 cm−1 depending on the host molecule to which H2O is attached. The absorption band appearing at 1414 cm−1 may be attributed to symmetric vibrations of surface hydroxyl groups. A medium shoulder appearing at 1282 cm−1 is attributed to the coupled bending and stretching vibrations of COO− of citric acid. Notably, this band appears in IR patterns of the samples calcined at 300 °C but not in the samples calcined at 1050 °C.
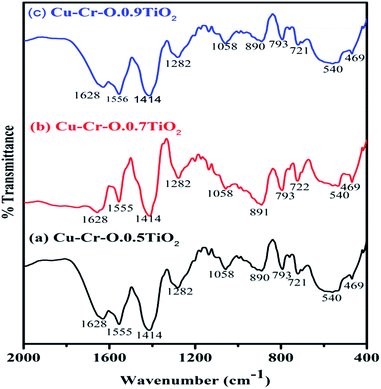 |
| | Fig. 4 FT-IR spectra of (a) Cu–Cr–O·0.5TiO2, (b) Cu–Cr–O·0.7TiO2 and (c) Cu–Cr–O·0.9TiO2 calcined at 300 °C. | |
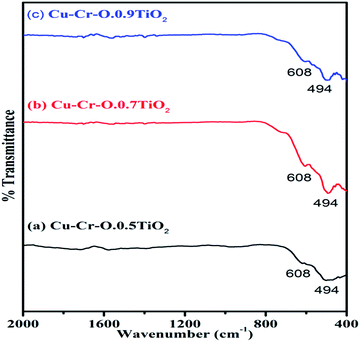 |
| | Fig. 5 FT-IR spectra of (a) Cu–Cr–O·0.5TiO2, (b) Cu–Cr–O·0.7TiO2 and (c) Cu–Cr–O·0.9TiO2 calcined 1050 °C. | |
The dramatic disappearance of this band in the samples fired at the higher temperature is due to the fact that citric acid has a melting point of 310 °C which is just above the 300 °C. The citric acid is not destroyed at 300 °C, however, at 1050 °C the citric acid is destroyed completely, resulting in the complete absence of the associated bands at this temperature. The peaks at 1058 cm−1 can be attributed to the C–H bending vibration of citric acid. However, a strong absorption band appearing at 1555 cm−1 is assigned to C–C or C–O stretching. These bands also appear at lower temperatures below crystallization Cu–Cr–O but not in the IR of the samples calcined at 1050 °C due to the destruction of citric acid at higher temperatures above crystallization. The absorption bands at 721, 793 and 890 cm−1 may be attributed to the different vibrational modes of TiO2. TiO2 exhibits strong absorption in the range 600–890 cm−1 when treated with some additive metal ions as in the present case. The broad band centered at 540 cm−1 in the IR spectra is attributed to the characteristic vibrational band (asymmetric) of Cu–O in CuO. This band also contains the contribution due to the stretching of Cu–O–Cr. These spectra show a strong shoulder at 469 cm−1 assigned to Ti–O asymmetric stretching vibrations. The amorphous and crystalline natures of the formed compounds are confirmed from the FT-IR spectra shown in Fig. 4(a–c) and Fig. 5(a–c) which are in line with the XRD results. IR spectroscopy confirms the formation of intermediate complex compounds in the amorphous phase as explained by the XRD results.
3.4 Scanning electron microscopic analysis
Fig. 6 shows the typical SEM images of the catalyst samples (a) Cu–Cr–O·0.5TiO2, (b) Cu–Cr–O·0.7TiO2 and (c) Cu–Cr–O·0.9TiO2 calcined at 300 °C for 3 h. As shown in the micrographs, the particles have grown in spherical and needle like structures with diameters of up to 180 nm and 50 nm respectively. However, Fig. 7(a–c) exhibits the respective microstructures of the same catalyst samples calcined at 1050 °C. Whilst the images shown in Fig. 6 exhibit the formation of nanoparticles of sizes 20–100 nm, the images in Fig. 7(a–c) show that cuboid-like particles of sizes below 1 μm have developed under heat treatment. The shape of the synthesized catalyst particles is more regular and the size is bigger compared with the samples calcined at 300 °C. This transformation of surface morphology with the increase in temperature (from 300 to 1050 °C) is very interesting. The particle size of the same samples has been changed from nano to micro order due to the diffusion of the particles to each other at the higher temperature (1050 °C) minimizing the surface energy. Moreover, the formation of some new phases occurred at 1050 °C. The bigger size of the particles leads to a reduction in specific surface area.
 |
| | Fig. 6 SEM images of (a) Cu–Cr–O·0.5TiO2, (b) Cu–Cr–O·0.7TiO2 and (c) Cu–Cr–O·0.9TiO2 calcined at 300 °C. | |
 |
| | Fig. 7 SEM images of (a) Cu–Cr–O·0.5TiO2, (b) Cu–Cr–O·0.7TiO2 and (c) Cu–Cr–O·0.9TiO2 calcined at 1050 °C. | |
3.5 Transmission electron microscopic analysis
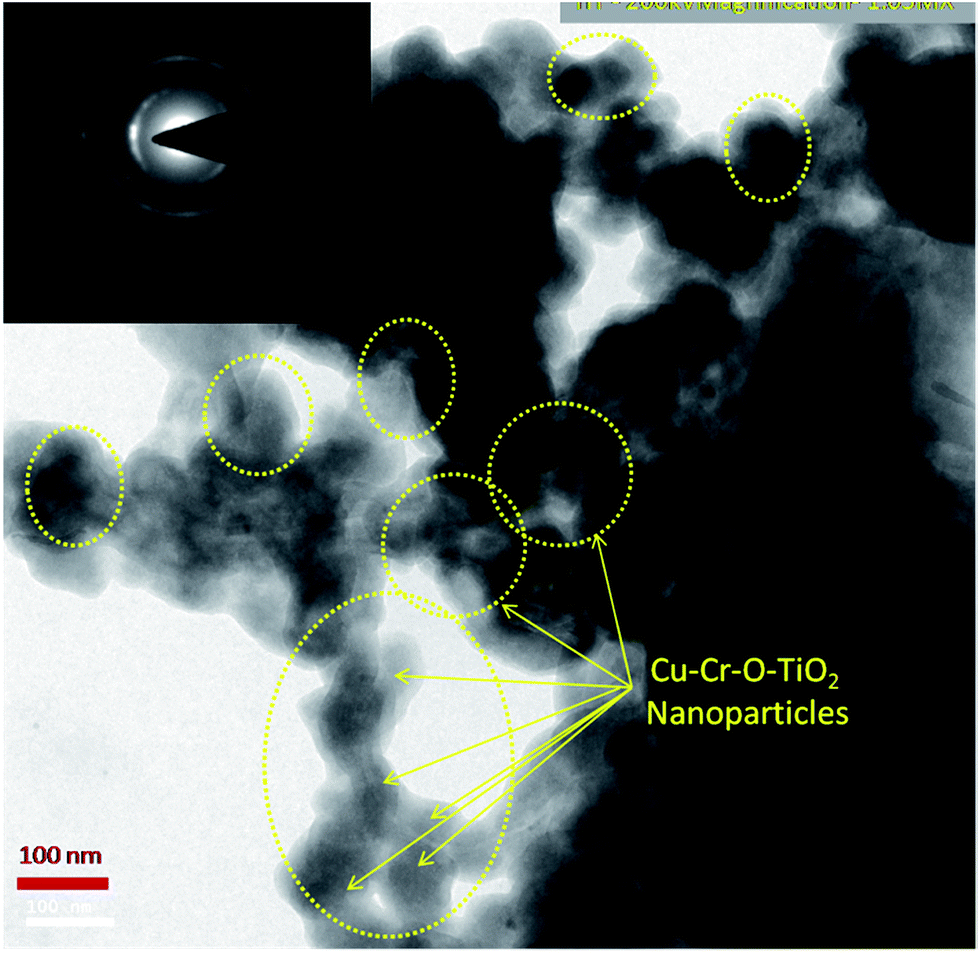
Fig. 8 shows the TEM image of Cu–Cr–O·0.7TiO2 calcined at 300 °C. TEM results are consistent with the previously observed crystallographic, spectroscopic and microstructural characterization results of all the compositions calcined individually at 300 °C and 1050 °C. The catalyst sample Cu–Cr–O–TiO2 calcined at 300 °C is structurally superb for further application in the desired experiment. The Cu–Cr–O–TiO2 particles sized around 100 nm can be seen easily in the yellow dotted rings as shown by yellow arrows. The TEM image confirms that the diffusion of particles does not occur at 300 °C, however it was happening at the higher temperature of 1050 °C as shown in Fig. 7.
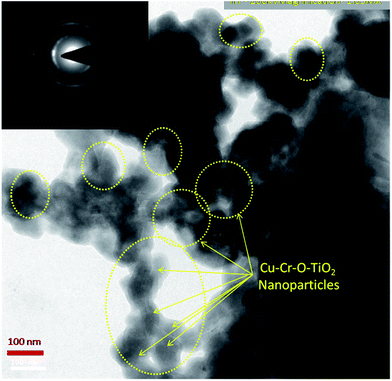 |
| | Fig. 8 TEM image of Cu–Cr–O·0.7TiO2 calcined at 300 °C; the selected area electron diffraction (SAED) pattern is shown in the inset image. | |
3.6 Electron diffraction X-ray analysis
Elemental compositions of catalysts calcined at temperatures of 300 and 1050 °C for 3 h were carried out using EDAX and are shown in Table 1. This analysis confirmed the presence of copper, chromium, titanium and oxygen. The increasing weight% of oxygen at the increased temperature may occur due to the transformation of intermediate compounds of Cu–Cr–O–citric acid into metal oxides. Interestingly, CuO has already shown its presence in XRD patterns of the samples calcined at 1050 °C but not in the samples calcined at 300 °C.
Table 1 Elemental composition (weight%) of different Cu–Cr–O–TiO2 composites calcined at 300 and 1050 °C
| Elements |
Elemental weight% of different Cu–Cr–O–TiO2 compositions at temperature |
| 300 °C |
1050 °C |
| Cu–Cr–O·0.5TiO2 |
Cu–Cr–O·0.7TiO2 |
Cu–Cr–O·0.9TiO2 |
Cu–Cr–O·0.5TiO2 |
Cu–Cr–O·0.7TiO2 |
Cu–Cr–O·0.9TiO2 |
| O |
15.02 |
39.72 |
14.93 |
34.39 |
41.81 |
33.2 |
| Ti |
08.09 |
13.28 |
19.37 |
08.51 |
14.03 |
15.28 |
| Cr |
17.49 |
30.67 |
44.91 |
34.25 |
27.37 |
30.64 |
| Cu |
59.40 |
16.33 |
20.79 |
22.85 |
16.08 |
20.88 |
3.7 Catalytic efficiency of the catalysts for the thermal decomposition of AP
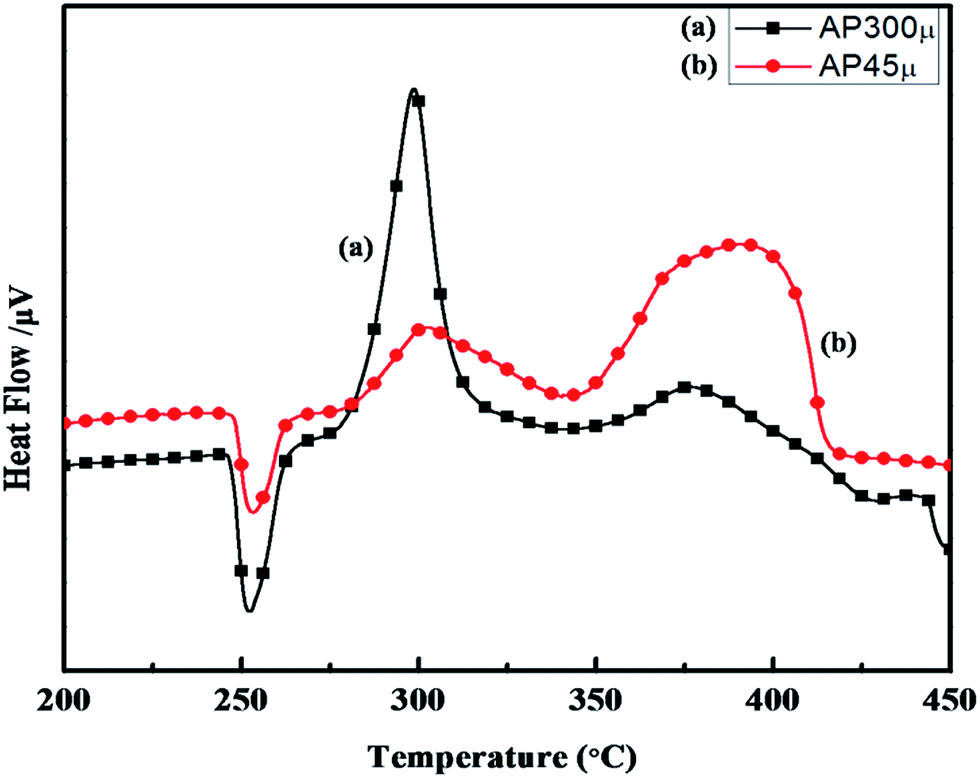
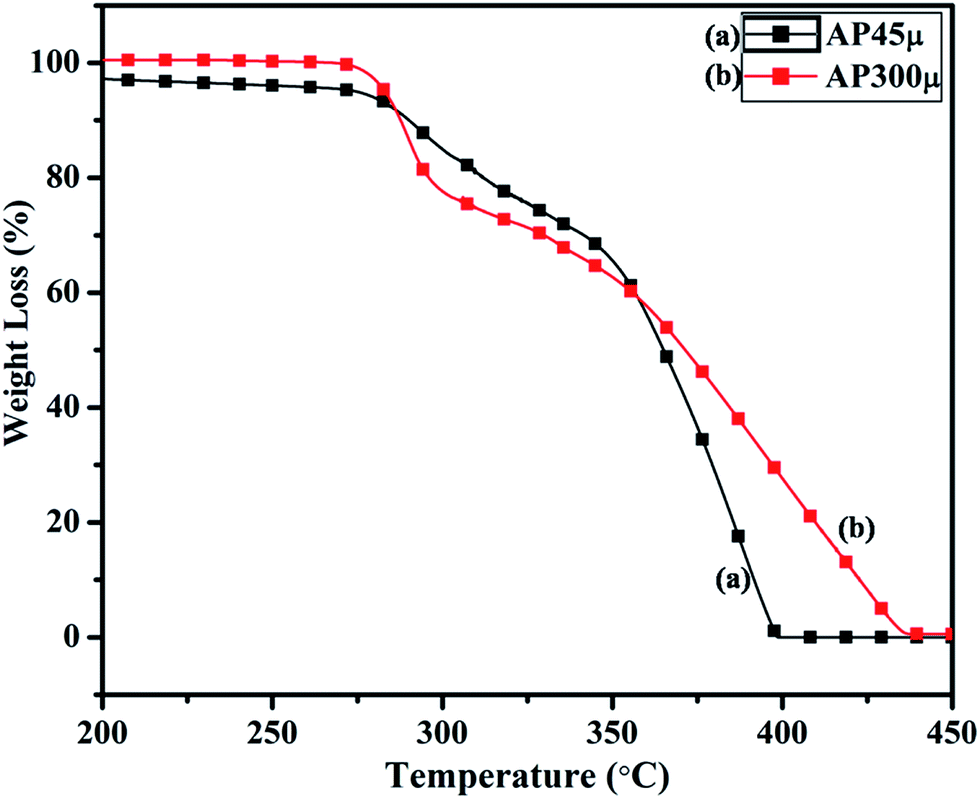
The synthesized nano-composites of Cu–Cr–O·nTiO2 with varying molar ratios of TiO2 were explored as additives to influence the thermal decomposition of AP. Fig. 9 shows the DTA curves of (a) AP300μ and (b) AP45μ, with particle sizes of 300 and 45 μm, respectively. Examination of the curves clearly shows that the decomposition pattern of AP strongly depends on particle size, resulting in step-wise energy release. Fig. 10 exhibits the corresponding % weight loss of the same samples. In Fig. 9, the endothermic peak for both the samples appears at 250 °C which may be due to the crystallographic transformation of AP from orthorhombic to cubic structure. Two exothermic peaks coinciding in both the samples are also observed at temperatures of 302 °C and 385 °C. The decomposition peak at 302 °C is attributed to the formation of intermediates such as NH4+ and HClO4− and is reported as LTD.2 The peak at 385 °C signifies the complete decomposition of volatile product into H2O, HCl, N2, H2 and O2 and is termed HTD.
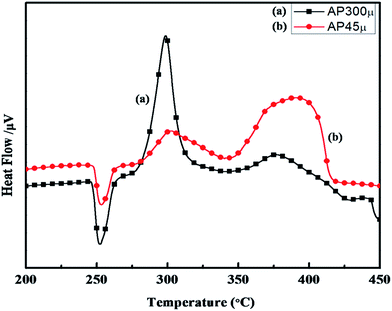 |
| | Fig. 9 DTA curves of (a) AP300μ and (b) AP45μ. | |
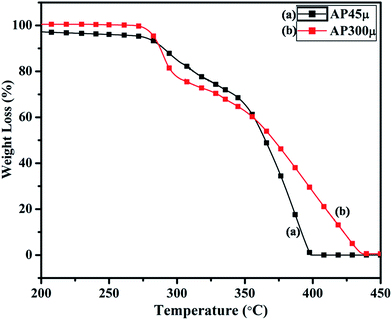 |
| | Fig. 10 TGA curves of (a) AP45μ and (b) AP300μ. | |
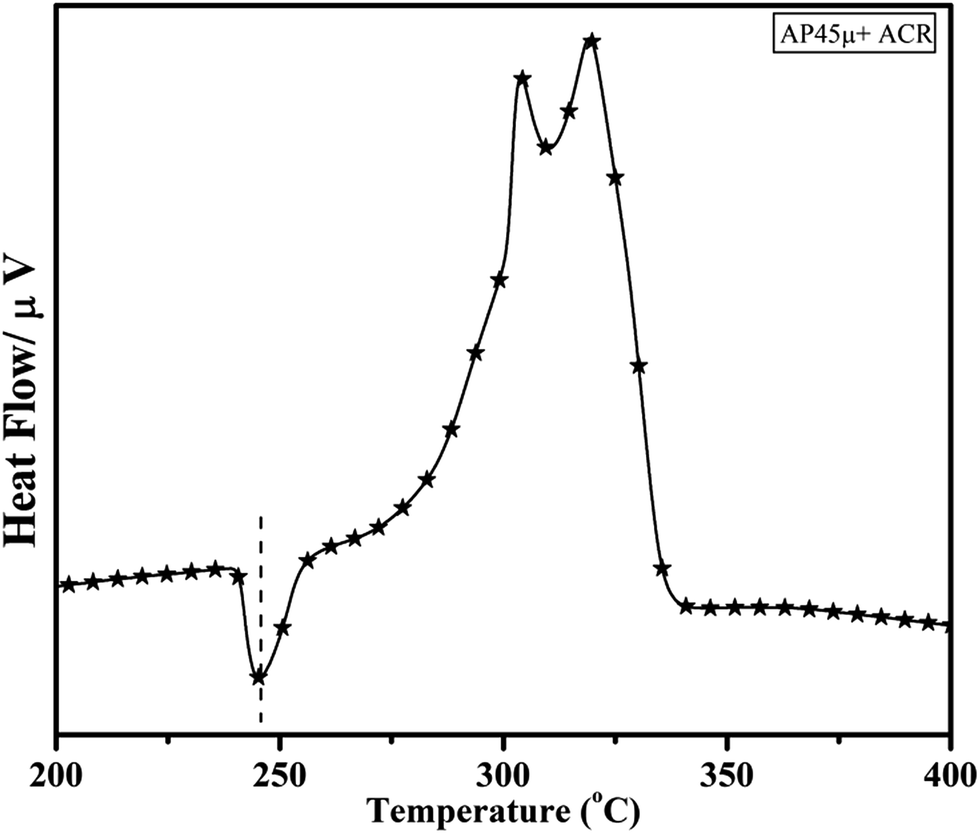
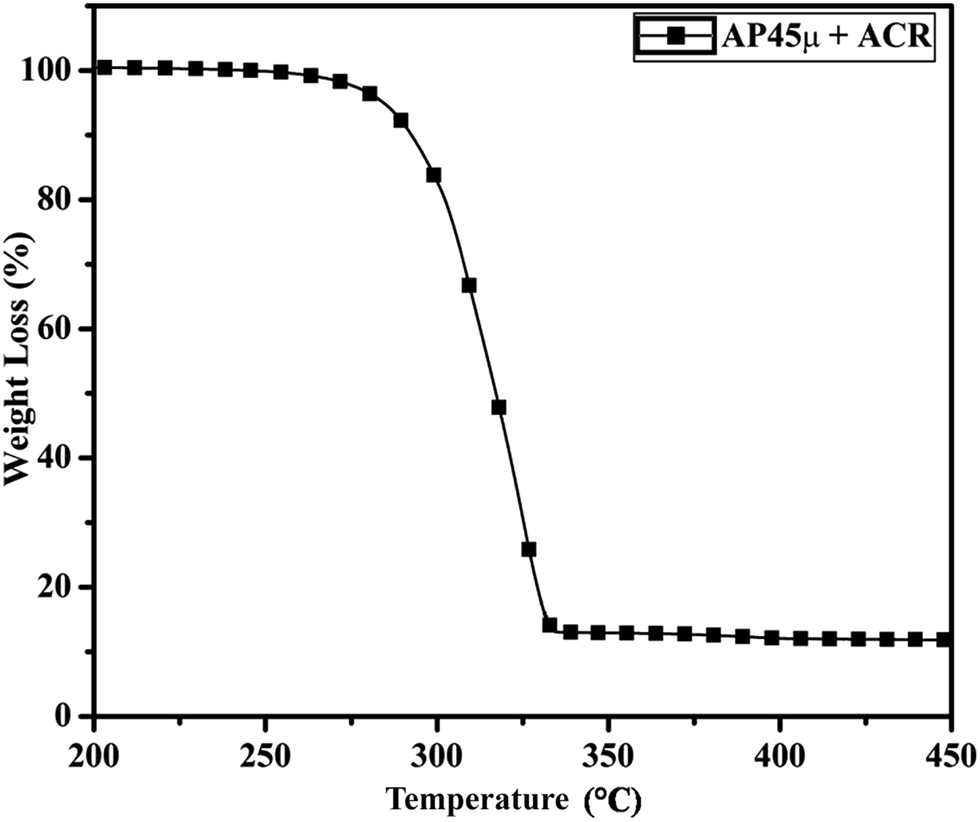
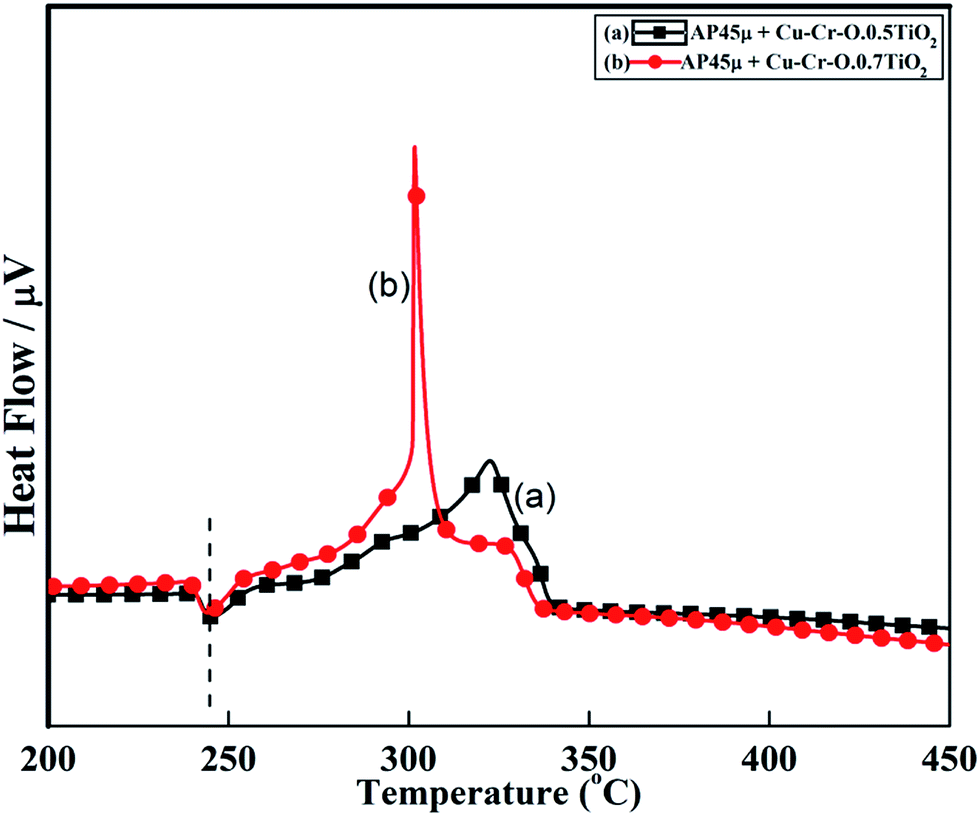
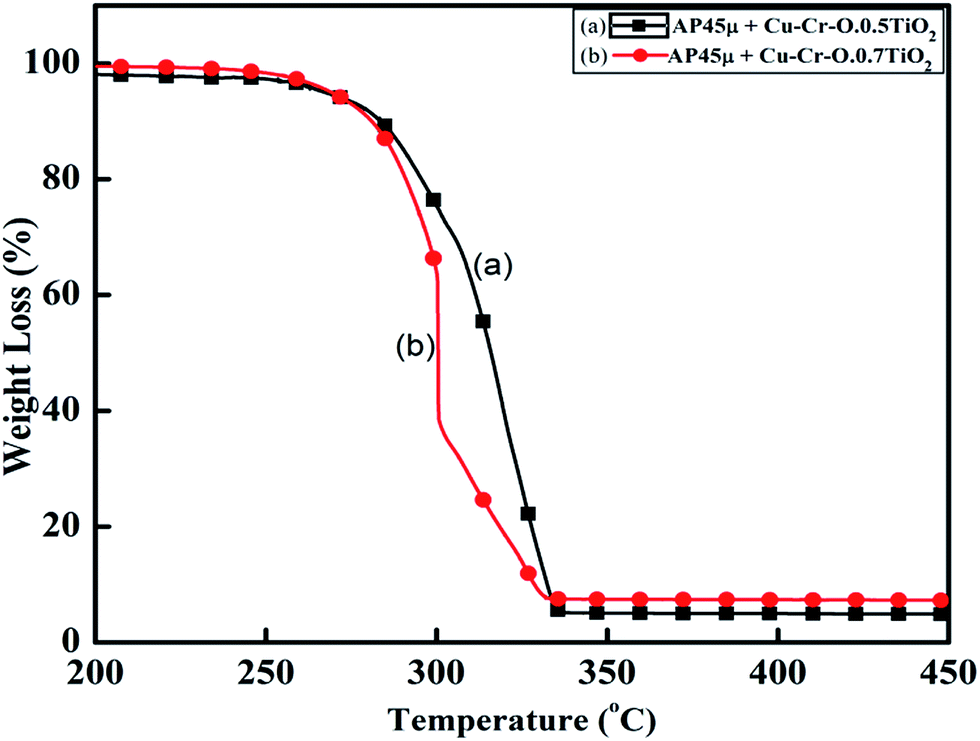
Fig. 11 and 12 show the decomposition (DTA) and % weight loss (TGA) patterns, respectively, of AP45μ modified with industrial ACR (Cu–Cr–O). A significant change can be observed in the thermogram shown in Fig. 11. An endothermic peak appearing at 250 °C in pure AP is shifted towards lower temperature and appears at 245 °C which is the well-known crystallographic change temperature for AP. The exothermic peak (LTD) appearing at 302 °C is shifted to 310 °C with a new proximal peak (HTD) at 330 °C probably due to the decomposition of additives and signifying the increase in decomposition energy. The TGA graph also shows significant changes. This thermogram shows three clear-cut stages of weight loss in which the major weight loss occurs in the second stage covering the range 285–335 °C. The thermograms shown in Fig. 13(a and b) and 14(a and b) represent the DTA (decomposition characteristic) and TGA (% weight loss), respectively, of AP modified with the prepared catalysts of Cu–Cr–O·nTiO2 (n= (a) 0.5 and (b) 0.7) calcined at 300 °C. The endothermic peak due to the crystallographic change appears again at 245 °C. A considerable improvement in the decomposition characteristic is observed with the introduction of TiO2 into the Cu–Cr–O with calcination at 300 °C. The 0.7 mol% addition of TiO2 into the Cu–Cr–O calcined at 300 °C results in a single step sharp decomposition of AP at 306 °C. The sharp and high intensity decomposition peak clearly depicts the significant improvement in decomposition energy as well as decomposition (burn) rate of the modified AP. However, the introduction of 0.5 mol% of TiO2 into the Cu–Cr–O calcined at 300 °C gives a wide decomposition peak centered at 320–322 °C. It is important to note that catalysts were calcined at 300 °C before using as a modifier in AP. Next, TiO2 added catalyst samples were calcined at 1050 °C before using them as modifiers in AP.
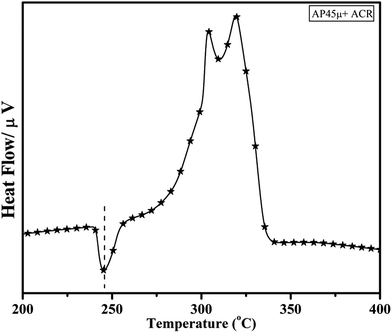 |
| | Fig. 11 DTA curve of AP45μ modified with industrial ACR (Cu–Cr–O). | |
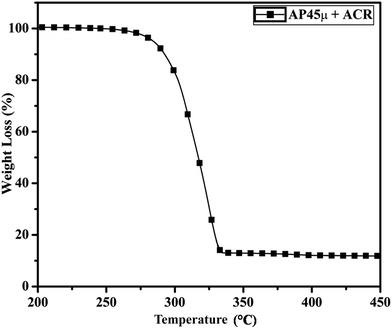 |
| | Fig. 12 TGA curve of AP45μ modified with industrial ACR (Cu–Cr–O). | |
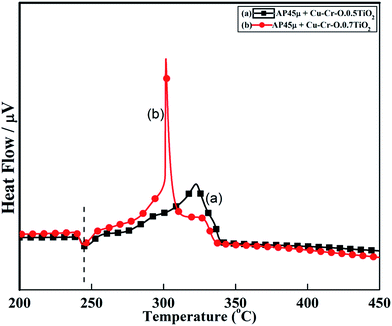 |
| | Fig. 13 DTA curves of AP45μ modified with (a) Cu–Cr–O–0.5TiO2 and (b) Cu–Cr–O–0.7TiO2 calcined at 300 °C. | |
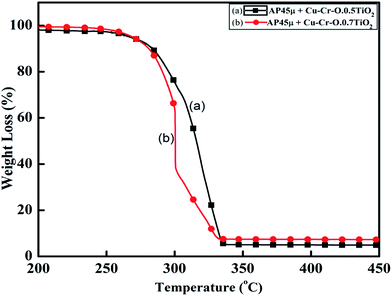 |
| | Fig. 14 TGA curves of AP45μ modified with (a) Cu–Cr–O·0.5TiO2 and (b) Cu–Cr–O·0.7TiO2 calcined at 300 °C. | |
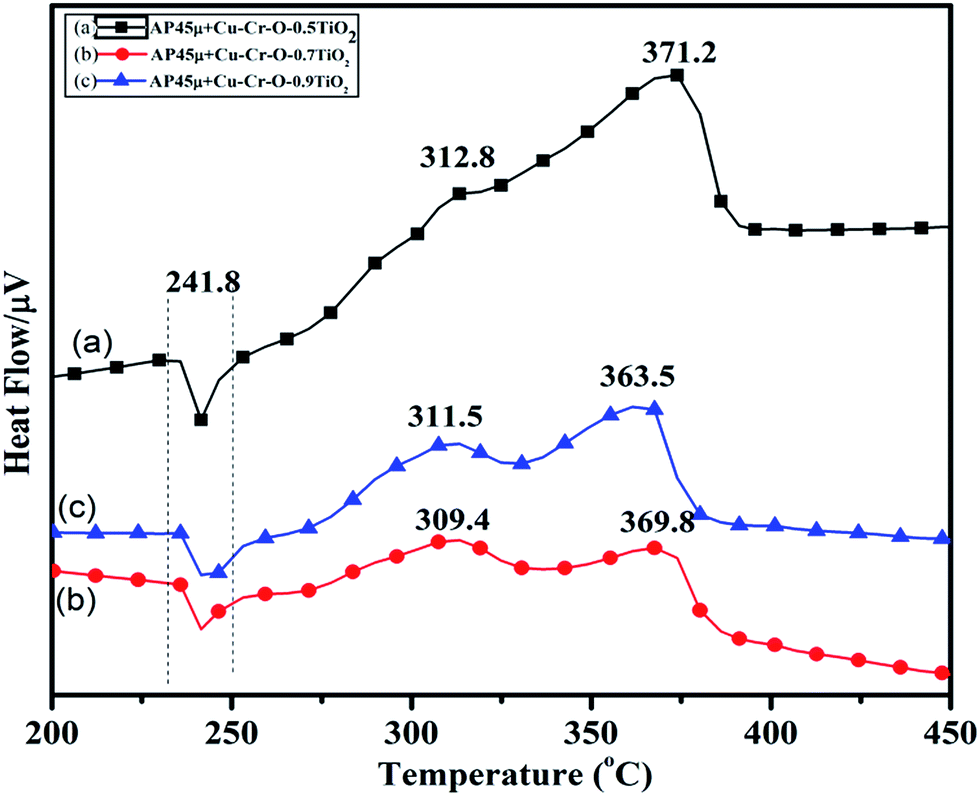
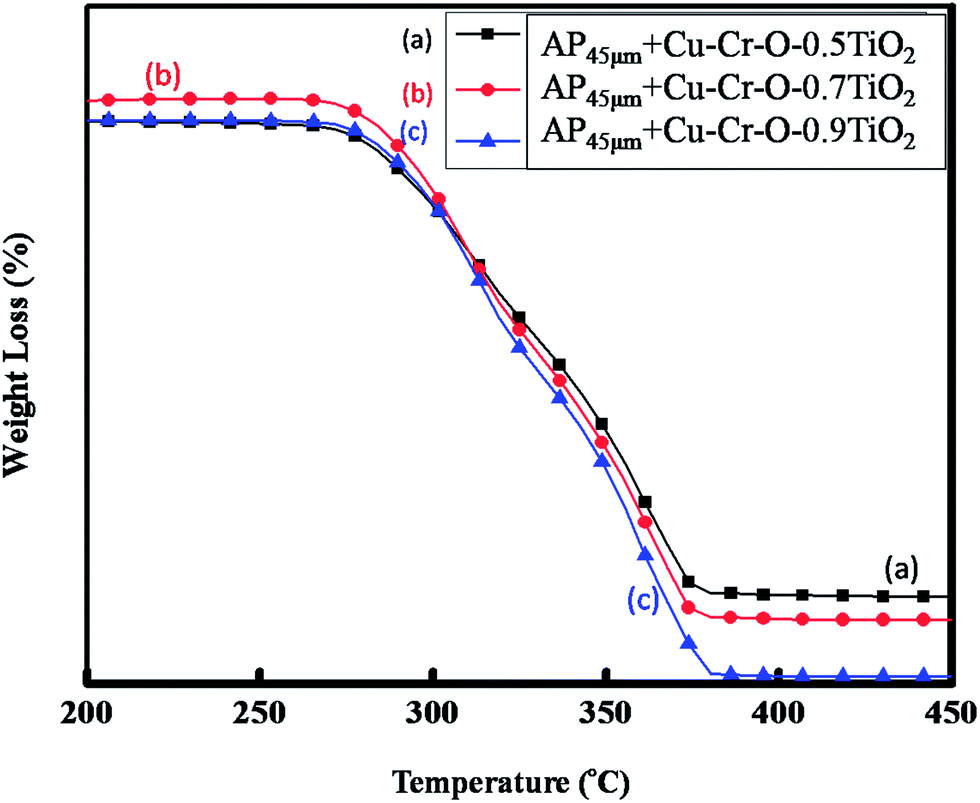
Fig. 15(a–c) and 16(a–c) exhibit the DTA and TGA patterns respectively of AP45μ modified with (a) Cu–Cr–O·0.5TiO2, (b) Cu–Cr–O·0.7TiO2 and (c) Cu–Cr–O·0.9TiO2 calcined at 1050 °C. In these samples the endothermic peak indicating the crystallographic change temperature for the AP appears at 241 °C rather than 245 °C.
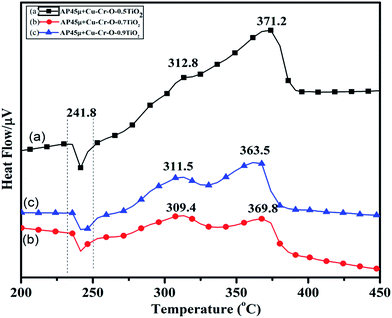 |
| | Fig. 15 DTA curves of AP45μ modified with (a) Cu–Cr–O·0.5TiO2, (b) Cu–Cr–O·0.7TiO2 and (c) Cu–Cr–O·0.9TiO2 calcined at 1050 °C. | |
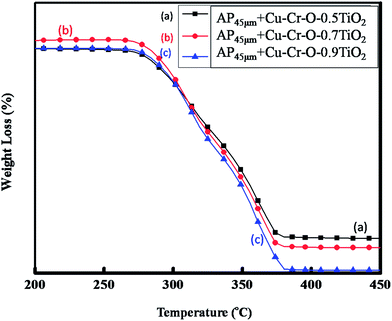 |
| | Fig. 16 TGA curves of AP45μ modified with (a) Cu–Cr–O·0.5TiO2, (b) Cu–Cr–O·0.7TiO2 and (c) Cu–Cr–O·0.9TiO2 calcined at 1050 °C. | |
The crystallographic change temperature for any allotropic/polymorphic material is constant. However, it may be recorded at an increased temperature (2–5 °C) in some certain circumstances like a change in pressure or impurity, a measurement error, or calibration fault. The lower shift of 3 °C in the crystallographic change temperature of pure AP may be due to one of these reasons. This endothermic peak attracts comparatively less interest than the exothermic peaks in the present study. The onset of thermal decomposition of all these samples was found within the temperature range of 300 °C to 313 °C. A brief description of decomposition temperature is shown in Table 2. The differential thermograph shown in Fig. 15 clearly depicts the least effect of the catalysts on the thermal decomposition of AP after calcination at 1050 °C. However, results obtained in Fig. 13 show the maximum effect of a catalyst (Cu–Cr–O·0.7TiO2 calcined at 300 °C) on the thermal decomposition of AP when compared with the results obtained in Fig. 11 and 15. The heats of reaction (i.e. calibrated delta (Δ)H values) are also estimated and are given in Table 3. A careful examination of the table shows the highest calibrated ΔH value (−1411.2097) for the AP modified with Cu–Cr–O0.7TiO2 calcined at 300 °C. Therefore, the conclusions may be verified by using Table 3. This table also shows the amount of the samples used in the characterization of thermal decomposition behavior. Thus the sample Cu–Cr–O·0.7TiO2 calcined at 300 °C is very clearly the most efficient (best) catalyst compared with other laboratory prepared samples calcined at 1050 °C as well as the industrial ACR (Cu–Cr–O).
Table 2 Decomposition temperatures of pure AP, AP modified with ACR and AP modified with different compositions of Cu–Cr–O–TiO2 calcined at 300 and 1050 °C
| Calcination temp. of Cu–Cr–O–TiO2 |
Composition |
Endothermic peak position (single peak) |
Exothermic peak positions |
| LTD (°C) |
HTD (°C) |
| As synthesized |
AP45μ/AP300μ |
250 |
302 |
385 |
| AP45μ + ACR (Cu–Cr–O) |
245 |
310 |
330 |
| 300 °C |
AP45μ + Cu–Cr–O–0.5TiO2 |
245 |
320 |
322 |
| AP45μ + Cu–Cr–O–0.7TiO2 |
245 |
306 |
306 |
| 1050 °C |
AP45μ + Cu–Cr–O–0.5TiO2 |
241.8 |
309.4 |
369.8 |
| AP45μ + Cu–Cr–O–0.7TiO2 |
241.8 |
311.5 |
363.5 |
| AP45μ + Cu–Cr–O–0.9TiO2 |
241.8 |
312.8 |
371.2 |
Table 3 Calibrated ΔH values for all the endothermic and exothermic peaks appearing in DTA of pure AP, AP modified with ACR and AP modified with different compositions of Cu–Cr–O–TiO2 calcined at 300 and 1050 °C
| Calcination temperature |
Sample composition |
Amount of sample |
ΔH (J g−1) |
| Wt (mg) |
Endoth. peak |
Exoth. peak-1 LTD |
Exoth. peak-2 HTD |
Exoth. peak-3 HTD (II) |
| As synthesized |
(a) AP300μ |
12.457 |
87.2360 |
−302.6432 |
−173.6290 |
−19.5203 |
| (b) AP45μ |
7.428 |
87.5459 |
−211.8264 |
−787.7451 |
N/A |
| AP45μ + ACR (Cu–Cr–O) |
10.373 |
80.4025 |
−27.3855 |
−112.2612 |
N/A |
| 300 °C |
(a) AP45μ + Cu–Cr–O–0.5TiO2 |
16.000 |
83.2412 |
−461.8651 |
−63.9416 |
N/A |
| (b) AP45μ + Cu–Cr–O–0.7TiO2 |
11.107 |
82.8426 |
−1411.2097 |
N/A |
N/A |
| 1050 °C |
(a) AP45μ + Cu–Cr–O–0.5TiO2 |
8.115 |
56.1904 |
−128.372 |
−452.176 |
N/A |
| (b) AP45μ + Cu–Cr–O–0.7TiO2 |
5.032 |
58.4718 |
−314.905 |
−180.880 |
N/A |
| (c) AP45μ + Cu–Cr–O–0.9TiO2 |
6.277 |
59.3275 |
−123.219 |
−186.66 |
N/A |
The decomposition behavior with and without the presence of the synthesized catalyst materials obtained from the DTA technique are shown in Fig. 9, 11, 13 and 15. The first peak in each figure is observed in the temperature range of 230 °C to 240 °C, which is endothermic in nature. This phenomenon can be attributed to the crystallographic change of AP structure from orthorhombic to cubic. The second decomposition is observed at the temperature of 310 °C and assigned as the LTD accompanied with an exothermic peak. The most probable associated chemical reaction is shown below in eqn (i):
| | |
NH4ClO4 → NH4+ (s) + ClO4− (s) ↔ NH3 (g) + HClO4 (g)
| (i) |
The third and final decomposition of AP occurs at the temperature of 380 °C, described as the HTD, and the peak is again exothermic in nature. This decomposition is caused by oxidation of NH3 by ClO4− in the gaseous phase and decomposition of AP on the solid surface of the catalyst. At the HTD, HClO4 is converted into small molecules like NO, O2, Cl2, and H2O as shown below in eqn (ii) and (iii):
| | |
NH4ClO4 → H2O + NOx + Cl2 + N2 + HCl
| (ii) |
| | |
HClO4 → H2 + Cl2 + O2 + H2O
| (iii) |
Thus, AP showed the best decomposition behavior when modified with Cu–Cr–O·0.7TiO2 calcined at 300 °C. In general, the modifier catalysts calcined at 300 °C exhibited better effects on the thermal decomposition of AP than the catalysts calcined at 1050 °C. Crystallinity, surface properties, and specific surface area are the three key factors which determine the catalytic activity of the catalyst. In the present case, the most plausible reason behind the superiority of the Cu–Cr–O samples calcined at 300 °C over the similar samples calcined at 1050 °C may be as follows: during calcination up to 300 °C, only about 20% of the precursor sample is decomposed as can be clearly seen in Fig. 1. Further decomposition of the remaining ∼80% precursor (above 300 °C) alters the AP decomposition as shown in Fig. 14, as the crystallization temperature of Cu–Cr–O is 700 °C. This is also reaffirmed by Fig. 4 and 5 (FT-IR). The other factors behind this may be that at the higher temperature (1050 °C) the particles diffuse to each other and form comparatively larger particles of different surface morphology (see Fig. 6 and 7) which results in the change in surface properties and a reduction in the specific surface area of the catalyst. Due to this reason, the efficiency of the catalysts was found to be reduced ultimately at the higher calcination temperature. However, the catalyst samples prepared at 300 °C showed tremendously enhanced thermal decomposition of AP. Among them, the catalyst sample with 0.7 mol% TiO2 (Cu–Cr–O·0.7TiO2) exhibited a unique decomposition (burn) rate of AP.
4. Conclusions
Nano sized Cu–Cr–O·nTiO2 (n = 0.5, 0.7 and 0.9) particles have been synthesized successfully via the sol–gel method. The nano sized composites of Cu–Cr–O·nTiO2 calcined at 300 °C for 3 h showed excellent catalytic activity on thermal decomposition of AP over the Cu–Cr–O·nTiO2 oxides calcined at 1050 °C for 3 h. Among the three compositions of Cu–Cr–O with their varying concentrations (n = 0.5, 0.7 and 0.9) of TiO2 calcined at 300 °C, 0.7TiO2 showed the best catalytic activity for decomposition of AP of particle size 45 μm. The Cu–Cr–O·0.7TiO2 composite calcined at 300 °C, when used as a catalyst in AP45μ, gave an excellent single step decomposition of AP45μ at 306 °C, which is 79 °C lower than that for the pure AP45μ, representing a significant change in HTD. Thus, the said catalyst achieved a remarkable enhancement in the thermal decomposition of AP which is very much larger than that produced by the industrial catalyst (ACR) as well as other laboratory prepared samples. The sharp peak of the decomposition reveals the faster rate of decomposition. This improved characteristic of synthesized Cu–Cr–O·0.7TiO2 would reduce the ignition delay of solid rocket motors and space vehicles utilizing AP based solid composite propellant. The obtained results of the present study show that the catalytic efficiency/activity of AP depends on the particle size of the AP and the presence of catalyst, and that both the calcination temperature of the catalyst and the molar concentration of TiO2 in the catalyst play a major role.
Conflict of interest
There is no conflict of interest for the authors.
Acknowledgements
H. Kumar is thankful to J. Ram Mohan, General Manager SF Complex, DRDO, for his techno-commercial support. The authors are thankful to Dr Nand Lal Singh, Department of Chemistry, Institute of Science, Banaras Hindu University, Varanasi-221005 for his valuable help in the characterization of the as synthesized samples.
Notes and references
- G. P. Shutton and O. Biblarge, Rocket Propulsion Elements, Wiley Inter-Science Publication, 7th edn, 2000 Search PubMed.
- A. Davenas, Solid Rocket Propulsion Technology, Pergamon Press, 1st edn, 1992 Search PubMed.
- P. W. M. Jacob and H. M. Whitehe, Chem. Rev., 1969, 69, 551–590 CrossRef.
- V. V. Boldyrev, Thermochim. Acta, 2006, 443, 1–36 CrossRef CAS.
- C. Huang, Q. Liu, W. Fan and X. Xiu, Sci. Rep., 2015, 5, 16736 CrossRef CAS PubMed.
- D. L. Reid, R. Draper, D. Richardson, A. Demko, T. Allen, E. L. Petersen and S. Seal, J. Mater. Chem. A, 2014, 2, 2313–2322 CAS.
- K. D. Grossman, T. S. Sakthivel, C. Dillier, E. L. Petersen and S. Seal, RSC Adv., 2016, 6, 89635–89641 RSC.
- D. L. Reid, A. E. Russo, R. V. caro, M. A. Stephens, A. R. L. Page, T. C. Spalding, E. L. Petersen and S. Seal, Nano Lett., 2007, 7, 2157–2161 CrossRef CAS.
- L. Liu, F. Li, L. Tan, L. Ming and Y. Yi, Propellants, Explos., Pyrotech., 2004, 29(1), 34–38 CrossRef CAS.
- L. Song, S. Zhang, B. Chen, J. Ge and X. Jia, Colloids Surf., A, 2010, 360, 1–5 CrossRef CAS.
- P. R. Patil, V. N. Krishnamurthy and S. S. Joshi, Propellants, Explos., Pyrotech., 2008, 33, 266–270 CrossRef CAS.
- H. Xu, X. Wang and L. Zhang, Powder Technol., 2008, 185, 176–180 CrossRef CAS.
- Y. Zhang, X. Liu, J. Nie, L. Yu, Y. Zhong and C. Huang, J. Solid State Chem., 2011, 184, 387–390 CrossRef CAS.
- R. A. Chandru, S. Patra, C. Oommen, N. Munichandraiah and B. N. Raghunandan, J. Mater. Chem., 2012, 22(14), 6536–6538 RSC.
- K. Kishore, V. R. P. Vernekar and R. R. Sunitha, AIAA J., 1980, 18(11), 1404–1405 CrossRef CAS.
- V. G. Dedgaonkar and D. B. Sarwade, J. Therm. Anal., 1990, 36, 223–229 CrossRef CAS.
- K. Kishore and M. R. Sunitha, AIAA J., 1979, 17, 1118–1125 CrossRef CAS.
- E. A. Gheshlaghi, B. Shaabani, A. Khodayari, Y. A. Kalandaragh and R. Rahimi, Powder Technol., 2012, 217, 330–339 CrossRef.
- T. Liu, L. Wang and B. Hu, Mater. Lett., 2008, 62, 4056–4058 CrossRef CAS.
- S. Zhao and D. Ma, J. Nanomater., 2010, 2010, 1–5 Search PubMed.
- L. Chen, P. Li and G. Li, J. Alloys Compd., 2008, 464, 532–536 CrossRef CAS.
- A. A. Said and R. Qasami, Thermochim. Acta, 1996, 275, 83–91 CrossRef CAS.
- R. Dubey, P. Srivastava, I. P. S. Kapoor and G. Singh, Thermochim. Acta, 2012, 549, 102–109 CrossRef CAS.
- M. A. Kawamoto, L. C. Pardini and L. C. Rezende, Aerosp. Sci. Technol., 2004, 8, 591–598 CrossRef.
- R. P. Fitzgeral and M. Q. Brewster, Combust. Flame, 2004, 136(3), 313–326 CrossRef.
- L. M. Song, S. J. Zhang, B. Chen, J. J. Ge and X. C. Xia, Colloids Surf., A, 2010, 360, 1–5 CrossRef CAS.
- J. Yan, L. Zhang, H. Yang, Y. Tang, Z. Lu, S. Guo, Y. Dai, Y. Han and M. Yao, Sol. Energy, 2009, 83, 1534–1539 CrossRef CAS.
- V. K. Mishra, B. N. Bhattacharjee, D. Kumar, S. B. Rai and O. Parkash, New J. Chem., 2016, 40, 5432–5441 RSC.
- V. K. Mishra, S. K. Srivastava, B. P. Asthana and D. Kumar, J. Am. Ceram. Soc., 2012, 95, 2709–2715 CrossRef CAS.
Footnotes |
| † Present address: Defence Materials and Stores Research and Development Establishment (DMSRDE), Defence Research and Development Organization, (DRDO) G. T. Road Kanpur-208013, India. |
| ‡ Present address: Endocrinology Division, CSIR-Central Drug Research Institute, Lucknow-226 031, India. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ab,
Prahalad N. Tenglic,
Vijay Kumar Mishra‡
*d,
Pankaj Tripathie,
Dan Bahadur Pala and
Pradeep Kumar Mishraa
ab,
Prahalad N. Tenglic,
Vijay Kumar Mishra‡
*d,
Pankaj Tripathie,
Dan Bahadur Pala and
Pradeep Kumar Mishraa