
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computational screening of phosphite derivatives as high-performance additives in high-voltage Li-ion batteries
Young-Kyu Han *a,
Jaeik Yooa and
Taeeun Yim*b
*a,
Jaeik Yooa and
Taeeun Yim*b
aDepartment of Energy and Materials Engineering, Advanced Energy and Electronic Materials Research Center, Dongguk University-Seoul, Seoul 100-715, Republic of Korea. E-mail: ykenergy@dongguk.edu
bDepartment of Chemistry, Incheon National University, Incheon 22012, Republic of Korea. E-mail: yte0102@inu.ac.kr
First published on 6th April 2017
Abstract
Phosphite derivatives are well known as cathode electrolyte interphase (CEI)-forming additives for greatly enhancing the performance of cathode materials for use in high-voltage Li-ion batteries. Our computational screening suggests four phosphite molecules as promising candidates for effective CEI-forming electrolyte additives on the basis of density functional theory (DFT)-based calculations of their redox potentials and chemical reactivities with a hydrogen fluoride (HF) molecule. Our computational screening protocol provides a fast and inexpensive method for the development of novel CEI-forming electrolyte additives for use in high-voltage Li-ion batteries.
Introduction
Researchers have examined high-voltage Li-ion batteries (LIBs) intensively in recent years as potential power sources for electric vehicles (EVs) and for use in energy storage systems (ESSs), both of which require high energy and high power density.1,2 Because the performance, cost, and safety of LIBs mainly depend on the properties of the cathode materials, many studies have been devoted to developing high capacity, inexpensive, and safe materials. Numerous layered transition-metal oxides, including LiNi1/3Co1/3Mn1/3O2, LiNi0.4Co0.4Mn0.2O2, and LiNi0.5Co0.2Mn0.3O2 (NCM) materials, have been explored as promising cathode materials for use in LIBs because of their lower cost, higher capacity, lower toxicity, and better thermal stability compared to commercial LiCoO2 materials.3,4 However, the use of batteries containing cathodes made of NCM materials leads to problems such as drastic capacity fading and poor cyclic performance, especially at high temperatures.5–8 The main cause of capacity fading is the dissolution of transition metal ions from the cathode surface due to the existence of hydrogen fluoride (HF) molecules, which are produced by the decomposition reactions of lithium hexafluorophosphate (LiPF6) salt with residual water in the electrolyte.9–12The use of cathode electrolyte interphase (CEI)-forming additives would be one of the most economic and efficient methods for improving the capacity retention and cyclic stability of high-voltage LIBs using NCM cathode materials. Song et al.13 and Han et al.,14 who examined LiNi0.5Mn1.5O4 and Li1.17Ni0.17Mn0.5Co0.17O2, respectively, both reported tris(trimethylsilyl) phosphite (TMSP) as a powerful electrolyte additive for improving the rate capability and cyclic stability of high-voltage cathode materials. Both studies proposed that the TMSP additive offers a stable protective surface layer on the electrode surface and effectively controls HF in electrolytes, thus resulting in improved electrochemical performances. Han et al.15 and Song et al.16 demonstrated why TMSP has a relatively high HF scavenging ability among the various phosphite-based compounds. Li et al.17 reported that trimethyl phosphite (TMP) as a CEI-forming additive can significantly improve the cycling performance and rate capability of lithium-rich layered oxide cathode for high-voltage LIBs. Pires et al.18 demonstrated that tris(2,2,2-trifluoroethyl) phosphite (TTFP) is a useful electrolyte additive for enhancing the long-term cycling stability of Li-rich cathode for high-voltage LIBs. Zhou et al.19 proposed triphenyl phosphite (TPPi) as a film-forming additive to significantly enhance the capacity retention and cycling performance of Li1.16Ni0.2Co0.1Mn0.54O2 cathode in high-voltage LIBs.
The use of a screening approach via computational evaluation is the basis of discovering novel materials, since this method allows material scientists to seek promising structures in an efficient manner.20 Computational searching becomes much less costly than experimental trial-and-error procedures when effective screening factors are known.21,22
The first requirement for achieving successful CEI-forming electrolyte additives is that these additives must have lower oxidation potentials (i.e., more facile oxidation) than the solvent molecules, since this allows the electrolyte additives to form a protective CEI layer on the cathode after their oxidation and before solvent oxidation. A previous study, however, has reported that phosphite derivatives usually have much lower oxidation potential (OP) values compared to ethylene carbonate (EC, 6.92 V).15 A synergistic effect observed when both the vinylene carbonate (VC) and TMSP additives are employed together has also been associated with a far smaller OP value of TMSP (4.29 V) than is the case with VC (5.46 V).15,23 Thus, an OP value that is lower than that of VC was chosen to satisfy the first requirement of this study. The second requirement is that the additives must possess a low reduction potential (RP). If the CEI-forming additive has a higher RP than the solvent, then the additive may exhibit problematic behavior on the anode surface, possibly due to the easier reduction and subsequent reductive decomposition. It should be noted that the most popular TMSP additive has a much lower RP value of −1.03 V compared to EC value of −0.32 V.15 The third requirement is that CEI-forming additives must have strong HF and LiF reactivity. LiF is highly resistive in permitting Li+ ion migration at the CEI region. Therefore, the strong LiF reactivity of phosphite additives can be an indicator of high-performance CEI-forming additives.12,24–26
TMSP, which is a representative CEI-forming additive, (1) has a positive effect on HF/LiF removal, (2) alleviates the dissolution phenomenon of transition metal ions and increases surface resistance on cathode, and (3) improves the rate capability and cycling stability of high-voltage cathode materials.13–16,27 The HF binding energy was calculated in a previous study.15 However, the reaction energies of phosphite additive candidates with HF were calculated in this study because earlier studies had reported that the decomposition reactions of phosphite molecules with HF are thermodynamically favorable.14,16
As noted above, we suggest three screening factors: (i) lower OP than VC value; (ii) lower RP than EC value (a smaller RP of the additive will result in a higher cathodic stability of the additive); and (iii) similar or higher HF/LiF reactivity to TMSP (higher HF/LiF reactivity values will result in more effective HF/LiF scavenging, thereby leading to high cell performance).
In this study, we examined 35 phosphites as potential CEI-forming additives for use in high-voltage LIBs. The molecular structures of phosphite candidates 1–35 that we considered are shown in Fig. 1. Each of the phosphites we considered in this study are available from Sigma Aldrich. Our computational screening suggests four phosphite molecules as promising candidates for high-performance CEI-forming additives using density functional theory (DFT)-based calculations of redox potentials and chemical reactivities with HF. We believe that our screening procedure can aid material scientists in efficiently searching for novel and effective CEI-forming additives for use in the high-voltage LIBs.
 | ||
| Fig. 1 2-D molecular structures of phosphite candidates 1–35. | ||
Computational details
Detailed simulated annealing procedure
After generation of the initial microstructure, the potential energy of the model was minimized using the COMPASS force-field. Annealing was then simulated by raising the temperature from 300 K to 1500 K and then allowing it to cool to 300 K at a rate of change of 100 K per 5 ps. The conformation was heated again at the same rate in order to jump to a different valley, and it was then allowed to cool again. A series of 10 cycles of annealing was then simulated between 300 and 1500 K for effective sampling of the conformational space. Each sampling involved 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200000 molecular dynamics steps in total for each molecule. The MD simulations were carried out in the microcanonical (NVE) ensemble. The velocity Verlet algorithm, with a time step of 1 fs, was used for the integration of the atom motion equations throughout all simulations.
200000 molecular dynamics steps in total for each molecule. The MD simulations were carried out in the microcanonical (NVE) ensemble. The velocity Verlet algorithm, with a time step of 1 fs, was used for the integration of the atom motion equations throughout all simulations.
The molecular simulations were performed with the Forcite Plus module of the commercial software Materials Studio 5.5 using the COMPASS forcefield. This forcefield enables accurate and simultaneous predictions of structural, conformational, vibrational, and thermophysical properties that exist for organic molecules, inorganic small molecules, and polymers, in isolation and in condensed phases, and under a wide range of conditions of temperature and pressure.28–30
Density functional theory (DFT) calculations
The Kohn–Sham DFT method is well known to be useful for studying the molecular properties of various organic compounds that are used in LIBs.31 We performed geometry optimizations using no symmetry restriction and the B3PW91 functional and 6-311G(d,p) basis sets of triple-ζ quality, which involves a three-parameter adiabatic connection exchange functional.32–34The solvent effect was addressed in the present study by using the conductor-variant polarized continuum model,35 which places the solute in a molecular-shaped cavity embedded in a continuum dielectric medium. We adopted a dielectric constant (31.9) as a weight-averaged value between the dielectric constants of ethyl methyl carbonate (EMC: 2.9) and EC: 89.2 since a combination of EC:EMC = 1:2 is often employed as the electrolyte solvent in LIBs.36 Several studies have shown that continuum model calculations are useful for assessing numerous electrochemical properties in the electrolyte used for LIBs.37–41
The energy cycle was used to calculate the oxidation potential of a molecule (M) relative to an electron at rest in vacuum ( ),42 as shown in Scheme 1.
),42 as shown in Scheme 1.

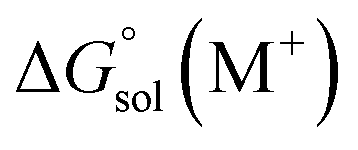 and
and  are the free energies of solvation of the oxidized and neutral molecules M+ and M, respectively. F is the Faraday constant. ΔGe has contributions from the adiabatic ionization energy, zero-point vibrational correction, and thermal energy correction, as well as the entropy term −TΔS. The redox potential (E°) of the electrolytes relative to the Li/Li+ reference electrode was determined using
are the free energies of solvation of the oxidized and neutral molecules M+ and M, respectively. F is the Faraday constant. ΔGe has contributions from the adiabatic ionization energy, zero-point vibrational correction, and thermal energy correction, as well as the entropy term −TΔS. The redox potential (E°) of the electrolytes relative to the Li/Li+ reference electrode was determined using  .42
.42
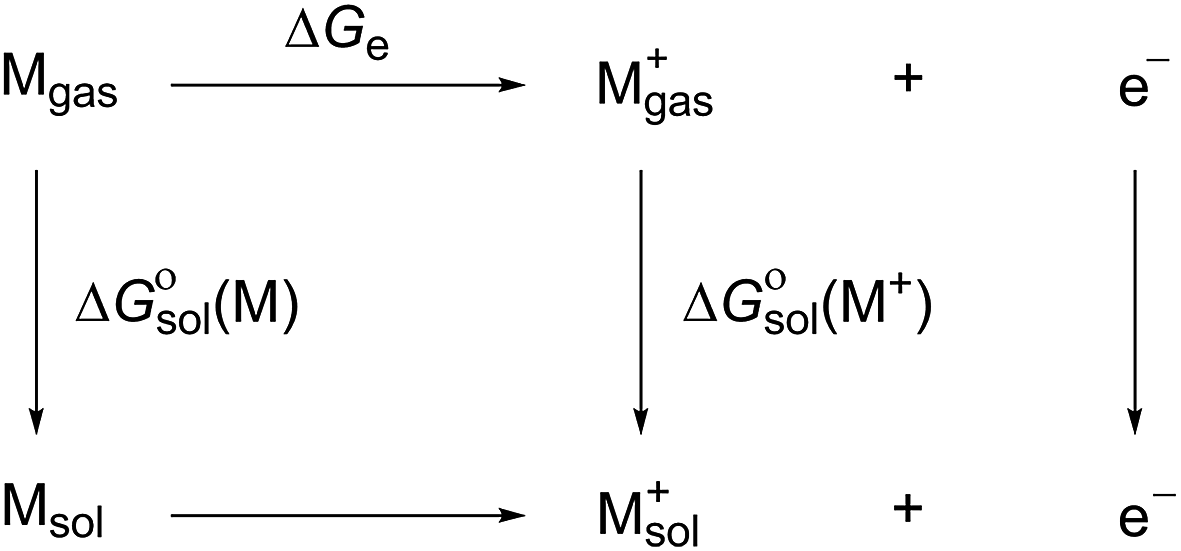 | ||
| Scheme 1 Free energy cycle for the redox reaction (M → M+ + e−), where Mgas denotes molecule M in the gas-phase and Msol denotes the solvated molecule. | ||
The ΔHF and ΔLiF values were calculated considering thermodynamic Gibbs free energies and solvent effects. The first-principles calculations were all carried out using the Gaussian09 package.43
Results and discussion
The calculated highest occupied molecular orbitals (HOMO), lowest unoccupied molecular orbitals (LUMO), and OP and RP values of the 35 phosphite molecules (1–35) are shown in Table 1. For comparison, we listed the HOMO, LUMO, and OP and RP values of EC, VC, and TMSP together. When the OP value is low, a molecule is easily oxidizable, and a molecule is difficult to be reduced when its RP value is low. It is worth noting that all of the phosphite derivatives that are considered (1–35) for this study have lower OP values than is the case with EC. That is, all the phosphites that we considered have the potential to be employed as electrolyte additives that are oxidized prior to EC. This means that the 35 phosphites can form a protective film on the positive electrode surface. The OP values of the 35 phosphite derivatives are in the 4.29–5.75 V range. The molecule corresponding to the OP value of 4.29 V is found to be tris(1-adamantyl) phosphite (20). Molecule 20 may oxidize better than other phosphite molecules and can be maintained in a more structurally stable manner than other phosphite structures in the CEI region. This can be attributed to a highly stable 1-adamantyl cation: one lobe of the empty p-orbital extends into the adamantine cage where an effective orbital overlap occurs with the backsides of the three sp3 bridgehead C–H orbitals.44 The molecule that has an OP value of 5.75 V is triacetyl phosphite (8). Molecule 8—P(OR)3, where R = acetyl—shows the highest OP value among the 35 phosphite molecules, because the R group has an electron-withdrawing nature. However, it is still lower compared to the OP value of EC (6.92 V).| EHOMO (eV) | ELUMO (eV) | OP (V vs. Li/Li+) | RP (V vs. Li/Li+) | ||
|---|---|---|---|---|---|
| (1) | Tetraethyl pyrophosphite | −6.42 | 0.29 | 4.50 | 0.59 |
| (2) | Tris ortho xenyl phosphite | −6.15 | −1.15 | 4.73 | 0.40 |
| (3) | Phosphorous acid 2-(1-oxo-1,3-dihydro-isoindol-2-yl)-ethyl ester dipropyl ester | −6.52 | −1.33 | 4.36 | 0.45 |
| (4) | 1,9-Ditert-butyl-3,7-dimethyl-11-phenoxy-5H-benzo [D][1,3,2]benzodioxaphosphocine | −6.16 | −0.62 | 4.65 | 0.00 |
| (5) | Diethyl 2,2,2-trichloroethyl phosphite | −6.95 | −1.29 | 4.97 | 2.22 |
| (6) | Methyl bis(trimethylsilyl) phosphite | −6.59 | 0.69 | 4.43 | −0.20 |
| (7) | 2-(Buta-1,3-dien-2-yloxy)-1,3,2-dioxaphospholane | −6.44 | −1.05 | 4.74 | 0.35 |
| (8) | Triacetyl phosphite | −7.62 | −0.81 | 5.75 | 0.94 |
| (9) | 4-Ethyl-2,6,7-trioxa-1-phosphabicyclo[2.2.2]octane | −7.45 | 0.67 | 5.71 | −0.60 |
| (10) | Triprop-2-enyl phosphite | −6.63 | −0.27 | 4.63 | −0.41 |
| (11) | Trineopentyl phosphite | −6.58 | 0.59 | 4.55 | −0.02 |
| (12) | 4,8-Di-tert-butyl-6-(4-tert-butyl-phenoxy)-2,10-dimethyl-12H-5,7-dioxa-6-phospha-dibenzo[a,d]cyclooctene | −6.13 | −0.56 | 4.80 | −0.25 |
| (13) | Diethyl hexyl phosphite | −6.51 | 0.76 | 4.49 | −0.54 |
| (14) | tert-Butyl diethyl phosphite | −6.36 | 0.78 | 4.54 | −0.22 |
| (15) | Phosphorous acid di-tert-butyl ester ethyl ester | −6.23 | 0.81 | 4.45 | −0.22 |
| (16) | 2-Ethoxy-4-methoxy-[1,3,2,4]dioxadiphosphinane 4-oxide | −7.17 | 0.01 | 5.01 | 1.05 |
| (17) | 4,8-Di-tert-butyl-2,10-dimethyl-6-phenoxy-5,7-dioxa-12-thia-6-phospha-dibenzo[a,d]cyclooctene | −5.90 | −0.86 | 4.55 | 0.13 |
| (18) | Trioleyl phosphite | −6.49 | 0.31 | 4.98 | −0.98 |
| (19) | 5,5-Dimethyl-2-oxotetrahydrofuran-3-yldihexadecyl phosphite | −6.59 | −0.36 | 4.75 | 0.27 |
| (20) | Tris(1-adamantyl) phosphite | −5.99 | 0.72 | 4.29 | −1.36 |
| (21) | 1,3,2-Dioxaphosphorinane,5-(1,1-dimethylethyl)-2-ethoxy-(9CI) | −6.54 | 0.34 | 4.67 | 0.01 |
| (22) | Bis(2-ethylhexyl) phenyl phosphite | −6.53 | −0.50 | 4.53 | −0.06 |
| (23) | 3,9-Bis(phenoxy)-2,4,8,10-tetraoxa-3,9-diphosphaspiro[5.5]undecane | −6.70 | −0.61 | 5.04 | −0.22 |
| (24) | 1,3,5-Cyclohexanetriyl phosphite | −7.51 | 0.64 | 5.49 | −0.64 |
| (25) | Tris(2-methylpropyl) phosphite | −6.56 | 0.69 | 4.60 | −0.09 |
| (26) | Tris(2-methylphenyl) phosphite | −6.43 | −0.57 | 4.78 | 0.34 |
| (27) | Tridecyl phosphite | −6.47 | 0.61 | 4.42 | −0.16 |
| (28) | Trioctyl phosphite | −6.65 | 0.40 | 4.50 | −0.17 |
| (29) | Distearyl pentaerythritol diphosphite | −6.60 | 0.19 | 4.91 | −0.36 |
| (30) | Trihexyl phosphite | −6.54 | 0.44 | 4.68 | −0.12 |
| (31) | Diethyl phenyl phosphite | −6.51 | −0.47 | 4.86 | 0.44 |
| (32) | Tritert-butyl phosphite | −6.12 | 0.84 | 4.39 | −0.52 |
| (33) | Tri-p-tolyl phosphite | −6.31 | −0.67 | 4.84 | 0.07 |
| (34) | Decyl diphenyl phosphite | −6.46 | −0.61 | 4.84 | 0.75 |
| (35) | Tris(4-nonylphenyl) phosphite | −5.99 | −1.43 | 4.49 | 1.11 |
| Tris(trimethylsilyl) phosphite (TMSP) | −6.52 | 0.70 | 4.29 | −1.03 | |
| Ethylene carbonate (EC) | −8.25 | 0.60 | 6.92 | −0.32 | |
| Vinylene carbonate (VC) | −7.23 | −0.36 | 5.46 | −0.12 |
The 32 phosphite molecules, with the exception of 8, 9, and 24, have smaller OP values than the 5.46 V value of VC (requirement i); in other words, they can form CEI at the cathode by oxidizing earlier than VC. We expect that the problem caused by the VC with a low oxidation stability—which oxidizes at the cathode of high-voltage LIBs (i.e., poor cycle performance)—can be solved by using VC together with these 32 molecules. The synergistic effects of the combination of VC with these molecules may also be expected, such as the improved cell performance of the TMSP + VC combination reported in a previous study.23
A previous report15 found that one advantage of TMSP is that it can form CEI at the cathode by oxidizing earlier than VC, thus making up for the disadvantage of VC with low oxidative stability; the present study also found that the synergy effect with VC may be expected for most phosphite materials.
Among the 35 molecules considered in the present work, only eight molecules (specifically, 9, 10, 13, 18, 20, 24, 29, and 32) are found to fulfill requirement ii, which states that compared to EC, the molecules must be more difficult to be reduced. It is difficult for these eight molecules to react at the negative electrode and to cause side effects between electrolyte and electrode. The RP values of these eight phosphites range from −1.36 V to −0.36 V. The molecule corresponding to an RP value of −1.36 V is tris(1-adamantyl) phosphite (20). Molecule 20 is more difficult to be reduced compared to other phosphite molecules, and the cathodic stability of 20 is higher than that of the other seven phosphite molecules. This value is even lower than that of TMSP (−1.03 V).
Fig. 2 shows the chemical reactivities of six phosphite molecules (10, 13, 18, 20, 29, and 32) with a HF and a LiF molecule which meet requirements (i) and (ii). The strong reactivity of additives for the HF- and LiF- scavenging reactions can efficiently remove the undesired molecules, HF and LiF in the electrolyte and on the cathode surface, respectively.
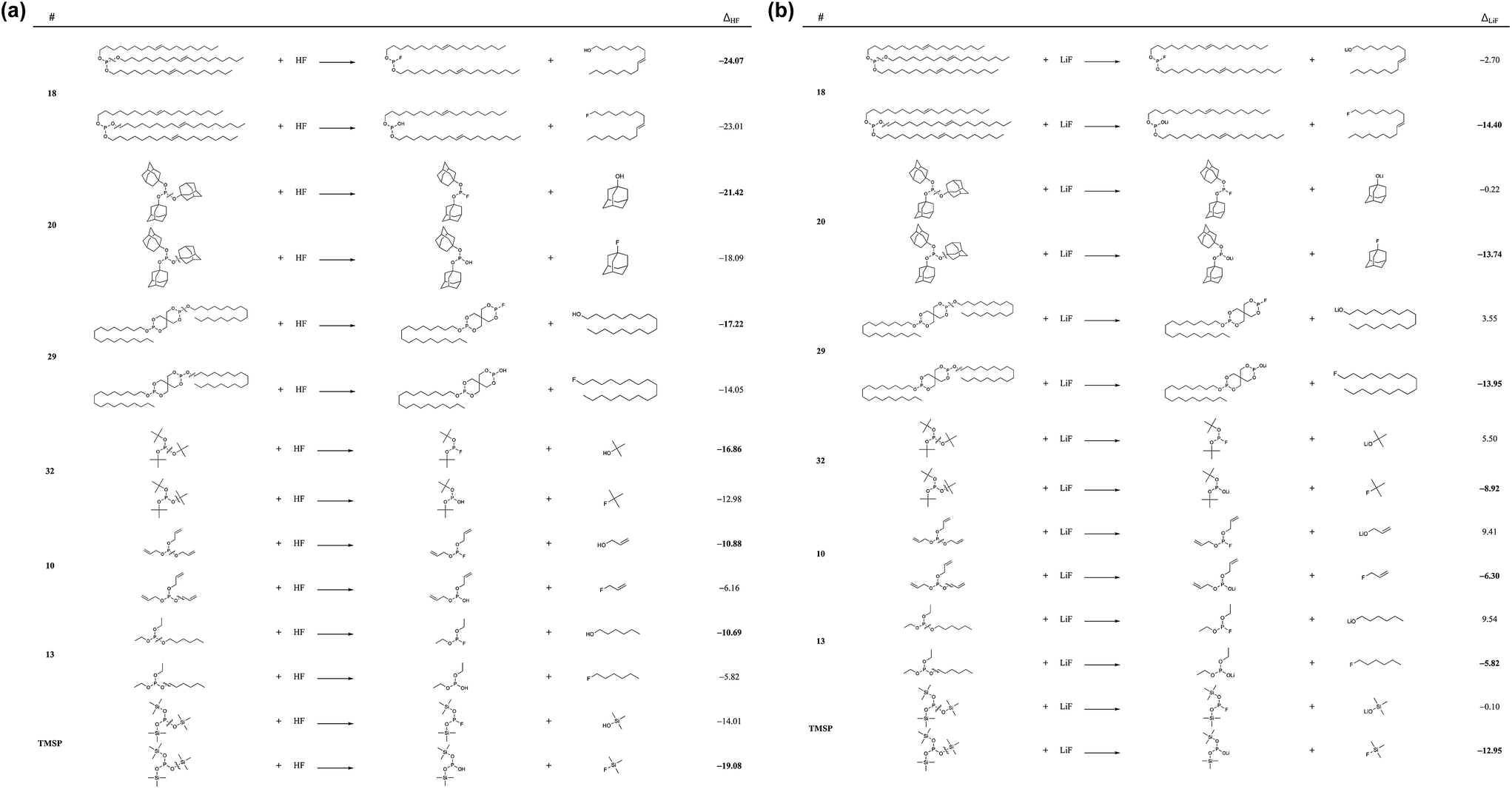 | ||
| Fig. 2 Reaction energies (ΔHF and ΔLiF in kcal mol−1) (a) with a HF molecule and (b) with a LiF molecule, respectively. | ||
We considered the reactions with HF and LiF via the P–O bond and O–C bond cleavages, as shown in Fig. 2. We sought to identify the molecules that would meet requirement (iii) (i.e., an HF reactivity that is similar to or better than TMSP) by comparing them to the HF reactivity of TMSP. We observed that the HF reactivities of these six molecules are greatly preferable thermodynamically (i.e., they are exoergic), and that P–O bond cleavage is a more preferable pathway for the HF scavenging reaction than O–C bond cleavage. The HF chemical reactivities of these six molecules are 18 > 20 > 29 > 32 > 10 > 13 in descending order. In particular, the HF reactivities of 18 and 20 are found to be −24.07 kcal mol−1 and −21.42 kcal mol−1, respectively, which are higher than that of TMSP (−19.08 kcal mol−1).
The reaction energies between LiF and phosphites show a quite different tendency when compared to the case of HF. In contrast to the HF case, the O–C bond cleavages are much preferred over the P–O bond cleavages. The P–O bond cleavage is thermodynamically unfavorable even for molecules 10, 13, and 32. This difference can be inferred from the much stronger O–Li ionic interaction of (RO)2PO–Li than of RO–Li. As shown in Fig. 3, the Li of the (RO)2PO–Li interacts with two electronegative O atoms to form a strong ionic bond, whereas the Li of RO–Li forms a bond with one O atom. By contrast, H binds only one O atom in either RO–H or (RO)2PO–H. That is, the bonding motif of O–Li differs significantly from that of O–H. Interestingly, despite the different bonding motif, the LiF chemical reactivities of the six molecules are similar to the HF reactivity, in descending order of 18 > 20–29 > 32 > 10 > 13. The LiF reactivities of 18 and 20 are −14.40 kcal mol−1 and −13.74 kcal mol−1, respectively, which are higher than that of TMSP (−12.95 kcal mol−1). We would expect 18 and 20 to be effective in removing HF and LiF molecules in the LIB electrolyte; we also propose 29 and 32 as promising candidates because of their comparable HF/LiF reactivities to that of TMSP.
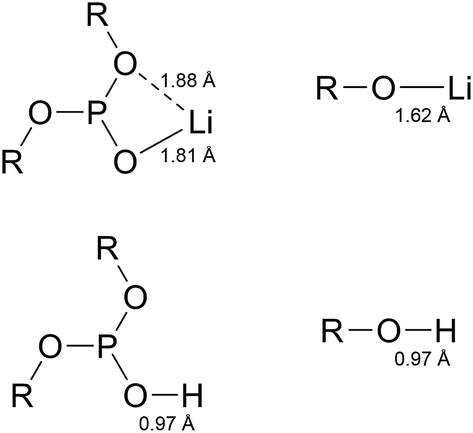 | ||
| Fig. 3 2-D bonding motifs of O–Li and O–H bonds. The bond lengths of molecule 18 are shown. | ||
The characteristics of the most promising molecules 18 and 20 are their low electron affinity values and their large ΔGsolv values compared to the phosphites we considered (Fig. 4). This suggests that these two molecules will be electronically reduced only with difficulty and that the stability of the anions in the solvent is relatively low. All phosphite anions are stabilized in the polar solvent because of the favorable interaction between charge and polar medium; however, the anions of 18 and 20 are relatively less stabilized. Actually, the same is true for TMSP, which is already recognized as a successful additive. Our examination of the additive–solvent interaction using electrostatic and non-electrostatic terms (Table 2) indicates that the large ΔGsolv values of 18 and 20 originate from only the electrostatic term (Fig. 4c). In other words, exposure of the bulky non-polar functional group to the outside lowers the stability of the ions in the polar medium. Therefore, controlling the solvent effect using relevant substituents can be a major issue in the design of additives.
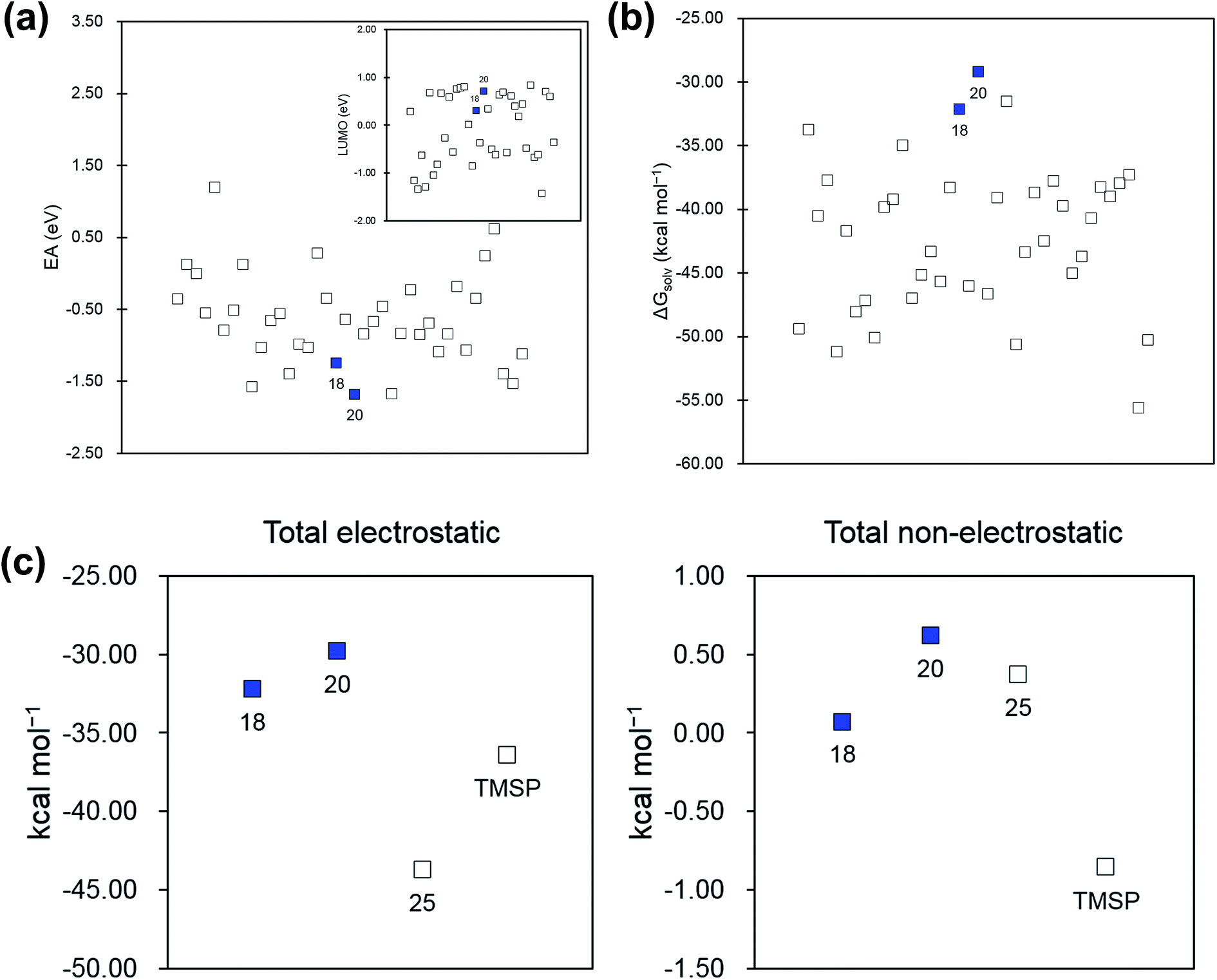 | ||
| Fig. 4 (a) Electron affinity and LUMO values (inset) in eV, (b) ΔGsolv(anion)–ΔGsolv(neutral) values in kcal mol−1, and (c) electrostatic and non-electrostatic terms are plotted. We included the results for molecule 25, which has an average behavior, for comparison. | ||
| 18 | 20 | TMSP | 25 | |
|---|---|---|---|---|
| a We included the results for molecule 25, which has an average behavior, for comparison. | ||||
| Total electrostatic | −32.22 | −29.83 | −36.41 | −43.73 |
| Total non-electrostatic | 0.07 | 0.62 | −0.86 | 0.37 |
| Cavitation | 0.23 | 0.61 | −0.87 | 0.20 |
| Dispersion | −0.20 | 0.01 | 0.01 | 0.23 |
| Repulsion | 0.04 | 0.00 | −0.01 | −0.06 |
Finally, for molecules that meet all three requirements (i–iii), our computational screening suggests four phosphite molecules as promising candidates for effective CEI-forming electrolyte additives: trioleyl phosphite (18), tris(1-adamantyl) phosphite (20), distearyl pentaerythritol diphosphite (29), and tritert-butyl phosphite (32).
In this study, we demonstrated a computational screening protocol for the development of high-performance CEI-forming electrolyte additives using DFT-based calculations of the redox potential and chemical reactivity with HF. Our computational screening included Gibbs free-energy calculations for neutral molecules, cations, and anions; we also considered the bulk solvent effects.
Simplicity is crucial for efficient computational screening; in this respect, researchers still generally use the frontier molecular orbital calculations. In many cases, however, such orbital energy calculations are not sufficiently accurate in screenings, particularly when conducting evaluations on numerous molecules of various sizes. Frontier molecular orbital calculations cannot properly account for structural changes due to redox reactions and bulk solvent effects. Fig. 5 shows the correlations of HOMO and LUMO with OP and RP values, respectively. Our results of the frontier orbital energies and redox potentials are often different qualitatively. We observed poor correlation coefficient (R2) values of 0.75 and 0.44 for oxidation and reduction, respectively.
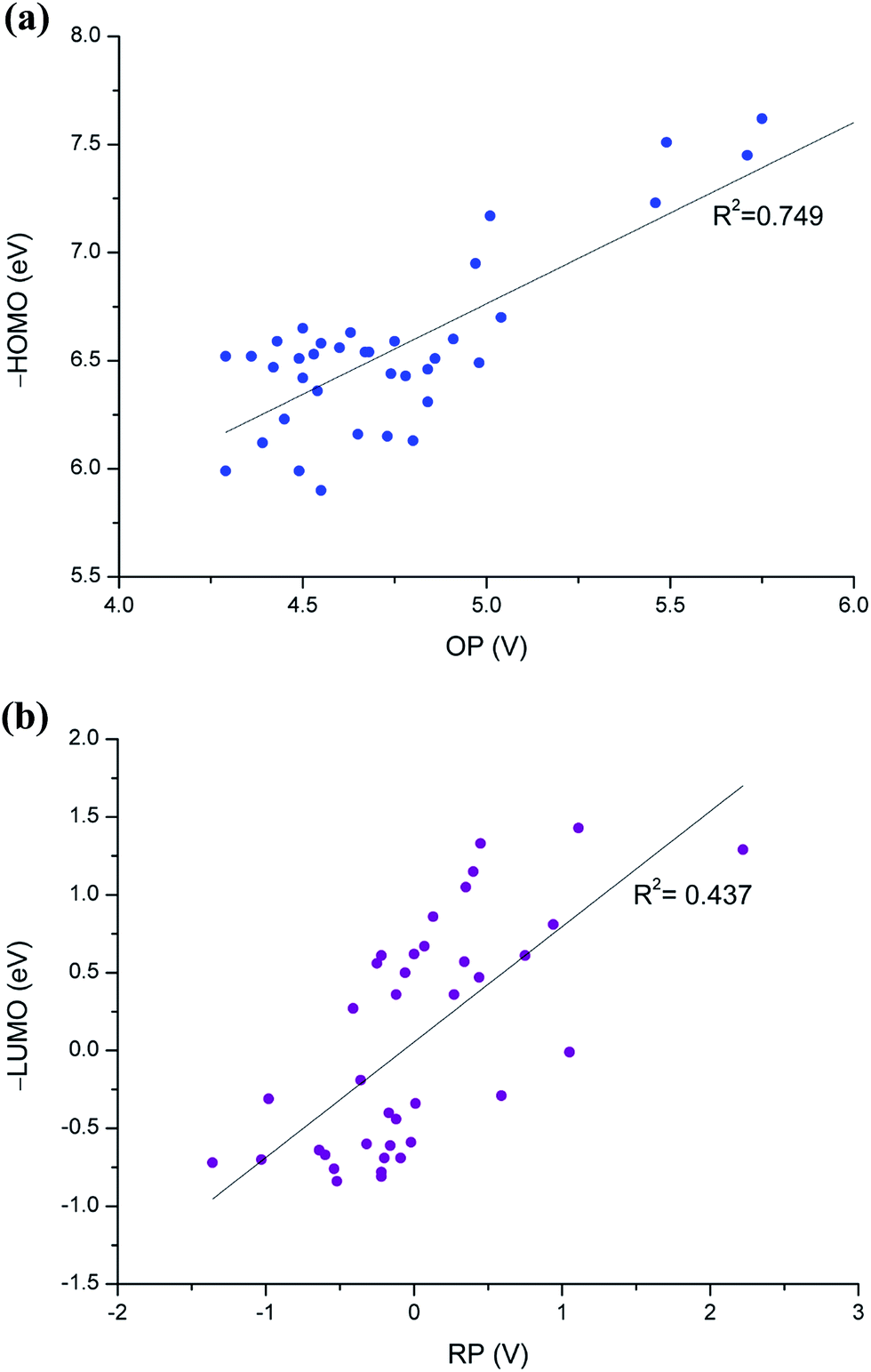 | ||
| Fig. 5 Correlation of HOMO and LUMO energies with OP and RP values, respectively, for the 35 phosphite candidate molecules. | ||
The two main sources of error between the orbital energies and redox potentials are: (1) neglect of structural relaxation when changing the number of electrons in the system; and (2) neglect of solvent effects in the orbital energy calculations. For instance, the EA values of molecules 18 and 20 are significantly low, but the LUMO energies do not correctly reflect this (Fig. 4a). In addition, the solvent effect calculations, which are not covered in the orbital energy calculations, show that the ΔGsolv variation between the various phosphites amounts to 20 kcal mol−1 for the phosphites, as shown in Fig. 4.
Conclusions
We have conducted a computational screening study based on the DFT method to search and develop novel CEI-forming electrolyte additives for use in high-voltage LIBs. Our computational screening suggests trioleyl phosphite, tris(1-adamantyl) phosphite, distearyl pentaerythritol diphosphite, and tritert-butyl phosphite as promising candidates for successful CEI-forming electrolyte additives; these additives will be further scrutinized in future experimental investigations. Our computational screening protocol can provide battery scientists with a faster and less expensive method for the development of novel CEI-forming electrolyte additives for use in high-voltage LIBs.Acknowledgements
The authors acknowledge the financial support by the National Research Foundation of Korea Grant funded by the Korean Government (NRF-2016R1A2B4013374 and 2016R1C1B1009452). This work was also supported by the Energy Efficiency & Resources Core Technology Program of the KETEP granted financial resource from the Ministry of Trade, Industry & Energy (No. 20152020106550).Notes and references
- M. Hu, X. Pang and Z. Zhou, J. Power Sources, 2013, 237, 229–242 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- A. Kraytsberg and Y. Ein-Eli, Adv. Energy Mater., 2012, 2, 922–939 CrossRef CAS.
- F. Lin, I. M. Markus, D. Nordlund, T.-C. Weng, M. D. Asta, H. L. Xin and M. M. Doeff, Nat. Commun., 2014, 5, 3529 Search PubMed.
- Y. Y. Xia, T. Sakai, T. Fujieda, X. Q. Yang, X. Sun, Z. F. Ma, J. McBreen and M. Yoshio, J. Electrochem. Soc., 2001, 148, 723–729 CrossRef.
- J. Shim, R. Kostecki, T. Richardson, X. Song and K. A. Striebel, J. Power Sources, 2002, 112, 222–230 CrossRef CAS.
- L. Yang, M. Takahashi and B. Wang, Electrochim. Acta, 2006, 51, 3228–3234 CrossRef CAS.
- L. Bodenes, R. Naturel, H. Martinez, R. Dedryvère, M. Menetrier, L. Croguennec, J.-P. Pérès, C. Tessier and F. Fischer, J. Power Sources, 2013, 236, 265–275 CrossRef CAS.
- H. Yamane, M. Saitoh, M. Sano, M. Fujita, M. Takada, E. Nishibori and N. Tanaka, J. Electrochem. Soc., 2002, 149, 1514–1520 CrossRef.
- C. J. Curtis, J. X. Wang and D. L. Schulz, J. Electrochem. Soc., 2004, 151, 590–598 CrossRef.
- J. B. Goodenough, J. Power Sources, 2007, 174, 996–10001 CrossRef CAS.
- N. P. W. Pieczonka, Z. Liu, P. Lu, K. L. Olson, J. Moote, B. R. Powell and J.-H. Kim, J. Phys. Chem. C, 2013, 117, 15947–15957 CAS.
- Y.-M. Song, J.-G. Han, S. Park, K. T. Lee and N.-S. Choi, J. Mater. Chem. A, 2014, 2, 9506–9513 CAS.
- J.-G. Han, S. J. Lee, J. Lee, J.-S. Kim, K. T. Lee and N.-S. Choi, ACS Appl. Mater. Interfaces, 2015, 7, 8319–8329 CAS.
- Y.-K. Han, J. Yoo and T. Yim, J. Mater. Chem. A, 2015, 3, 10900–10909 CAS.
- Y.-M. Song, C.-K. Kim, K.-E. Kim, S. Y. Hong and N.-S. Choi, J. Power Sources, 2016, 302, 22–30 CrossRef CAS.
- Z. D. Li, Y. C. Zhang, H. F. Xiang, X. H. Ma, Q. F. Yuan, Q. S. Wang and C. H. Chen, J. Power Sources, 2013, 240, 471–475 CrossRef CAS.
- J. Pires, A. Castets, L. Timperman, J. Santos-Peña, E. Dumont, S. Levasseur, C. Tessier, R. Dedryvère and M. Anouti, J. Power Sources, 2015, 296, 413–425 CrossRef CAS.
- Z. Zhou, Y. Ma, L. Wang, P. Zuo, X. Cheng, C. Du, G. Yin and Y. Gao, Electrochim. Acta, 2016, 216, 44–50 CrossRef CAS.
- L. Cheng, R. S. Assary, X. Qu, A. Jain, S. P. Ong, N. N. Rajput, K. Persson and L. A. Curtiss, J. Phys. Chem. Lett., 2015, 6, 283–291 CrossRef CAS PubMed.
- M. D. Halls and K. Tasaki, J. Power Sources, 2010, 195, 1472–1478 CrossRef CAS.
- M. H. Park, Y. S. Lee, H. Lee and Y.-K. Han, J. Power Sources, 2011, 196, 5109–5114 CrossRef CAS.
- N. N. Sinha, J. C. Burns and J. R. Dahn, J. Electrochem. Soc., 2014, 161, A1084–A1089 CrossRef CAS.
- D. Aurbach, B. Markovsky, Y. Talyossef, G. Salitra, H.-J. Kim and S. Choi, J. Power Sources, 2006, 162, 780–789 CrossRef CAS.
- J.-H. Kim, N. P. W. Pieczonka and L. Yang, ChemPhysChem, 2014, 15, 1940–1954 CrossRef CAS PubMed.
- Y.-K. Han, J. Yoo and T. Yim, Electrochim. Acta, 2016, 215, 455–465 CrossRef CAS.
- S. Mai, M. Xu, X. Liao, J. Hu, H. Lin, L. Xing, Y. Liao, X. Li and W. Li, Electrochim. Acta, 2014, 147, 565–571 CrossRef CAS.
- H. Sun, P. Ren and J. R. Fried, Comput. Theor. Polym. Sci., 1998, 8, 229–246 CrossRef CAS.
- H. Sun, J. Phys. Chem. B, 1998, 102, 7338–7364 CrossRef CAS.
- X. P. Chen, C. A. Yuan, C. K. L. Wong, S. W. Koh and G. Q. Zhang, Mol. Simul., 2011, 37, 990–996 CrossRef CAS.
- P. Johansson, J. Phys. Chem. A, 2006, 110, 12077–12080 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404–417 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS PubMed.
- L. Xing, C. Wang, W. Li, M. Xu, X. Meng and S. Zhao, J. Phys. Chem. B, 2009, 113, 5181–5187 CrossRef CAS PubMed.
- L. Xing, W. Li, C. Wang, F. Gu, M. Xu, C. Tan and J. Yi, J. Phys. Chem. B, 2009, 113, 16596–16602 CrossRef CAS PubMed.
- J. Jeon, S. Yoon, T. Park, J.-J. Cho, S. Kang, Y.-K. Han and H. Lee, J. Mater. Chem., 2012, 22, 21003–21008 RSC.
- H. M. Jung, S.-H. Park, J. Jeon, Y. Choi, S. Yoon, J.-J. Cho, S. Oh, S. Kang, Y.-K. Han and H. Lee, J. Mater. Chem. A, 2013, 1, 11975–11981 CAS.
- J. Self, D. S. Hall, L. Madec and J. R. Dahn, J. Power Sources, 2015, 298, 369–378 CrossRef CAS.
- O. Borodin, W. Behl and T. R. Jow, J. Phys. Chem. C, 2013, 117, 8661–8682 CAS.
- M. J. Frisch, et al., Gaussian 09 (Revision D.01), Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- P. von R. Schleyer, R. C. Fort, W. E. Watts, M. B. Comisarow and G. A. Olah, J. Am. Chem. Soc., 1964, 86, 4195–4197 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |