
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Surface-enhanced Raman scattering of pyrazine on Au5Al5 bimetallic nanoclusters†
Quanjiang Lia,
Qianqian Dingab,
Weihua Linb,
Jiangcai Wangb,
Maodu Chen *a and
Mengtao Sun*bc
*a and
Mengtao Sun*bc
aKey Laboratory of Materials Modification by Laser, Electron, and Ion Beams, Ministry of Education, School of Physics and Optoelectronic Technology, Dalian University of Technology, Dalian 116024, People's Republic of China. E-mail: mdchen@dlut.edu.cn
bBeijing Key Laboratory for Magneto-Photoelectrical Composite and Interface Science, School of Mathematics and Physics, University of Science and Technology Beijing, Beijing 100083, China. E-mail: mengtaosun@ustb.edu.cn
cBeijing National Laboratory for Condensed Matter Physics, Beijing Key Laboratory for Nanomaterials and Nanodevices, Institute of Physics, Chinese Academy of Sciences, Beijing, 100190, People's Republic of China
First published on 20th February 2017
Abstract
In this study, we theoretically investigated the Raman and absorption spectra of pyrazine adsorbed on Au5Al5 bimetallic nanoclusters by a time-dependent density functional theory (TD-DFT) method. The surface-enhanced resonance Raman scattering (SERRS) spectra of pyrazine absorbed on different isomers and sites of the Au5Al5 cluster were simulated. The visualization of orbital transitions in electronic transitions was used to analyze the enhancement mechanism of SERRS spectroscopy. Compared with those of isolated pyrazine excited at 598 nm, the SERRS of pyrazine–Au–Au4Al5-a excited at the same incident light can be enhanced on the order of 104, which is a typical charge transfer (CT) resonance excitation and charge transfer from substrate to pyrazine. Due to the fact that the intensity of ultraviolet SERRS can be significantly enhanced to 1.2 × 106 A4 per amu for pyrazine–Au–Au4Al5-a model at 280 nm, the Au5Al5 cluster may be a good candidate for research of the ultraviolet SERRS materials. Other key factors that can change the intensity of SERRS include the resonance excitation wavelength, oscillator strength of the electronic excited state, metal–molecule binding site and structure of the substrate cluster. Hence, the optical properties of complexes can be tuned by varying these factors.
1. Introduction
Raman signals can be greatly enhanced when the molecules are absorbed on rough metallic surfaces.1–4 As a molecular detection tool with high sensitivity, surface-enhanced Raman scattering (SERS) overcomes the disadvantages of conventional Raman spectra, and plays an important role in the studies of spectroscopic properties and electronic structures of molecules.5–11 There are two generally accepted enhanced mechanisms for SERS: the electromagnetic field enhancement and the charge transfer enhancement. For the electromagnetic field enhancement mechanism, the excitations in the metal cluster can cause collective oscillation of electrons, which results in local surface plasmon resonance (LSPR), and then the molecular Raman signals can be strongly enhanced by a factor of 1011.7,9,12–14 However, for the charge transfer enhancement, the incident light radiates on the nanostructures causing charge transfer (CT) between the metal clusters and adsorbate molecules, which results in some Raman peaks being selectively enhanced by a factor of 105.15–18 The method of visualization of orbital transitions in electronic excitations of a molecule–nanostructure system has been developed in recent years,16,19 where detailed information about the electronic transitions can be easily obtained and analyzed. Then, the contributions of intra-metallic excitation and charge transfer excitation can be distinguished.In principle, all metals can enhance local electromagnetic fields, and the intensity of SERS mainly depends on the ability of metals to support strong LSPR. Since the discovery of rough sliver substrates that strongly enhance signals of Raman spectra, extensive research of SERS has focused on coinage metal.1,2,5,8,12,15,20,21 With the exploration of metal substrates, transition metal substrates have been theoretical designed and experimentally synthesized to enhance the SERS of adsorbate molecules, which are mainly based on the electromagnetic field enhancement mechanism, such as Pt, Ru, Rh, Pd, Fe, Co, Ni.3,22–25 In addition, bimetallic nanoparticles have been widely investigated due to their unique properties on spectroscopic and electronic structures. In recent years, numerous studies on SERS have shown that the optical properties of molecules can be affected by the multicomponent substrates. Moreover, the SERS of molecules absorbed on different bimetallic nanoclusters were simulated and analyzed, such as planar nanocluster Ac@Au7,26 and core@shell nanoclusters of Cr@Au12, W@Ag12, Mo@Ag12, and Th@Au14.27–30
In this study, using a time dependent density functional theory method, we theoretically investigated the enhancement mechanism of surface-enhanced resonance Raman scattering (SERRS) spectroscopy of pyrazine absorbed on Au5Al5 bimetallic nanoclusters. We select the CT excitations and the intra-metallic excitations, which can strongly enhance the SERS of pyrazine in the visible or the ultraviolet regions, to calculate the SERRS of complexes. The electromagnetic enhancements and the chemical enhancements for this model system are consistently treated based on a short-time approximation for the Raman scattering cross-section. Due to the inert and hypo-toxic properties of Au, gold can be used as an appropriate substrate in medical applications. Investigations on gold substrates have shown excellent enhancement of SERRS. A study on the absorption and SERS spectra of pyridine interacting with Au20 by Schatz et al. showed that the Au20 nanocluster can significantly enhance the intensity of SERS up to about 103 to 104.31 In addition, aluminum is a good metal substrate for ultraviolet SERRS. An investigation on resonance Raman spectroscopy of pyrazine absorbed on two Al3 clusters or one Al4 cluster has been reported by Sun et al.32 It revealed that the enhancements can respectively reach up to about 104 and 105, compared with the Raman intensity of pyrazine excited at the same incident light, due to intra-metallic excitation and charge transfer excitation. The models of Au5Al5 clusters can be seen in Fig. 1(a) and (b). The structures of Au5Al5-a and Au5Al5-b have been demonstrated to be stabilized,33 and Au5Al5-a is the lowest energy structure of Au5Al5 clusters. The Au5Al5 bimetallic nanoclusters protected by appropriate ligands are expected to have a favorable influence on the enhancement mechanism of SERRS for probe molecules. Pyrazine is a suitable probe molecule to investigate the SERRS enhancement mechanism of substrates in metal–molecule models8,32 owing to the bonding interaction between the nitrogen and metal nanoparticles. The metal–molecule models, used in this study, are shown in Fig. 1(c–f), which can be considered as models of simplified, tip-enhanced Raman scattering.34,35
 | ||
| Fig. 1 The models of (a) Au5Al5-a (C4V), (b) Au5Al5-b (C4V), (c) pyrazine–Au–Au4Al5-a (C2V), (d) pyrazine–Al–Au5Al4-a (CS), (e) pyrazine–Au–Au4Al5-b (C2V), (f) pyrazine–Al–Au5Al4-b (CS). | ||
2. Methods
All calculations discussed here were performed with the 2014.07 version of the Amsterdam density functional (ADF) suite.36 The structures of complexes were optimized at the density functional theory (DFT)37 level of theory using the exchange–correlation functional BP86.38,39 A triple-ζ with polarization functions and a (TZP)40 Slater type basis set were used, where the Au(1s2–4f14), Al(1s2–2p) are the frozen core, and C, N and H are full electrons. Relativistic corrections of elements (Au, Al) at the zeroth order regular approximation (ZORA)41–44 level have been taken into account. The electronic transitions were calculated by a TD-DFT method,45 using the same functional and basis sets. The adiabatic local density approximation (ALDA) was employed in all calculations of polarizability. The SERRS intensities were calculated by differential Raman scattering cross-section (DRSC), which is given by eqn (1):46
 | (1) |
For the corresponding physical quantities, h is the Planck constant, c is the speed of light, kB is the Boltzmann constant, and T is the Kelvin temperature. The terms  and
and 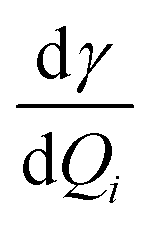 are the derivatives of the isotropic and anisotropic polarizability of the ith vibrational mode, respectively, and ω0 and ωi respectively denote the frequencies of the incident light and the ith vibrational mode. The finite lifetime was included using a damping parameter of 0.004 a.u., which was determined by the dephasing time of the conduction electrons and reasonable for metal clusters and resonant Raman intensities.20
are the derivatives of the isotropic and anisotropic polarizability of the ith vibrational mode, respectively, and ω0 and ωi respectively denote the frequencies of the incident light and the ith vibrational mode. The finite lifetime was included using a damping parameter of 0.004 a.u., which was determined by the dephasing time of the conduction electrons and reasonable for metal clusters and resonant Raman intensities.20
3. Results and discussion
Raman scattering in ground state
The stabilizations for the structures of Au5Al5-a (C4V) and Au5Al5-b (C4V) have been demonstrated in a recent report,33 and the calculations have shown that Au5Al5-a is the lowest energy structure, and Au5Al5-b is a typical stabilized local minima for the Au5Al5 cluster. The unique structural symmetry of the Au5Al5 cluster allowed us to study two very different tip binding sites, which we denoted as the pyrazine–Au–Au4Al5 model and the pyrazine–Al–Au5Al4 model. The two tip sites are presented in Fig. 1, which have been chosen for their quite different local chemical environments. Similar to most previous studies on pyridine/pyrazine adsorbed on a gold cluster or aluminum cluster,26,27,31,32 we assumed that pyrazine binds to the Au5Al5 cluster through the nitrogen atom. The structures of complexes were optimized by density functional theory at the BP86/TZP level, and more than ten different initial conformations were designed and optimized for each complex. Herein, the orientation of pyrazine on a cluster was adjusted so that every representative local minimum could be obtained for each complex. The lowest energy structure of each complex was chosen as the representative model (see Fig. 1), and there were no imaginary frequencies for each structure.Fig. 1(c–f) show the stable configurations of pyridine adsorbed on an Au5Al5 cluster in our calculations, and pyrazine binds to bimetallic cluster through the interaction between the nitrogen atom and the tip atom of the bimetallic electrode. In order to explore the SERS enhancement mechanism of pyrazine binding to Au5Al5 clusters, the static electronic polarizability components of the four models were calculated (Table 1). These calculated results of complexes were significantly increased compared to those of pyrazine, particularly in the z-direction, which resulted in strong enhancement of the static Raman intensity of complexes. These changes in the polarizabilities are due to the interactions between the molecular and metal clusters. To analyze the charge density redistribution during the formation of the complexes, atomic charges were computed within the Voronoi deformation density (VDD) scheme.36 The VDD charges correspond to the quantity of electronic charge, which is entering or leaving a region of space around the nucleus. The calculated values of the bonding energy, Al/Au–N bond length, and VDD charge transfer for the complexes are shown in Table 2. The VDD charges denote the electronic charge transfers from molecule to metal, which agrees with the visual graph of the calculated deformation density for each complex (Fig. 2). A visual graphical representation of the calculated deformation density for complexes can be seen in Fig. 2(a–d). It was found that the direction of density flow agrees well with the overall transfer of charge from molecule to substrate. The enhanced, static electronic polarizability and the large charge transfer resulted in the enhancement of SERS in the ground state.
| αxx | αyy | αzz | |
|---|---|---|---|
| Pyrazine | 72.5 | 30.9 | 63.1 |
| Pyrazine–Au–Au4Al5-a | 442.9 | 412.1 | 554.9 |
| Pyrazine–Al–Au5Al4-a | 447.2 | 420.7 | 768.7 |
| Pyrazine–Au–Au4Al5-b | 469.3 | 442.5 | 490.6 |
| Pyrazine–Al–Au5Al4-b | 475.7 | 425.3 | 667.1 |
| Pyrazine–Au–Au4Al5-a | Pyrazine–Al–Au5Al4-a | Pyrazine–Au–Au4Al5-b | Pyrazine–Al–Au5Al4-b | |
|---|---|---|---|---|
| ΔEelectrostatic | −39.46 | −98.44 | −31.11 | −42.59 |
| ΔEPauli | 49.7 | 140.97 | 42.6 | 64.34 |
| ΔEorbital | −18.22 | −57.38 | −14.58 | −24.79 |
| Etotal binding energy | −7.98 | −14.85 | −3.08 | −3.04 |
| Au–N bond length | 2.469 | 2.034 | 2.568 | 2.367 |
| VDD charge (molecule → metal) | 0.13 | 0.128 | 0.121 | 0.131 |
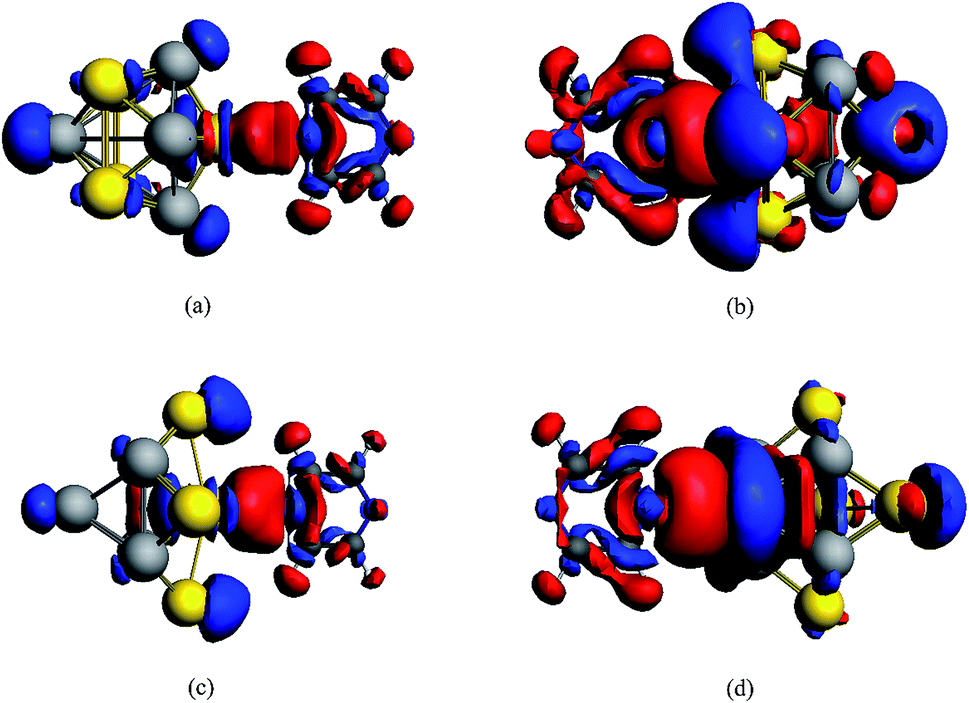 | ||
| Fig. 2 Calculated deformation density (Δρ) isosurfaces and contour plots for complexes. An isosurface value of 0.0004 a.u. with enhanced density is blue and the depletion density is red. Δρ = ρtotal − ρpyrazine − ρAu5Al5. (a) Pyrazine–Au–Au4Al5-a, (b) pyrazine–Al–Au5Al4-a, (c) pyrazine–Au–Au4Al5-b, (d) pyrazine–Al–Au5Al4-b. | ||
Then, the static Raman spectra of pyrazine and complexes in the ground state were simulated and analyzed, as shown in Fig. 3 and S5,† respectively. The well-known seven vibrational modes of pyrazine at 591, 697, 1006, 1213, 1326, 1519 and 1554 cm−1 have been analyzed in detail, which have significant signals in the prior experiments on SERS. The vibrational motions and frequencies of those important modes are displayed in Fig. S6.† We found that the completely symmetric modes at 591 (ring-stretching), 1006 (ring breathing), 1213 (C–H bending), and 1554 cm−1 (ring-stretching) were strongly enhanced in the static Raman spectra of the four complexes. For the structure of pyrazine–Au–Au4Al5-a (C2V), the Raman peak of pyrazine at 1554 cm−1 was enhanced 15 times and showed a blue shift of about 6 cm−1, compared with that of pyrazine alone. In addition, the Raman peak at 1006 cm−1 was separated into two Raman peaks due to the break of molecular vibrational symmetry. For the pyrazine–Al–Au5Al4-a (CS) structure, the Raman peak of pyrazine at 1006 cm−1 was enhanced about 7 times than that of pyrazine, and some new vibrational peaks appeared in the spectrum. For the pyrazine–Au–Au4Au5-b (C2V) structure, the Raman peak of pyrazine at 1006 cm−1 was weakly enhanced about 2 times. Due to the break in molecular vibrational symmetry, this peak was also separated into two Raman peaks. For the pyrazine–Al–Au5Al4-b (CS) model, the Raman intensity was strongly and selectively enhanced due to charge redistribution between the molecule and substrate. The Raman peak at 1554 cm−1 was enhanced by a factor of 3.6 × 102, which is the strongest static Raman intensity in these four complexes. All of these results were induced by the charge redistribution of the molecular–metal complex due to non-resonant interaction between the substrate and the adsorbate.
 | ||
| Fig. 3 Static Raman spectra of pyrazine in the ground state. (a) Pyrazine, (b) pyrazine–Au–Au4Al5-a, (c) pyrazine–Al–Au5Al4-a, (d) pyrazine–Au–Au4Al5-b, (e) pyrazine–Al–Au5Al4-b. Spectra were broadened by a Lorentzian having a width of 20 cm−1. | ||
The frequency shifts of Raman peaks were observed for the four complexes, and the different shifts for the complexes were in agreement with the different bonding interactions of the four complexes. The largest shifts were observed for the modes at 591 (ring-stretching), 1006 (ring breathing), and 1554 cm−1 (ring-stretching). The modes at 1554 cm−1 mainly involved C–C stretching with the carbon next to the nitrogen atom vibrating towards the metal cluster, and the 591 and 1006 cm−1 modes consisted of vibrational motion of the N atom along the Al/Au–N bond. Therefore, these shifts are expected to be influenced more by the interaction with the metal cluster. In addition, the structure of the substrate and the binding site can also affect the frequency shift of Raman peaks in complexes.
Electronic transition properties
Fig. 4 shows the simulated UV-vis absorption spectra of four complexes and two metal clusters in this study. The wavelength range was from 1.7 eV to 5 eV, and the spectra have been broadened by a Lorentzian with a width of 0.1 eV. It was found that the metal clusters Au5Al5-a and Au5Al5-b both have strong optical absorption peaks in the ultraviolet region around 4.7 eV, which was not discussed in the prior study.33 For pyrazine absorbed on the Au5Al5 cluster, the absorption strength in the ultraviolet region decreased, and several new absorption peaks appeared in the region from 400 to 700 nm, compared with the bare clusters. The detailed electronic excitations of pyrazine are shown in ESI part 1.† It is clear that the electronic transitions of pyrazine were in the region of deep ultraviolet region. The absorption band of complexes can be red shifted to the visual regions, even near the IR region, because of the interaction between the molecule and metal nanoclusters. Therefore, the electronic transitions that can strongly enhance the SERS of pyrazine in the visible and the ultraviolet regions were selected for the four complexes. Table 3 demonstrates the detailed properties of electronic transitions selected in this study. | ||
| Fig. 4 Simulated UV-vis absorption spectra. (a) The absorption spectra of pyrazine absorbed on Au5Al5-a, (b) the absorption spectra of pyrazine absorbed on Au5Al5-b. Spectra have been broadened by a Lorentzian having a width of 0.1 eV. | ||
| Models | E | f | Components of orbital transitions |
|---|---|---|---|
| Pyrazine–Au–Au4Al5-a | 2.07 | 0.0235 | 98.6% (15b2 → 16b2) |
| 4.43 | 0.5131 | 22.6% (29a1 → 35a1), 8.6% (14b2 → 18b2), 6.6% (19b1 → 22b1), 6.0% (27a1 → 32a1) | |
| Pyrazine–Al–Au5Al4-a | 4.47 | 0.2277 | 78.4% (47a′ → 59a′), 2.8% (47a′ → 58a′), 2.7% (38a′ → 48a′) |
| Pyrazine–Au–Au4Al5-b | 3.68 | 0.2178 | 26.5% (20b1 → 11a2), 12.8% (31a1 → 19b2), 12.8% (30a1 → 18b2), 10.5% (15b2 → 34a1) |
| 4.66 | 0.2122 | 27.3% (27a1 → 33a1), 11.1% (27a1 → 34a1), 7.0% (14b2 → 18b2), 6.8% (15b2 → 19b2) | |
| Pyrazine–Al–Au5Al4-b | 4.11 | 0.1620 | 28.4% (26a′′ → 30a′′), 17.4% (40a′ → 49a′), 13.1% (39a′ → 49a′), 4.6% (27a′′ → 30a′′) |
| 4.56 | 0.1793 | 23.1% (37a′ → 30a′′), 11.5% (39a′ → 30a′′), 4.7% (24a′′ → 50a′), 4.4% (35a′ → 30a′′) |
A method of visualization for electronic transitions was used to analyze the components of electronic transitions shown in Fig. 5 and ESI part 2.† According to the information of electronic transitions and the visual graphics of molecular orbitals in different electronic transitions, we can select the appropriate electronic resonance excitation for SERRS. The excitations which can strongly enhance the SERS of pyrazine in the visible and the ultraviolet regions were selected for the four complexes. The absorption spectrum of pyrazine–Au–Au4Al5-a was dominated by a broad peak with a maximum at 280 nm, which was associated with the Au5Al5 cluster. In addition to the excitations associated with Au5Al5 cluster, the presence of pyridine also created a new charge-transfer excitation at 598 nm. The molecular orbitals involved in the two electronic transitions are shown in Fig. 5. It is clearly shown that the electronic resonance excitation at 598 nm was a charge transfer excitation and charge transfer from substrate to pyrazine, while intra-metallic charge redistribution and charge transfer transitions were all included in the excitation at 280 nm. The absorption spectrum of pyrazine–Al–Au5Al4-a was dominated by a broad peak with a maximum at 277 nm as shown in Fig. 4(a). For the cases in Fig. S2,† the visual graphics of molecular orbitals in the electronic transition at 277 nm show that the excitation included intra-metallic charge redistribution and charge transfer from metal to molecule, and the dominant contribution was the intra-metallic excitation. The absorption spectrum of pyrazine–Au–Au4Al5-b was dominated by two broad peaks with maximum at 266 and 337 nm, respectively, as shown in Fig. 4(b). The electronic resonance excitations at 337 and 266 nm were the mixture of intra-cluster excitation and metal to pyrazine charge transfer excitation, as shown in Fig. S3,† and the dominant contribution was the intra-cluster charge redistribution. For pyrazine–Al–Au5Al4-b, Raman spectra excited at the incident wavelengths of 301 and 272 nm were resonance Raman spectra. The electronic transition at 301 nm was an intra-metallic excitation, and the dominant contribution for the electronic transition at 272 nm was also the intra-cluster charge redistribution, as shown in Fig. S4.† As a result, the absorption spectra of four pyrazine–Au5Al5 complexes were all dominated by the intra-cluster electronic transitions. It is expected that the intensity and profile of SERRS can be significantly influenced by the orientation and degree of intra-metallic charge redistribution. If the metal cluster is large enough, intra-metallic excitation and charge transfer excitation will be the mechanisms of electromagnetic and chemical enhancement, respectively.
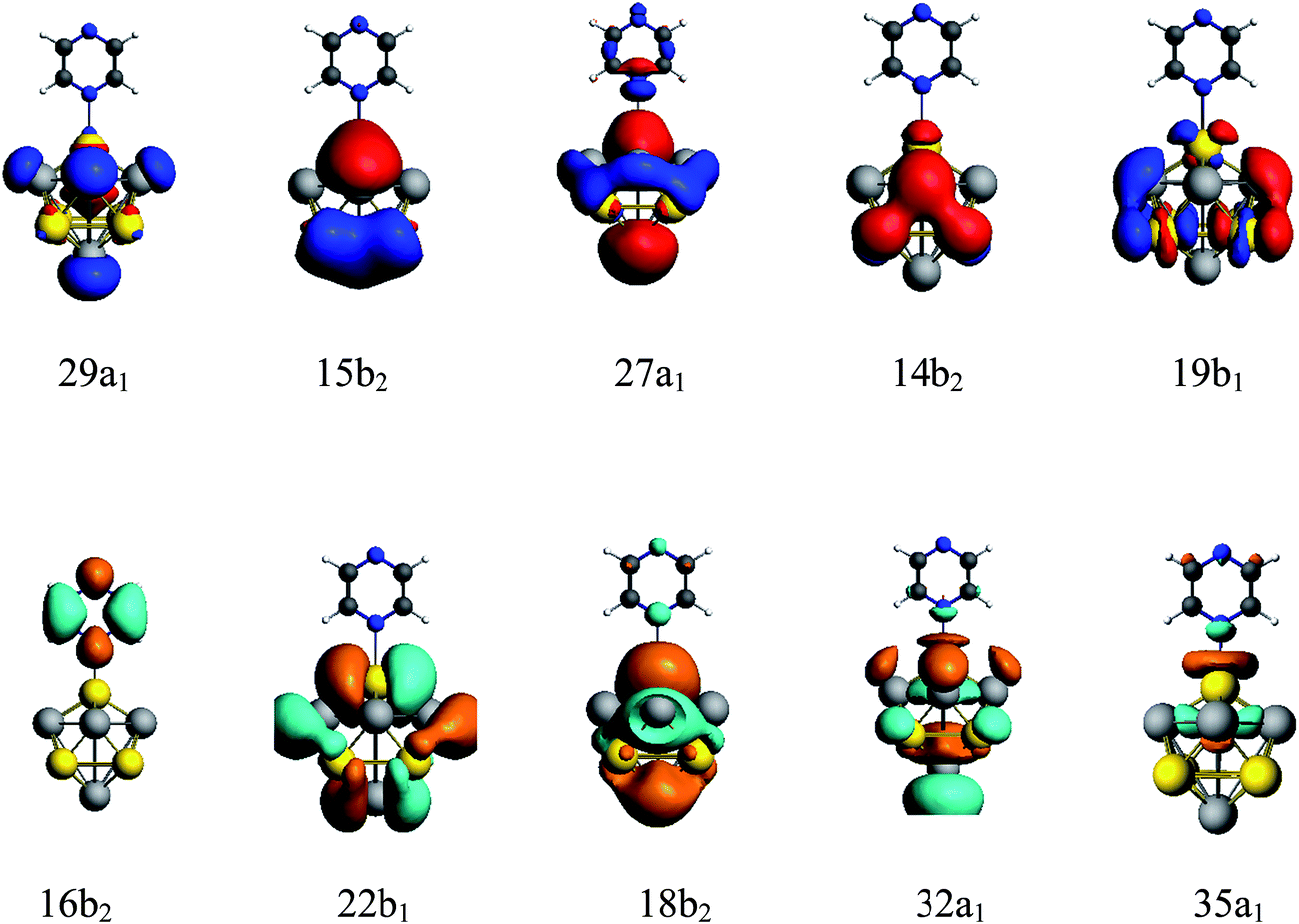 | ||
| Fig. 5 Molecular orbitals of pyrazine–Au–Au4Al5-a complex in electronic transitions at 598 and 280 nm. | ||
Resonant Raman scattering
Due to the interaction between the molecular and substrate clusters, there were new CT excitation transitions that involved charge transfer from the bimetallic cluster to pyrazine. The pure CT transitions were mainly limited in the range of 500–800 nm for the models in this study, and the variations in the adsorption site and structure of metal cluster could tune the energy of the CT excitations. In addition, some electronic excitations in the ultraviolet region were the intra-metallic charge redistribution, as seen in Fig. 5 and ESI part 2,† and the electromagnetic enhancement mechanism was significant when the incident light was resonated with those excitations. We selected the CT excitations and the intra-metallic excitations, which can strongly enhance the SERS of pyrazine in the visible or the ultraviolet regions, to calculate the SERRS of pyrazine absorbed on the Au5Al5 cluster.For pyrazine, the absorption peaks were in the deep ultraviolet region as shown in Fig. S1.† Therefore, the Raman spectra excited at wavelengths near the absorption peaks of pyrazine were near-resonant excitation Raman spectra as shown in Fig. S5(b–g).† The Raman spectra of pyrazine excited in the visible region were normal Raman spectra, which were almost the same as observed by comparing Fig. 3(a) and S5(a).†
We calculated the resonance Raman spectra of pyrazine–Au–Au4Al5-a at 598 and 280 nm, as shown in Fig. 6(a) and (b). The Raman intensity at 598 nm was enhanced 1.2 × 104 times (see Fig. 6(a)), compared with the static normal Raman spectra of isolated pyrazine excited at 598 nm. There was a weak electronic transition at 598 nm for the pyrazine–Au–Au4Al5-a structure, which was a charge transfer absorption peak and the charge transfer from Au5Al5-a to pyrazine with the oscillator strength of 0.023 a.u., as shown in Fig. 4(a) and 5. Therefore, the SERRS spectrum excited at 598 nm was strongly enhanced by the charge transfer mechanism. The Raman intensity excited at 280 nm was selectively enhanced 1.6 × 104 times, compared to the Raman spectra of pyrazine excited at the same wavelength (see Fig. 6(b)). It was found that the electronic transition at 280 nm was the mixture of intra-metallic excitation and charge transfer excitation, but the visual graphics of molecular orbitals in the electronic transition demonstrated that the dominant contribution was the electromagnetic mechanism.
 | ||
| Fig. 6 SERRS of pyrazine adsorbed on different substrates, excited with different wavelength lasers. Spectra were broadened by a Lorentzian having a width of 20 cm−1. | ||
For the pyrazine–Al–Au5Al4-a structure, strong electronic excited state existed at 277 nm, and the resonance Raman spectroscopy excited at this wavelength is presented in Fig. 6(c). It was found that the intensity of SERRS at 277 nm was selectively enhanced about 104 times, compared with the corresponding normal Raman spectra of pyrazine. The intra-metallic excitation and charge transfer excitation were all included in this resonance excitation. The visual graphics of molecular orbitals in the electronic transition at 277 nm denoted that the dominant contribution was the intra-cluster charge redistribution, which was evidence for electromagnetic enhanced mechanism. Hence, according to the results of pyrazine–Au–Au4Al5-a and pyrazine–Al–Au5Al4-a models, the variation of adsorption sites can greatly affect the intensity of SERRS.
We compared the resonance Raman spectra of the pyrazine–Au–Au4Al5-b structure with the static normal Raman spectra of pyrazine excited at 337 and 266 nm as shown in Fig. 6(d) and (e). It was found that the SERRS enhancement can be approximately 3 × 103 times from the contributions of the excitation at 337 nm, compared with the normal Raman spectra. The visual graphics of molecular orbitals in electronic resonance excitation at 337 nm are presented in Fig. S3,† which revealed that the Raman spectra excited at 337 nm should be electromagnetically enhanced SERRS. At 266 nm, the SERRS intensity of pyrazine–Au–Au4Al5-b was of the order of 106 A4 per amu, which was 2 × 103 times that of the Raman spectrum of pyrazine at 266 nm. In addition, the electronic transition at 266 nm for pyrazine–Au–Au4Al5-b included an intra-metallic excitation and charge transfer excitation, and the dominant contribution was the intra-metallic excitation. As a result, the enhancement of Raman spectrum excited at 266 nm was due to the electromagnetic field mechanism.
Fig. 4 and S4† show that the absorption peak at 301 nm for pyrazine–Al–Au5Al4-b structure was a strong broad peak, and the dominant contribution was the intra-cluster excitation. The SERRS intensity was enhanced 3 × 102 times (see Fig. 6(f)) due to the resonance with the electronic excitation at 301 nm, whose oscillator strength was 0.162. We also compared the SERRS excited at 272 nm (see Fig. 6(g)) with the normal Raman spectrum of pyrazine, and the SERRS intensity was selectively enhanced 1.3 × 103 times. Fig. S4† demonstrated that the electronic transition at 272 nm was the intra-metallic excitation, and the visual graphics of molecular orbitals in the electronic transition revealed that the electromagnetic mechanism had a strong effect on the SERRS enhancement of pyrazine–Al–Au5Al4-b at 272 nm.
As discussed above, the Au5Al5 clusters can strongly enhance the SEERS spectra of pyrazine in the visible and ultraviolet regions. The selectivity of enhancement is shown in the calculated results of SERRS, as shown in Fig. 6, which is due to the significant contributions of the electromagnetic and CT enhancement mechanism. Pyrazine–Au–Au4Al5-a was the best structure to observe the strongest SERS enhancement in the visible and the ultraviolet region. The SERRS intensity of pyrazine–Au–Au4Al5-a model excited at 598 and 280 nm was of the order of 105 and 106 A4 per amu, and the dominant contribution was the CT enhancement and electromagnetic enhancement mechanisms, respectively. In addition, all the four complexes had strong SERRS in the ultraviolet region, among which Au5Al5 clusters may be good candidates in studies of ultraviolet SERRS materials. These results indicated strong dependence on the incident excitation wavelength and the oscillator strength of the electronic excited state. The metal–molecule binding site of the adsorbate molecule absorbed on the substrate cluster also had an important effect on the SERRS enhancement, and the different isomers of Au5Al5 were the most important factor that affected the energy and the profile of the absorption peaks. It can be concluded that the energy of the transitions and the intensity of SERRS can be tuned by varying the structure of the substrate and the binding site of the molecule absorbed on the metal cluster.
4. Conclusion
In this study, a detailed analysis of the quantum chemical results explained the enhancement mechanism of SERRS for bimetallic nanoclusters, particularly focused on the Au5Al5 clusters. The electromagnetic enhancement and the chemical enhancement for our model systems were consistently treated based on a short-time approximation for the Raman scattering cross-section. The visualization of charge transfer provided conclusive evidence for the enhancement mechanism of SERRS spectroscopy. The static chemical enhancement can reach up to a value that is 3.6 × 102 times that for the pyrazine–Al–Au5Al4-b complex due to the contributions of charge transfer between the probe molecule and bimetallic nanocluster in the ground state. Due to the interactions between molecular and substrate clusters, new CT excitation transitions appeared in the visible region. We selected the CT and the intra-cluster excitations, which can strongly enhance the SERS of pyrazine in the visible or the ultraviolet regions, to calculate the SERRS of complexes. The intensity of SERRS can be significantly enhanced in the visible region for the pyrazine–Au–Au4Al5-a model, which can reach the order of magnitude of 106 A4 per amu at the incident light of 598 nm due to CT-SERRS enhancement mechanism. All the four complexes had strong SERRS in the ultraviolet region, and the SERRS enhancement of pyrazine–Al–Au5Al4-a is the strongest and can reach 107 A4 per amu at an incident wavelength of 280 nm. Therefore, Au5Al5 clusters may be good candidates in the search for ultraviolet SERRS material. The results also indicated the key factors that affect the intensity of SERRS, including the incident excitation wavelength, oscillator strength of the electronic excited state, metal–molecule binding site and the structure of substrate cluster. As a result, the optical properties of complexes and the wavelengths of excitation can be tuned by varying these factors. We hope our research will be useful to characterize excellent multi-composition SERS substrates, and promote the understanding of the SERRS enhancement mechanism of bimetallic substrate systems.Acknowledgements
This study was supported by the NSFC (Grant No. 11374045, 91436102, 11374353 and 11274149) and Program for New Century Excellent Talents in University (Grant No. NCET-12-0077).References
- M. G. Albrecht and J. A. Creighton, J. Am. Chem. Soc., 1977, 99, 5215–5217 CrossRef CAS.
- D. L. Jeanmaire, R. P. V. Duyne, D. L. Jeanmaire and R. P. V. Duyne, J. Electroanal. Chem., 1977, 84, 1–20 CrossRef CAS.
- Z. Q. Tian, B. Ren and D. Y. Wu, J. Phys. Chem. B, 2002, 106, 9463–9483 CrossRef CAS.
- D. Y. Wu, J. F. Li, B. Ren and Z. Q. Tian, Chem. Soc. Rev., 2008, 37, 1025–1041 RSC.
- M. Moskovits, Rev. Mod. Phys., 1985, 57, 783 CrossRef CAS.
- I. R. Nabiev, R. G. Efremov and G. D. Chumanov, Soviet Physics–Uspekhi, 1988, 31, 241–262 CrossRef.
- K. Kneipp, H. Kneipp, I. Itzkan, R. R. Dasari and M. S. Feld, Chem. Rev., 1999, 99, 2957–2976 CrossRef CAS PubMed.
- L. L. Zhao, L. Jensen and G. C. Schatz, Nano Lett., 2006, 6, 1229–1234 CrossRef CAS PubMed.
- M. Sun, Z. Zhang, H. Zheng and H. Xu, Sci. Rep., 2012, 2, 647 Search PubMed.
- K. Kneipp, H. Kneipp, V. B. Kartha, R. Manoharan, G. Deinum, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1998, 57, R6281–R6284 CrossRef CAS.
- D. Senapati, S. S. R. Dasary, A. K. Singh, T. Senapati, H. Yu and P. C. Ray, Chem.–Eur. J., 2011, 17, 8445–8451 CrossRef CAS PubMed.
- M. Sun, Z. Zhang, P. Wang, Q. Li, F. Ma and H. Xu, Light: Sci. Appl., 2013, 2, e112 CrossRef CAS.
- Y. Huang, Y. Fang, Z. Zhang, L. Zhu and M. Sun, Light: Sci. Appl., 2014, 3, e199 CrossRef CAS.
- M. Moskovits, J. Raman Spectrosc., 2005, 36, 485–496 CrossRef CAS.
- A. Otto, I. Mrozek, H. Grabhorn and W. Akemann, J. Phys.: Condens. Matter, 1992, 4, 1143 CrossRef CAS.
- M. T. Sun, S. S. Liu, M. D. Chen and H. X. Xu, J. Raman Spectrosc., 2009, 40, 137–143 CrossRef CAS.
- E. Hao and G. C. Schatz, J. Chem. Phys., 2004, 120, 357–366 CrossRef CAS PubMed.
- L. Jensen, C. M. Aikens and G. C. Schatz, Chem. Soc. Rev., 2008, 37, 1061–1073 RSC.
- L. Xia, M. Chen, X. Zhao, Z. Zhang, J. Xia, H. Xu and M. Sun, J. Raman Spectrosc., 2014, 45, 533–540 CrossRef CAS.
- L. Zhao, L. Jensen and G. C. Schatz, J. Am. Chem. Soc., 2006, 128, 2911–2919 CrossRef CAS PubMed.
- Q. Ding, Y. Shi, M. Chen, H. Li, X. Yang, Y. Qu, W. Liang and M. Sun, Sci. Rep., 2016, 6, 32724 CrossRef CAS PubMed.
- B. Ren, X. F. Lin, J. W. Yan, B. W. Mao and Z. Q. Tian, J. Phys. Chem. B, 2002, 107, 899–902 CrossRef.
- P. G. Cao, J. L. Yao, B. Ren, B. W. Mao, R. A. Gu and Z. Q. Tian, Chem. Phys. Lett., 2000, 316, 1–5 CrossRef CAS.
- Z. Q. Tian, A. B. Ren and B. W. Mao, J. Phys. Chem. B, 1997, 101, 1338–1346 CrossRef CAS.
- B. Ren, D. Wu, B. Mao and Z. Tian, J. Phys. Chem. B, 2003, 107, 2752–2758 CrossRef CAS.
- Y. Gao, L. Chen, X. Dai, R. Song, B. Wang and Z. Wang, RSC Adv., 2015, 5, 32198–32204 RSC.
- Y. Gao, B. Wang, Y. Lei, B. K. Teo and Z. Wang, Nano Res., 2016, 9, 622–632 CrossRef CAS.
- Y. Gao and Z. Wang, Chin. Phys. B, 2016, 25, 83102–083102 CrossRef.
- L. Chen, Y. Gao, Y. Cheng, Y. Su, Z. Wang, Z. Li and R. Q. Zhang, J. Phys. Chem. C, 2015, 119, 17429–17437 CAS.
- L. Chen, Y. Gao, Y. Cheng, H. Li, Z. Wang, Z. Li and R. Q. Zhang, Nanoscale, 2016, 14, 4086–4093 RSC.
- C. M. Aikens and G. C. Schatz, J. Phys. Chem. A, 2007, 110, 13317–13324 CrossRef PubMed.
- M. Sun, S. Zhang, Y. Fang, Z. Yang, D. Wu, B. Dong and H. Xu, J. Phys. Chem. C, 2009, 113, 19328–19334 CAS.
- R. G. Aztatzi, E. Formoso, J. M. Mercero, J. M. Matxain, S. J. Grabowski and J. M. Ugalde, J. Chem. Phys., 2016, 144, 137–168 Search PubMed.
- Y. Fang, Z. Zhang and M. Sun, Rev. Sci. Instrum., 2016, 87, 033104 CrossRef PubMed.
- Z. Zhang, S. Sheng, R. Wang and M. Sun, Anal. Chem., 2016, 88, 9328–9346 CrossRef CAS PubMed.
- G. t. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- R. M. Dreizler and E. K. U. Gross, Density functional theory: an approach to the quantum many-body problem, Springer-Verlag, 1990 Search PubMed.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS.
- E. Van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- E. van Lenthe, E.-J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- E. van Lenthe, E.-J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- E. Van Lenthe, J. Snijders and E. Baerends, J. Chem. Phys., 1996, 105, 6505–6516 CrossRef CAS.
- E. van Lenthe, A. Ehlers and E.-J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS.
- E. Gross and W. Kohn, Phys. Rev. Lett., 1986, 57, 923 CrossRef.
- J. Neugebauer, M. Reiher, C. Kind and B. A. Hess, J. Comput. Chem., 2002, 23, 895–910 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28240g |
| This journal is © The Royal Society of Chemistry 2017 |