
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effects of microwaves on molecular arrangements in potato starch†
Huijie Shena,
Daming Fan *a,
Luelue Huangc,
Yishu Gaoa,
Huizhang Lianb,
Jianxin Zhaoa and
Hao Zhanga
*a,
Luelue Huangc,
Yishu Gaoa,
Huizhang Lianb,
Jianxin Zhaoa and
Hao Zhanga
aState Key Laboratory of Food Science and Technology, School of Food Science and Technology, Jiangnan University, Wuxi 214122, China. E-mail: fandm@jiangnan.edu.cn; Fax: +86 0510 85912155; Tel: +86 0510 85884620
bWuxi Huashun Minsheng Food Co. Ltd., Wuxi 214218, China
cSchool of Applied Chemistry and Biological Technology, Shenzhen Polytechnic, Shenzhen 518055, China
First published on 3rd March 2017
Abstract
The effects of microwave heating on the molecular arrangements in potato starch, including single and double helices and amorphous structures, were studied using a 13C CP/MAS NMR method combined with X-ray diffraction. Rapid heating in an oil bath and conventional slow heating were used as controls. During the microwave heating process, the double helical structures, the V-single and the crystallinity of potato starch exhibited similar changes to those observed when heated using conventional methods, although there were some differences. The effects on the structures were most pronounced when using conventional slow heating, followed by microwave heating, then conventional rapid heating. Both the rapid heating and electromagnetic effects of microwaves on potato starch, influenced the nature of the amorphous and double helical structures; although the rapid heating effect was greater. Conventional slow heating resulted in the thorough gelatinisation of starch.
Introduction
Starch is made of amylose and amylopectin, and is considered to be a semicrystalline polymer, in which the crystalline regions are composed of double helical structures of highly branched amylopectin molecules. Amylopectin side chains with branching points and amylose are located in amorphous domains.1,2 X-ray diffraction methods provide information on the long-range molecular order (the degree of crystallinity). 13C CP/MAS NMR is used to determine the shorter-range structures, including single and double helices and amorphous structures, due to its high sensitivity.3,4Microwave radiation is an important method used to physically modify the structure and properties of starch, with lower shear than conventional heating methods. Lewandowicz et al. studied the effects of different microwave energies on various starches. They found that the molecular structure was rearranged and properties such as water absorbing capacity, solubility and swelling power were altered.5 The granule morphology of potato starch appeared to be unaffected by microwave radiation, however, the relative crystallinity was increased and the crystalline type changed from B-type to A-type.6 Luo et al. reported that microwave radiation did not affect the external shape of normal and waxy corn starch. However, cavities were formed in the structure of the granules, and the crystalline type of high-amylose corn starch was changed from A + B to B.7 The chemical structure of starch and its derivatives are similar. Derivatives differ only in the number of repeating units or the order of atomic arrangement. However, few studies have examined the changes to the amorphous, single and double helix structures within starch granules, during gelatinisation when heated with microwave radiation.
Microwave heating differs from traditional convection heating: materials absorb microwave energy and this energy is subsequently converted into heat through molecular vibration and friction.8 The mechanisms about how microwave heating affects starch granules still remain unclear. There may be some special microwave effects in addition to rapid heating effect according to a series of phenomena found in the current study of microwave reaction, and these two effects may also act on the material and then influence physical and chemical properties and structure of materials.9 However, some researchers deny the existence of special effect of microwave.10
In this study, we evaluated the effect of different heating methods on the amorphous, single and double helix structures and the relative crystallinity of potato starch using 13C CP/MAS NMR and X-ray diffraction. The starch samples that underwent rapid heating were compared with samples heated using a conventional slow heating method to examine rapid heating effect of microwave. Rapid heating in an oil bath was used to simulate the special effect of a microwave, and thereby to investigate the thermal and electromagnetic effects of microwave heating on the structure of starch.
Materials and methods
Materials
Potato starch (protein content 0.23%, starch content 94.22%, amylose content of 13.62%, water content 5.5%, particle size of 38.55 ± 2.48 μm, weight-average molar mass of 5.048 × 107 g mol−1) was from Fengning Shuangxin Agricultural Development Co., Ltd. The onset gelatinization temperature (To) of potato starch was 57.16 + 0.11 °C.The water content would affect the dielectric properties of starch samples and further enhance the effect of microwave on starch.11 The permittivity (ε′) and dielectric loss factor (ε′′) of native starch with 5.5% water content is 3.04 and 0.23 respectively and the ε′ and ε′′ of 3% concentration of starch suspensions (97% water content) is 70.76 and 9.75 respectively, which indicates that the water significantly improves the dielectric properties of the system.
Methods
The starch samples were heated by microwave or oil bath to 45 °C, 55 °C, 60 °C, 65 °C and 75 °C in quartz reactor under mechanical agitation with a stirrer to ensure uniform heating before being cooled with ice bath and lyophilised. Each dried starch sample was crushed, and then powdered by passing it through a 75 μm sieve.
Analytical methods
Results and discussion
Comparing the three heating methods
As shown in Fig. 1A, the temperature curve resulting from microwave heating (1000 W) does not correlate with the temperature curve obtained from rapid heating in a 200 °C oil bath. A turning point was observed at 60 °C in the samples heated in the oil bath, and above this temperature the heating rate decreased. The microwave power level was altered such that the temperature curves of both methods were identical. The final temperature achieved through rapid heating in an oil bath was 200 °C. The microwave apparatus was set at 1000 W for 70 s, then 350 W for 50 s and 650 W for 125 s. The heating curves obtained with the rapid oil bath and modified microwave methods are shown in Fig. 1B. The correlation coefficient of the two curves was calculated to be 0.9981, and the heating rate of both methods was approximately 27.2 °C min−1. The temperature curve of the samples treated with the conventional slow heating method (with a heating rate of 4.68 °C min−1) is shown in Fig. 1C. | ||
| Fig. 1 (A) Temperature curves of the samples heated using a microwave (1000 W) and an oil bath (200 °C); (B) temperature curves of the samples heated using a microwave (1000 W for 70 s; 350 W for 50 s; 650 W for 125 s) and an oil bath (200 °C); (C) temperature curve of the heated with the slow heating method. | ||
Changes in crystallinity of starch samples measured by XRD
As shown in Fig. 2, the crystallinity of each sample declined as soon as the heating processes were initiated. At temperatures below the gelatinisation temperature, the heat absorbed by the starch had little effect on the crystalline regions, as indicted by the slow decline in crystallinity below 60 °C. Furthermore, when the gelatinisation temperature was reached the mobility of the amorphous segments was increased, leading to instability in the starch granules. As heating continued, microcrystalline starch melted and the starch granules broke up, accompanied by amylose leaching and double helices being opened, leading to amorphous paste formation.16 The gelatinisation temperature of potato starch is 60–65 °C, which was confirmed by the significant reduction in crystallinity at this temperature, as shown in Fig. 2. Beyond this temperature the crystallinity declined far more slowly. | ||
| Fig. 2 Changes in the relative crystallinity of three potato starch samples with different heat treatment (RS, conventional rapid heating (◆); SS, conventional slow heating (■); MS, microwave heating (▲)) were determined by X-ray diffractometry. | ||
The SS exhibited a maximum decreases in crystallisation of 31.0%, the MS showed a decrease of 30.8% and the minimum relative changes in crystallinity of RS decreased by 30.6%.
Structural changes determined by 13C CP/MAS NMR
 | ||
| Fig. 3 13C CP/MAS NMR spectra of potato starch samples at different temperatures ((A) MS; (B) RS; (C) SS). Spectra of amorphous and raw starch (NS) samples included for comparison. | ||
| Starch sample | Chemical shift (ppm) | |||
|---|---|---|---|---|
| C1 | C2, 3, 5 | C4 | C6 | |
| Raw (NS) | 94.42, 98.18, 100.94, 102.87 | 71.3, 72.5, 73.9 | 82.6 | 61.9 |
| Amorphous | 103.13 | 72.4, 74.3 | 82.3 | 61.5 |
| MS45 °C | 94.39, 98.68, 101.57, 103.55 | 71.2, 72.4, 73.9 | 82.6 | 61.5 |
| MS55 °C | 94.33, 98.65, 99.37, 100.95, 103.67 | 71.1, 72.4, 74.0 | 82.0 | 61.2 |
| MS60 °C | 94.35, 98.57, 99.32, 100.57, 103.52 | 71.1, 72.4, 74.0 | 82.3 | 61.2 |
| MS65 °C | 103.55 | 71.2, 72.5, 73.9 | 82.2 | 61.8 |
| MS75 °C | 103.51 | 71.4, 72.5, 74.0 | 82.4 | 61.5 |
| RS45 °C | 94.39, 98.68, 101.57, 103.55 | 72.4, 74.1 | 81.9 | 61.4 |
| RS55 °C | 94.31, 98.45, 99.57, 100.98, 103.62 | 72.4, 74.0 | 82.6 | 61.5 |
| RS60 °C | 94.57, 98.17, 99.49, 100.47, 103.54 | 71.4, 72.5, 73.6 | 82.7 | 61.5 |
| RS65 °C | 102.78 | 71.2, 72.5, 73.9 | 82.5 | 61.7 |
| RS75 °C | 103.17 | 71.1, 72.5, 74.2 | 82.5 | 61.6 |
| SS45 °C | 94.52, 98.68, 101.67, 103.41 | 71.2, 72.4, 74.0 | 82.5 | 61.5 |
| SS55 °C | 94.28, 98.48, 99.39, 100.20, 103.64 | 71.2, 72.4, 74.0 | 82.0 | 61.9 |
| SS60 °C | 94.76, 98.20, 99.47, 100.71, 103.57 | 72.4, 74.1 | 82.1 | 61.5 |
| SS65 °C | 102.95 | 72.4, 74.0 | 82.6 | 61.8 |
| SS75 °C | 103.37 | 71.1, 72.4, 74.0 | 82.5 | 61.5 |
Morrison et al. reported the 13C CP/MAS NMR signal data of several C regions. The signals at 99–102 ppm in the C1 region (90–110 ppm) were found to contain information on the double helices, and the signals at 93–99 ppm were found to be several signals originating from the amorphous regions. The broad peak at 103 ppm contained information on the amorphous state of the starch. The C2, C3 and C5 regions (70–79 ppm) mainly originated from B-type double helices, whereas the C4 region (80–84 ppm) contained information on the amorphous regions and a small proportion of left-handed single-stranded V-type single helices.19
In A-type starch, maltotriose is the smallest repeating unit, and double helical structures are formed from two-fold axisymmetric helical structures, which produce a triplet in the C1 peak. In B-type starch, maltose is the smallest repeating unit, and double helical structures are formed from three-fold screw axes, which produce a doublet in the C1 peak.18 As shown in Fig. 3, double peaks were observed in the C1 region, indicating that the double helical structures in potato starch are B-type double helices. This was consistent with the results of previously reported studies.20,21 As the heating duration increased, the crystalline type of potato starch transformed gradually from A-type to B-type, which is consistent with previous reports.20
The signal from the triplet peak gradually diminished with heating, and only a single peak around 103 ppm in the C1 region was observed in the spectrum of the highest temperature sample, suggesting that potato starch had started the gelatinisation process. With all three heating methods, at temperatures above 65 °C the spectra were similar to the spectrum of fully gelatinised amorphous starch. This finding indicated that the gelatinisation temperature range of the potato starch was consistent between all three heating methods. The relative intensities of the signals in the C4 region increased with increasing temperature. These data suggest that the amorphous content in the starch granules substantially increased with temperature, which resulted in a higher level of gelatinisation.
We found that the temperature points at which the triplet peaks changed into doublet peaks, the weak signal intensity of the triplet peaks and the increase in the signal strength in the C4 region were the same in all three sets of samples.
Changes in the single and double helical and amorphous structures of the starch samples
Ihwa et al. improved the 13C NMR method, and used it to determine the proportion of double helices, amorphous structures and V-type single helices.15 In this study, the Solver data analysis tool was used to obtain the subspectra of the amorphous content in the samples at different temperatures, based on the 13C NMR spectra. An ordered subspectrum was obtained by subtracting the subspectrum of the amorphous component from the original spectrum of the sample. For example, Fig. 4 shows the three subspectra of the MS sample at different temperatures. Sub-peak fitting of ordered and disordered structures was performed on the subspectra, using PeakFit software. A combination (50/50) of Lorentzian and Gaussian profiles produced acceptable curve fitting (r2 ≥ 0.9990). | ||
Fig. 4 Deconvolution of the MS 13C CP/MAS NMR spectra (![[thick line, graph caption]](https://www.rsc.org/images/entities/char_e117.gif) ) into the amorphous ( ) into the amorphous (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) and ordered (—) phases. The Solver data analysis tool in Excel was used to calculate the deconvolution. An ordered subspectrum was obtained by subtracting the subspectrum of the amorphous component from the original spectrum of the sample. The spectra of the other two samples are contained in the ESI.† ) and ordered (—) phases. The Solver data analysis tool in Excel was used to calculate the deconvolution. An ordered subspectrum was obtained by subtracting the subspectrum of the amorphous component from the original spectrum of the sample. The spectra of the other two samples are contained in the ESI.† | ||
As shown in Fig. 4, the intensity and area of the spectra of the amorphous structures gradually increased with temperature, while the intensity and area of the spectra of the ordered structures declined. The subspectra of the ordered structures were weak and highly unstable at temperatures higher than 65 °C.
The peak fitting results for the ordered subspectra of MS are shown in Fig. 5, and the calculated proportions of amorphous, single and double helix structures are summarised in Table 2.
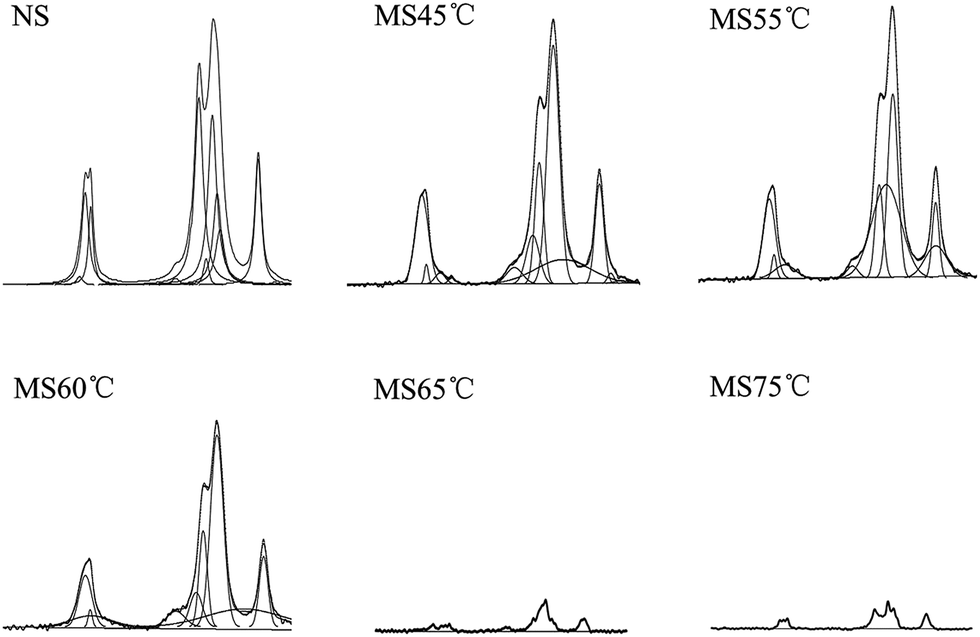 | ||
| Fig. 5 Peak fitting results of the subspectra of ordered structures in MS samples. The number following ‘MS’ indicates the temperature that the samples were heated to. PeakFit version 4 for Win 32 (Jandel Scientific Software, CA) was used to fit the peaks of the ordered spectra shown in Fig. 4. The proportions of double and single helical structures in the starch samples were calculated using the method of Tan et al. The spectra of the other two samples are in the ESI.† | ||
| Sample | Amorphous content (%) | Single helix content (%) | Double helix content (%) |
|---|---|---|---|
| Raw starch | 58.71 | 7.08 | 34.21 |
| MS45 °C | 61.82 | 5.93 | 32.25 |
| MS55 °C | 65.67 | 3.40 | 30.93 |
| MS60 °C | 77.94 | 1.30 | 20.76 |
| MS65 °C | 85.81 | 0 | 14.19 |
| MS75 °C | 87.99 | 0 | 12.01 |
| RS45 °C | 58.06 | 6.27 | 35.67 |
| RS55 °C | 62.83 | 3.70 | 33.47 |
| RS60 °C | 74.47 | 1.83 | 23.70 |
| RS65 °C | 83.96 | 0 | 16.04 |
| RS75 °C | 85.57 | 0 | 14.43 |
| SS45 °C | 65.43 | 5.54 | 29.03 |
| SS55 °C | 68.22 | 2.13 | 29.65 |
| SS60 °C | 84.77 | 0.87 | 14.36 |
| SS65 °C | 87.45 | 0 | 12.55 |
| SS75 °C | 89.93 | 0 | 10.07 |
As shown in Table 2, the samples exhibited similar trends in the changing proportions of amorphous, double helices and V-type single helices, regardless of the heating method and all changed with the rising temperature, especially when the temperature was near the gelatinization point. At temperatures below 60 °C, the proportions of amorphous regions and double helices changed very slowly; however, the changes in the SS samples were 6% and 10%, respectively, slightly greater than those in the MS (around 4% and 7%) and RS (around 1% and 3.5%) samples. In all of the samples, when the temperature rose above 60 °C the proportion of double helices decreased rapidly, while the proportion of amorphous regions increased rapidly and the proportion of V-type single helices decreased to zero before the end of the experiment. At 75 °C, the proportion of double helices in the RS, MS and SS samples decreased by 20%, 22% and 24%, respectively. The amorphous content of RS, MS and SS increased by 27%, 29% and 30%, respectively.
Using these results, we can predict both the thermal and special effects of microwaves on the proportion of amorphous region, double helices and V-type single helices in potato starch. At the beginning of the microwave treatment, the destruction of the internal structure of the starch had not commenced, and thus the structure was relatively stable. When the temperature exceeded 60 °C, the hydration and particle swelling rates of the starch rapidly increased, as a large quantity of water molecules entered the starch granules. During this period, the heating rates of the MS and RS samples slowed down, but remained greater than the heating rate of the SS sample. The heating time of the MS and RS samples were correspondingly short. This may be the cause of the lower degrees gelatinisation in the MS and RS samples.
Conclusions
Three heating methods were established as a model to study the effects of microwaves on molecular arrangements. We carried out 13C CP/MAS NMR combined with X-ray to detect the proportions of amorphous structures, double helices and V-type single helix structures changes in potato starch. Compared with native potato starch, the amorphous content of microwave heating samples increased 29%, the double helix structure decreased by 22%, and the relative crystallinity of potato starch after microwave heating decreased by 30.8%, which were all between rapid heating and slow heating sample. From these results, we determined that both the rapid heating and electromagnetic effects of microwaves influenced the proportion of amorphous and double helical structures, and that rapid heating had a greater effect than the electromagnetic effects. Additionally, microwave special effects may strengthen and accelerate the change from ordered to disordered structures.Conflicts of interest
The authors declare that they have no conflicts of interest.Abbreviations used
| NS | Native potato starch sample |
| MS | Microwave-heated potato starch sample |
| RS | Rapidly heated potato starch sample |
| SS | Slowly heated potato starch sample |
Acknowledgements
This research was supported by “Five-Twelfth” National Science and Technology Support Program (Grant No. 2014BAD04B03), National Natural Science Foundation of China (Grant No. 31301504, 31571879), and Technological Innovation and Industrial Upgrading Program of Wuxi (Grant No. WX03-02B0105-041500-07).References
- J. L. Jane, K. S. Wong and A. E. McPherson, Carbohydr. Res., 1997, 300, 219–227 CrossRef CAS.
- P. A. M. Steeneken and A. J. J. Woortman, Carbohydr. Res., 1994, 258, 207–221 CrossRef CAS.
- M. J. Gidley and S. M. Bociek, J. Am. Chem. Soc., 1985, 107, 7040–7044 CrossRef CAS.
- M. J. Gidley and S. M. Bociek, J. Am. Chem. Soc., 1988, 110, 3820–3829 CrossRef CAS.
- G. Lewandowicz, J. Fornal and A. Walkowski, Carbohydr. Polym., 1997, 34, 213–220 CrossRef CAS.
- A. Szepes, M. Hasznos-Nezdei, J. Kovacs, Z. Funke, J. Ulrich and P. Szabo-Revesz, Int. J. Pharm., 2005, 302, 166–171 CrossRef CAS PubMed.
- Z. G. Luo, X. W. He, X. Fu, F. X. Luo and Q. Y. Gao, Starch-Stärke, 2006, 58, 468–474 CrossRef CAS.
- D. M. Fan, W. R. Ma, L. Y. Wang, J. L. Huang, F. M. Zhang, J. X. Zhao, H. Zhang and W. Chen, Carbohydr. Polym., 2013, 92, 1395–1401 CrossRef CAS PubMed.
- A. Loupy, F. Maurel and A. Sabatie-Gogova, Tetrahedron, 2004, 60, 1683–1691 CrossRef CAS.
- E. J. Dominic, M. Lopez and B. Thomas, J. Chem. Sci., 2007, 119, 47–51 CrossRef.
- R. Torrealbameléndez, M. E. Sosamorales, J. L. Olveracervantes and A. Coronachávez, Int. J. Food Prop., 2015, 19, 564–577 CrossRef.
- G. E. Vandeputte, R. Vermeylen, J. Geeroms and J. A. Delcour, J. Cereal Sci., 2003, 38, 43–52 CrossRef CAS.
- J. A. Putseys, C. J. Gommes, P. Van Puyvelde, J. A. Delcour and B. Goderis, Carbohydr. Polym., 2011, 84, 1141–1150 CrossRef CAS.
- P. H. Hermans and A. Weidinger, J. Appl. Phys., 1948, 19, 491–506 CrossRef CAS.
- I. Tan, B. M. Flanagan, P. J. Halley, A. K. Whittaker and M. J. Gidley, Biomacromolecules, 2007, 8, 885–891 CrossRef CAS PubMed.
- T. A. Waigh, M. J. Gidley, B. U. Komanshek and A. M. Donald, Carbohydr. Res., 2000, 328, 165–176 CrossRef CAS PubMed.
- M. Urbanova, A. Sturcova, J. Brus, H. Benes, E. Skorepova, B. Kratochvil, J. Cejka, I. Sedenkova, L. Kobera, O. Policianova and A. Sturc, J. Pharm. Sci., 2013, 102, 1235–1248 CrossRef CAS PubMed.
- F. Delval, G. Crini, S. Bertini, N. Morin-Crini, P. M. Badot, J. L. Vebrel and G. Torri, J. Appl. Polym. Sci., 2004, 93, 2650–2663 CrossRef CAS.
- W. R. Morrison, R. V. Law and C. E. Snape, J. Cereal Sci., 1993, 18, 107–109 CrossRef CAS.
- W. Blaszczak, S. Valverde and J. Fornal, Carbohydr. Polym., 2005, 59, 377–383 CrossRef CAS.
- S. Mathur, R. Vyas, K. Sachdev and S. K. Sharma, J. Non-Cryst. Solids, 2011, 357, 1632–1635 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28048j |
| This journal is © The Royal Society of Chemistry 2017 |