
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Detection of phosphatidylserine in the plasma membrane of single apoptotic cells using electrochemiluminescence†
Rui Wang and
Danjun Fang *
*
School of Pharmacy, Nanjing Medical University, Jiangsu 210000, China. E-mail: djf@njmu.edu.cn
First published on 24th February 2017
Abstract
Phosphatidylserine (PS) in the plasma membrane of single apoptotic cells was detected using luminol electrochemiluminescence for the first time. Membrane PS was converted by aqueous phospholipase D and L-amino acid oxidase to generate hydrogen peroxide, inducing electrochemically generated luminescence. The successful observation of PS in the cellular membrane provided a special strategy for the observation of cellular apoptosis of single cells.
Apoptosis is programmed cell death that is highly controlled in the cells.1 An internal program initiated by the cell, including the expression of a series of proteins in apoptosis and the activation of related signal transduction pathways, leads to cell death.2 Therefore, the usage of chemotherapy to induce apoptosis in cancer cells is one of the effective ways to overcome these deadly diseases.3 However, any critical defect in the apoptotic signalling pathways may result in uncontrolled proliferation and growth of cells, which ultimately induces cancers.4 As a result, the assay of cellular apoptosis is significant for the accurate diagnosis and treatment of cancers.5 Due to high cellular heterogeneity, the assay at the single cell level is preferred to elucidate the apoptotic process.6 In the past years, considerable efforts have been made to develop fluorescent probes for the imaging of intracellular DNA damage in single cells for the characterization of cell apoptosis.7 Although these fluorescence assays are popular and robust for biological studies, the staining of the cells with the fluorescent probe needs multiple steps and is not favourable for clinical tests.
Phosphatidylserine (PS) is a quantitatively minor component of cell membranes, which plays important roles in biological processes such as apoptosis.8 In healthy cells, PS is located in the inner leaflet of the plasma membrane, which flip-flops to the outer leaflet of the plasma membrane in early apoptosis.9 Since the flip-flop movement of PS in this process was observed to increase the resistance of the plasma membrane, an electrochemical impedance method was applied to record the impedance change of the cells at the electrode during the apoptotic process.10 This platform provides detailed information of this process using a relatively low-cost electrochemical station. The further fabrication of multiple electrodes connected with a multichannel electrochemical system enables the monitoring of the individual cells’ apoptosis in parallel. However, the further increase in the throughput requires more microelectrodes in one chip and more electrochemical recording channels, which raises the difficulty in the chip fabrication and the cost.
Recently, our group developed a strategy to analyse cholesterol in the plasma membrane of single cells using luminol electrochemiluminescence (ECL).11 ECL is a light-emitting phenomenon whereby the species electrochemically generated on the electrodes form excited products to create the luminescence.12 Luminol/hydrogen peroxide is a classic ECL system that produces the luminescence under positive potential, which is applied to quantify the concentration of hydrogen peroxide.13 In our strategy, aqueous cholesterol oxidase was introduced in a cell-cultured medium and reacted with cholesterol in the plasma membrane to produce hydrogen peroxide, which induced the electrochemically generated luminescence in the presence of aqueous luminol. After the insertion of a cell-sized pinhole between the indium tin oxide (ITO) electrode and the photomultiplier tube (PMT), the cholesterol in the plasma membrane of single cells was measured. Since the luminescence was produced locally near the cell, a relatively high throughput analysis of the membrane cholesterol of single cells could easily be achieved by the collection of luminescence using a charge-coupled device (CCD).14 The ECL-based assay does not need the complicated chip fabrication and could be utilized to monitor the alteration in the amount of the membrane molecule in single cells.
In this communication, luminol ECL is applied to monitor the membrane PS of single cells in an early apoptotic state for the first time. The detection procedure scheme is shown in Fig. 1. Aqueous phospholipase D (PLD) hydrolyzes membrane PS into phosphatidic acid (PA) and serine.15 Then, serine is oxidized by aqueous L-amino acid oxidase (LAAO) to generate ammonia, 2-oxo-3-hydroxypropionic acid, and hydrogen peroxide. Finally, hydrogen peroxide induces electrochemically generated luminescence with L012, a luminol analog with enhanced ECL,16 for the monitoring of the PS change in the cellular membrane during the apoptosis process. The successful observation of the alteration of membrane PS in this apoptotic process should provide a special method for the study of cellular apoptosis.
 | ||
| Fig. 1 A schematic demonstration of the analysis of PS in the plasma membrane of a single apoptotic cell. | ||
The detection of PS in the plasma membranes of apoptotic cells through a serial reaction with PLD and LAAO has been validated using fluorescence assay.15 In our work, the same condition was adapted to attempt the assay of aqueous PS using luminol ECL. As compared with background luminescence from 10 mM phosphate buffer saline (PBS) with aqueous PS, more luminescence was observed in Fig. 2A after the introduction of the cocktail of PLD and LAAO into PBS. The removal of either the PLD or the LAAO from the cocktail did not induce the luminescence increase. All these results confirmed the ECL detection of PS in the presence of PLD and LAAO. Fig. 2B shows that the luminescence ratio before and after the introduction of PLD and LAAO was linearly correlated with the concentration of aqueous PS from 5 to 50 μM, which supported the quantitative measurement of PS. The detection limit was determined to be 5 μM, and the relative standard deviation of three independent measurements was less than 8.7%.
 | ||
| Fig. 2 (A) The luminescence traces from 10 mM PBS and PBS with 10 μM aqueous PS in the presence of 30 U PLD and 1 U LAAO. (B) The dependence of the luminescence ratio on the concentration of aqueous PS. The error bar represents the relative standard deviation from three independent measurements. The PMT voltage was set at 600 V. | ||
It is known that PS moves from the inner leaflet to the outer leaflet of cellular plasma membranes during the apoptotic process. Therefore, in the presence of luminol, PLD, and LAAO, enhanced electrochemically generated luminescence should be observed for apoptotic cells. To induce the cellular apoptosis, 106 MCF-7 cells cultured at an ITO electrode were exposed to 0.1 μg mL−1 doxorubicin at 37 °C for 24 h. The other group of healthy cells was cultured at the ITO electrode in 10 mM PBS as the control group. For the ECL analysis, both of the groups were exposed to the cocktail of PLD and LAAO for 1.5 minutes and the luminescence was recorded subsequently. As expected, a significant increase in luminescence was observed from the apoptotic group in Fig. 3A. The luminescence increase from the apoptotic group should be attributed to PS in the outer leaflet of the plasma membrane. The statistical data in Fig. 3B reveal an average 1.3 fold luminescence increase that corresponds to ∼5 μM PS according to the calibration curve. Taking into account the solution volume (50 μL), ∼0.25 nmole PS from 106 cells was estimated. As a result, ∼0.25 fmole PS was collected from one cell, which was consistent with the previous reports.17 To exclude the effect of doxorubicin possibly remaining at the cellular surface on the luminescence intensity, the luminescence intensities from 200 μM luminol in the absence and presence of 0.1 μg mL−1 doxorubicin were compared. As shown in Fig. S1 (ESI†), no obvious increase in the luminescence intensity was observed, suggesting that there is no effect of doxorubicin on ECL detection in our assay.
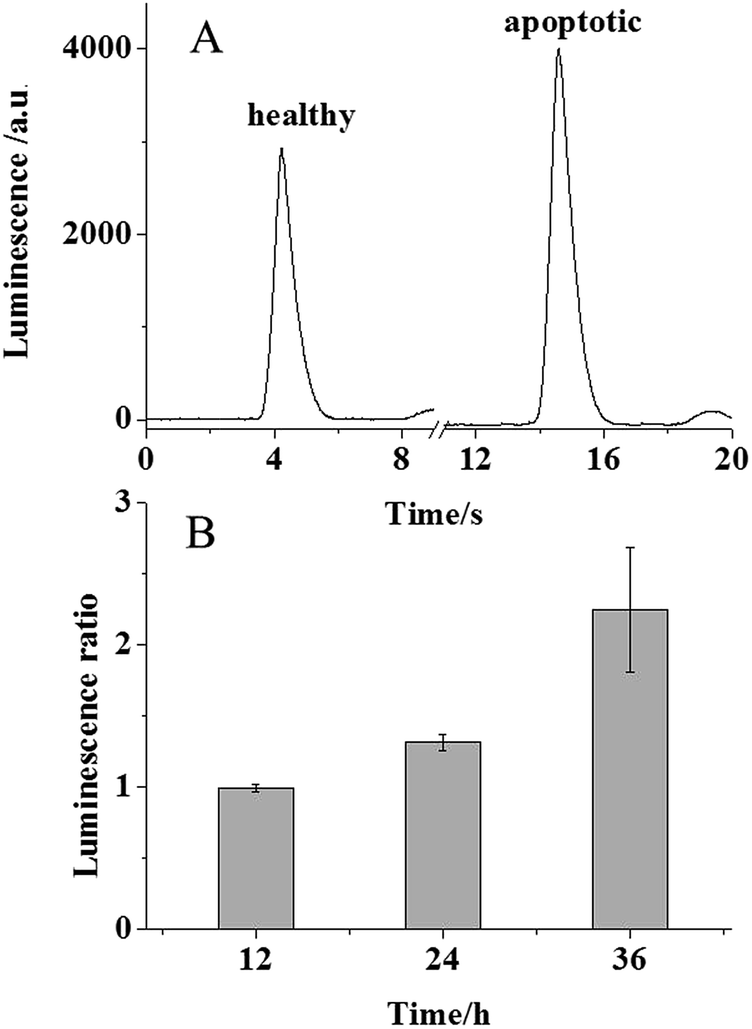 | ||
| Fig. 3 (A) The luminescence traces from healthy and apoptotic cells in 10 mM PBS with 30 U PLD, 1 U LAAO, and 200 μM luminol. (B) The luminescence ratio from healthy and apoptotic cells after the treatment with 0.1 μg mL−1 doxorubicin at 37 °C for 12, 24, and 36 h. The error bar represents the relative standard deviation from three independent measurements. The PMT voltage was set at 600 V. | ||
The variation of the doxorubicin treatment time of the cells regulates the apoptotic process. When the cells were exposed to doxorubicin for 12 h and analysed using our ECL assay, no significant luminescence increase was observed for the cells. This phenomenon suggested undetectable PS in this cellular state. Once the treatment time was extended to 24 and 36 h, the luminescence ratio increased 2.25 fold in Fig. 3B, which confirmed more PS movement to the outer leaflet of the cellular membrane. This result revealed that our ECL assay could be applied to monitor the apoptotic process quantitatively.
After the validation of the membrane PS analysis in the apoptotic cell population, single cell PS analysis was attempted. Following the previous protocol,11a a pinhole (100 μm in diameter) was placed between the ITO electrode and the PMT so that only one cell was exposed to the PMT for the analysis. Since the signal from the single cell was too weak, the previous PMT voltage of 600 V for the detection of aqueous PS was not sufficient for the detection of membrane PS in single cells. Therefore, a higher voltage of 900 V was applied on the PMT to amplify the signal. 20 individual cells were randomized into 10 groups. In each group, one cell was healthy and the other cell was treated using 0.1 μg mL−1 doxorubicin at 37 °C to induce cellular apoptosis. The two cells in the group were analysed using our ECL assay and the peak luminescence intensity was ratioed. The typical luminescence traces collected for single healthy and apoptotic cells are shown in Fig. 4A. An increase in luminescence for the apoptotic cell indicated that the membrane PS was measurable in a single apoptotic cell. The luminescence ratios between the apoptotic and healthy cells in these 10 groups are shown in Fig. 4B. All the ratios were larger than 1, supporting our successful ECL analysis of membrane PS at the single cell level. The average 2.29 fold increase was close to the result collected at the cell population level. The relative standard deviation from these groups was calculated to be 28.2%. As compared with the relative standard deviation of 8.7% from the detection of aqueous PS, this large deviation from the single cell analysis was likely associated with high cellular heterogeneity. A similar difference in membrane PS in individual cells during the apoptotic process was observed using impedance monitoring and flow cytometry-based assay.18 All of these results gave direct evidence of the temporal effects of cell death, which was significant for the study of cellular apoptosis.
 | ||
| Fig. 4 (A) The luminescence traces from single healthy and apoptotic cells in 10 mM PBS with 30 U PLD, 1 U LAAO, and 200 μM luminol. (B) The luminescence ratios from 10 groups of single apoptotic and healthy cells. The PMT voltage was set at 900 V. | ||
In conclusion, PS in the plasma membrane of single apoptotic cells was analysed for the first time using luminol ECL, which could provide a special strategy to observe the apoptosis state in single cells. In future chemotherapy, single cancer cells could be examined using this method after treatment with antitumor medication so that an optimized dosage could be figured out. As compared with the current new techniques for cellular apoptotic assessment, including lateral flow immunoassay, atomic force microscopy, microelectroanalysis, and the fluidic based cell-on-a-chip approach,19 the future coupling of luminescence analysis of membrane PS with an ECL imaging system should provide the advantages of a relatively high throughput and low cost in the investigation of cellular apoptosis.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21575068).References
- D. Heiser, V. Labi, M. Erlacher and A. Villunger, Exp. Gerontol., 2004, 39, 1125 CrossRef CAS PubMed.
- V. Sharma, D. Anderson and A. Dhawan, Apoptosis, 2012, 17, 852 CrossRef CAS PubMed.
- F. Lordick, W. Allum, F. Carneiro, E. Mitry, J. Tabernero, P. Tan, E. Van Cutsem, C. van de Velde and A. Cervantes, Cancer Treat. Rev., 2014, 40, 692 CrossRef PubMed.
- X. S. Deng, S. Wang, A. Deng, B. Liu, S. M. Edgerton, S. E. Lind, R. Wahdan-Alaswad and A. D. Thor, Cell Cycle, 2012, 11, 367 CrossRef CAS PubMed.
- V. Almendro, A. Marusyk and K. Polyak, Annu. Rev. Pathol.: Mech. Dis., 2013, 8, 277–302 CrossRef CAS PubMed.
- X. Qiu, J. R. Johnson, B. S. Wilson, S. T. Gammon, D. Piwnica-Worms and E. M. Barnett, PLoS One, 2014, 9, e88855 Search PubMed.
- A. B. Chinen, C. M. Guan, J. R. Ferrer, S. N. Barnaby, T. J. Merkel and C. A. Mirkin, Chem. Rev., 2015, 115, 10530 CrossRef CAS PubMed.
- J. E. Vance, J. Lipid Res., 2008, 49, 1377 CrossRef CAS PubMed.
- J. Suzuki, D. P. Denning, E. Imanishi, H. R. Horvitz and S. Nagata, Science, 2013, 341, 403–406 CrossRef CAS PubMed.
- I. Giaever and C. R. Keese, IEEE Trans. Biomed. Eng., 1986, 33, 242 CrossRef CAS PubMed.
- (a) G. Z. Ma, J. Y. Zhou, C. X. Tian, D. C. Jiang, D. J. Fang and H. Y. Chen, Anal. Chem., 2013, 85, 3912 CrossRef CAS PubMed; (b) C. X. Tian, J. Y. Zhou, Z. Q. Wu, D. J. Fang and D. C. Jiang, Anal. Chem., 2014, 86, 678 CrossRef CAS PubMed.
- W. Miao, Chem. Rev., 2008, 108, 2506–2553 CrossRef CAS PubMed.
- (a) H. Cui, G. Z. Zou and X. Q. Lin, Anal. Chem., 2003, 75, 324 CrossRef CAS PubMed; (b) H. Jiang and H. X. Ju, Anal. Chem., 2007, 79, 6690 CrossRef CAS PubMed.
- J. Y. Zhou, G. Z. Ma, Y. Chen, D. J. Fang, D. C. Jiang and H. Y. Chen, Anal. Chem., 2015, 87, 8138 CrossRef CAS PubMed.
- (a) M. Liscovitch, M. Czarny, G. Fiucci and X. Tang, Biochem. J., 2000, 345, 401 CrossRef CAS PubMed; (b) S. Y. Morita, S. Shirakawa, Y. Kobayashi, K. Nakamura, R. Teraoka, S. Kitagawa and T. Terada, J. Lipid Res., 2012, 53, 325 CrossRef CAS PubMed.
- Y. Nishinaka, Y. Aramaki, H. Yoshida, H. Masuya, T. Sugawara and Y. Ichimori, Biochem. Biophys. Res. Commun., 1993, 193, 554 CrossRef CAS PubMed.
- P. A. Leventis and S. Grinstein, Annu. Rev. Biophys., 2010, 39, 407 CrossRef CAS PubMed.
- F. Asphahani, M. Thein, K. Wang, D. Wood, S. S. Wong, J. Xu and M. Q. Zhang, Analyst, 2012, 137, 3011 RSC.
- P. Shah, A. Kaushik, X. N. Zhu, C. X. Zhang and C. Z. Li, Analyst, 2014, 139, 2088 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra28031e |
| This journal is © The Royal Society of Chemistry 2017 |