
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Effect of lanthanum doping on the far-infrared emission property of vanadium–titanium slag ceramic
Kewei Zhang†
a,
Yi Deng† a,
Yuanyi Yangd,
Yongkang Liaob,
Bozhi Wangc,
Bing Gonga and
Weizhong Yang*a
a,
Yuanyi Yangd,
Yongkang Liaob,
Bozhi Wangc,
Bing Gonga and
Weizhong Yang*a
aSchool of Materials Science and Engineering, School of Chemical Engineering, Sichuan University, Chengdu 610065, China. E-mail: scuywz@139.com
bSichuan Baita Xinlianxing Ceramic Co., Ltd., Neijiang 642450, China
cCollege of Architecture and Environment, Sichuan University, Chengdu 610065, China
dDepartment of Materials Engineering, Sichuan College of Architectural Technology, Deyang 618000, China
First published on 28th February 2017
Abstract
In the present study, a series of far-infrared ceramics were successfully synthesized using vanadium–titanium slag solid waste and some ordinary minerals as main raw materials with lanthanum (La) as an additive. The phase composition and microstructure of the prepared samples were characterized by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM). Besides, the far-infrared emission and absorption properties of the ceramics were determined via Fourier transform infrared spectroscopy (FT-IR). The results indicated that the doped La3+ could efficiently promote the transformation of Fe2+ to Fe3+, which decreased crystallite size and increased lattice strain in the orthopyroxene-like structure. When doped with 9 wt% La, the vibration absorption intensities of Si–O–Mg and Si–O–Fe in orthopyroxene's irregular polyhedron and inerratic octahedron sites were strongest, which consequently contributed to enhanced far-infrared emissivity in 8–14 μm wavebands, reaching as high as 0.927. Moreover, the product exhibited a high bending strength of 30.45 ± 0.54 MPa for the La-doped groups, satisfying the requirement of ISO 13006 standard (>18 MPa). These results indicated that our prepared La3+-doped vanadium–titanium slag ceramics hold great promise for practical infrared applications due to their high far-infrared emissivity and excellent physical performances.
1. Introduction
Far-infrared ceramics, which can spontaneously launch intense infrared rays at specific wavelengths, are extensively employed in many advanced fields, such as energy, health care, military areas, etc., owing to their excellent radiometric force and permeability.1–3 For instance, far-infrared ceramics have the ability to energize water activation by stimulating resonance among water molecules,4 which is conducive to penetration, mineralization, and biochemistry processes in organisms. On the other hand, a far-infrared ceramic powder has been incorporated into low-density polyethylene for the preparation of an antibacterial packaging material.5 In these applications, far-infrared ceramic exhibits excellent thermal stability, antibacterial properties, and good corrosion resistance, as well as high mechanical strength. Therefore, far-infrared ceramics have attracted considerable attention in current scientific research and technological applications for protecting human health and saving energy.Vanadium–titanium slag (VTS) is a type of solid waste from vanadium–titanium magnetite. With the huge increasing of industrial production, the management of VTS has become a tough challenge for environment conservation. The resource utilization of VTS is becoming a big tendency to solve the problem of VTS pollution. Over the past few decades, numerous research studies mainly focused on the recovery of titanium, vanadium, iron, chromium from VTS through various approaches, including water leaching after NaOH molten salt roasting,6 liquid oxidation technologies,7 acidic solution leaching,8,9 and pressure acid leaching.10 It was also reported that VTS could be used as a raw material to fabricate a gamma-ray shielding material,11 sorbents,12,13 and Ti-rich materials.14 To the best of our knowledge, nevertheless, there is rare work regarding the application of VTS in manufacturing far-infrared ceramics. Using VTS to prepare the far-infrared ceramic arises from the following advantageous factors: compositions and costs. Firstly, the selected VTS is iron-rich rather than low-titanium VTS. Fe element presents various valences in the VTS, and influences crystallization behaviour of special phases, which plays a role in the infrared radiation properties of the ceramic.15 Additionally, the VTS contains some oxides including SiO2, Al2O3, Na2O, MgO and CaO, conforming to the composition of silicate ceramic matrix. Secondly, far-infrared materials derived from purified oxides or expensive rare minerals enhance the production cost, while VTS with cheap price are easily obtained. Thus, using TVS as a secondary resource for the preparation of far-infrared ceramic can substantially decrease the production cost. More importantly, this approach turns wastes into assets.
Rare earths (RE) own the reputation of “industrial vitamin”, which are extensively used in the field of ceramic in order to enhance their performances. Furthermore, rare earths play a critical role in improving the luminescent properties of materials.16,17 Single RE ions doped or co-doped infrared emission materials have been widely investigated.18–20 Wu et al.21 reported an enhancement in infrared radiation properties for CoFe2O4 through single Ce3+ doping. Zhao et al.22 studied the infrared emission from Er3+/Y3+ co-doped oxyfluoride glass-ceramic, and found that intensities of the band emissions at 980 nm and 2730 nm were greatly increased due to the RE co doping. Zhang et al.23 improved the near-infrared luminescence attributes of Zn2SnO4 ceramic discs with the incorporation of Eu3+. Previous efforts indicated that RE ions doping would be a valid way to improve the infrared emission property of materials. Two main advantages of using RE ions in various systems are summarized here: (1) the powerful oxidization of RE ions causes the variation of cationic valences, radius and distribution, resulting in enhancing lattice distortion and lattice asymmetric vibration.24,25 (2) RE ions tend to occupy lattice or interstitial site in crystal structure to further introduce internal defect.26,27 In a word, RE ions can effectively break interatomic equilibrium state so that they have an ability to modulate far-infrared emission properties of material through above two effects. From the quantum mechanics viewpoint, whether single doping or co-doping, the probabilities of multi-phonon relaxation would be reduced in energy level transition system, which contributes to promoting far-infrared radiation property.28,29 In our experiment, representative rare earth La was selected as a dopant in a bran-new VTS ceramic system considering the following reasons. (1) according to Hund rules, La with 5d16s2 electronic configuration is apt to lose three electrons to become La3+, displaying strong reaction activities and stable trivalent state. (2) Considering the lanthanide contraction effect, La3+ has largest ionic radius (0.1061 nm) among lanthanide elements and the movement of La3+ in lattice structure mostly depends on its large radius. La3+ may form an isolating secondary phase in ceramic matrix or dissolve into particular phases to form solid solution.30 Coincidentally, both of the aspects are closely associated with far-infrared radiation performance. Therefore, an in-depth investigation on the far-infrared emission properties of La-doped VTS ceramic is still desired.
Hence, in this paper, VTS were chosen as the main raw materials to prepare the far-infrared ceramic with La doping. The effect of La element on the far-infrared radiation property was systematically studied. It is extra noteworthy that compared with expensive pure materials, our raw materials including VTS solid waste and other normal minerals used in the work are more suitable for large-scale production, because of their low production cost and rich source. Overall, our results revealed that the as-obtained far-infrared ceramic displayed both high far-infrared emissivity and strong bending strength. The aim of our current study resides in promoting the functional application of VTS and accelerating the introduction of VTS into IR flied.
2. Materials and methods
2.1. Materials
VTS wastes were provided from Pangang Group Company (Panzhihua, China), and other mineral materials including clay 1, clay 2, dolomite (CaMg(CO3)2), aluminum (Al), and sand were obtained from the Sichuan Baita Xinlianxing Ceramic Co., Ltd. (Neijiang, China). Table 1 showed the main chemical compositions of all raw materials tested by X-ray fluorescence spectrometer (XRF, XRF-1800, Shimadzu, Japan). Lanthanum nitrate hexahydrate (La(NO3)3·6H2O, ≥99.5 wt%) was purchased from Chendu Kelong Reagent Company (Chengdu, China). All other chemicals were of analytical reagent grade and were used as received without further purification. All aqueous solutions were prepared with de-ionized water (D.I. water).| Raw material | SiO2 | Al2O3 | Fe2O3 | TiO2 | CaO | MgO | K2O | Na2O | |
|---|---|---|---|---|---|---|---|---|---|
| Test 1 | VTS | 17.16 | 3.70 | 40.06 | 10.35 | 2.41 | 1.92 | 0 | 6.51 |
| Clay 1 | 59.85 | 25.49 | 1.36 | 1.16 | 0.19 | 0.78 | 3.13 | 0.28 | |
| Clay 2 | 68.29 | 18.16 | 1.22 | 0.79 | 0.69 | 1.28 | 2.76 | 0.30 | |
| Dolomite | 5.35 | 1.65 | 0.80 | 0.06 | 38.10 | 11.53 | 0.85 | 0.01 | |
| Aluminum | 42.71 | 33.01 | 2.40 | 4.76 | 1.39 | 0.47 | 0.30 | 0.52 | |
| Sand | 83.91 | 8.81 | 0.60 | 0.27 | 0.35 | 0.14 | 1.98 | 0.66 | |
| Test 2 | VTS | 16.75 | 4.20 | 42.10 | 9.46 | 1.51 | 1.44 | 0.05 | 7.02 |
| Clay 1 | 56.91 | 24.56 | 1.79 | 1.08 | 0.09 | 0.53 | 4.31 | 0.10 | |
| Clay 2 | 72.32 | 16.29 | 0.85 | 0.32 | 0.51 | 2.34 | 4.02 | 0.38 | |
| Dolomite | 6.79 | 1.97 | 0.52 | 0.02 | 40.33 | 10.87 | 1.45 | 0 | |
| Aluminum | 39.56 | 29.47 | 1.03 | 6.79 | 0.84 | 0.33 | 0.11 | 1.04 | |
| Sand | 85.03 | 7.79 | 0.21 | 0.45 | 0.09 | 0.42 | 3.02 | 0.40 | |
| Test 3 | VTS | 18.03 | 3.25 | 43.01 | 11.27 | 2.79 | 2.65 | 0 | 5.98 |
| Clay 1 | 61.17 | 26.79 | 2.02 | 0.94 | 0.31 | 0.66 | 4.22 | 0.15 | |
| Clay 2 | 69.93 | 16.94 | 1.91 | 0.52 | 0.83 | 2.09 | 2.35 | 0.20 | |
| Dolomite | 5.69 | 1.84 | 1.12 | 0.03 | 40.95 | 12.34 | 0.94 | 0.01 | |
| Aluminum | 43.25 | 30.98 | 1.94 | 6.34 | 1.71 | 0.50 | 0.19 | 0.86 | |
| Sand | 82.62 | 9.03 | 0.72 | 0.55 | 0.18 | 0.39 | 2.25 | 0.43 |
2.2. Preparation of far-infrared vanadium–titanium slags ceramic
In the present work, La(NO3)3 were used as dopant, and the weight percentages of La(NO3)3 in the experimental group were 3 wt%, 5 wt%, 7 wt%, 9 wt%, and 11 wt%, denoted as La3, La5, La7, L9, and La11, respectively; while the control group was noted as La0 (0 wt%). The formulas of far-infrared ceramic bodies obtained from orthogonal experiment were presented in Table 2.| Sample | La(NO3)3 | VTS | Total amount of clay (Clay 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Clay 2 = 1:1) :Clay 2 = 1:1) |
Dolomite | Aluminum | Sand |
|---|---|---|---|---|---|---|
| La0 | 0 | 20 | 50.00 | 11.0 | 4.0 | 15.0 |
| La3 | 3 | 19.4 | 48.5 | 10.67 | 3.88 | 14.55 |
| La5 | 5 | 19.0 | 47.5 | 10.45 | 3.80 | 14.25 |
| La7 | 7 | 18.6 | 46.5 | 10.23 | 3.72 | 13.95 |
| La9 | 9 | 18.2 | 45.5 | 10.01 | 3.64 | 13.65 |
| La11 | 11 | 17.8 | 44.5 | 9.97 | 3.56 | 13.35 |
The procedural details of experiment were given below. The raw materials and dopants were first mixed completely through wet milling in a high-density nylon pot for 30 min using zirconia balls using water as grinding medium. The resultant slurry was screened via 104 μm sieve, dried at 110 °C, powdered to crush the agglomerate, homogenized with 7–9 wt% water for pelleting (granules size around 1 mm). The humidified granules were pressed at 50 MPa into 40 × 6 × 5 mm strip body, and subsequently dried at 110 °C for 2–3 h. The sintering process involved in a step-by-step heating and cooling approach as follows: (1) ceramic body was put into a muffle furnace (SX-G03163, Tianjin Zhonghuan Lab Furnace Company, China) and heated from room temperature to 300 °C for 2 h to remove the residual moisture. (2) Then the temperature was slowly raised to 800 °C with heating rate of 2.5 °C min−1. (3) The temperature was continually elevated to 1100 °C for 2 h and just kept for 5 min, and then naturally cooled to room temperature to obtain the far-infrared ceramics. The prepared ceramics were crushed into fine powder with the help of an agate mortar for material characterization.
2.3. Characterization
The crystalline phases of the as-obtained samples were examined by X-ray diffraction analysis (XRD, Empyrean, PANalytical B.V, Netherlands) using graphite monochromatized Cu Kα radiation (λ = 1.54056 Å). The diffraction angles (2θ) were set between 10° and 60° with an incremental step size of 0.02°. The phase identification was achieved by comparing the sample diffraction pattern with standard cards in ICDD-JCPDS database. The elemental states of Fe ions were characterized by X-ray photoelectron spectroscopy (XPS, XSAM800, Kratos, UK) and the spectral deconvolution was conducted using the XPSPEAK software (Version 4.1). The microstructure and elemental analysis of the samples was examined by scanning electron microscope (SEM, JSM-7500F, JEOL, Japan) equipped with X-ray energy dispersive spectrometer (EDS). All samples were coated by gold for 1 min before SEM observation.2.4. Far-infrared absorption and emission properties evaluation
Fourier transform infrared spectra of these VTS ceramic powders were recorded on an infrared spectrometer (FT-IR, NEXUS 670, Thermo Electron Corporation, USA) detected in the form of pellets (KBr pellet). The spectra were recorded from 400 cm−1 to 1600 cm−1. The far-infrared emissivity of samples in 8–14 μm wavebands was tested by an FT-IR emission tester (Bruker 80V, Bruker, Germany).2.5. Physical performances tests
The strip samples were reserved for the physical performance test. The linear shrinkage (LS%) was calculated by the formula of LS = (La − Lb)/La (La = initial length of ceramic body before sintering, and Lb = final length of ceramic after sintering). The water absorption (WA%) were measured according to the ISO-EN 10545-3 standard. Samples were vacuumized for 30 min and soaked in D.I. water for 15 min. The values of water absorption were calculated using the equation of WA = (M2 − M1)/M1, where M1 is the initial weight of ceramic, and M2 denotes the ceramic weight after soaking. The bending strength (BS) was tested through a universal material testing machine (XDL-50000N, Xinhong Test Factory, Jiangdu, China) with the loading rate of 12 mm min−1. Three pieces of samples were used to improve the statistics. All quantitative data expressed as mean ± standard deviations were derived from experiments.3. Result and discussion
3.1. XRD analysis
The chemical compositions of the slag and mineral raw materials were examined by XRF. It was clear in Table 1 that these raw materials from different batches contained nearly identical inorganic components including SiO2, Al2O3, Fe2O3, TiO2, CaO, MgO, K2O and Na2O, although there were some tiny differences in the amount, indicating the good quality consistency of all raw materials. We first performed powder XRD analysis to identify the crystalline phase of these ceramics after sintering as shown in Fig. 1. It could be seen that as-prepared samples were predominantly consisted of quertz (SiO2, JCPDS no. 79-1906) and orthopyroxene (Ca0.043Mg0.254Fe0.703)(Mg0.896Fe0.104)[Si2O6], (JCPDS no. 86-0162), coupled with anorthite (Na0.48Ca0.53Al1.52Si2.48, JCPDS no. 85-0878), iron titanium (Fe9TiO15, JCPDS no. 24-0072) and lanthanum oxide (La2O3, JCPDS no. 83-1349) as minor phases. Comparing doped groups with the undoped counterpart, the obvious diffraction peaks of La2O3 indicated that La3+ formed isolating crystalline phases during sintering process. The sharp typical diffraction peaks of orthopyroxene implied good crystallinity with the augment of La amounts. The variation of orthopyroxene's crystallite size was further calculated according to Debye Scherrer's formula:31,32where D is the crystallite size (nm), k is the shape factor (k = 0.89), λ is the X-rays wavelength (λ = 1.54056 Å), θ is Bragg diffraction angle (°), and β is the full width at half maximum of an estimated peak (rad). The result of crystallite sizes in various planes and their averages (

![[D with combining macron]](https://www.rsc.org/images/entities/i_char_0044_0304.gif) ) are shown in Table 3.
) are shown in Table 3.
 | ||
| Fig. 1 XRD patterns of vanadium–titanium slags ceramic doped with different La contents. | ||
| Code | D211 | D420 | D202 | D502 | (nm) |
|---|---|---|---|---|---|
| La3 | 59.457 | 21.594 | 43.327 | 24.995 | 37.343 |
| La5 | 54.978 | 23.606 | 25.870 | 30.859 | 33.828 |
| La7 | 26.582 | 21.421 | 33.370 | 37.065 | 29.609 |
| La9 | 42.472 | 20.715 | 30.542 | 21.342 | 28.768 |
| La11 | 37.334 | 24.982 | 29.349 | 25.077 | 29.186 |
It could be found that descended firstly and then rebounded mildly, reaching its minimum (28.768 nm) when doping 9 wt% La. This result has a certain correlation with the far-infrared emissivity of samples. The decrease of average crystallite size might cause from the addition of La3+, and the main reason for the change of average crystallite size was that Fe elements were oxidized by La3+ transforming from Fe2+ to Fe3+, and then entered into orthopyroxene's structure.15 Given that the radius of Fe3+ (0.0645 nm) is smaller than that of Fe2+ (0.078 nm), the shrinkage of unit cell volume was inevitable.
3.2. XPS spectra with high resolution of Fe2p
To confirm the predication that Fe2+ was oxidized to Fe3+ by La3+, the high-resolution XPS spectra of Fe2p for La0, La7, La9 and La11 were presented in Fig. 2. The Fe2p spectrum exhibited a doublet at approximate 710.0 eV and 722.5 eV, which was consistent with the published work.33,34 The overlapped peaks of Fe2+ and Fe3+ were resolved by XPSPEAK software, and the test curves were divided into four parts corresponding to the peaks of Fe3+ (Fe2p1/2 = 723.2 ± 0.5 eV and Fe2p3/2 = 710.4 ± 0.3 eV) and Fe2+ (Fe2p1/2 = 721.0 ± 0.4 eV and Fe2p3/2 = 707.9 ± 0.3 eV). Simultaneously, the peak area of corresponding valence could be automatically generated by XPSPEAK software, and the calculated peak area ratio (Fe3+ area/Fe2+ area) of the each sample had been marked in the spectra. It could be intuitively found that the peak area ratio of Fe3+/Fe2+ was gradually increasing from 1.30 to 2.11 with the La concentration varying from 0 wt% to 11 wt%. Because of a positive correlation between the content and the peak area per Gaussian–Lorentzian function,24,35 we might safely draw a conclusion that the content ratio of Fe3+/Fe2+ was improved as the increasing of La doping. It demonstrated that the powerful oxidization of La3+ could strengthen the transformation of Fe2+ to Fe3+, which provided a proof for the conjecture of XRD analysis. | ||
| Fig. 2 XPS high resolution spectra of Fe2p peaks in La0, La7, La9 and La11 ceramic powders. | ||
3.3. SEM analysis
The SEM micrographs of all samples were presented in Fig. 3, where Fig. 3(A) and (B) were element information derived from EDS. Evidently, the fractured surface of ceramics appeared to be relative smooth. The significant distinction between non-doped one and the doped groups was that some granular phases with submicron level were dispersed in original amorphous ceramic matrix, and the amounts of granular phases were mounting with the increasing of La content. In consideration of XRD results and EDS consequences in Fig. 3(A) and (B), the granular phases containing La could be verified to be La2O3. The EDS results conducted on granular phase (A) and ceramic matrix (B) suggested that elements of Fe, Ca, Mg were concentrated in La2O3 phases. These concomitant elements were identical with the main compositions of orthopyroxene. The results illustrated that there was a tight connection between La2O3 and orthopyroxene. | ||
| Fig. 3 SEM micrographs of vanadium–titanium slags ceramic samples (La0–La11), and EDS spectra of representative 9 wt% La-doped sample. (A) The red circle was marked for granular phase, and (B) the blue box was marked for ceramic matrix. | ||
The movement process of La3+ was shown in Fig. 4. The evolution of La2O3 could be detailed as follows: the foregoing consequence showed that the La3+ primarily entered into orthopyroxene. Then La3+ would migrate into its crystal structure or interstitial site, giving rise to internal defects. With increment of La dopant, the vacancy and interstitial site in orthopyroxene would be filled as far as possible. Redundant La ions had a tendency to diffuse toward grain boundary.21 Once the limitation of energy barriers was overcome, the excrescent La3+ would separate out of orthopyroxene, and La2O3 was generated around grain boundary, resulting in the junction of La2O3 and orthopyroxene. It suggested that the excess La2O3 phases might hamper the formation of orthopyroxene phases, because the competition for crystal nucleation and growth between La2O3 and orthopyroxene was inevitable. To summarize, the effect of La was prone to a balance between generating crystal defect and limiting the development of sosoloid.
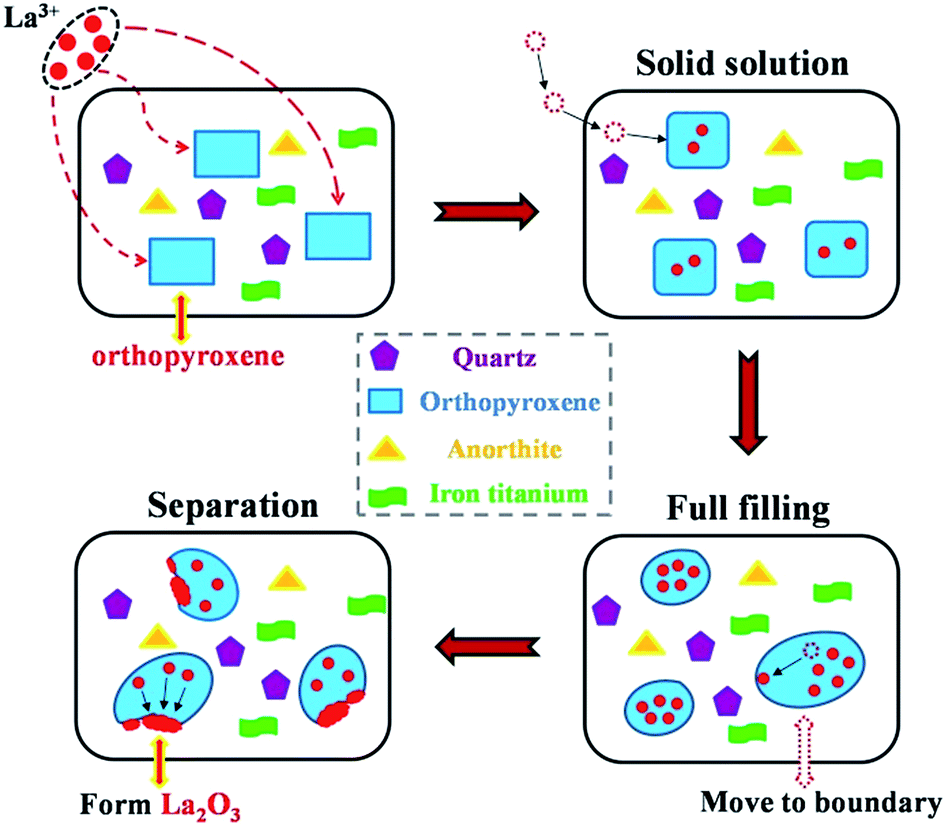 | ||
| Fig. 4 The proposed motion of La3+ in the VTS ceramics. | ||
3.4. Local microstructure analysis
To further explore the impact of cation distribution on the microstructure of orthopyroxene, the results from XRD, XPS and SEM were analysed together. Based on the equation of| Δr = (ra − rb)/ra(ra > rb),36 |
It is well-known that orthopyroxene belongs to orthorhombic system and possesses pyroxene-type construction (XY[Si2O6]). In XY[Si2O6], larger cation X (six coordination) tends to occupy irregular polyhedron sites (A-site), and smaller cation Y (eight coordination) is inclined to enter into inerratic octahedron sites (B-site).37,38 In detail, because of the restriction of coordination polyhedron and the selection of potential energy, the smaller Fe3+ ions was much easier to move into B-site than larger Fe2+ ions. Due to the powerful oxidization of La3+, the content of Fe2+ would be reduced drastically, whereas Fe3+ gradually increased and occupied the main position in the B-site. It meant that lattice distortion from shrinkage of the unit cell might be introduced. To quantify the degree of its lattice distortion, the lattice strain of orthopyroxene was analysed via the Williamson–Hall equation:39
 sinθ (x-axis) and βcosθ (y-axis) fits straight line and its positive slope represents the lattice strain (ε). As seen in Fig. 5, the value of lattice strain reached its maximum (0.27%) when doping 9 wt% La, corresponding to the smallest crystallite size (28.768 nm). The rising trend of the lattice strain was synchronized with the downtrend of crystallite size, and vice versa. In the range of 3–9 wt% doping, Fe3+ played a dominant role in lattice distortion and leaded to a decrease of atoms order degree. Simultaneously, it should not be ignored that a part of La ions dissolved into orthopyroxene, which also made a contribution to the increased lattice strain. Nevertheless, lattice strain fell back to 0.24% when doped contents reached 11 wt%. Based on SEM analysis, it seemed to be that the excessive second phase (La2O3) around crystal boundary would inhibitthe growth of orthopyroxene-like phases and reduce its lattice strain. It has been reported that the lattice strain of materials is of great relevance to the infrared radiation property.21,40
sinθ (x-axis) and βcosθ (y-axis) fits straight line and its positive slope represents the lattice strain (ε). As seen in Fig. 5, the value of lattice strain reached its maximum (0.27%) when doping 9 wt% La, corresponding to the smallest crystallite size (28.768 nm). The rising trend of the lattice strain was synchronized with the downtrend of crystallite size, and vice versa. In the range of 3–9 wt% doping, Fe3+ played a dominant role in lattice distortion and leaded to a decrease of atoms order degree. Simultaneously, it should not be ignored that a part of La ions dissolved into orthopyroxene, which also made a contribution to the increased lattice strain. Nevertheless, lattice strain fell back to 0.24% when doped contents reached 11 wt%. Based on SEM analysis, it seemed to be that the excessive second phase (La2O3) around crystal boundary would inhibitthe growth of orthopyroxene-like phases and reduce its lattice strain. It has been reported that the lattice strain of materials is of great relevance to the infrared radiation property.21,40
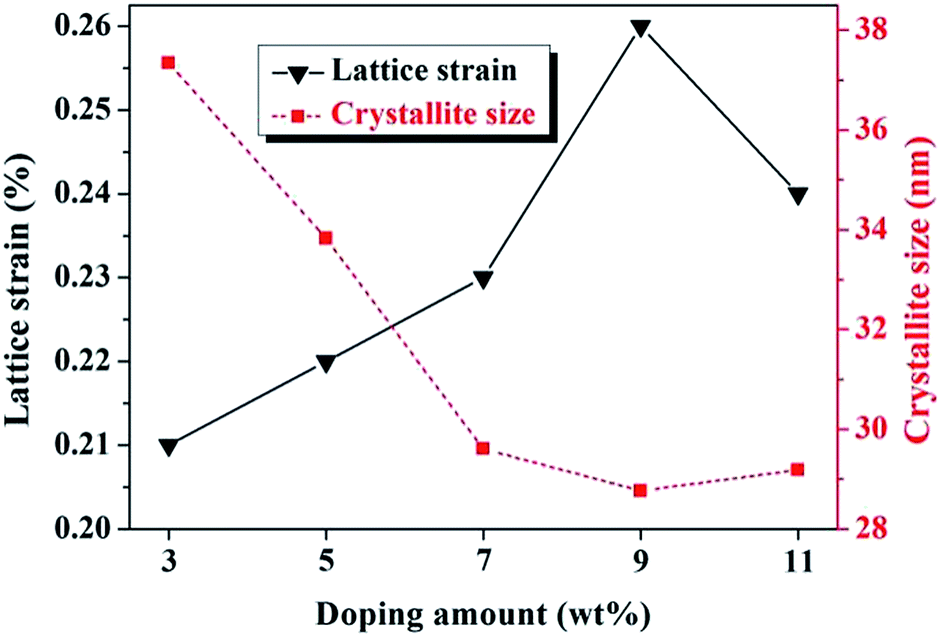 | ||
| Fig. 5 Alteration of orthopyroxene's lattice strain and crystallite size with different La doping. | ||
3.5. Infrared absorption spectra
Fig. 6 showed the FT-IR spectra of the samples in the range of 400–1600 cm−1. There were two broad absorption bands ν1 and ν2 in the range of 400–1600 cm−1 wavenumber. These bands ν1 and ν2 were ascribed to the stretching vibration of multifarious bridge bonds in A- and B-site, respectively. Combined with the analytic results of local microstructure, the existence of a large number of Fe3+ ions in B-site might push Mg2+ and Fe2+ ions towards A-site. On account of being oxidized, the influences of Fe2+ seemed to be small, even negligible in A-site. Meanwhile, the larger Ca2+ originally tended to occupy A-site, which should not alter the original mode of interatomic vibration.38 Mg2+ and Fe3+ were considered as newly-introduced ions to substitute for quondam ions in the A-site and B-site respectively, which necessarily affected the stretching vibration of conterminal Si–O. Thus, the band ν1 at 1080 cm−1 might be linked with the bridging vibration of Si–O–Mg in A-site, and the band ν2 at 465 cm−1 was related with the bending vibration of Si–O–Fe in B-site.41,42 These bridge bonds were crucial to infrared properties. | ||
| Fig. 6 IR spectra of vanadium–titanium slags ceramic samples doped with different La contents. | ||
In general, with the addition of La, the alterations of cations distribution in A-site and B-site decrease the metal–oxygen bond lengths, enhance the lattice asymmetric vibration, and ultimately increase IR absorption vibration intensity of sample.43 As for La9 group, the absorption vibration of Si–O–Mg in A-site and Si–O–Fe in B-site were strengthened greatly.
According to the kirchhoff's law of thermal radiation, the ratio of emissivity and absorption is irrelevant to property of substances, which is the universal function of temperature and wavelength. It shows a positive correlation with the absorption coefficient.29 In brief, the superior absorption ability means the excellent emission capacity, and the position of vibration absorption is just the location of radiation release. The basic reason on the improvement of the far-infrared absorption properties of materials derived from appropriate doping, which strengthened the infrared radiation capabilities of materials synchronously.
3.6. Far-infrared emission property
Infrared emissivity is significant to evaluate the infrared emission property, especially in the wavelength range of 8–14 μm.20 The average infrared emissivity of all samples in the 8–14 μm wavelength range was listed in Fig. 7. It witnessed an evident increase in the far-infrared emissivity with the doping La from 0 wt% to 7 wt%, reaching the maximum (0.927) at 9 wt%. Then, it decreased dramatically to 0.916 for La11 sample. The infrared emission spectrum of La9 was displayed in the upper left inset of Fig. 7. Because the highest emissivity was 0.927, the as-prepared ceramics belonged to the category of high far-infrared emissivity materials, based on the comparison with reported studies.20,44,45 Our experiments demonstrated that infrared emission properties could be effectively stimulated by appropriate La additives, and the optimal doped amount of La for the specimen was 9 wt% in this work. The generation of high far-infrared emissivity resulted from oxidation of Fe2+ to Fe3+ by La3+, which caused alteration of cations distribution in A-site and B-site, leading to the shrinkage of unit cell and lattice strain. From Fig. 5 and 7, it was worthy to note that the IR emissivity was changed from 0.854 to 0.927 (increased by 7.1%) as the reduction of crystallite size of orthopyroxene from 37.343 nm to 28.768 nm and the increasing of its lattice strain from 0.21% to 0.26%. The severer lattice strain is and the lower lattice vibration symmetry has. When the lattice strain achieved the maximum, the vibration intensity of Si–O–Mg and Si–O–Fe in A-site and B-site were strongest. Thereby, the sample displayed the greatest far-infrared emission properties. The profound reasons might was attributed to aggrandizing the lattice asymmetrical vibrations, and largely changing the dipole matrix, and improving the impacts of a harmonic oscillation of polar lattice and phonon coupling, and promoting the action of phonon combination radiation.18,28 Hence, the far-infrared radiation property was strongly strengthened in our prepared La-doped VTS ceramics.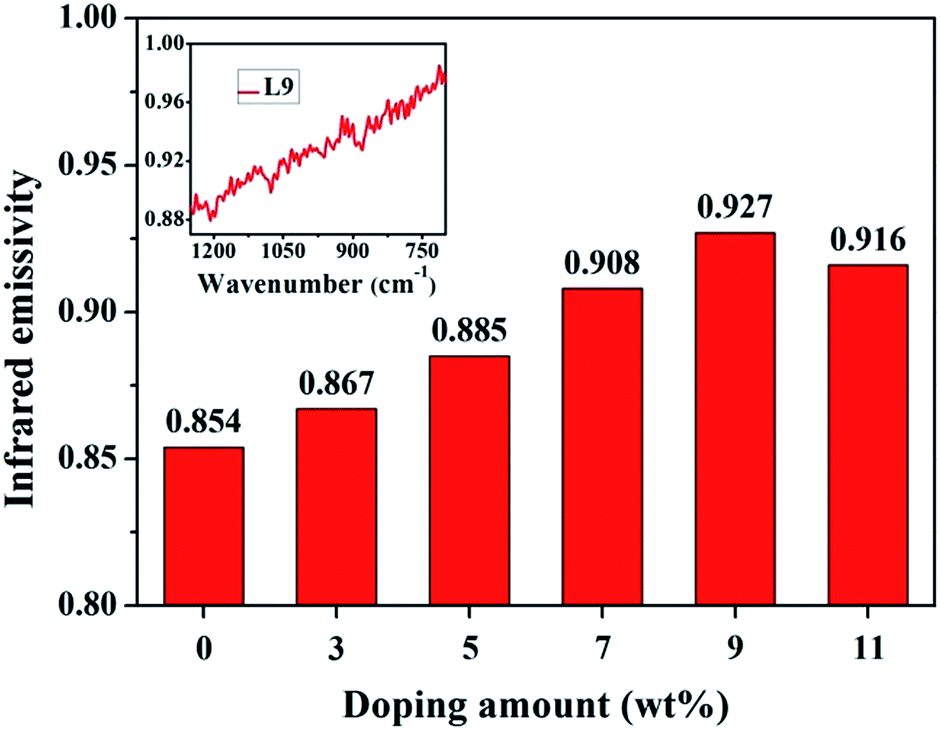 | ||
| Fig. 7 Far-infrared emissivity of vanadium–titanium slags ceramic powder samples. | ||
3.7. Physical properties
The linear shrinkage (LS%), water absorption (WA%), and bending strength (BS) were further measured. As reported in Table 4, the LS% of all experimental and control ceramic samples of was around 1.6% within a controllable range, and the bending strength of the all samples was higher than 25 MPa. According to ISO 13006 standard-Group BIIIb (6% < WA% < 10%), this kind of far-infrared ceramic meets the requirement of ISO 13006 standard (>18 MPa). It was noted that bending strength of the doped groups was slightly higher than that of pure VTS ceramics, and it peaked at 30.63 MPa for La11 group.| Sample | LS% | WA% | BS (MPa) |
|---|---|---|---|
| L0 | 1.62 ± 0.10 | 6.75 ± 0.55 | 26.38 ± 2.43 |
| L3 | 1.59 ± 0.05 | 6.50 ± 0.84 | 28.91 ± 1.04 |
| L5 | 1.66 ± 0.04 | 6.49 ± 0.58 | 29.57 ± 3.27 |
| L7 | 1.63 ± 0.09 | 6.58 ± 0.87 | 30.47 ± 3.70 |
| L9 | 1.65 ± 0.12 | 6.61 ± 0.72 | 30.45 ± 2.28 |
| L11 | 1.66 ± 0.06 | 6.63 ± 0.34 | 30.63 ± 2.25 |
Form XRD analysis, we could see that the quartz and anorthite phases were co-existed in as-burnt production. The chemical structure of quartz is stable, and the anorthite possessed high mechanical strength and low sintering temperature. The existences of quartz and anorthite phases would ensure the fundamental physical strength and chemical stability of the ceramics. From the SEM images, the doped production with smoother fracture surface displayed more glass phase during sintering. The addition of La could accelerate the rate of mass transfer and facilitate the densification process of ceramics.4 Moreover, the dispersive distribution of La2O3 phase also contributed to the augment of bending strength via crack deflection toughening. When cracks extended to the surface of dispersive granular phases (La2O3), the direction of cracks propagation would deflect and slope, which could enhance energy consumption of crack propagation and improve toughness. Consequently, these factors jointly contributed to the higher bending strength of doped groups, especially for La11 samples.
4. Conclusions
In conclusion, VTS has been successfully used as a secondary resource for the preparation of far-infrared ceramic doped with 0–11 wt% rare earth La. The effect of doped La on the far-infrared radiation properties of VTS ceramics have been studied detailedly. When doped with 9 wt% La, the VTS ceramics possessed the best far-infrared radiation attribute, and its emissivity reached 0.927. Our finding showed that La3+ ions made Fe2+ oxidize to Fe3+ in the orthopyroxene-type phases, causing the shrinkage of its unit cell volume. Simultaneously, La3+ dissolved into orthopyroxene-type phases to form solid solution with a complex structure. The superposed effects of La3+ work together to cause lattice distortion and improve the vibration of two bridge bonds Si–O–Mg and Si–O–Mg, which ultimately result in the enhancement of the far-infrared emission properties of VTS ceramics. Despite of the uncontrollability of vast quantities for mineral raw materials, this study may provide some suggestive guidance for the bulk-production of far-infrared ceramics and the resource utilization of VTS.Acknowledgements
This work was supported by the Science and Technology Support Program of Sichuan Province (2015GZ0175), and the Special Fund of Science and Technology Incubation and Achievement Transformation of Neijiang (2015-QT-03).Notes and references
- A. Dolgorsuren, K. Yamashita and S.-O. Dalkhsuren, J. Hard Tissue Biol., 2014, 23, 423–434 CrossRef CAS.
- G. Shao, X. Wu, Y. Kong, S. Cui, X. Shen, C. Jiao and J. Jiao, Surf. Coat. Technol., 2015, 270, 154–163 CrossRef CAS.
- Y. M. Wang, H. Tian, X. E. Shen, L. Wen, J. H. Ouyang, Y. Zhou, D. C. Jia and L. X. Guo, Ceram. Int., 2013, 39, 2869–2875 CrossRef CAS.
- L. Jie, M. Junping, L. Jinsheng and H. Xiaoli, J. Rare Earths, 2014, 32, 890–894 CrossRef.
- P. Ketsuk, M. Lim, S. H. Baek, J. Jung, D. Kim, R. G. Malegowd, J. Seo and S. B. Khan, J. Appl. Polym. Sci., 2016, 133, 43102 CrossRef.
- D. Chen, L. Zhao, Y. Liu, T. Qi, J. Wang and L. Wang, J. Hazard. Mater., 2012, 244, 588–595 Search PubMed.
- Z. H. Wang, S. L. Zheng, S. N. Wang, B. Liu, D. W. Wang, D. U. Hao and Y. Zhang, Trans. Nonferrous Met. Soc. China, 2014, 24, 1273–1288 CrossRef CAS.
- Z. Liu, Y. Li, M. Chen, A. Nueraihemaiti, J. Du, X. Fan and C. Y. Tao, Hydrometallurgy, 2015, 159, 1–5 CrossRef.
- L. Zhao, L. Wang, T. Qi, D. Chen, H. Zhao and Y. Liu, Hydrometallurgy, 2014, 149, 106–109 CrossRef CAS.
- X. Li, H. Yu and X. Xue, Procedia Environ. Sci., 2016, 31, 582–588 CrossRef.
- M. Dong, X. Xue, H. Yang, D. Liu, C. Wang and Z. Li, J. Hazard. Mater., 2016, 318, 751–757 CrossRef CAS PubMed.
- Y. Shi, S. Deng, H. Wang, J. Huang, Y. Li, F. Zhang and X. Shu, RSC Adv., 2016, 6, 15999–16009 RSC.
- D. Tang, D. Zhou, J. Zhou, P. Zhang, L. Zhang and Y. Xia, Hydrometallurgy, 2015, 157, 90–96 CrossRef CAS.
- S. S. Liu, Y. F. Guo, G. Z. Qiu, T. Jiang and F. Chen, Trans. Nonferrous Met. Soc. China, 2013, 23, 1174–1178 CrossRef CAS.
- S. M. Wang, F. H. Kuang, Q. Z. Yan, C. C. Ge and L. H. Qi, J. Alloys Compd., 2011, 509, 2819–2823 CrossRef CAS.
- M. Chang, Y. Sheng, Y. Song, K. Zheng, X. Zhou and H. Zou, RSC Adv., 2016, 6, 52113–52121 RSC.
- V. Mahalingam and J. Thirumalai, RSC Adv., 2016, 6, 80390–80397 RSC.
- Y. Zhang, J. Ma, J. Lu and D. Wen, Ceram. Int., 2014, 40, 4437–4443 CrossRef CAS.
- Y. Zhang and D. Wen, J. Chin. Ceram. Soc., 2008, 36, 743–747 CAS.
- Y. Zhang and D. Wen, Mater. Sci. Eng., B, 2010, 172, 331–335 CrossRef CAS.
- X. Wu, H. Yu, H. Dong and L. Geng, Ceram. Int., 2014, 40, 5905–5911 CrossRef CAS.
- Z. Zhao, C. Liu, Y. Jiang, J. Zhang, H. Tao, J. Han, X. Zhao and J. Heo, J. Non-Cryst. Solids, 2014, 404, 37–42 CrossRef CAS.
- Y. Zhang, R. Huang, Z. Lin, J. Song, X. Wang, Y. Guo, C. Song and Y. Yu, J. Alloys Compd., 2016, 686, 407–412 CrossRef CAS.
- J. Liu, J. Meng, J. Liang, X. Duan, X. Huo and Q. Tang, Mater. Res. Bull., 2015, 66, 26–31 CrossRef.
- X. Yu, Z. Zhang and J. Xie, Mater. Lett., 2016, 184, 294–297 CrossRef CAS.
- M. V. D. S. Rezende, M. E. G. Valerio and R. A. Jackson, Opt. Mater., 2011, 34, 109–118 CrossRef CAS.
- Y. Xu, R. Fu, S. Agathopoulos, X. Wang, Y. Yang and J. Cai, J. Alloys Compd., 2017, 693, 454–461 CrossRef CAS.
- S. Vijay, K. R. Vineet, V. Venkatramu, R. P. S. Chakradhar and H. K. Sang, J. Lumin., 2013, 134, 396–400 CrossRef.
- Y. Zhang and D. Wen, Mater. Chem. Phys., 2012, 131, 575–580 CrossRef CAS.
- A. Noviyanto, S. W. Han, H. W. Yu and D. H. Yoon, J. Eur. Ceram. Soc., 2013, 33, 2915–2923 CrossRef CAS.
- P. K. Baitha and J. Manam, J. Rare Earths, 2015, 33, 805–813 CrossRef CAS.
- A. Lukowiak, R. J. Wiglusz, R. Pazik, K. Lemanski and W. Strk, J. Rare Earths, 2009, 27, 564–568 CrossRef.
- J. Conradie and E. Erasmus, Polyhedron, 2016, 119, 142–150 CrossRef CAS.
- J. A. R. Guivar, E. A. Sanches, F. Bruns, E. Sadrollahi, M. A. Morales, E. O. López and F. J. Litterst, Appl. Surf. Sci., 2016, 389, 721–734 CrossRef.
- E. A. Skryleva, I. V. Kubasov, P. V. Kiryukhantsev-Korneev, B. R. Senatulin, R. N. Zhukov, K. V. Zakutailov, M. D. Malinkovich and Y. N. Parkhomenko, Appl. Surf. Sci., 2016, 389, 387–394 CrossRef CAS.
- H. Wang, S. Feng and W. Yang, Mater. Res. Bull., 2014, 54, 268–271 CrossRef.
- A. L. Huber, M. Heuer, K. T. Fehr, K. Bente, E. Schmidbauer and G. D. Bromiley, Phys. Chem. Miner., 2004, 31, 67–79 CrossRef CAS.
- S. P. Thompson, K. Demyk, S. J. Day, A. Evans, H. Leroux, C. Depecker, J. E. Parker, L. Connor, H. Wilhelm and G. Cibin, J. Non-Cryst. Solids, 2016, 447, 255–261 CrossRef CAS.
- C. Sujatha, K. V. Reddy, K. S. Babu, R. C. Reddy and K. H. Rao, Ceram. Int., 2013, 39, 3077–3086 CrossRef CAS.
- D. Zou, X. Chu and F. Wu, Ceram. Int., 2013, 39, 3585–3589 CrossRef CAS.
- J. Zhu, P. Shi, F. Wang, T. Zhao and H. Jiang, Ceram. Int., 2015, 42, 5250–5257 CrossRef.
- P. Makreskia, G. Jovanovskia and T. Biljanc, J. Mol. Struct., 2006, 788, 102–114 CrossRef.
- A. M. Shaikh, S. A. Jadhav, S. C. Watawe and B. K. Chougule, Mater. Lett., 2000, 44, 192–196 CrossRef CAS.
- D. Wang, M. Han, M. Li and X. Yin, Mater. Lett., 2016, 183, 223–226 CrossRef CAS.
- J. Ye, C. Bu, Z. Han, F. Wang, X. Li, Y. Chen and J. Li, J. Eur. Ceram. Soc., 2015, 35, 3111–3118 CrossRef CAS.
Footnote |
| † These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2017 |