
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Copolymers of aniline and 2-aminoterephthalic acid as a novel cathode material for hybrid supercapacitors†
Yi Gao‡
a,
Yu Li‡acd,
Haoran Ana,
Yiyu Fengacd and
Wei Feng *abcd
*abcd
aSchool of Materials Science and Engineering, Tianjin University, Tianjin 300072, P. R China. E-mail: weifeng@tju.edu.cn
bCollaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300072, P. R China
cKey Laboratory of Advanced Ceramics and Machining Technology, Ministry of Education, Tianjin 300072, P. R China
dTianjin Key Laboratory of Composite and Functional Materials, Tianjin 300072, P. R China
First published on 27th January 2017
Abstract
Hybrid supercapacitors (HSCs) have attracted increasing attention as they can deliver energy densities sufficient for batteries and power densities of supercapacitors. The exploration of novel types of capacitor cathode materials with higher energy densities than conventional active carbons is in demand. Here, a series of copolymers of aniline (ANI) and 2-aminoterephthalic acid (ATA) were synthesized by the chemical oxidation polymerization at different molar ratios of monomers (ANI/ATA). The morphologies of the products varied from nanoparticles to nanorods and then returned to granular nanoparticles with the increase of the ATA amount. The copolymer nanorods synthesized at an ANI/ATA ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 exhibited a high specific capacitance (198 F g−1 at the current density of 20 mA g−1), excellent rate capability (the maximum current density of 50 A g−1), and excellent cycling stability (capacitance retention of 78.5% after 1000 cycles at 2 A g−1). The appropriate amount of carboxyl groups enabled the delocalization of charge along the polymer backbone and the one-dimensional nanostructures facilitated the anion diffusion to the electrode, which enhanced the electrochemical activity of the copolymer significantly. The excellent rate capability as well as the anion doping/dedoping energy storage mechanism impelled the copolymer to be employed as the supercapacitor cathode of HSCs. The energy density of 153.9 W h kg−1 and the power density of 3011.5 W kg−1 of HSCs are obtained with commercial meso-carbon microbeads (MCMBs) and Li4Ti5O12 (LTO) as anode materials, respectively.
:2 exhibited a high specific capacitance (198 F g−1 at the current density of 20 mA g−1), excellent rate capability (the maximum current density of 50 A g−1), and excellent cycling stability (capacitance retention of 78.5% after 1000 cycles at 2 A g−1). The appropriate amount of carboxyl groups enabled the delocalization of charge along the polymer backbone and the one-dimensional nanostructures facilitated the anion diffusion to the electrode, which enhanced the electrochemical activity of the copolymer significantly. The excellent rate capability as well as the anion doping/dedoping energy storage mechanism impelled the copolymer to be employed as the supercapacitor cathode of HSCs. The energy density of 153.9 W h kg−1 and the power density of 3011.5 W kg−1 of HSCs are obtained with commercial meso-carbon microbeads (MCMBs) and Li4Ti5O12 (LTO) as anode materials, respectively.
1. Introduction
With the development of various technological applications, including portable electronic devices and electric vehicles, much attention has been paid to create new-types of energy storage devices with both high energy density and power density. Recently, a novel supercapacitor-battery hybrid energy storage system, also called hybrid supercapacitors (HSCs), was proposed to combine the fast charging rate of a supercapacitor with the high energy density of lithium ion batteries (LIBs).1 HSCs is usually coupled with a capacitive cathode and a Li-insertion anode. In 2001, Amatucci et al. first fabricated HSCs by using active carbon (AC) as the cathode and nanostructured Li4Ti5O12 (LTO) as the anode. The energy density of the packaged device was nearly three times higher than conventional supercapacitors.2,3To the scope of capacitive cathode, based on the mechanism of anions adsorption/desorption onto/from the surface of cathode, carbonaceous materials with high specific surface areas and micro/mesoporous structures are considered as ideal candidates. In addition to typical electrochemical double layer capacitance (EDLC) cathode materials like AC, some novel carbonaceous materials are also explored to be a capacitive cathode for HSCs, such as three-dimensional graphene,4–6 trigol-reduced graphene oxide (TRGO),7 oligomer derived carbon (ODC),8 activated polyaniline-derived carbon (NAC),9 hierarchical nitrogen-doped carbon (HNC),10 etc. However, EDLC cathode materials exhibit limited capacitance properties due to the physical energy storage mechanism. The microporous structure further undermines the capacitive reaction at high current densities. Therefore, it is still urgent to explore new kinds of HSC's cathode materials with high specific capacitances and rate capabilities.
Conducting polymers (CPs) have been applied as the electrode materials for rechargeable batteries because of their redox activities since 1980s.11 The energy storage for CPs is usually realized by the anions doping/dedoping into/from the backbone, regardless the routes of polymerization.12 However, CPs are usually used as the conductive additives in the composite electrodes rather than the electrode materials in the field of rechargeable batteries due to the lower energy densities of CPs than those of commercialized inorganics.13 Nowadays, in addition to be used in the aqueous supercapacitors, CPs are also the important cathode materials of HSCs for their pristine high electronic conductivities and the special energy store mechanism derived from the doping/dedoping anions with favorable kinetics.14–16
Among the kinds of CPs, polyaniline (PANI) has been received research interests consecutively because of its oxidative stability, relatively high theoretical capacity, convenient synthesis, and electrochemical reversibility between many redox states.3,17 In fact, PANI lithium battery is one of few examples of polymer-based energy storage that have been commercialized, but the relatively low energy density and the undesirable cycling stability blocked its further application.18 In order to solve these problems, PANI with special nanostructures or combined with nanostructured carbons was designed. For example, PANI nanotubes doped with HClO4 exhibited higher specific capacity (75.7 mA h g−1) than commercial PANI powders (65.8 mA h g−1) due to the higher surface area and faster diffusion kinetics of anions.19 The nanocomposite of PANI with carbon nanotubes prepared by the layer-by-layer self-assembly and electrodeposition methods exhibited the high specific capacity and superior rate capability due to the synergetic effects of EDLC and faradaic mechanism,20–22 though the synthesis methods were time consuming and hardly to accommodate with the industrial manufacture. Recent advances in research have exploited some promising strategies, such as being modified with redox active groups or incorporated with redox polymers to enhance the storage capacity and rate capability of PANI. Yang et al.23 prepared the copolymer of aniline (ANI) and o-nitroaniline that exhibited a high redox potential as the cathode material of sodium ion batteries. Sulfide-containing and ferrocene-containing aniline copolymers were also synthesized as the novel cathode materials with significantly improved cycling stability,24,25 though the rate capability of these copolymers was not further investigated.
In this study, copolymers of ANI and 2-aminoterephthalic acid (ATA) were synthesized by the chemical oxidation polymerization and the molecular structures, crystallinities, morphologies, and electrochemical performances of copolymers with different ratios of ANI to ATA (ANI/ATA) were characterized. It was found that the copolymer obtained at ANI/ATA ratio of 8:2 exhibited significantly improved electrochemical performances due to the appropriate charge delocalization and the special nanostructure for the introduction of carbonyl compared with pristine PANI. The maximum charge/discharge current density of the copolymer reached to 50 A g−1, with the specific capacitance of 18.3 F g−1 and the rate performance is excellent in the polyaniline based supercapacitors tested in the organic electrolyte. The HSCs fabricated by the synthesized copolymer as the cathode and commercial meso-carbon microbeads (MCMBs) as the anode yielded the energy density of 153.9 W h kg−1 at the power density of 42 W kg−1 while the energy density of the HSC with Li4Ti5O14 (LTO) as the anode material reached 130.2 W h kg−1 at power density of 39.2 W kg−1, which also remained at 26.1 W h kg−1 even at high power density of 3011.5 W kg−1. Besides, differ from the traditional nanorod preparation process where the organic acids work as the soft templates and doping aids at the same time, the dimers of monomers serve as the soft template. The molecular weight of HCl is relatively lower than those organic doping acids and this is beneficial to the specific capacitance.
2. Experimental
2.1 Preparation of P(ANI-co-ATA)
P(ANI-co-ATA) was synthesized via the chemical oxidative polymerization. In a typical experiment, some amount of ATA (Aldrich, 98%) was dissolved in 500 ml 1 M HCl solution, followed by the adding of aniline (ANI, Acros, 99.8%) with specific volume. The monomers were dissolved in the HCl medium with the help of an ultrasonic treatment completely until the solution becoming transparent. Then ammonium persulfate (APS), in the equimolar ratio with the sum of monomers, solved in 20 ml 1 M HCl solution was added dropwise in 1 h. The polymerization was processed in the ice bath for 12 h. The product was collected by filtration and washed by ethanol and water for several times to remove the residual monomers and APS. After being dried at 60 °C under vacuum for 24 h, the dark green powder of copolymers was obtained finally. A series of copolymers with different molar ratios of ANI to ATA were polymerized, which were denoted as P(ANI-co-ATA, 9:1), P(ANI-co-ATA, 8:2), P(ANI-co-ATA, 7:3), and P(ANI-co-ATA, 6:4), respectively. PANI was also synthesized in the same procedure so as to investigate the effects of ATA segment on the electrochemical performances.
2.2 Characterization
FT-IR was carried out on Bruker Tensor 27 by using KBr pellets. Raman spectroscopy was get from Thermo Electron with 514 nm laser excitation. UV-vis spectra was measured using a Persee/tU1901 spectrometer in the wavelength range of 200 to 900 nm and XRD analyses were performed on a Perkin Elmer PHI 3056 diffractometer with Cu Kα radiation (λ = 1.5406 Å) and the diffraction data were recorded for 2θ angles between 10° and 50°. The SEM was conducted using Hitachi S-4800 field emission scanning electron microscope with acceleration voltage of 10 kV. TEM was carried out on a Philips Tecnai G2 F20 electron microscopy operated at 200 kV.2.3 Fabrication of half cell
The copolymer powder (90 wt%), as the cathode material, was mixed directly with 5 wt% acetylene black and 5 wt% polytetrafluorethylene (PTFE), rolled into thin sheets, then punched into 1 cm diameter electrode and pressed onto aluminum steel foil coated by carbon (10 μm). The mass of materials loading in each electrode was around 2 mg. Half cells were firstly assembled so as to evaluate the electrochemical performances of copolymer cathodes by using CR-2016 type coin cells. Batteries were assembled in an argon-filled glove box (Mikrouna Co., Advanced 2440/750) with Li metal foil as the counter and reference electrode, Celgrad 3500 as the separator and 1 M LiClO4 in a mixture of ethylene carbonate and dimethyl carbonate (EC:DMC = 1:1 by volume) as the electrolyte.
2.4 Electrochemical measurement
All the electrochemical tests were carried out at the constant 25 °C. The cyclic voltammetry (CV) and electrical impedance spectroscopy (EIS) studies were performance by using an electrochemical workstation (Advanced Electrochemical System Parstat 2263). The galvanostatic charge/discharge measurements of half cells and HSCs at various current densities with potential windows for specific materials were tested using a cell test instrument (Land CT2001A, Wu Han JinNuo Electronics Co., China).The specific capacitance of an electrode can be calculated according to the following equation from CV curves
| Cg = S/(2vmΔU) | (1) |
The specific capacitance Cg (F g−1) of copolymers in the half cells could also be calculated from galvanostatic charge–discharge profiles as the following:
| Cg = IΔt/(mΔU) | (2) |
The energy density values E (W h kg−1) of half cells and HSCs were calculated by numerically integrating the t–V graph area during the discharge process according to the following formula:
 | (3) |
The power density values P (W kg−1) of half cells and HSCs were calculated according to equation:
| P = E/t | (4) |
In which t is the discharge time.
3. Results and discussion
The copolymers were synthesized by the conventional chemical oxidation polymerization at different ANI/ATA values, detonated as P(ANI-co-ATA, ANI:ATA). PANI was also polymerized under the same condition for comparison. The detailed synthesis procedure is exhibited in the ESI.† The Fourier transform infrared spectroscopy (FT-IR) spectra of copolymers and PANI are shown in Fig. 1a. The peak at 3341 cm−1 in the copolymers and PANI are attributed to the N–H stretching vibrations of secondary amines.23 The peak at 822 cm−1 is assigned to the out-of-plane of vibration para-disubstituted benzene.26 The peaks lie at 1570 and 1500 cm−1 in all the samples are attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching of the quinoid and benzenoid rings, respectively. The intensity ratio of peaks at 1570 and 1500 cm−1 implies the oxidation degree of the main chain.27,28 It can be found out that copolymers and PANI are all in the half-oxidized state due to the approximately same peak intensities in all curves. In addition, compared with PANI, the two additional peaks at 1700 and 1250 cm−1 in the copolymers, ascribed to the CO and C–O stretching vibration of carboxylic groups in ATA, confirm the successful copolymerization of ANI and ATA. The intensities of these two peaks enhance with the decrease of ANI/ATA value. Besides, compared with the CO stretching vibration of free-state carboxyl that exhibits the absorption peak at 1760 cm−1 normally, the red shift of this peak in copolymers is caused by the formation of hydrogen bonds between carboxyl groups.29 Furthermore, two small peaks at 1445 and 1414 cm−1 observed in the samples of P(ANI-co-ATA, 7:3) and P(ANI-co-ATA, 6:4) are stemmed from the CC stretching vibration of the substituted aromatic ring and symmetric stretching in the phenazine ring, respectively, which indicate the partial formation of oligomers in the copolymers with the increase of ATA amount,30,31 because the steric effects of the carboxyl substituent in ATA tends to prohibit the polymerization of the polymer.32
C stretching of the quinoid and benzenoid rings, respectively. The intensity ratio of peaks at 1570 and 1500 cm−1 implies the oxidation degree of the main chain.27,28 It can be found out that copolymers and PANI are all in the half-oxidized state due to the approximately same peak intensities in all curves. In addition, compared with PANI, the two additional peaks at 1700 and 1250 cm−1 in the copolymers, ascribed to the CO and C–O stretching vibration of carboxylic groups in ATA, confirm the successful copolymerization of ANI and ATA. The intensities of these two peaks enhance with the decrease of ANI/ATA value. Besides, compared with the CO stretching vibration of free-state carboxyl that exhibits the absorption peak at 1760 cm−1 normally, the red shift of this peak in copolymers is caused by the formation of hydrogen bonds between carboxyl groups.29 Furthermore, two small peaks at 1445 and 1414 cm−1 observed in the samples of P(ANI-co-ATA, 7:3) and P(ANI-co-ATA, 6:4) are stemmed from the CC stretching vibration of the substituted aromatic ring and symmetric stretching in the phenazine ring, respectively, which indicate the partial formation of oligomers in the copolymers with the increase of ATA amount,30,31 because the steric effects of the carboxyl substituent in ATA tends to prohibit the polymerization of the polymer.32
 | ||
| Fig. 1 (a) FT-IR, (b) Raman, (c) UV-vis spectras and (d) XRD pattern for PANI, P(ANI-co-ATA, 9:1), P(ANI-co-ATA, 8:2), P(ANI-co-ATA, 7:3), P(ANI-co-ATA, 6:4) and PANI. | ||
The molecular structures of copolymers and PANI were further characterized by Raman spectroscopy (Fig. 1b). The C–C stretching vibration at 1390 cm−1, C–N˙+ stretching at 1331 cm−1, and CC stretching vibration at 1571 cm−1 are observed in all the samples, demonstrating the presence of the doped state. The bands lie at 1466 cm−1 and 1500 cm−1 in all samples are attributed to the CN stretching mode in the unprotonated and protonated segment of quinonimine, respectively.33,34 The significantly decreased intensity of band at 1466 cm−1 and the gradually enhanced intensity of band at 1500 cm−1 with the decrease of ANI/ATA value demonstrate the protonation effect of carboxyl groups in ATA segment to the copolymer backbone. Besides, the band around 580 cm−1 observed in P(ANI-co-ATA, 6:4) associated with a substituted phenazine segment is the signal of the existence of oligomers, which is in agreement with the FTIR spectra.29
The different conformations and structures of copolymers and PANI were further characterized by ultraviolet-visible (UV-vis) absorption spectroscopy in N-methyl pyrrolidone (Fig. 1c). There are two distinct absorption bands located at 340 and 560 nm in the UV-vis spectra of PANI. The former is ascribed to the π–π* transition of the benzenoid ring, which is related to the extended conjugation between adjacent rings in the polymeric chains, and the latter is assigned to the π–polaron transition.33 The copolymers show the similar adsorption spectra with PANI. The wavelengths of the absorption bands of copolymers are larger than that of PANI because of the protonation effect of carboxyl groups in ATA, revealed in Raman spectra.35 Moreover, the tiny band at 450 nm in the spectra of P(ANI-co-ATA, 9:1) and P(ANI-co-ATA, 8:2) represents the polaron–π* transition, indicating these copolymers in the doped state.36 When the ratio of ANI/ATA decreases to 7:3 and 6:4, however, an additional peak lies at 420 nm appears in the spectra of these copolymers, which is associated with the benzoid rings in the polymer chain.37 The appearance of this peak and the decreasing band intensity above 600 nm indicate the lowered conjugation level and the formation of oligomers, which is in accordance with the results of FTIR and Raman spectra.
The crystal structures of copolymers and PANI are characterized by X-ray diffraction (XRD) (Fig. 1d). PANI shows three broad diffraction peaks with relatively high intensity indicating the semicrystalline nature of PANI. The diffraction peak of PANI located at 25.0° is attributed to the (200) plane, which is periodicity perpendicular to the polymer chains, and the peak centered at 20.5° is correspond to the (100) plane that are periodicity parallel to the polymer chain.38,39 The broad band around 15.1° is on account of the dopant.28 Similar diffraction peaks are also observed in the XRD patterns of copolymers, which indicates the existence of continuous PANI segment in the copolymers backbone. However, additional diffraction peaks at 2θ = 27.8°, 17.7° and 11.9° demonstrate the oligomers existence in the copolymers further.40 The increased intensities of these peaks with the decrease of ANI/ATA value indicate the more existent oligomer components in the copolymer. The higher degree of crystallinity of copolymer polymerized at lower ANI/ATA value is generated by the more polymer cation–anion complexes between carboxyl groups of ATA and polycation PANI, which enhances the inter-chain packing and the structural order in the solid state.41
The morphologies of copolymers were characterized by scanning electron microscopy (SEM) at first. From SEM images of copolymers shown in Fig. 2a–d, it can be found that the ANI/ATA value has a remarkable influence on the morphology of the synthesized copolymers. P(ANI-co-ATA, 9:1) and P(ANI-co-ATA, 8:2) are both in the regular rod-like shapes with almost uniform diameters in the range of 50–100 nm and lengths about hundreds nanometers, and these nanorods are twinkled each other. However, except nanorods, some agglomerates can still be observed in the SEM image of P(ANI-co-ATA, 9:1). Although the shape of P(ANI-co-ATA, 7:3) is still rod-like, the SEM image reveals that these rods have some irregular shapes. As the ANI/ATA value further decrease to 6:4, granular nanoparticles with size about 50–80 nm, rather than nanorods, are observed. The transmission electron microscopy (TEM) images of these copolymers, shown in the insets of corresponding SEM images, reveal the same result. Irregular PANI agglomerates are observed from SEM and TEM images shown in Fig. S1 (ESI†), which is consistent with the conventional PANI powder polymerized in the aqueous solution at low pH values.42 The different morphologies of PANI and copolymers demonstrate that the amount of ATA have a great impact on the growth mechanism of copolymers. The dimers of ANI (in the yellow frame) and ATA (in the green frame) tends to form micelles due to the coexistent hydrophilic groups (carboxyl and protonated amino) and hydrophobic group (benzene ring).43 The micelles can be regarded as the soft temples in the formation of different morphologies through the self-assembly process.44 When the value of ANI/ATA is higher than 8:2, at beginning of the polymerization, the excessive protonated ANI monomers are absorbed on the external of the micelles and enhance the linear growth of the copolymer. At the same time, the free ANI monomers diffuse into the micelles and form the solid core subsequently.45 However, PANI nanoparticles also form in the initial period of the polymerization due to the much abundant protonated ANI monomers in the polymerization system when ANI/ATA value reaches to 9:1. These agglomerates are likely to be absorbed on the surface of nanorods through hydrogen bonds or π–π stacking interaction,45,46 represented by the SEM and TEM images shown in Fig. 2a. On the other hand, when ANI/ATA value reduces to 7:3, more dimers form and hence aggregate to spherical micelles. The rest of ANI monomers are free to diffuse into the micelles to form granular nanoparticles after the beginning of polymerization.34 The different growth mechanism at varied ANI/ATA values is represented in Fig. 2e.
 | ||
| Fig. 2 SEM images of P(ANI-co-ATA, 9:1) (a), P(ANI-co-ATA, 8:2) (b), P(ANI-co-ATA, 7:3) (c) and P(ANI-co-ATA, 6:4) (d). The insets in each SEM images are the corresponding TEM images of copolymers with the scale bar represents 50 nm. (e) The proposed growth mechanism for copolymers synthesized at different ANI/ATA values. | ||
The electrochemical characters of copolymers and PANI are evaluated through Li-half cells at first and the detailed information is provided in the ESI.† The cyclic voltammograms (CV) of all the copolymers as well as PANI cathodes with the potential window from 2.0 to 4.0 V (vs. Li+/Li) are shown in Fig. 3a. By comparing the CV curves of all the copolymers, it can be seen that the area surrounded by the CV curve of P(ANI-co-ATA, 8:2) is the largest, indicating the highest specific capacitance of the copolymer. Therefore P(ANI-co-ATA, 8:2) was taken as the typical one for analysis and three couples of redox peaks are observed in the CV curve. The main two couples of redox peaks around 3.40/3.32 V and 3.05/2.98 V are attributed to the doping/dedoping reaction of ClO4− anions into/from the para-disubstituted benzene ring and quinone diimine structure of the copolymer, respectively.47,48 The small pair of redox peaks located at 2.70/2.40 V is probably accompanied by the Li+ extraction/insertion from/into the carbonyl groups of benzenoid and quinonoid ATA segment due to the absence of this redox peak in the CV curve of PANI.49 The calculated specific capacitances of copolymers, synthesized at ANI/ATA value of 9:1, 8:2, 7:3, and 6:4, are 137, 191, 129 and 45 F g−1 from CV curves at the scan rate of 0.5 mV s−1, respectively, while the specific capacitance of PANI is calculated as 110 F g−1 at the same test condition.
 | ||
| Fig. 3 The electrochemical measurements of copolymers and PANI electrodes. (a) CV curves of copolymers and PANI electrodes scanned between 2.0 to 4.0 V (vs. Li+/Li) at the rate of 0.5 mV s−1. (b) Galvanostatic charge–discharge profiles of copolymers and PANI electrodes tested under the current density of 20 mA g−1 at room temperature. (c) Specific capacitances of copolymers and PANI electrodes versus current densities. (d) The comparison of specific capacitance versus current density for P(ANI-co-ATA, 8:2) with other carbonaceous cathode materials for HSCs reported in the literatures, and the abbreviations represent: AC: activated carbon, NPG: nitrogen-doped porous carbon, TRGO: trigol-reduced graphene oxide, PGM: porous graphene macroform, N-GMCs: nitrogen-enriched mesoporous carbon nanospheres/graphene, HNC: porous 3D hierarchical N-doped carbon nanosheet, ODC: oligomer derived carbon, BTCA: 1,2,4,5-benzene tetracarboxylic acid, PHPNC: pyridine derived hierarchical porous nitrogen-doped carbon. (e) Discharge–charge capacitances of copolymers and PANI electrodes versus cycle numbers tested under the current density of 20 mA g−1. (f) EIS spectra presented as Nyquist plots measured from copolymers and PANI electrodes after 100 cycles and the inset shows the equivalent circuit for the spectra fitting. | ||
The galvanostatic charge–discharge profiles of copolymers and PANI at the current density of 20 mA g−1 were shown in Fig. 3b, all profiles exhibit almost symmetric charge–discharge curves, representing the typical electrochemical redox process of p-type CPs. The gradient of the charge–discharge profile is caused by the gradual change of the doping level, and the abrupt transition of the profile at 2.9 V (vs. Li+/Li) represents the full-reduction state of the copolymer. P(ANI-co-ATA, 8:2) shows the smallest IR drop than those of the other copolymers and PANI, reflecting the lowest internal resistance of this copolymer, which is consistent with the result of electronic conductivity test (Table S1, ESI†).50 The charge–discharge duration of P(ANI-co-ATA, 8:2) is much longer than those of the other cathode materials, implying the highest specific capacitance. The calculated specific capacitances of copolymers, synthesized at ANI/ATA value of 9:1, 8:2, 7:3, 6:4, and PANI are 140, 198, 129, 41, and 106, respectively, which are close to the values obtained from the CV curves.
The rate capability of cathode material is the crucial factor for being used in the HSCs energy storage system. Fig. 3c shows the specific capacitances changes of copolymers and PANI as a function of discharge current densities. The corresponding charge–discharge profiles of copolymers and PANI at different current densities are shown in Fig. S2 (ESI†). It can be observed that P(ANI-co-ATA, 8:2) exhibits the best rate capability because of the higher capacitance retention than those of other cathode materials at the same current density. The capacitance retention of P(ANI-co-ATA, 8:2) is 69.4% (137.5 F g−1) at the current density of 1 A g−1, while the capacitance retentions of copolymers, synthesized at ATA/ANI value of 9:1, 7:3, 6:4, and PANI are 45.3%,20.1%,19.5%, and 30.1%, respectively. The maximum charge/discharge current density of P(ANI-co-ATA, 8:2) cathode reaches to 50 A g−1, with the specific capacitance of 18.2 F g−1. The excellent rate capability of P(ANI-co-ATA, 8:2) gives it the potential of being applied as the cathode materials of HSCs. By comparing P(ANI-co-ATA, 8:2) with other reported cathode materials (Fig. 3d), it can be claimed that the specific capacitance of P(ANI-co-ATA, 8:2) is higher than commercial AC and even comparable to some nanostructured carbons. The more detailed comparisons are listed in Table S2 (ESI†) and it can be found that P(ANI-co-ATA, 8:2) delivers higher energy density than the majority of carbonaceous cathode materials due to the pseudocapacitance offered by anions doping/dedoping mechanism. Although some carbonaceous cathode materials with extremely large surface area like three-dimensional porous graphene or heteroatoms doped graphene shows higher specific capacitance than P(ANI-co-ATA, 8:2), the synthesis of the copolymer nanocomposites with nanostructured carbons is expected to improve the specific capacitance further because of the great successes in aqueous supercapacitors through the same approach.
The cycling stability is usually a drawback for electrochemical energy storage electrodes based on CPs. The cycling performances of copolymers and PANI were evaluated by repeating the charge–discharge tests for 100 cycles at the current density of 20 mA g−1, and the specific capacitance as the function of cycle number is shown in Fig. 3e. The slightly increased specific capacitance at initial cycles for each tested cathode is correlated to activated process, which has been observed in several CPs electrodes previously.50 The specific capacitance of P(ANI-co-ATA, 8:2) holds steady in the subsequent cycles and the capacitance retention is 85.5% after 100 cycles, which is higher than that of PANI (42.2%). The capacitance retentions of copolymers synthesized at ANI/ATA value of 9:1, 7:3, and 6:4 are 68.8, 48.1, and 45.5%, respectively. The alternating swell and shrinkage of CPs during the doping and dedoping process deteriorate the electronic conductivity and charge storage capability.51 The interaction between anionic carboxyl group of ATA and polyanionic PANI, revealed by XRD patterns, enhances the structural stability during the doping/dedoping process and hence improves the cycling stability efficiently. In addition, the one-dimensional nanostructure of P(ANI-co-ATA, 8:2) also contributes to the improvement of the cycling stability due to the relatively high flexibility of nanorods.18 The specific capacitance of P(ANI-co-ATA, 8:2) retains about 78.5% after 1000 cycles at the current density of 2 A g−1 (Fig. S3, ESI†), accounting for the superior cycling stability of the copolymer even at relatively high current densities. After this cycling test, the maintained one-dimensional morphology of P(ANI-co-ATA, 8:2) observed from the SEM and TEM images (Fig. S4, ESI†), confirms the excellent structural stability of the copolymer. However, the slightly enlarged size of P(ANI-co-ATA, 8:2) nanorods still represents the negative effects of alternative volume change and lead to the capacitance fading during the cycling test.
In order to further investigate the mechanism of the improved electrochemical performances, the electrochemical impedance spectra (EIS) of copolymers and PANI, after 100 cycles at the current density of 20 mA g−1, were performed, which are shown in the Fig. 3f, the analysis of the impedance spectra is based on the equivalent circuit model shown inset. Re represents the total resistance of electrolyte, electrode, and separator. Rct and CPE represent the charge-transfer resistance and the double layer capacitance, respectively. Zw is the Warburg impedance which is derived from to the diffusion of ions into the bulk electrode. The Nyquist plots consist of a semicircle at high frequency that represents the value of Rct and a straight sloping line at low frequency associated with the ions diffusion process within the electrode. By fitting the impedance spectroscopy, the fitted impedance parameters are listed in Table S3 (ESI†). The Re values of all the cathodes are close to each other because of the same cathode fabrication process and the employed electrolyte. P(ANI-co-ATA, 8:2) features the smallest Rct value, indicating the most feasible charge transportation through the electrode. Moreover, the almost vertical line of P(ANI-co-ATA, 8:2) in the low frequency region, represented by the phase angle close to 90°, indicates the capacitive behavior and high ion diffusion coefficient.52 Therefore, the synthesis of ANI and ATA copolymer with appropriate components is an effective approach to improve the electrochemical performances of PANI electrode.
Because P(ANI-co-ATA, 8:2) exhibited the best electrochemical performances, the electrode process kinetics of this copolymer was further analyzed by EIS. The Nyquist plots of P(ANI-co-ATA, 8:2) electrode charged/discharged to different potentials in the 1st cycles were shown in Fig. 4a. The Rct increases slightly during the first charge process which is attributed to the partially forming pernigraniline caused by the doped ClO4− into the copolymer backbones. The reversion of Rct to the original value during the subsequent discharge process is attributed to the dedoping process. To confirm the structural stability, the FT-IR analysis before and after cycles was performed (Fig. 5). Before the test, all the electrodes had been discharge to 2 V. Compared with original curve shown in the Fig. 1a, the same bands could also be found here. Besides, the two similar curves indicate the excellent structural stability. The detailed analysis and ex-situ FT-IR spectra of the P(ANI-co-ATA, 8:2) electrode in the first and one hundredth cycle taken at different cut-off potentials have been shown in the ESI (Fig. S5, ESI†). The FT-IR spectra of P(ANI-co-ATA, 8:2) at the same cut-off potentials in the first cycle reveal the increased amount of quinoid rings during the charge process and the gradual transformation to benzenoid rings during the discharge process. The reduced Rct at different potentials after 100 cycle indicates the more efficient doping–dedoping process to the copolymer. The more vertical line in the low frequency region indicates the more capacitive behavior of the copolymer. The FT-IR spectra of P(ANI-co-ATA, 8:2) (Fig. S5b, ESI†) at the same cut-off potentials in the 100th cycle demonstrated the well maintained chemical structure after the cycling test. The analysis of CV curves at different scan rates after 100 cycles (Fig. 4c) demonstrates the capacitive behavior of P(ANI-co-ATA, 8:2) further. The total stored charge of P(ANI-co-ATA, 8:2) can be separated into the capacitive process and the diffusion-limited redox reaction. Approximately 96.10% fraction of the total charge is contributed from the capacitive process (Fig. 4d) and the detailed analysis process is shown in Fig. S6 (ESI†), which verifies the ultrahigh rate capability and the long cycling stability of P(ANI-co-ATA, 8:2) cathode. Although the energy storage of P(ANI-co-ATA, 8:2) is realized by the pseudocapacitance generated from the anions doping/dedoping mechanism, the capacitive controlled electrochemical reaction rather than the diffusion controlled process of the copolymer is attributed to the synergetic effects of ATA and ANI monomers. The alternating electron donating (–NH–) and withdrawing (–COOH) groups in the copolymer enhance the charge delocalization on the copolymer backbone.22 At the low concentration of ATA in the co-monomers feed, the delocalization effect is not effective enough, resulting the copolymer with lower electrochemical activity; whereas, at high concentration of ATA in the copolymer, steric effects of the substituent predominates over the electronic effect and degrades electrochemical performances subsequently. The similarly electrochemical activation of copolymers by introducing the electron withdrawing groups into PANI backbone has also been reported.53,54 In addition, the negative charged carboxyl in ATA, as intramolecular dopant anions, avoids the loss of electrical conductivity during the charging/discharging cycles,55,56 which usually deteriorates the power density and cycling stability of electrode based on CPs. Moreover, the presence of hydrogen bonding and inter chain linking, revealed by XRD patterns, maintain the copolymer structure not to collapse during the charging/discharging cycles. The one dimension geometry of P(ANI-co-ATA, 8:2) also improves the utilization of cathode material and accommodates the volume change during cycling, which further increases the charge storage and capacity retention. Therefore, P(ANI-co-ATA, 8:2) exhibit the best electrochemical performances because of the synergistic effects provided by the introduction of appropriate ATA amount into the copolymer backbone (Fig. 6).
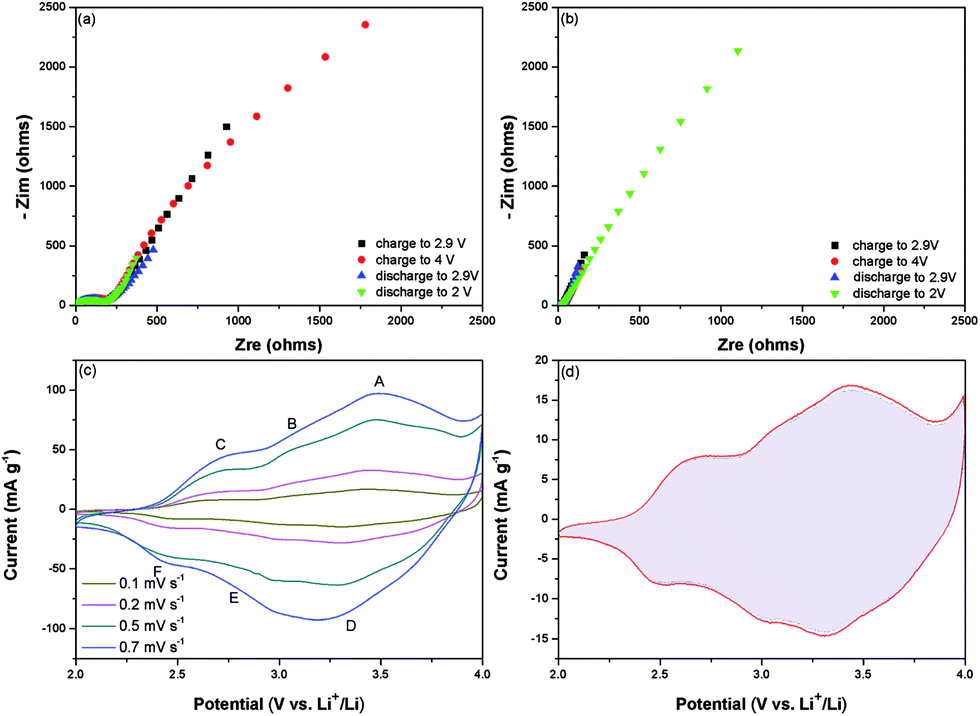 | ||
| Fig. 4 Kinetics characterization of P(ANI-co-ATA, 8:2) electrode. The Nyquist plots of the impedance at the first (a) and at the one hundredth (b) cycles in different charged and discharged states. (c) CV curves at different scan rates after 100 cycles. (d) The CV curve tested at the scan rate of 0.5 mV s−1 with the capacitive contribution to the total current shown by the shaded region. | ||
 | ||
| Fig. 5 The FT-IR spectra of the P(ANI-co-ATA, 8:2) electrodes in the first and one hundredth cycles. | ||
 | ||
| Fig. 6 Ragone plots of P(ANI-co-ATA, 8:2)//MCMBs and P(ANI-co-ATA, 8:2)//LTO HSCs with the comparison with other reported HSCs in literature: AC: activated carbon, TRGO: trigol-reduced graphene oxide, N-GMCs: nitrogen-enriched mesoporous carbon nanospheres/graphene, ODC: oligomer derived carbon, BTCA: 1,2,4,5-benzene tetracarboxylic acid, PLMG: prelithiated microcrystalline graphite. The calculated energy and power densities are based on total active mass in both electrodes. | ||
The excellent rate capability as well as the anion doping/dedoping energy storage mechanism of P(ANI-co-ATA, 8:2) promotes it to be employed as the cathode material for HSCs. Herein, two kinds of commercial LIBs anode materials, which were MCMBs and LTO, were coupled with P(ANI-co-ATA, 8:2) to fabricate HSCs. Before the fabrication, both P(ANI-co-ATA, 8:2) cathode and MCMBs (or LTO) anode were preactivated for three cycles at 20 mA g−1 in the Li half-cell to achieve the high efficiency. Based on the tested electrochemical results of MCMBs and LTO (Fig. S7 and S8, ESI†), the mass ratios of P(ANI-co-ATA, 8:2) to MCMBs and LTO were optimized to be 5:2 and 3:2 to match the charge balance between the cathode and anode respectively. The potential windows of P(ANI-co-ATA, 8:2)∥MCMBs and P(ANI-co-ATA, 8:2)∥LTO HSCs were chosen from 1.0 V to 3.8 V and 2.8 V, respectively, based on the charging/discharging profiles of individual electrodes, and their electrochemical performances are shown in Fig. S9 and S10 (ESI†), respectively. The Ragone plot of the fabricated HSCs is shown in Fig. 5. The energy density and power density are calculated based on the galvanostatic discharge profiles at different current densities (Fig. S9b and S10b, ESI†). At the low power density (100 W kg−1), the energy density of P(ANI-co-ATA, 8:2)∥MCMBs is higher than that of P(ANI-co-ATA, 8:2)∥LTO due to the wider potential window of the former one. However, the energy density of P(ANI-co-ATA, 8:2)∥MCMBs drastically decreases with the increase of the power density due to the low kinetics of Li+ diffusion into MCMBs anode. On the other hand, P(ANI-co-ATA, 8:2)∥LTO HSC exhibits better rate capability than P(ANI-co-ATA, 8:2)∥MCMBs, which delivers the maximum power density of 3011.5 W kg−1, associated with the energy density of 26.1 W h kg−1. By comparing with previous HSCs fabricated by carbonaceous cathode and graphitic or LTO anode, HSCs using P(ANI-co-ATA, 8:2) exhibit higher energy densities at the low power density region because of the superior energy density of P(ANI-co-ATA, 8:2). It is worthy of attention that for commercial energy storage devices, the P(ANI-co-ATA, 8:2)∥MCMBs reaches the energy density of commercial LIBs, and P(ANI-co-ATA, 8:2)∥LTO delivers the power density comparable to commercial electrochemical capacitors, associated with the energy density compared with Li–metal hydrides or LIBs (Fig. S11, ESI†).57 These values demonstrate that P(ANI-co-ATA, 8:2) features the energy and power density requirements (about 5 W h kg−1 at 5000 W kg−1) 58of HSCs cathode, and the optimization of the component of the copolymers cathode as well as the coupled anode materials can further improve the electrochemical performances of HSCs.
4. Conclusion
Copolymers of ANI and ATA with different ratios of ANI/ATA were prepared by chemical oxidation polymerization. The morphologies of the products vary from nanoparticle to nanorod and then convert back to nanoparticle with the decrease of ANI/ATA value. The dimers of monomers serve as the soft template in the formation of the special morphologies and P(ANI-co-ATA, 8:2) nanorods form due to the appropriate micelle concentration. The optimized delocalization and steric effects lead P(ANI-co-ATA, 8:2) to exhibit the best electrochemical performances among all the copolymers, especially the rate capability, which works steadily until the charge–discharge current density up to 50 A g−1. The excellent rate capability as well as the anion doping/dedoping mechanism enables it as a suitable cathode material for HSCs. The fabricated HSCs of P(ANI-co-ATA, 8:2)∥MCMBs exhibits the high energy density of 153.9 W h kg−1 at 42 W kg−1, and the HSCs of P(ANI-co-ATA, 8:2)∥LTO delivers the high power density of 3011.5 W kg−1 accompanied by the energy density of 26.1 W h kg−1. These results demonstrate that the incorporation of P(ANI-co-ATA, 8:2) cathode is an effective strategy to enhance the energy densities of HSCs, and the further optimization component will improve the power density.
Acknowledgements
This work was financially supported by the National Natural Science Funds for Distinguished Young Scholars (51425306), the National Key Research and Development Program of China (No. 2016YFA0202302), the State Key Program of National Natural Science Foundation of China (No. 51633007), and National Natural Science Foundation of China (No. 51573125).Notes and references
- I. Plitz, A. DuPasquier, F. Badway, J. Gural, N. Pereira, A. Gmitter and G. G. Amatucci, Appl. Phys. A, 2006, 82, 615–626 CrossRef CAS.
- G. Glenn, Z. Amatucci, F. Badway, A. D. Pasquier and T. Zheng, J. Electrochem. Soc., 2001, 148, A930–A939 CrossRef.
- A. D. Pasquier, I. Plitz, J. Gural, S. Menocal and G. Amatucci, J. Power Sources, 2003, 113, 62–71 CrossRef.
- F. Zhang, T. Zhang, X. Yang, L. Zhang, K. Leng, Y. Huang and Y. Chen, Energy Environ. Sci., 2013, 6, 1623 CAS.
- K. Leng, F. Zhang, L. Zhang, T. Zhang, Y. Wu, Y. Lu, Y. Huang and Y. Chen, Nano Res., 2013, 6, 581–592 CrossRef CAS.
- T. Zhang, F. Zhang, L. Zhang, Y. Lu, Y. Zhang, X. Yang and Y. Huang, Carbon, 2015, 92, 106–118 CrossRef CAS.
- V. Aravindan, D. Mhamane, W. C. Ling, S. Ogale and S. Madhavi, ChemSusChem, 2013, 6, 2240–2244 CrossRef CAS PubMed.
- R. Gokhale, V. Aravindan, P. Yadav, S. Jain, D. Phase, S. Madhavi and S. Ogale, Carbon, 2014, 80, 462–471 CrossRef CAS.
- R. Wang, J. Lang, P. Zhang, Z. Lin and X. Yan, Adv. Funct. Mater., 2015, 25, 2270–2278 CrossRef CAS.
- M. Yang, Y. Zhong, J. Ren, X. Zhou, J. Wei and Z. Zhou, Adv. Energy Mater., 2015, 5, 1500550 CrossRef.
- P. J. Nigrey, D. MacInnes, D. P. Nairns, A. G. MacDiarmid and A. J. Heeger, J. Electrochem. Soc., 1981, 128, 1651–1654 CrossRef CAS.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 CAS.
- J. F. Mike and J. L. Lutkenhaus, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 468–480 CrossRef CAS.
- S. P. Palaniappan and P. Manisankar, Electrochim. Acta, 2011, 56, 6123–6130 CrossRef CAS.
- S. Radhakrishnan, S. Prakash, C. R. Rao and M. Vijayan, Electrochem. Solid-State Lett., 2009, 12, A84–A87 CrossRef CAS.
- S. P. Palaniappan, S. R. P. Gnanakan, Y. S. Lee and P. Manisankar, Ionics, 2011, 17, 603–606 CrossRef CAS.
- Y. Cao and T. E. Mallouk, Chem. Mater., 2008, 20, 5260–5265 CrossRef CAS.
- P. Novak, K. Muller, K. S. Santhanam and O. Haas, Chem. Rev., 1997, 97, 207–282 CrossRef CAS PubMed.
- F. Cheng, W. Tang, C. Li, J. Chen, H. Liu, P. Shen and S. Dou, Chem.–Eur. J., 2006, 12, 3082–3088 CrossRef CAS PubMed.
- M. N. Hyder, S. W. Lee, F. C. Cebeci, D. J. Schmidt, Y. Shao-Horn and P. T. Hammond, ACS Nano, 2011, 5, 8552–8561 CrossRef CAS PubMed.
- M. N. Hyder, R. Kavian, Z. Sultana, K. Saetia, P. Y. Chen, S. W. Lee and P. T. Hammond, Chem. Mater., 2014, 26, 5310–5318 CrossRef CAS.
- H. Zhang, G. Cao, Z. Wang, Y. Yang, Z. Shi and Z. Gu, Electrochem. Solid-State Lett., 2008, 11, A223–A225 CrossRef CAS.
- R. Zhao, L. Zhu, Y. Cao, X. Ai and H. X. Yang, Electrochem. Commun., 2012, 21, 36–38 CrossRef CAS.
- C. Su, Y. Ye, L. Xu and C. Zhang, Chem. Phys., 2011, 212, 2487–2492 CAS.
- C. Su, L. Ji, L. Xu, X. Zhu, H. He, Y. Lv and C. Zhang, RSC Adv., 2015, 5, 14053–14060 RSC.
- C. Gong, F. Deng, C. P. Tsui, Z. Xue, Y. S. Ye, C. Y. Tang and X. Xie, J. Mater. Chem. A, 2014, 2, 19315–19323 CAS.
- S. Quillard, G. Louarn, S. Lefrant and A. G. MacDiarmid, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 12496–12508 CrossRef.
- G. E. Asturias, A. G. MacDiarmid, R. P. McCall and A. J. Epstein, Synth. Met., 1989, 29, 157–162 CrossRef.
- B. Nie, J. Stutzman and A. Xie, Biophys. J., 2005, 88, 2833–2847 CrossRef CAS PubMed.
- Y. Zhao, E. Tomšík, J. Wang, Z. Morávková, A. Zhigunov, J. Stejskal and M. Trchová, Chem.–Asian J., 2013, 8, 129–137 CrossRef CAS PubMed.
- M. Trchová, I. Šedenková, E. N. Konyushenko, J. Stejskal, P. Holler and G. Ciric-Marjanovic, J. Phys. Chem. B, 2006, 110, 9461–9468 CrossRef PubMed.
- J. Arias-Pardilla, H. J. Salavagione, C. Barbero, E. Morallón and J. L. Vázquez, Eur. Polym. J., 2006, 42, 1521–1532 CrossRef CAS.
- C. Kvarnström, A. Petr, P. Damlin, T. Lindfors, A. Ivaska and L. Dunsch, J. Solid State Electrochem., 2002, 6, 505–512 CrossRef.
- C. A. Amarnath, J. Kim, K. Kim, J. Choi and D. Sohn, Polymer, 2008, 49, 432–437 CrossRef CAS.
- Y. Şahin, K. Pekmez and A. Yıldız, J. Appl. Polym. Sci., 2003, 90, 2163–2169 CrossRef.
- S. I. A. Razak, A. L. Ahmad and S. H. S. Zein, J. Phys. Sci., 2009, 20, 27–34 CAS.
- A. Malinauskas and R. Holze, J. Appl. Polym. Sci., 1999, 73, 287–294 CrossRef CAS.
- L. Zhang and M. Wan, Adv. Funct. Mater., 2003, 13, 815–820 CrossRef CAS.
- J. Bhadra and D. Sarkar, Mater. Lett., 2009, 63, 69–71 CrossRef CAS.
- Z. D. Zujovic, C. Laslau, G. A. Bowmaker, P. A. Kilmartin, A. L. Webber, S. P. Brown and J. Travas-Sejdic, Macromolecules, 2009, 43, 662–670 CrossRef.
- A. A. Athawale and V. V. Chabukswar, J. Appl. Polym. Sci., 2001, 79, 1994–1998 CrossRef CAS.
- C. Laslau, Z. Zujovic and J. Travas-Sejdic, Prog. Polym. Sci., 2010, 35, 1403–1419 CrossRef CAS.
- Z. Zhang, L. Wang, J. Deng and M. Wan, React. Funct. Polym., 2008, 68, 1081–1087 CrossRef CAS.
- Z. Zhang, Z. Wei and M. Wan, Macromolecules, 2002, 35, 5937–5942 CrossRef CAS.
- H. Xia, J. Narayanan, D. Cheng, C. Xiao, X. Liu and H. S. O. Chan, J. Phys. Chem. B, 2005, 109, 12677–12684 CrossRef CAS PubMed.
- Z. Wei and M. Wan, J. Appl. Polym. Sci., 2003, 87, 1297–1301 CrossRef CAS.
- Z. Wang, C. Xu, P. Tammela, P. Zhang, K. Edström, T. Gustafsson and L. Nyholm, Energ. Tech., 2015, 3, 563–569 CrossRef CAS.
- L. Yang, W. Qiu and Q. Liu, Solid State Ionics, 1996, 86, 819–824 CrossRef.
- Y. Liang, Z. Tao and J. Chen, Organic electrode materials for rechargeable lithium batteries, Adv. Energy Mater., 2012, 2, 742–769 CrossRef CAS.
- Y. R. Chen, K. F. Chiu, H. C. Lin, C. Y. Hsieh, C. B. Tsai and B. T. T Chu, Mater. Sci. Eng., B, 2014, 190, 59–65 CrossRef CAS.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- Y. A. Ismail, F. Mohammad and A. Ahmad, J. Macromol. Sci., Part A: Pure Appl.Chem., 2011, 952–961 CrossRef CAS.
- M. Zhou, W. Li, T. Gu, K. Wang, S. Cheng and K. Jiang, Chem. Commun., 2015, 51, 14354–14356 RSC.
- C. Li, H. Bai and G. Shi, Chem. Soc. Rev., 2009, 38, 2397–2409 RSC.
- H. R. Ghenaatian, M. F. Mousavi and M. S. Rahmanifar, Electrochim. Acta, 2012, 78, 212–222 CrossRef CAS.
- S. Dong, L. Shen, H. Li, P. Nie, Y. Zhu, Q. Sheng and X. Zhang, J. Mater. Chem. A, 2015, 3, 21277–21283 CAS.
- H. S. Choi, J. H. Im, T. Kim, J. H. Park and C. R. Park, J. Mater. Chem., 2012, 22, 16986–16993 RSC.
- A. Du Pasquier, I. Plitz, S. Menocal and G. Amatucci, J. Power Sources, 2003, 115, 171–178 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27900g |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |