
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The water–iodine oxide system: a revised mechanism for hydration and dehydration†
Dylan K. Smitha,
Michelle L. Pantoya *a,
Jeffrey S. Parkeyb and
Mehmet Kesmezb
*a,
Jeffrey S. Parkeyb and
Mehmet Kesmezb
aDepartment of Mechanical Engineering, Texas Tech University, Lubbock, TX 79409, USA. E-mail: michelle.pantoya@ttu.edu; Tel: +1-806-834-3733
bLynntech, Inc., College Station, TX 77845, USA
First published on 6th February 2017
Abstract
Iodic acids are widely studied in atmospheric and biological applications but their inherent hydrophilic properties introduce complexities that affect their functionality and reactivity. We have shown that iodic acid (HIO3) dehydrates directly into iodine pentoxide (I2O5) in contradiction to the generally accepted multi-step dehydration mechanism where HIO3 dehydrates into HI3O8 first, then dehydrates into I2O5. The generally accepted mechanism is used to determine the concentration of iodic acid by TGA and is only valid for special conditions. The revised mechanism allows for the determination of concentrations of iodic acids under all conditions, and the more specific conditions where the accepted mechanism is valid are shown. The determination of concentration of iodic acid with the revised dehydration mechanism is dependent on assumptions of residual water and initial concentration of HI3O8. The validity of these assumptions is established by studying the absorption and hydration behavior of I2O5 from atmospheric water. These results will have an impact on the handling and use of iodine.
Introduction
Iodine(V) oxides have biocidal potential in neutralizing bacterial agents released in certain weapon systems. Handling iodine(V) oxides is one of the biggest challenges toward their effective utilization in this application. Specifically, iodine(V) oxides are hygroscopic to the point of being deliquescent. A pile of di-iodine pentoxide (I2O5) powder, for example, will absorb so much water from the surrounding atmosphere over time, that a puddle is produced. This property has limited iodine(V) oxides usefulness in energetic applications because an abundance of water can greatly consume liberated energy from a chemical reaction between iodine(V) oxide and a metal fuel, such as aluminum, thereby inhibiting combustion performance. Understanding, absorption, hydration, desorption and dehydration mechanisms associated with iodine(V) oxides may enable more tailored synthesis and control over this property for engineering future prompt-agent-defeat technologies (or bio-agent defeat systems).Iodine(V) oxide water absorption behavior is studied extensively with inconsistent results that may stem from a limited understanding of the fundamental kinetics and dynamics of water absorption and reaction with iodine(V) oxides. The inconsistencies highlighted here involve iodine(V) oxides reaction with water and include: different humidity levels where absorption begins; variations in concentration of iodic acids (HIO3, HI3O8); crystal structure; onset temperature for dehydration and decomposition; and, absorption rates. All of the inconsistencies discussed below are affected by the hydration and dehydration mechanism.
The accepted mechanism for hydration of I2O5 into iodic acids (i.e., HIO3 and HI3O8) and dehydration of iodic acids into I2O5 (i.e., HIO3 and HI3O8) was first proposed by Selte et al.1 and shown in eqn (1a) and (1b), respectively.1
Hydration:
| 3I2O5 + 3H2O → 2HI3O8 + 2H2O → 6HIO3 | (1a) |
Dehydration:
| 6HIO3 → 2HI3O8 + 2H2O → 3I2O5 + 3H2O | (1b) |
In dehydration, six moles of iodic acid in the form of HIO3 form three moles of I2O5 and three moles of water with an intermediate step involving the formation of two moles of HI3O8. In this reaction, water comprises 5.11 wt% of the total mass. The mass loss from dehydration of water at each step is shown in eqn (2).
| 3HIO3 → HI3O8 + H2O: 3.41% water | (2a) |
| 2HI3O8 → 3I2O5 + H2O: 1.76% water | (2b) |
Using mass loss from water calculated from eqn (2a) and (2b), concentrations of iodic acids can be determined from thermal equilibrium analysis using differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA).1,2 The equations used to calculate concentration of HIO3 (XHIO3) and HI3O8 (XHI3O8) are shown in eqn (3a) and (3b), respectively.
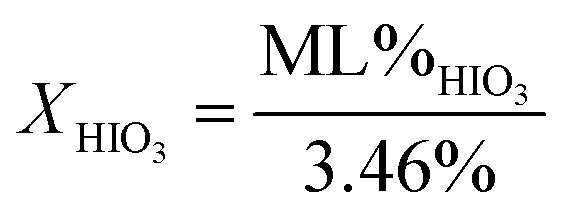 | (3a) |
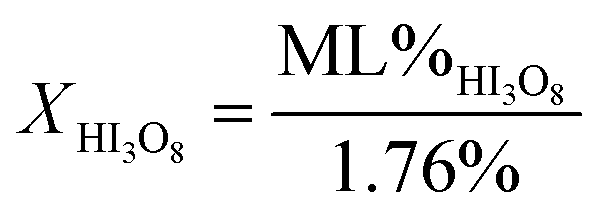 | (3b) |
In eqn (3), ML% is measured mass loss from TGA. The mass loss percent is divided by the calculated maximum mass loss from eqn (2a) and (2b) for HIO3 and HI3O8 respectively, to obtain the concentration of each iodic acid. The DSC/TGA data shows an endotherm and mass loss at 110 °C, where HIO3 dehydrates into HI3O8, followed by an endotherm and mass loss at 210 °C, where HI3O8 dehydrates into I2O5. Dehydration of HI3O8 has always been observed when HIO3 dehydration was observed, indicating an intermediate step where HIO3 dehydrates into HI3O8.1,2 In Selte et al.1 and multiple references within, determination of iodic acid concentration by TGA is confirmed with XRD analysis. The temperatures where the onset of iodic acid dehydration begins is widely varied and often overlooked.
A review of the publications over the thermochemical properties of iodine oxides3 summarized data available for reported onset temperatures of each dehydration step and decomposition of I2O5. The onset temperatures reported for I2O5 decomposition are between 300 and 450 °C.3 Selte et al. report a lower onset temperature of 280 °C for amorphous I2O5, with onset temperatures between 300 and 420 °C for crystalline I2O5.1 Iodic acids such as HI3O8 onset temperatures are reported between 195 and 220 °C (ref. 1) and between 100 and 130 °C for HIO3.1 The reason for the variation in temperature is not known. Formation of hydrated species of iodine oxides is a complex, dynamic process that can depend on variables such as relative humidity (RH), particle size, hydration and desorption rates, available water, and crystal structure. These variables can be dependent on each other and could affect properties of the final material including dehydration temperature.
Relative humidity (RH) has a significant influence on the oxidation state and kinetics of iodine(V) oxides. Selte et al.1 report that crystalline HI3O8 powder can be stored in open vessels for an indefinite period independent of atmospheric humidity but fine-powdered samples completely convert to HIO3 in moist air. Kumar et al.4 studied the deliquescence and efflorescence behavior of I2O5 and HIO3. They describe deliquescence as the transformation from a solid crystal phase to an aqueous solution and efflorescence as the reverse, when an aqueous solution crystallizes to a solid phase; and, define these differences by changes in the optical properties of the sample. Deliquescence and efflorescence are seen at specific threshold RH levels. Also, I2O5 and HIO3 crystals with an average size of 10–50 μm were studied at different temperatures. Deliquescence was reported at 85% RH and room temperature for HIO3, and at 80% RH and room temperature for I2O5.4 Efflorescence was not seen in iodine(V) oxide samples with RH as low as 1.0%. Little et al.2 observed hydration according to the steps in the hydration mechanism in eqn (1a). They explain different hydrated species of iodine(V) oxide can precipitate out when exposed to water and recrystallize depending on impurities, rate of evaporation, and crystal structure.2 This finding is in contrast to Selte et al.,1 where all samples were exclusively HIO3. Little et al.2 note that multiple iterations of this method produced varying concentrations of iodic acid. Also, a threshold value of 60% RH was found by Little et al.2 to hydrate commercially available samples without dissolving the solid in water and recrystallization. When commercially available samples are exposed to air above 60% RH threshold, the product is HIO3 and when exposed to air below 60% RH, no hydration is observed. Because the commercial sample was entirely HI3O8, a threshold of 60% RH was assumed to be the RH that HI3O8 started absorbing water.2 To obtain I2O5, a sample initially HI3O8 was ball milled, and produced a sample 75 wt% I2O5 and 25 wt% HI3O8. For hydration from I2O5 to HI3O8, Little et al.2 exposed the ball milled samples to 40% RH over a period of several days. The TG analysis indicated increased mass loss at 210 °C such that samples with higher concentrations of I2O5 were hydrated at 40% RH. It is interesting to note that the observation of an endotherm with an onset temperature of 170 °C remained constant with increased hydration. An endotherm with an onset temperature of 170 °C is outside of the range of generally accepted dehydration temperatures for hydrated species of iodine oxides. Over time with exposure to 40% RH, the endothermic peak indicating HI3O8 increased and formed a double peak. Commercially available I2O5 was used to study the hydration step from HI3O8 to HIO3 (eqn (1a)). No hydration was seen below 50% RH over a period of weeks, however, hydration was seen when RH increased to 70% RH. Hydration rates were measured at 70% RH for HI3O8 to HIO3 (eqn (1a)) and at 40% RH for I2O5 to HI3O8 (eqn (1a)). At 40% RH, the hydration rate went to 0 before 80 wt% of the sample was HI3O8, but at 70% RH hydration continued until the sample was solely HIO3. In summary, a discrepancy from previous research is that water absorption thresholds for any iodic acid widely range from 0 to 85% RH. The reason for the dramatic variation may be due to limited understanding of the fundamental mechanism of hydration and dehydration. A better understanding of the fundamental hydration and dehydration kinetics is needed to reconcile these discrepancies.
Our goal is to improve the fundamental mechanism of hydration and dehydration of I2O5 by studying hydration and dehydration from atmospheric water (i.e. relative humidity). The objectives are to understand the fundamental absorption mechanism of I2O5; understand how I2O5 reacts with water to form iodic acids; and, identify how the reactions between I2O5 and water affect the kinetics of hydration and dehydration. It is noted that hydration and absorption are not interchangeable terms. For the purpose of this study, absorption refers to water that is weakly bonded and not yet reacted to hydrate I2O5 into HIO3. Hydration refers to absorbed water that has reacted to form an IO3− solution and will form an HIO3 crystal structure upon desorption. Desorption is when mass loss occurs without heating and dehydration is mass loss as a result of HIO3 forming I2O5 from heating.
Experimental
I2O5
Two synthesis procedures are used to produce I2O5 powders. One method is based on the thermal dehydration of iodic acid to form I2O5.5,6 These samples were supplied by Sigma Aldrich (St. Louis, MO) and come as crystals that require the sample to be crushed using a mortar and pestle. This sample is referred to as 17 μm I2O5 because it has an average particle diameter of 16.9 μm.7 The 17 μm I2O5 is the same as commercial I2O5 used in Smith et al.7 where characterization of these samples was performed. The second method is a “dry” method that combines elemental iodine and oxygen using a non-thermal plasma energy source to form spherical I2O5 particles with an average diameter of 4.7 μm.7 These samples were labeled nano C I2O5 when used in Smith et al.7 because they were initially measured erroneously to have an average particle diameter of 400 nm.7 This sample is now referred to as 5 μm I2O5 here. The two distinct synthesis methods enable production of particles with discretely different properties inherent from the synthesis methods and tailored for experimental designs that focus on deriving a more general hydration/dehydration mechanism.Humidity chamber
A humidity chamber was designed and built to control relative humidity, RH. The chamber was designed from a 60 × 30 × 30 cm acrylic air tight box. A 1.27 cm hole was drilled in the wall of the chamber and a variable speed fan was placed outside the chamber so the current of air from the fan passed over a hole to draw air out of the inside the chamber. A heating pad was placed inside the chamber and a Pyrex™ pan filled with water positioned on top of the heating pad. Humidity was controlled by adjusting the temperature on the heating pad and the speed of the air from the fan. Relative humidity was measured with an AMPROBE TH-3 relative humidity temperature meter and was held above 80% but below the dew point at a temperature of 23 °C so drops of water did not condense inside the chamber. The 80% RH threshold was selected because it is the highest RH without having droplets form and thus a reasonable upper bound for this study.Thermal equilibrium analysis
Simultaneous thermal analysis (STA) was performed using a Netzsch model STA 449 differential scanning calorimeter (DSC) with thermo-gravimetric (TG) analyzer. All powder samples measured approximately 10 mg and were placed inside alumina crucibles designed for the STA 449 DSC-TGA. To ensure the samples were initially comprised of only I2O5 and not another form of iodic acid, all samples were heated in a Neytech QEX oven to 250 °C, above the dehydration temperature of HIO3 (110 °C) and HI3O8 (210 °C) but below the dissociation temperature of I2O5 (350 °C), and held for 5 minutes. The heat flow and mass loss showing that samples are pure I2O5 after heat treatment are shown in ESI Fig. S1.† The sample covers were removed and the samples were placed inside the humidity controlled chamber for varying intervals. The samples were weighed before and after they were placed inside the humidity controlled chamber and the mass change is reported as a percentage in Tables 1 and 2. It is important to note that the starting material was purposefully heated such that the material is initially solely I2O5 as shown in Fig. S1.† Most other studies reporting iodine(V) compounds may not start with pure I2O5 as the precursor because a heat treatment procedure as described above is not mentioned.2,4,8,9 Each sample was covered and loaded into the STA manually to minimize exposure time to the ambient held at an average 20% RH. Samples were heated to 600 °C at 10 °C min−1 in an argon atmosphere. The argon atmosphere was created by a constant flow of argon into the STA at 100 ml min−1. The control software for the STA requires a 1 min equilibration period between sample loading and heating that ensures the atmosphere inside the STA is argon. This equilibration period is included in the time interval between when the samples were removed from 80% RH and measurements.| Exposure time (min) | Mass gain, % | HIO3 mass loss, % | HI3O8 mass loss, % | Total mass loss | HIO3/HI3O8 |
|---|---|---|---|---|---|
| 1 | 1.0% | 0.0% | 0.02% | 0.02% | 0.00 |
| 5 | 1.5% | 0.0% | 0.08% | 0.08% | 0.00 |
| 10 | 3.2% | 0.0% | 0.06% | 0.06% | 0.00 |
| 15 | 9.0% | 1.63% | 0.23% | 1.86% | 7.09 |
| 30 | 13.7% | 2.70% | 0.10% | 2.80% | 27.00 |
| 180 | 34.8% | 5.36% | 0.22% | 5.58% | 24.36 |
| 900 | 32.7% | 7.41% | 0.17% | 7.58% | 43.59 |
| 2680 | 23.1% | 8.49% | 0.29% | 8.78% | 29.28 |
| Exposure time (min) | Mass gain, % | HIO3 mass loss, % | HI3O8 mass loss, % | Total mass loss | HIO3/HI3O8 |
|---|---|---|---|---|---|
| 1 | 0.7% | 1.12% | 0.42% | 1.54% | 2.67 |
| 5 | 4.7% | 2.01% | 0.57% | 2.58% | 3.53 |
| 10 | 6.9% | 4.63% | 0.29% | 4.92% | 15.97 |
| 30 | 9.1% | 4.74% | 0.32% | 5.06% | 14.81 |
| 85 | 6.1% | 4.55% | 0.50% | 5.05% | 9.10 |
| 220 | 4.7% | 4.81% | 0.49% | 5.30% | 9.82 |
| 2880 | 4.8% | 4.87% | 0.42% | 5.29% | 11.60 |
Results
Fig. 1a and b are the heat flow from DSC and mass change from TG measurements as a function of exposure time to 80% RH from 1 to 3060 minutes for 17 μm I2O5, respectively. Fig. 1a shows endothermic reactions starting at an exposure time of 10 minutes. At an exposure time of 15 minutes, there are two distinct endotherms below 160 °C. The second endotherm, with onset at 110 °C, increases in magnitude with time while the first endotherm reduces in magnitude until 900 minutes exposure time where it is no longer detectable. To the authors' knowledge, two distinct peaks in the range of HIO3 dehydration have not been reported previously. However, similar endothermic activity reported in Smith et al.10 observed a single endotherm with a double peak before 110 °C. They proposed the multiple endothermic activity in this temperature range was latent water or IO3− solution formed when iodine oxides react with water, and that theory is supported by the results shown here. The first endotherm in samples that show two endotherms is assumed to be evaporation of latent water and only mass loss from the second endotherm is used for calculation of mass loss from dehydration of HIO3. Additionally, most samples showed slight endothermic activity at 210 °C with corresponding mass loss (Fig. 1b). The beginning and ending points for mass loss in Fig. 1b are taken from endotherm onset and ending temperatures in Fig. 1a. All mass loss for HIO3 and HI3O8 dehydration corresponding to the endotherms in Fig. 1a are shown in Fig. 1b and tabulated along with the mass increase from absorption and mass loss from hydration when exposed to 80% RH in Table 1.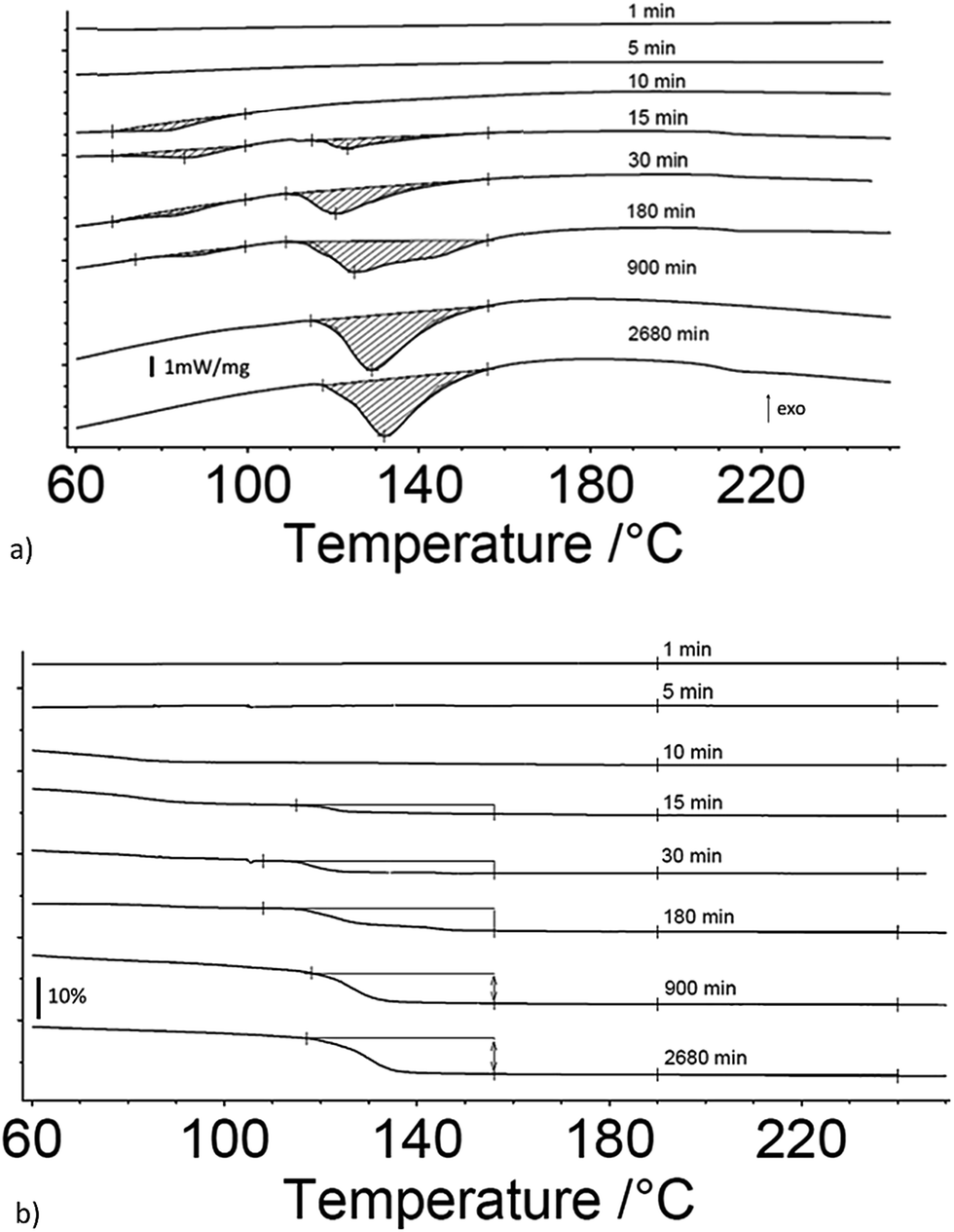 | ||
| Fig. 1 (a) DSC measurements of heat flow for 17 μm I2O5 at different exposure time intervals to 80% RH as indicated on the curves; and, (b) TG measurements of mass change for 17 μm I2O5 at different exposure time intervals to 80% RH as indicated on the curves. Onset and end temperature for measurements of mass loss are determined by endotherm onset and end temperatures. | ||
Fig. 2a and b are the heat flow from DSC and mass change from TG measurements as a function of exposure time to 80% RH from 1 to 2880 minutes for 5 μm I2O5. Fig. 2a shows two distinct endotherms with onset temperature below 110 °C at an exposure time of 1 minute. With an exposure time of 5 minutes, there is only one distinct endotherm with an onset temperature of 90 °C. These endotherms increase in magnitude and onset temperature through the entire range tested. Fig. 2a shows most 5 μm I2O5 samples produce an endotherm at 210 °C with an associated mass loss (Fig. 2b). The beginning and ending temperatures for mass loss in Fig. 2b are taken from endotherm onset and end temperatures in Fig. 2a. Table 2 summarizes the data in Fig. 2b along with the mass increase from absorption.
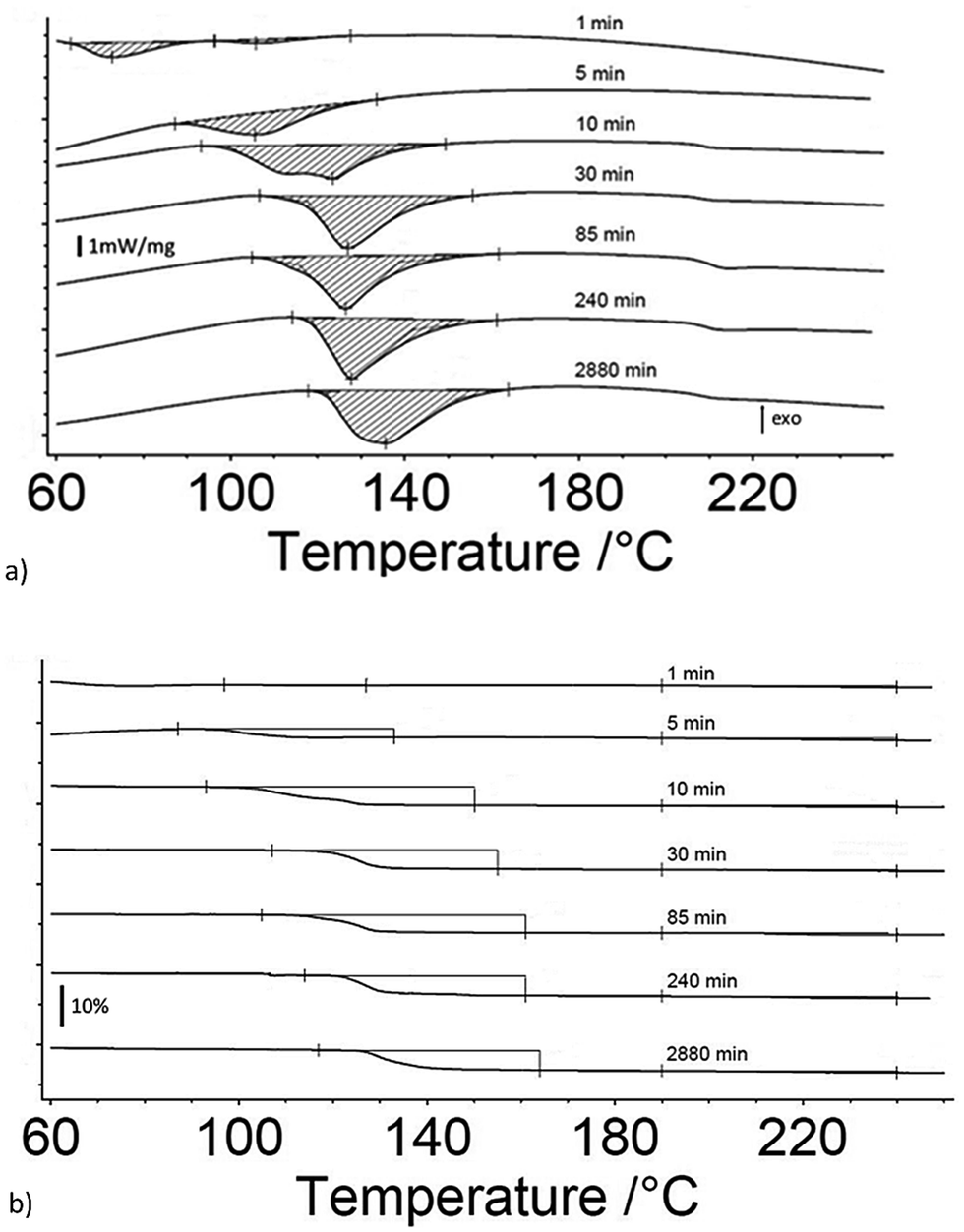 | ||
| Fig. 2 (a) DSC measurements of heat flow for 5 μm I2O5 at different exposure time intervals to 80% RH as indicated on the curves; and, (b) TG measurements of mass change for 5 μm I2O5 after different exposure time intervals to 80% RH as indicated on the curves. Onset and end temperature for measurements of mass loss are determined by endotherm onset and end temperatures. | ||
Fig. 3a and b show the mass gain for 17 μm I2O5 and 5 μm I2O5 respectively, determined by weighing the samples before and after exposure to 80% RH at 23 °C for the indicated exposure times. The mass gain for 17 μm I2O5 increases to a maximum of 34.8% at an exposure time of 180 minutes and then decreases to 7.0% at an exposure time of 3060 minutes. The mass gain for 5 μm I2O5 increases to a maximum of 9.1% at an exposure time of 30 minutes then decreases to 4.8% at an exposure time of 2880 minutes. An opposite trend is seen in the mass gain rate in Fig. 3a and b after the mass gain rate starts to decrease, the decrease in the 5 μm I2O5 mass gain after an exposure time of 30 min is initially fast and slows with increased exposure time, while 17 μm I2O5 is initially slow and increases with exposure time.
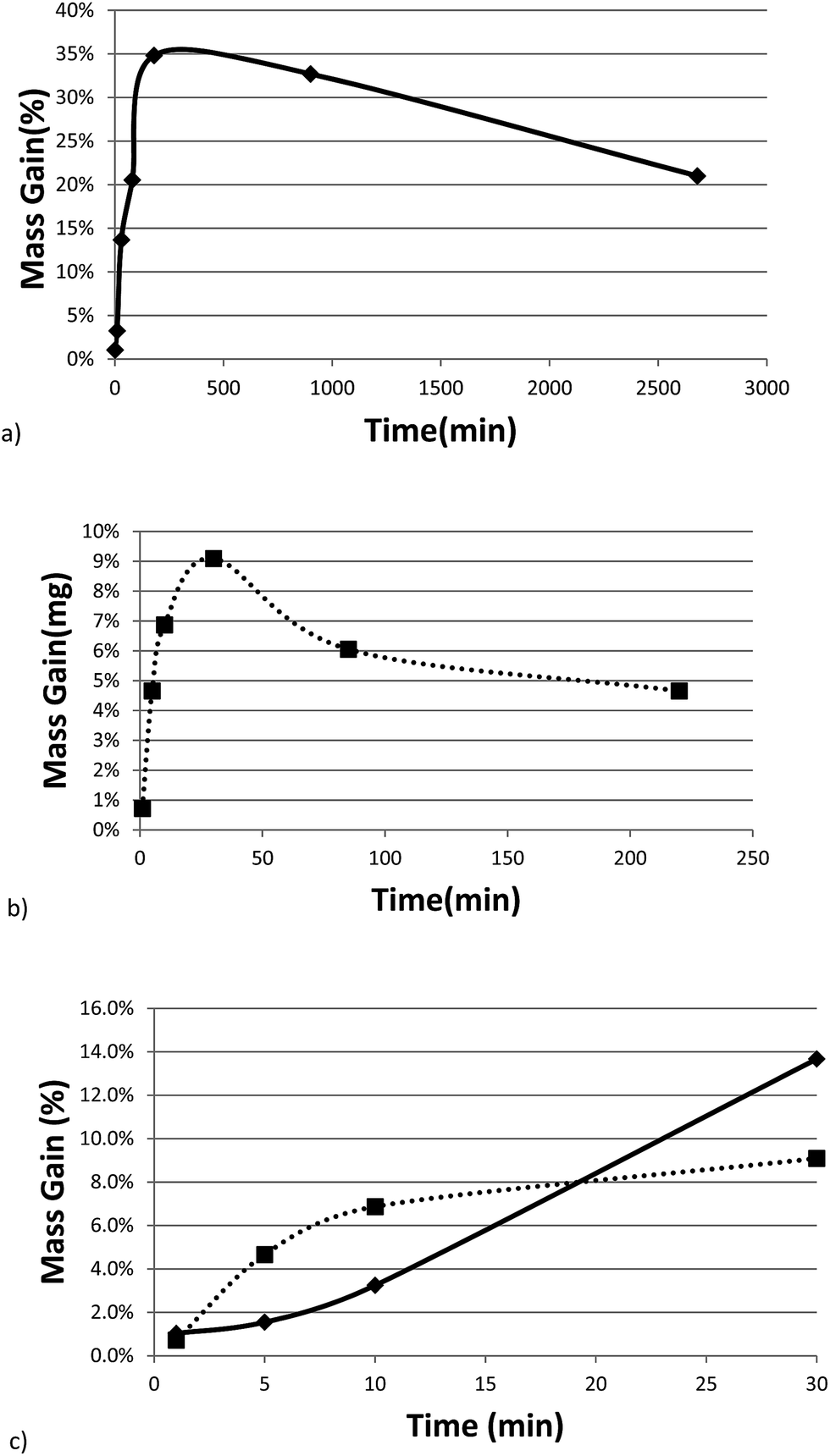 | ||
| Fig. 3 All graphs provide information on the relative rates of absorption, hydration or dehydration as a function of particle size. Samples shown are exposed to 80% RH (23 °C) throughout the entire duration. (a) Mass gain percentage as a function of time for 17 μm I2O5. (b) Mass gain percentage as a function of time for 5 μm I2O5 up to 220 minutes (i.e., 2880 minute data excluded from graph for clarity of presentation but all data shown in Table 2 and Fig S2†). (c) Mass gain for both 5 μm I2O5 (dashed line) and 17 μm I2O5 (solid line) for the first 30 minutes of exposure to 80% RH. | ||
Fig. 3c shows the mass gain for both 5 μm I2O5 and 17 μm I2O5 for exposure time periods less than 30 minutes. The mass gain rate for the 5 μm I2O5 is linear until an exposure time of 5 minutes, then starts plateauing at an exposure time of 5 minutes. In contrast, the mass gain for 17 μm I2O5 is negligible until an exposure time of 5 minutes, then starts to increase.
Discussion
Mechanism
The mechanism in eqn (1a) and (1b) assumes that one mole of water and two moles of I2O5 initially react to form HI3O8, and then two moles of water and two moles of HI3O8 react to form HIO3. This intermediate step (i.e., formation of HI3O8 before HIO3) in the formation of HIO3 from I2O5 is assumed because in almost all reported cases, there is an endothermic peak at 210 °C, the dehydration temperature of HI3O8, and an associated mass loss that corresponds to the mechanism in eqn (1a) and (1b).1,2 Eqn (1) and TG mass loss (Tables 1 and 2 along with Fig. 1b and 2b) can be used to calculate the initial concentration of HIO3. Eqn (2a) and (2b) show the dehydration steps of both iodic acids along with the mass percent of water in each step. If the mechanism accepted and described in eqn (1a) and (1b) is correct, the mass loss in the HIO3 dehydration temperature range cannot be greater than 3.41% if there is no residual water. Mass loss from HIO3 greater than 3.41% will give a concentration of HIO3 greater than 100% when calculated using eqn (3a).The mechanism in eqn (1) also has a condition that HIO3 dehydrates directly into HI3O8. This condition is represented by the ratio of mass loss of HIO3 to HI3O8. The maximum ratio of mass loss percentage at 110 °C for HIO3 to mass loss percentage at 210 °C for HI3O8 should be 3.41![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.76 (i.e., a ratio of 1.94). A ratio higher than 1.94 indicates that a portion of HIO3 is dehydrating directly into I2O5 (not HI3O8) and is seen for multiple samples (highlighted in Tables 1 and 2). These results indicate a new mechanism is needed and presented in eqn (4a) and (4b) for hydration and dehydration of I2O5 that does not have an intermediate step resulting in the formation of HI3O8.
:1.76 (i.e., a ratio of 1.94). A ratio higher than 1.94 indicates that a portion of HIO3 is dehydrating directly into I2O5 (not HI3O8) and is seen for multiple samples (highlighted in Tables 1 and 2). These results indicate a new mechanism is needed and presented in eqn (4a) and (4b) for hydration and dehydration of I2O5 that does not have an intermediate step resulting in the formation of HI3O8.
Hydration:
| I2O5 + H2O → 2HIO3 | (4a) |
Dehydration:
| 2HIO3 → I2O5 + H2O | (4b) |
The mechanism in eqn (4a) and (4b) is proposed because the accepted mechanism (eqn (1)) is valid only under the condition that all HIO3 forms HI3O8 (eqn (2a)) during heating. The initial concentrations of HIO3 and I2O5 can be calculated from TG analysis of mass loss as tabulated in Tables 1 and 2 and eqn (4). The equations to calculate initial concentration of HIO3 and I2O5 from eqn (4) are shown in eqn (5a) and (5b).
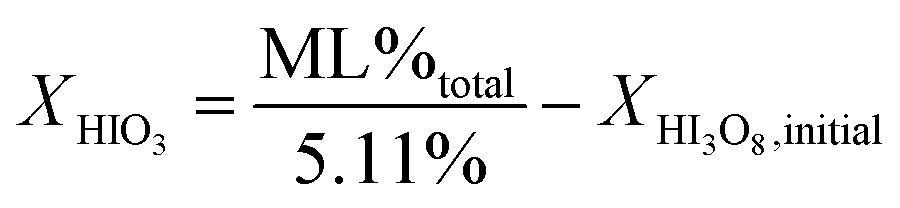 | (5a) |
| XI2O5 = 1 − XHIO3 | (5b) |
In eqn (5), X is the concentration of HIO3 or I2O5 as indicated, ML%total is the measured mass loss for the entire iodic acid dehydration temperature range, and XHI3O8,initial is the initial concentration of HI3O8. The total measured mass loss is divided by the calculated mass loss percent of total water (5.11%), not including residual water, calculated from eqn (4b).
Eqn (1)–(3) represent a model for the previously accepted hydration/dehydration mechanism that enables determination of the concentration of iodine species by TGA mass change measurements. A new model is presented in eqn (4)–(7). The major difference between the models shown is seen by comparing eqn (5) to (3) and is that HIO3 dehydrates into I2O5 instead of HI3O8, and that HI3O8 is formed during heating. The amount of HI3O8 that is formed during heating is accounted for by using the total mass loss in the iodic acid (HIO3 and HI3O8) dehydration temperature range instead of the individual mass loss from HIO3 and HI3O8 dehydration respectively (e.g., used in eqn (3a) and (3b)). The model in eqn (5) includes a term for initial concentration of HI3O8. The DSC cannot differentiate between initial HI3O8 and HI3O8 that is formed during heating. To accurately determine the concentration of HIO3 and I2O5 using eqn (5), the initial concentration of HI3O8 is needed. In this study, the initial concentration of HI3O8 is assumed to be 0.0% because the samples are initially dehydrated before hydration in 80% RH. The validity of this assumption is discussed below. The amount of HIO3 that dehydrates into I2O5 (eqn (6b)) and the amount of HIO3 that forms HI3O8 during heating (eqn (6a)) is shown in eqn (6).
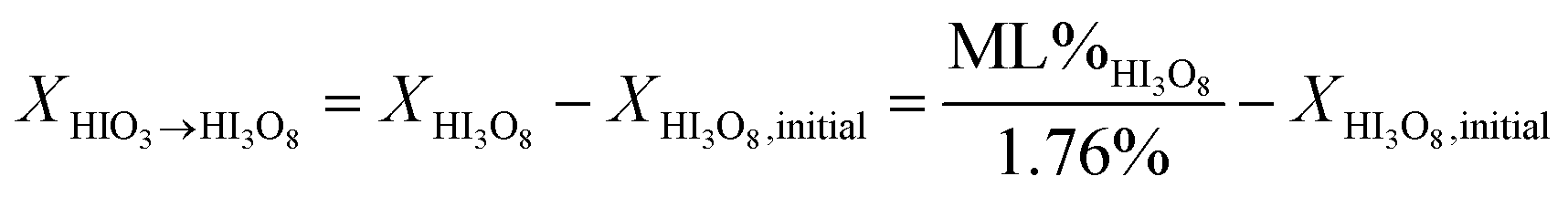 | (6a) |
| XHIO3→I2O5 = XHIO3 − XHI3O8 | (6b) |
In eqn (6), ML%HI3O8 is the measured mass loss from TG analysis for HI3O8, XHIO3→I2O5 is the concentration of HIO3 that dehydrates directly into I2O5, XHI3O8 is the concentration of HI3O8 used in eqn (3b) and is also the concentration of HIO3 that forms HI3O8 during heating (XHIO3→HI3O8) using eqn (4) assuming the initial concentration of HI3O8 is 0%. The mechanism in eqn (4) is proposed because the accepted mechanism (eqn (1)) is a special condition but eqn (7) shows the conditions where the mechanism in eqn (4) is equivalent to the mechanism in eqn (1).
| XHIO3→HI3O8 = XHIO3 | (7a) |
| XHIO3→I2O5 = 0 | (7b) |
The concentrations of HIO3 and HI3O8 calculated using the mechanism in eqn (1) and (4) are shown in ESI Tables S1 and S2.† The samples for which eqn (1) is not valid, because the concentration of HIO3 is greater than 100% (XHIO3 > 100%), are shaded in ESI Tables S1 and S2.† The mechanism in eqn (4a) and (4b) may not have been observed by others because the methods previously used to study hydration of I2O5 are similar and thus produce similar results1,4,8,11,12 that follow eqn (1) and (7) where HIO3 dehydrates directly into HI3O8. The variables that determine whether the conditions in eqn (7) are met are not known. Multiple different experimental conditions (DSC heating rate, water impurities, hydration method (e.g., atmospheric vs. dissolution in water), particle size, I2O5 synthesis method (e.g., dry or dehydration of HIO3)) have been tested in an effort to determine the condition when eqn (1) is valid; however, a direct relation has not been found.
In previous studies,1,2 concentration of iodic acid has been verified by XRD analysis. The mechanism in eqn (4a) and (4b) does not affect the validity of these results and the XRD analysis from previous studies show that TGA is an accurate technique for determination of iodic acids. The XRD analysis happens over 1–2 hours for accurate measurements and RH cannot be controlled during measurements. For this study, the samples exposure to 20% RH was limited to less than 5 minutes. The limitation on exposure time in 20% RH and the time needed for XRD analysis did not allow XRD data collection.
Assumptions
The model described in eqn (4)–(7) is based on assumptions that may not have been considered previously. We have shown that the accepted mechanism for dehydration of iodic acids is a special condition that is violated when the ratio of mass loss from HIO3 to HI3O8 is greater than 1.9 or when the mass loss of water from HIO3 is greater than 3.14%. These are general conditions and are only valid when the only mass loss is water from dehydration of HIO3 (not residual water). When I2O5 is hydrated by exposure to 80% RH, this assumption does not always hold and is seen by a total mass loss greater than 5.11%. Samples that have a greater mass loss then 5.11% are assumed to have residual water and are underlined in ESI Tables S1 and S2.† For accurate calculation of initial HIO3 and I2O5 concentration, it is assumed that if total mass loss is greater than 5.11%, the sample is initially pure HIO3. The validity of the assumption that residual water in the sample indicates the sample is pure HIO3 is discussed below. The calculated concentrations for each sample using eqn (3) and the assumption that the concentration of HIO3 is 100% for samples with a greater total mass loss than 5.11% is shown in Tables 3 and 4.| Exposure time (min) | Residual water (g) | XHIO3 | XHI3O8 | XI2O5 | XHIO3→I2O5 |
|---|---|---|---|---|---|
| 1 | 0 | 0.4% | 1.1% | 99.6% | −0.7% |
| 5 | 0 | 1.6% | 4.5% | 98.4% | −3.0% |
| 10 | 0 | 1.2% | 3.4% | 98.8% | −2.2% |
| 15 | 0 | 36.4% | 13.0% | 63.6% | 23.3% |
| 30 | 0 | 54.8% | 5.7% | 45.2% | 49.1% |
| 180 | 0.47 | 100.0% | 12.5% | 0.0% | 87.5% |
| 900 | 0.24 | 100.0% | 9.6% | 0.0% | 90.4% |
| 2680 | 0.36 | 100.0% | 16.4% | 0.0% | 83.6% |
| Exposure time (min) | Residual water (g) | XHIO3 | XHI3O8 | XI2O5 | XHIO3→I2O5 |
|---|---|---|---|---|---|
| 1 | 0 | 30.1% | 23.8% | 69.9% | 6.3% |
| 5 | 0 | 50.5% | 32.3% | 49.5% | 18.2% |
| 10 | 0 | 96.2% | 16.4% | 3.8% | 79.8% |
| 30 | 0 | 99.0% | 18.1% | 1.0% | 80.8% |
| 85 | 0 | 98.8% | 28.3% | 1.2% | 70.4% |
| 220 | 0.019 | 100.0% | 27.8% | 0.0% | 72.2% |
| 2880 | 0.018 | 100.0% | 23.8% | 0.0% | 76.2% |
In Tables 3 and 4, the concentration of HIO3 that dehydrates into I2O5 is negative for the 17 μm sample after an exposure time of 1, 5 and 10 minutes (underlined). The negative value for the concentration of HIO3 that dehydrates into I2O5 is a result of the assumption that the initial concentration of HI3O8 is 0.0%. The new mechanism (i.e., eqn (4)) has an inherent assumption that during hydration, I2O5 hydrates directly into HIO3. Any I2O5 that hydrates directly into HI3O8 will show up as initial HI3O8 in the model shown in eqn (4)–(7) because it is not possible to differentiate between HI3O8 that is formed during heating and HI3O8 that is formed during hydration. This leads to the small error (<1.6%) in HIO3 concentration calculated using eqn (5a). Since HI3O8 is an adjunct of HIO3 and I2O5,13 the small error in HIO3 calculated using total mass in eqn (5a) is the amount of HIO3 in HI3O8 from hydration. The model in eqn (4)–(7) can be improved by determining the amount of HI3O8 formed during hydration, but a method to determine the amount of HI3O8 formed during hydration has not been developed. Examining the absorption behavior of these samples can help verify the validity of the assumptions that initial concentration of HI3O8 is 0.0% and that mass loss over 5.11% indicates the samples in 100% HIO3. By weighing samples before and after exposure to 80% RH, differences in absorption and hydration were measured.
HI3O8 formation from hydration
Tables 1 and 2 show the mass gain from absorption in 80% RH and the mass loss from HIO3 dehydration. In both samples, the mass gained from absorption is higher than the total iodic acid mass loss from dehydration. After exposure to 80% RH in a humidity chamber, the samples were removed from the camber and exposed to 20% RH ambient environment for weighing and placement in the STA. Since only the RH of the environment is changed after the samples are removed from the humidity chamber, the difference between mass gain and mass loss is assumed to be caused by rapid desorption of residual water and/or IO3− solution. The time of exposure to low humidity was kept as constant as possible and was less than 30 seconds for weight measurements and less than 5 minutes for DSC loading. This limitation to the experimental setup inadvertently helps explain the differences between absorption and the hydration mechanism. The difference in mass gain from absorption and mass loss from dehydration of HIO3 indicates that hydration of I2O5 into HIO3 is a time dependent reaction. The time dependence of this reaction could influence the hydration mechanism and allow HI3O8 to form during hydration.In Table 1 for 17 μm I2O5, there is no mass loss from dehydration of HIO3 until 15 minutes exposure time, but a mass gain of 1.0%, 1.5%, and 3.2%, and a total mass loss of 0.02%, 0.08%, and 0.06% is seen at 1, 5, and 10 minutes exposure time, respectively. The results in Table 1 indicate the mass loss is due to dehydration of HI3O8, indicating that initially, HI3O8 is forming instead of HIO3 for the 17 μm I2O5. Fig. 3c shows that mass gain in the first 30 minutes of exposure time for both 17 μm I2O5 and 5 μm I2O5. The 17 μm I2O5 initially has a fast rate of absorption with an inflection point at 10 minutes where the absorption rate decreases (Fig. 3c). After 10 minutes exposure time, mass loss in the HIO3 dehydration temperature range is seen. The inflection point at 10 minutes and mass loss in the HIO3 dehydration temperature range after 10 minutes exposure suggests that the increased rate of absorption in the 17 μm I2O5 is due to I2O5 forming directly into HI3O8, and after the rate of absorption slows, water that is absorbed starts to hydrate I2O5 into HIO3. The 5 μm I2O5 does not have an inflection point where the rate of absorption slows and does not have exposure times where mass loss from HI3O8 and no mass loss from HIO3 is seen. We speculate that the differences in absorption behavior and formation of HI3O8 are a result of the different particle size or processing methods. Decreased specific surface area of 17 μm I2O5 could slow absorption. If hydration of I2O5 into HIO3 is dependent on absorption rate, the slower absorption rate of 17 μm I2O5 could slow the hydration rate of I2O5 into HIO3. HI3O8 is an adjunct of HIO3 and I2O5 and HIO3 and I2O5 must both be present to form HI3O8. If the absorption rate of 17 μm I2O5 slows the hydration rate of I2O5 to the point where both I2O5 and HIO3 are present, then formation of HI3O8 could be a result of particle size. Alternatively, the processing method between 17 μm I2O5 and 5 μm I2O5 could result in conditions where the formation of HI3O8 is favored over the formation of HIO3 in 17 μm I2O5, and the difference in absorption is a result of formation of HI3O8. The 17 μm I2O5 were created by dehydration of commercial I2O5 that has been shown to be composed mostly of HI3O8.2,7,10 The dehydration of HI3O8 during the heating process could leave discontinuities in the crystal lattice that could favor the formation of HI3O8 over HIO3 during hydration. The initial formation of HI3O8 may not be seen in the 5 μm I2O5 because it is created using a “dry” method and may have more limited discontinuities in the crystal lattice formed from initial dehydration. HI3O8 that is formed during hydration cannot be distinguished from HI3O8 that is formed during heating and leads to the error shown in Tables 3 and 4. Because of this, further studies are need to determine the effects of particle size and processing methods on absorption and formation of HI3O8.
Residual water
The other major assumption used in the calculation of iodic acid concentration from TGA is that any mass loss over 5.11% indicates that the sample is entirely composed of HIO3. In both 17 μm I2O5 and 5 μm I2O5, more water is absorbed than is needed to hydrate I2O5 into HIO3. The maximum mass gained for 5 μm I2O5 is 9.1% at an exposure time of 30 minutes and the maximum mass gain for the 17 μm I2O5 is 34.8% at an exposure time of 180 minutes. Also, the first exposure time where the mass loss is over 5% for 5 μm I2O5 is at an exposure time of 30 minutes and the first exposure time where the mass loss is over 5% for 17 μm I2O5 is at an exposure time of 180 minutes. The exposure time where maximum mass gain is seen and mass loss first reaches 5% suggests that at these exposure times, all I2O5 has reacted with water to form HIO3. Residual water is shown in Tables 3 and 4 and there is no residual water until after the exposure times where maximum mass gain is seen and mass loss first reaches 5%. This suggests that residual water is not formed until all I2O5 has reacted with water to form HIO3 and the assumption that mass loss over 5.11% indicates the sample is pure HIO3 is reasonable. Fig. 3a and b also show that the mass gained starts to decrease after all I2O5 has reacted to form HIO3. We propose that maximum mass gain is related to the particle size and the extra water reacts with I2O5 to form an IO3− solution. When the samples are removed from 80% RH to 20% RH for weighing and loading into the DSC, rapid desorption of extra water occurs forming an HIO3 crystal structure. The decrease in mass gain after 180 minutes exposure for 17 μm I2O5 and 30 minutes for 5 μm I2O5 indicates that an HIO3 crystal structure is forming while still exposed to 80% RH. The HIO3 crystal structure that is formed while the samples are still exposed to 80% RH acts as a diffusion barrier that slows the rapid desorption of water when the samples are taken from 80% RH to 20% RH. The diffusion barrier traps the extra water/IO3− solution in the sample. When the samples are heated in the DSC, discontinuities in the crystal structure are formed by dehydration of the HIO3, reducing the effects of the diffusion barrier and allowing the residual water to be released in the dehydration temperature range of HIO3. Further studies with a greater range of particle sizes and synthesis methods will allow us to determine the effects that these variables have on the formation of HI3O8 and residual water. A better understanding of these variables could eliminate the need for the assumptions that introduce error, but are necessary to calculate concentration of iodic acid by TG mass change and eqn (4)–(7).Conclusion
We have shown that the accepted dehydration mechanism is a special condition of a more general mechanism presented here. The condition where the accepted dehydration mechanism is equivalent to the new mechanism is when all HIO3 dehydrates directly into HI3O8. The new dehydration mechanism for I2O5 was proven by detecting reduced concentrations of HI3O8 in two different I2O5 powder samples. When HIO3 partially dehydrates directly into I2O5, the previously accepted mechanism breaks down because the concentration of HIO3 calculated is greater than 100%. The model presented here more accurately predicts concentrations of iodic acids when assumptions about HI3O8 formation from hydration and residual water are made. The validity of the assumptions is shown by comparisons of mass gained during hydration and mass lost during heating. The data presented confirms the validity of the assumptions, but also show that these assumptions introduce small error in the iodic acid concentrations calculated. Further studies are needed that would allow us to expand the hydration/dehydration model that would allow the concentration of iodic acid to be calculated without assumptions that lead to error.Acknowledgements
The authors Smith and Pantoya are grateful for partial support from DTRA under award HDTRA1-15-1-0029. The authors Parkey and Kesmez are grateful for support from DTRA under award HDTRA1-15-P-0037. We are all grateful for helpful discussion and encouragement from our program manager, Dr Douglas Allen Dalton.References
- K. Selte and A. Kjekshus, Iodine Oxides Part II on the System H2O I2O5, Acta Chem. Scand., 1968, 22, 3309–3320 CrossRef CAS.
- B. K. Little, S. B. Emery, J. C. Nittinger, R. C. Fantasia and C. M. Lindsay, Physiochemical Characterization of Iodine(V) Oxide, Part 1: Hydration Rates, Propellants, Explos., Pyrotech., 2015, 40, 595–603 CrossRef CAS.
- M. W. Chase, NIST-JANAF Thermochemical Tables for the Iodine Oxides, J. Phys. Chem. Ref. Data, 1996, 25(5), 1297–1340 CrossRef CAS.
- R. Kumar, R. W. Saunders, a. S. Mahajan, J. M. C. Plane and B. J. Murray, Physical Properties of Iodate Solutions and the Deliquescence of Crystalline I2O5 and HIO3, Atmos. Chem. Phys., 2010, 10(24), 12251–12260 CrossRef CAS.
- J. Feng, G. Jian, Q. Liu and M. R. Zachariah, Passivated Iodine Pentoxide Oxidizer for Potential Biocidal Nanoenergetic Applications, ACS Appl. Mater. Interfaces, 2013, 5(18), 8875–8880 CAS.
- A. B. Lamb, W. C. Bray and W. J. Geldard, The Preparation of Iodic Acid and Its Anhydride, J. Am. Chem. Soc., 1920, 42(8), 1636–1648 CrossRef CAS.
- D. K. Smith, K. Hill, M. L. Pantoya, J. S. Parkey and M. Kesmez, Reactive Characterization of Anhydrous Iodine(V) Oxide (I2O5) with Aluminum: Amorphous versus Crystalline Microstructures, Thermochim. Acta, 2016, 641, 55–62 CrossRef CAS.
- K. Selte and A. Kjekshus, Iodine Oxides Part III. The Crystal Structure of I2O5, Acta Chem. Scand., 1970, 6(24), 1913–1924 Search PubMed.
- A. Wikjord, P. Taylor, D. Torgerson and L. Hachkowski, Thermal Behaviour of Corona-Precipitated Iodine Oxides, Thermochim. Acta, 1980, 36(3), 367–375 CrossRef CAS.
- D. K. Smith, J. McCollum and M. L. Pantoya, Effect of Environment of Iodine Oxidation State on Reactivity with Aluminum, Phys. Chem. Chem. Phys., 2016, 18, 11243–11250 RSC.
- B. K. Little, E. J. Welle, S. B. Emery, M. B. Bogle, V. L. Ashley, A. M. Schrand and C. M. Lindsay, Chemical Dynamics of Nano-Aluminum/iodine(V) Oxide, J. Phys.: Conf. Ser., 2014, 500(5), 52025 CrossRef.
- B. Little, S. Emery and C. Lindsay, Physiochemical Characterization of Iodine(V) Oxide Part II: Morphology and Crystal Structure of Particulate Films, Crystals, 2015, 5(4), 534–550 CrossRef CAS.
- A. Fischer, Redetermination of HI3O8, an Adduct of HIO3 I2O5, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2005, 61, 278–279 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27854j |
| This journal is © The Royal Society of Chemistry 2017 |