
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solution copolymerization of ethylene and propylene by salicylaldiminato-derived [O-NS]TiCl3/MAO catalysts: synthesis, characterization and reactivity ratio estimation
Zhen Yaob,
Da-Feng Mab,
Zhi-xian Xiaob,
Wen-long Yangb,
Yu-Xia Tub and
Kun Cao*ab
aState Key Laboratory of Chemical Engineering, College of Chemical and Biological Engineering, Zhejiang University, Hangzhou 310027, China. E-mail: kcao@che.zju.edu.cn; Fax: +86 571 87951612; Tel: +86 571 87951832
bInstitute of Polymerization and Polymer Engineering, Zhejiang University, Hangzhou 310027, China
First published on 6th February 2017
Abstract
Salicylaldiminato-derived [O-NS]TiCl3 is used in the copolymerization of ethylene and propylene in toluene solution with methylaluminoxane as the co-catalyst. The effects of temperature, Al/Ti molar ratio and feed ratio of ethylene and propylene on the solution copolymerization and its resulting copolymer structure are investigated. It is revealed that the ethylene content in the resultant copolymers and the copolymerization activity decrease with the increase of propylene addition. The 13C-NMR analysis demonstrates that the copolymers are essentially composed of long ethylene sequences with some isolated propylene units and smaller amount of PP diad. Moreover, the triad distribution data are elaborated in a statistical strategy to determine the reactivity ratios, which are also compared with the first-order direct fit method and the first-order/second-order Markovian methods. The product of monomer reactivity ratio reaches a minimum value of 0.37 at 60 °C, indicating the best temperature for ethylene–propylene copolymerization in this system. The results also indicate that there are hardly penultimate unit effects.
Introduction
Ethylene–propylene copolymer is a commercially important material, and has been widely studied in the past decades for its insensitivity to oxygen, ozone, acids, and alkaline with no double bonds in the backbone of the polymer chain. In most technical processes for the production of ethylene–propylene copolymers, soluble or highly dispersed vanadium compounds such as VCl4, VOCl3 and VO(OR)3, co-catalyzed by alkylaluminium chloride in the presence of an organic halogen promoter, are commonly used.1,2 However, it is inconvenient because the residual vanadium content in the polymer above 10 ppm can cause discoloration, aging, toxicity, etc.3In 1985 Kaminsky first synthesized the ethylene–propylene copolymer using a metallocene catalyst.4 During the last three decades, the single-site catalytic systems based on the metallocene complexes of group IV metals have been extensively reported.5–10 A well-known distinct feature of the single-site metallocene catalysts is their ability to copolymerize ethylene and α-olefins with both narrow molecular weight distribution and narrow chemical composition distribution, which offers a desirable control over the physical and mechanical properties. Waymouth et al.11 studied the copolymerization of ethylene and propylene with metallocenes containing different substituent and bridge. The results indicated that metallocenes having heterotopic active sites yielded alternating, isotactic copolymers, and silicon-bridged metallocenes produced copolymers with higher activity and molecular weight but lower propylene incorporation at similar feeds than the carbon-bridged analogues. Recently, the research of Mitsui Chemicals on bis(phenoxyimine) group 4 transition metal catalysts known as FI catalysts revealed that the appropriate choice and design of phenoxyimine ligands, metal, co-catalysts, and polymerization condition made the catalysts very selective in almost all aspects of polymerization and polymer structure.12–14 The development of well-defined catalysts from coordination compounds could create new opportunities for synthesis of desired polymers with tailored structure and relevant properties. Fujita et al.15 synthesized a FI catalyst for producing amorphous ethylene–propylene copolymer with ultrahigh molecular weight. The resultant copolymer had a weight-average molecular weight of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 200 kg mol−1, which represents the highest molecular weight known for linear, synthetic copolymers to date. Moreover, Redshaw and Tang systematically reviewed the development of tridentate ligands which incorporate a functionality capable of weakly binding to a metal and thereby altering either the electronics, steric or both.16 It is proving to be a fruitful strategy for controlling the properties of complexes in polymerization catalysis and elsewhere.
200 kg mol−1, which represents the highest molecular weight known for linear, synthetic copolymers to date. Moreover, Redshaw and Tang systematically reviewed the development of tridentate ligands which incorporate a functionality capable of weakly binding to a metal and thereby altering either the electronics, steric or both.16 It is proving to be a fruitful strategy for controlling the properties of complexes in polymerization catalysis and elsewhere.
The reactivity ratio is an important parameter in the copolymer composition equation, and it is widely used to describe the features of ethylene–propylene copolymerization. Most authors have already commented on the ability of metallocenes to prepare ethane and propene copolymers with random distribution of the comonomers.5,17 Zhu et al.18 developed a simple method to determine the Markoff model and the monomer reactivity ratio through the two component sequence of the copolymer determined by 13C-NMR and the feed ratio of ethylene–propylene copolymerization.
In this work, a salicylaldiminato-derived titanium complex containing a tridentate [O-NS] ligand ([O-NS] = N-(3,5-di-tert-butylsalicylidene)-2-methylsulfanylanilinato) is used for the copolymerization of ethylene and propylene in toluene solution with methylaluminoxane as the cocatalyst. The effects of polymerization conditions such as polymerization temperature, cocatalyst concentration and feed ratio of ethylene and propylene are investigated. Moreover, taking into account the effect of the penultimate monomer unit, the first-order Markovian model and second-order Markovian model are both used to describe the monomer reactivity ratios and resultant structure are also discussed.
Experimental
Materials
Toluene was dried over a sodium/potassium alloy and distilled for 6 hours before use. All other organic solvents were freshly distilled from sodium benzophenone ketyl immediately prior to use. Polymerization grade gases of ethylene (E) (purity 99.95%) and propylene (P) (purity 99.95%) were dried by passing through the columns filled with 3 Å molecular sieve and Cu catalyst before entering the reactor. The salicylaldiminato-derived complex, [O-NS]TiCl3, was synthesized according to the reference.19 The co-catalyst, methylaluminoxane (MAO), was provided by AKZO NOBEL in the form of 10 wt% solution in toluene.Polymerization procedure
All polymerization runs were carried out in a 100 mL clean glass flask with a heating magnetic stirrer under atmospheric pressure. Before polymerization, the reactor was evacuated and charged with nitrogen alternatively for three times. The reactor was heated to the required temperature and remained constant. Then, the certain amounts of toluene solution, MAO/toluene suspension and gaseous mixture of ethylene and propylene were added to the reactor sequentially. After the solution was saturated with ethylene and propylene, the polymerization reaction was started by injection of the [O-NS]TiCl3/toluene solution. During the polymerization process, the mixture of ethylene and propylene was fed to the reactor continuously to maintain the atmospheric pressure. After 15 min, the copolymerization was terminated by the addition of acidified ethanol. The reaction mixture was poured into dilute HCl/ethanol solution. The precipitated product was filtered and washed with ethanol and then was dried under vacuum at 60 °C.The concentration of ethylene in toluene during the polymerization reaction was estimated with the equation provided by Kissin:20
|
CE = KEPEexp[QE/(RT)]
| (1) |
The concentration of propylene in toluene during the polymerization reaction was calculated with the equation obtained by Galland.21
|
CP = KPPPexp[QP/(RT)]
| (2) |
Characterization
The molecular weight and its distribution were determined by a Viscotek 350A HT-GPC System. 1,2,4-Trichlorobenzene was used as solvent and the analysis were performed at 120 °C and 1.0 mL min−1 flow rate. The thermal properties of copolymers were measured using a DSC Q200 instrument with a heating/cooling/heating rate of 10 °C min−1 from −90 °C to +150 °C under a dry nitrogen atmosphere after eliminating thermal history and the second heat curve was adopted.The microstructure of copolymer was characterized with Varian Unity-300 nuclear magnetic resonance (NMR) instrument. The samples were dissolved in deuterated o-dichlorobenzene with a concentration about 10%. At least 3000 scans at 120 °C were applied for each acquisition to obtain a good signal-to-noise ratio.
Results and discussion
Copolymerization features and copolymer characteristics
The copolymerization of ethylene and propylene was carried out using salicylaldiminato-derived [O-NS]TiCl3 (Scheme 1) activated by MAO. The effects of polymerization temperature, Al/Ti molar ratio and monomer feed ratio on the copolymerization behavior were investigated and the relevant experimental results are listed in Table 1. The polymerization activity increases with higher reaction temperature, but further increase in the temperature led to activity reduction. In this copolymerization system, not only the reaction rate constant, but also the solubility of monomer in toluene and the stability of the catalyst are affected by the reaction temperature. This behavior can be ascribed to the competition between the decreasing of monomer concentration, partial deactivation of catalyst and the increasing of propagation rate. And at high temperature, the former one becomes the main factor affecting the reaction activity. An activity peak is obtained at a mole ratio of Al/Ti = 3000. At much higher concentration of MAO, the polymerization activity is slightly declined. This phenomenon is possible due to excessive complexation of MAO with the active centers. Therefore, the vacant coordination positions at these active centers become unavailable for monomer complexation. Ethylene/propylene (E/P) feed ratios of 75/25, 67/33, 50/50 and 33/67 mol% are also employed for the copolymerization process. The activity of catalyst is reduced with decreasing E/P feed ratio due to the greater steric hindrance of propylene and the variety chain transfer reaction with propylene incorporation, which should be a common behavior in a similar polymerization process.13 | ||
| Scheme 1 The structure of salicylaldiminato-derived [O-NS]TiCl3 catalyst. | ||
| Samples | E/Pb | Al/Tic | Temperature (°C) | Activity (kg P per mol per Ti per h) | Tg/Tmd (°C) | Tcd (°C) | Xce% | MWf | MWD | EEE | PEE + EEP | EPE | PEP | EPP + PPE | Pg mol% | nEh | nPi |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Polymerization conditions: [Cat.] = 2 μmol Ti, pressure = 1 atm, time = 15 min, toluene = 50 mL.b E/P feed ratio (mol mol−1) in gas phase.c MAO/Cat. feed ratio (mol mol−1).d Melt temperature, crystallization temperature and glass transition temperature measured by DSC.e Percentage of crystallinity estimated from the DSC data and the reference ΔHf value of the orthorhombic PE crystal (287.3 J·g−1).f Weight average molar mass measured by high-temperature GPC.g Propylene content (in mol%) in polymers estimated from 13C-NMR data.h Average sequence length of ethylene in the copolymer.i Average sequence length of propylene in the copolymer. | |||||||||||||||||
| 1 | 50:50 |
1500 | 40 | 328.4 | —/113.4 | 98.5 | 7.7 | 74400 |
2.21 | 0.704 | 0.174 | 0.097 | 0.011 | 0.014 | 10.8 | 9.1 | 1.1 |
| 2 | 50:50 |
1500 | 50 | 637.0 | —/114.1 | 99.9 | 6.0 | 55300 |
2.09 | 0.529 | 0.267 | 0.152 | 0.029 | 0.023 | 17.4 | 5.1 | 1.1 |
| 3 | 50:50 |
1500 | 60 | 873.0 | —/104.8 | 89.1 | 3.7 | 35200 |
1.94 | 0.503 | 0.260 | 0.171 | 0.028 | 0.039 | 19.3 | 5.0 | 1.1 |
| 4 | 50:50 |
1500 | 70 | 721.8 | —/104.3 | 88.2 | 5.0 | 27900 |
1.82 | 0.619 | 0.239 | 0.123 | 0.019 | — | 13.1 | 6.3 | 1.0 |
| 5 | 50:50 |
1500 | 80 | 386.4 | —/110.9 | 94.9 | 6.7 | 18100 |
1.76 | 0.688 | 0.188 | 0.109 | 0.015 | 11.8 | 8.3 | 1.1 | |
| 6 | 67:33 |
1500 | 40 | 377.5 | —/119.4 | 99.5 | 38.3 | 146700 |
2.62 | 0.820 | 0.110 | 0.070 | — | — | 6.2 | 16.9 | 1.0 |
| 7 | 67:33 |
1500 | 50 | 684.3 | —/112.7 | 95.3 | 9.2 | 64000 |
2.63 | 0.714 | 0.172 | 0.103 | 0.012 | — | 10.0 | 9.2 | 1.0 |
| 8 | 67:33 |
1500 | 60 | 925.2 | —/112.8 | 96.0 | 5.8 | 45700 |
2.08 | 0.656 | 0.196 | 0.125 | 0.023 | — | 12.3 | 7.3 | 1.0 |
| 9 | 67:33 |
1500 | 70 | 774.3 | —/113.1 | 100.2 | 4.6 | 35100 |
2.60 | 0.729 | 0.156 | 0.115 | — | — | 9.7 | 11.3 | 1.0 |
| 10 | 67:33 |
1500 | 80 | 398.9 | —/118.8 | 106.0 | 46.5 | 21000 |
1.92 | 0.739 | 0.163 | 0.098 | — | — | 9.0 | 11.1 | 1.0 |
| 11 | 75:25 |
1500 | 40 | 472.3 | —/120.3 | 94.2 | 15.2 | 173500 |
2.79 | 0.879 | 0.077 | 0.043 | — | — | 4.1 | 24.8 | 1.0 |
| 12 | 75:25 |
1500 | 50 | 800.6 | —/117.4 | 91.3 | 12.0 | 120800 |
2.44 | 0.793 | 0.127 | 0.079 | — | — | 7.1 | 14.5 | 1.0 |
| 13 | 75:25 |
1500 | 60 | 1044.8 | —/116.3 | 89.8 | 9.1 | 62400 |
2.24 | 0.729 | 0.162 | 0.102 | 0.007 | — | 9.5 | 10.2 | 1.0 |
| 14 | 75:25 |
1500 | 70 | 901.0 | —/109.4 | 87.5 | 6.5 | 35800 |
2.25 | 0.820 | 0.110 | 0.070 | — | — | 6.2 | 16.9 | 1.0 |
| 15 | 75:25 |
1500 | 80 | 457.8 | —/110.7 | 93.9 | 6.7 | 26500 |
2.12 | 0.886 | 0.115 | — | — | — | 2.9 | 17.5 | — |
| 16 | 33:67 |
1500 | 40 | 217.8 | —/84.6 | 62.2 | 1.5 | 64700 |
2.10 | 0.611 | 0.229 | 0.138 | 0.022 | — | 13.7 | 6.3 | 1.0 |
| 17 | 33:67 |
1500 | 50 | 522.6 | −64.7/83.7 | 60.6 | 1.3 | 58200 |
1.99 | 0.440 | 0.283 | 0.059 | 0.165 | 0.053 | 22.3 | 2.9 | 1.3 |
| 18 | 33:67 |
1500 | 60 | 625.0 | −66.9/82.2 | 60.4 | 1.4 | 31300 |
1.82 | 0.310 | 0.307 | 0.202 | 0.075 | 0.106 | 29.5 | 3.0 | 1.2 |
| 19 | 33:67 |
1500 | 70 | 537.4 | −64.2/86.1 | 66.7 | 1.8 | 23100 |
1.81 | 0.379 | 0.256 | 0.203 | 0.068 | 0.094 | 27.0 | 3.6 | 1.2 |
| 20 | 33:67 |
1500 | 80 | 330.0 | —/90.9 | 73.0 | 1.8 | 11900 |
1.89 | 0.494 | 0.279 | 0.185 | 0.043 | — | 18.3 | 4.5 | 1.0 |
| 21 | 50:50 |
500 | 60 | 551.6 | —/109.9 | 86.9 | 4.6 | 86300 |
2.05 | 0.722 | 0.165 | 0.105 | — | — | 9.4 | 10.7 | 1.0 |
| 22 | 50:50 |
1000 | 60 | 732.4 | —/101.1 | 81.9 | 4.2 | 47200 |
1.94 | 0.605 | 0.219 | 0.131 | 0.019 | 0.026 | 14.9 | 6.6 | 1.1 |
| 23 | 50:50 |
2000 | 60 | 948.4 | −61.3/101.6 | 83.0 | 3.5 | 30900 |
1.94 | 0.440 | 0.275 | 0.178 | 0.047 | 0.061 | 22.7 | 4.1 | 1.1 |
| 24 | 50:50 |
3000 | 60 | 1084.4 | −62.6/100.7 | 84.0 | 2.8 | 26900 |
2.00 | 0.367 | 0.341 | 0.179 | 0.056 | 0.057 | 24.6 | 3.4 | 1.1 |
| 25 | 50:50 |
4000 | 60 | 1014.2 | −64.1/100.7 | 81.3 | 2.7 | 19500 |
1.87 | 0.394 | 0.287 | 0.192 | 0.056 | 0.071 | 24.9 | 3.7 | 1.2 |
| 26 | 50:50 |
5000 | 60 | 1026.0 | −65.6/100 | 83.3 | 3.4 | 19900 |
1.82 | 0.389 | 0.292 | 0.191 | 0.055 | 0.073 | 25.1 | 3.7 | 1.2 |
It is observed that the propylene content of the obtained copolymer can be dependent on different polymerization parameters, such as temperature, Al/Ti ratio and E/P feed ratio etc. Propylene content of the copolymers declines with increasing E/P feed ratio. The variety of propylene content in the copolymers versus polymerization temperature reveals typical mountain shape with maximum at 60 °C. This behavior may be attributed to the greater thermal stability of the propylene π-complex of Ti+P compared to the ethylene π-complex of Ti+E and the increment of E/P concentration ratio in toluene though lower monomer concentration by temperature increase.22 Propylene content in the copolymers increases with increasing the Al/Ti molar ratio up to 3000, and remains stable beyond this ratio.
Table 1 also shows the weight-average molecular weight (MW) and its distribution (MWD) of the copolymers obtained at different conditions. For this catalyst system, the temperature increases, the MW and MWD decrease. These should be attributed to the increase of chain transfer rate and faster monomer diffusion at higher temperature, respectively. Besides, MW of the obtained copolymers decreases with the boosting of Al/Ti ratio. This means the chain transfer to aluminium is a prominent chain termination route.23 Meanwhile, the Al/Ti ratio almost has no effect on MWD. Most E–P copolymers have polydispersity values close to a Schulz–Flory distribution.
The melt temperature (Tm) and crystallization temperature (Tc) are found in the DSC curves of all samples listed in Table 1. It is observed that Tm and Tc increase with increasing average sequence length of ethylene in the resultant copolymers under the same E/P ratio in feed due to the crystallization of EEE block segments. Moreover, the variation tendency of the percentage of crystallinity (Xc) agrees with the Tc. The thermal properties such as Tm, Tc, Xc seem to remain constant while changing the Al/Ti molar ratio beyond 1000. When the propylene content is more than 22.3 mol%, the glass transition temperature (Tg) becomes visible. The variety of average sequence length of ethylene in the copolymers is negligible with the mutation of Al/Ti ratio. Furthermore, the triad fraction of EPP + PPE segments is increased when increasing the Al/Ti molar ratio.
Reactivity ratios
Fig. 1 presents the assignments of the characteristic signals in the 13C-NMR spectrum of sample,18 which is selected as a representative obtained copolymer. The results indicate that there are no significant head-to-head and tail-to-tail structures (inverted propylene sequence). Moreover, it is advisable to neglect the signal of PPP triad which is too feeble to being detected. Therefore, the quantitative evaluation of the sequence distribution at triad level from 13C-NMR can be allowed. The chemical shift assignments and calculation follow the analysis method of Randall24 and Carman et al.25 Table 2 lists most of the peaks found in the spectrum and their chemical shift, which allows to estimate relative percentages of every triad present in the copolymer.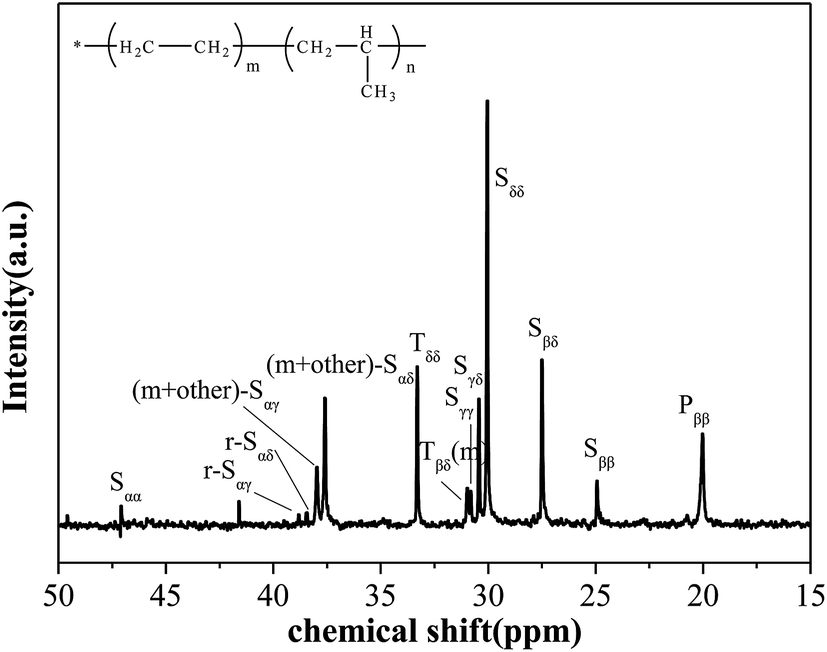 | ||
| Fig. 1 13C-NMR spectrum of sample 8 showing the peek assignments. | ||
| Carbon assignment | Chemical shifts (ppm) | Sequence |
|---|---|---|
| a Herein, P, S and T denote primary, secondary, and tertiary carbons, respectively. The letters m and r refer to meso and racemic, respectively. The position of a carbon relative to its nearest tertiary carbon was expressed by two Greek subscripts.26,27 | ||
| Sαα | 48.1–45.3 | PP |
| r-Sαγ | 38.8 | EPEP + PEPE |
| r-Sαδ | 38.5 | PEE |
| (m + other)-Sαγ | 37.9 | EPEP + PEPEγ |
| (m + other)-Sαδ | 37.6 | PEE + EEP |
| Tδδ | 33.3 | EPE |
| Tβδ(m) | 30.95 | PPE + EPP |
| Sγγ | 30.8 | PEEP |
| Sγδ | 30.4 | PEEE + EEEP |
| Sδδ | 30 | (EEE)n |
| Sβδ | 27.5 | EPEE + EEPE |
| Sββ | 24.7 | PEP |
| Pββ(rr) | 22.0–19.5 | mm, mr, rr |
From these triads determination, it is possible to calculate the average sequence length of the monomeric blocks as following:
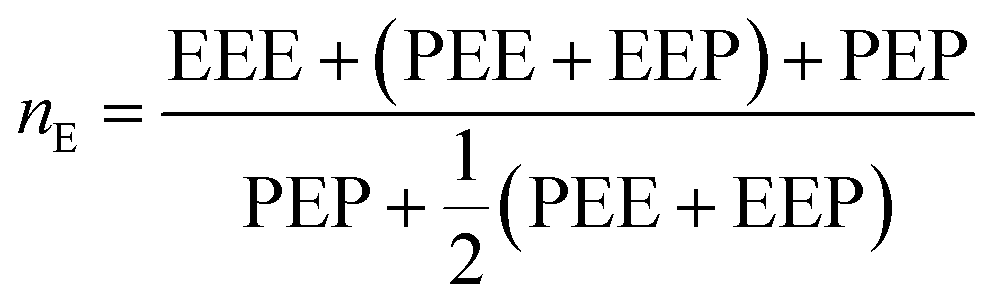 | (3) |
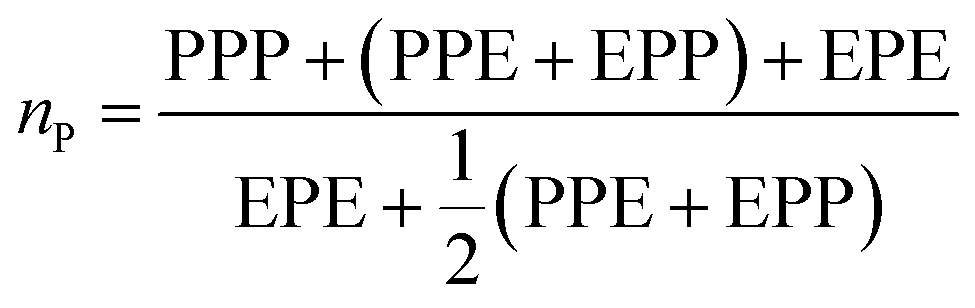 | (4) |
Herein, a first-order Markovian statistical model is adopted to determine the reactivity ratios of ethylene and propylene in E/P copolymerization.26 According the first-order Markovian method, the first-order reactivity ratios of ethylene and propylene copolymerization base on the input of diad fractions [EE], [EP] and [PP] and the molar ratio of ethylene to propylene in the liquid phase (F = CE/CP):
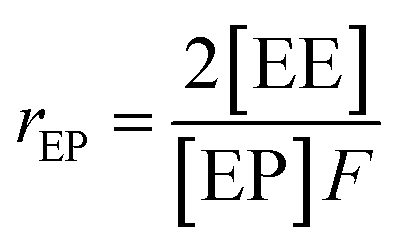 | (5) |
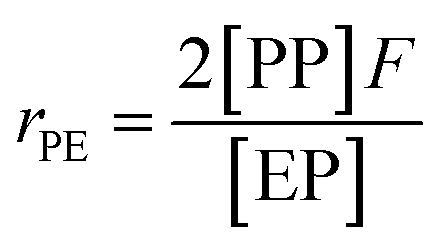 | (6) |
It is suggested that the second-order Markovian model should be more appropriate, taking into account not only the effect of the ultimate monomer unit but also that of the penultimate one. According the second-order Markovian method, the second-order reactivity ratios base on the input of the triad fractions [EEE], [EEP], [PEP], [EPE], [EPP] and [PPP] and the reactor feed F:
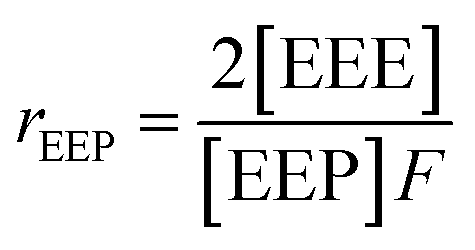 | (7) |
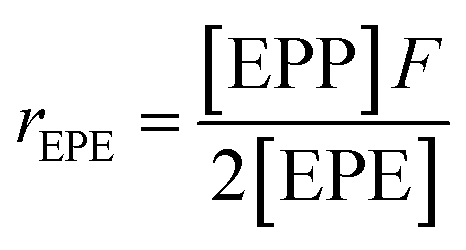 | (8) |
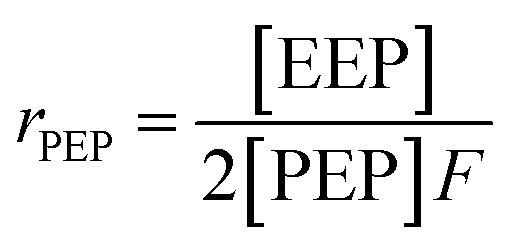 | (9) |
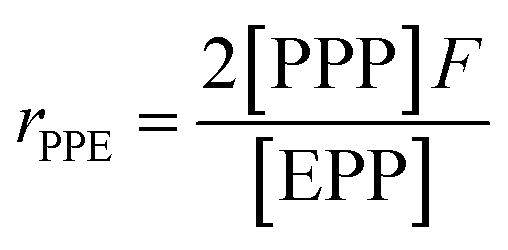 | (10) |
Table 3 presents the reactivity ratios calculated by the first-order and second-order Markovian, and the first-order direct fit method including error margins. For both Markovian methods, the reactivity ratios are the average of individual results of the copolymerization and the error margins are determined by:
 | (11) |
| Temperature | Markovian method (first-order) | Direct peak method (first-order) | Markovian method (second-order) | ||
|---|---|---|---|---|---|
| 40 °C | rEP | 22.78 ± 1.28 | 21.74 ± 0.12 | rEEP | 25.89 ± 3.98 |
| rEPE | 0.024 ± 0.001 | ||||
| rPE | 0.0228 ± 0.0001 | 0.0224 ± 0.0001 | rPEP | 28.05 ± 3.83 | |
| rPPE | — | ||||
| 50 °C | rEP | 10.89 ± 0.14 | 11.13 ± 0.02 | rEEP | 12.53 ± 2.50 |
| rEPE | 0.028 ± 0.001 | ||||
| rPE | 0.0284 ± 0.0004 | 0.0283 ± 0.0001 | rPEP | 11.72 ± 1.44 | |
| rPPE | — | ||||
| 60 °C | rEP | 8.10 ± 1.22 | 7.99 ± 0.02 | rEEP | 8.61 ± 1.00 |
| rEPE | 0.051 ± 0.004 | ||||
| rPE | 0.0356 ± 0.0003 | 0.0358 ± 0.0001 | rPEP | 8.89 ± 2.27 | |
| rPPE | — | ||||
| 70 °C | rEP | 9.73 ± 0.55 | 9.64 ± 0.01 | rEEP | 11.24 ± 1.00 |
| rEPE | 0.042 ± 0.012 | ||||
| rPE | 0.0485 ± 0.0001 | 0.0441 ± 0.0004 | rPEP | 10.90 ± 2.74 | |
| rPPE | — | ||||
| 80 °C | rEP | 16.75 ± 3.23 | 16.90 ± 0.06 | rEEP | 11.86 ± 2.45 |
| rEPE | — | ||||
| rPE | 0.0572 ± 0.0001 | 0.0506 ± 0.0002 | rPEP | 12.28 ± 0.42 | |
| rPPE | — |
For the first-order direct fit method, Fineman–Ross method and Kelen–Tüdös method can be applied to calculate the monomer reactivity ratios.27 The corresponding methods at different temperature are shown in Fig. 2. For the Fineman–Ross method, X(Y − 1)/Y and X2/Y have a good linear relationship, X(Y − 1)/Y = rEP(X2/Y) − rPE, where X and Y represent the molar concentration ratio of ethylene and propylene in the feed and copolymer, respectively. With regard to the Kelen–Tüdös method, constant α is introduced to correct the Fineman–Ross method in consideration of the penultimate effect.  and
and 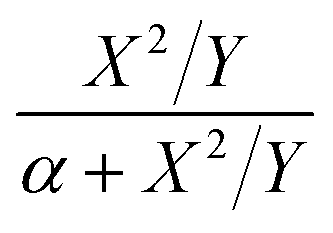 also have a reasonable linear relationship,
also have a reasonable linear relationship,  , where the constant α is the geometric mean of minimum and maximum of the X2/Y in a group of experimental data. Comparing the results obtained from different methods, there is a gap between the two sets of rEP and rPE (Fig. 3). Furthermore, the Fineman–Ross method matches with the first-order Markovian faultlessly. So the better fitting between the Fineman–Ross method and the first-order Markovian method indicates that the copolymerization may be just influenced by the terminal monomer unit. And the gap between the first-order direct fit method and the second-order Markovian method denotes that the effect of the penultimate monomer unit turns out to be small.
, where the constant α is the geometric mean of minimum and maximum of the X2/Y in a group of experimental data. Comparing the results obtained from different methods, there is a gap between the two sets of rEP and rPE (Fig. 3). Furthermore, the Fineman–Ross method matches with the first-order Markovian faultlessly. So the better fitting between the Fineman–Ross method and the first-order Markovian method indicates that the copolymerization may be just influenced by the terminal monomer unit. And the gap between the first-order direct fit method and the second-order Markovian method denotes that the effect of the penultimate monomer unit turns out to be small.
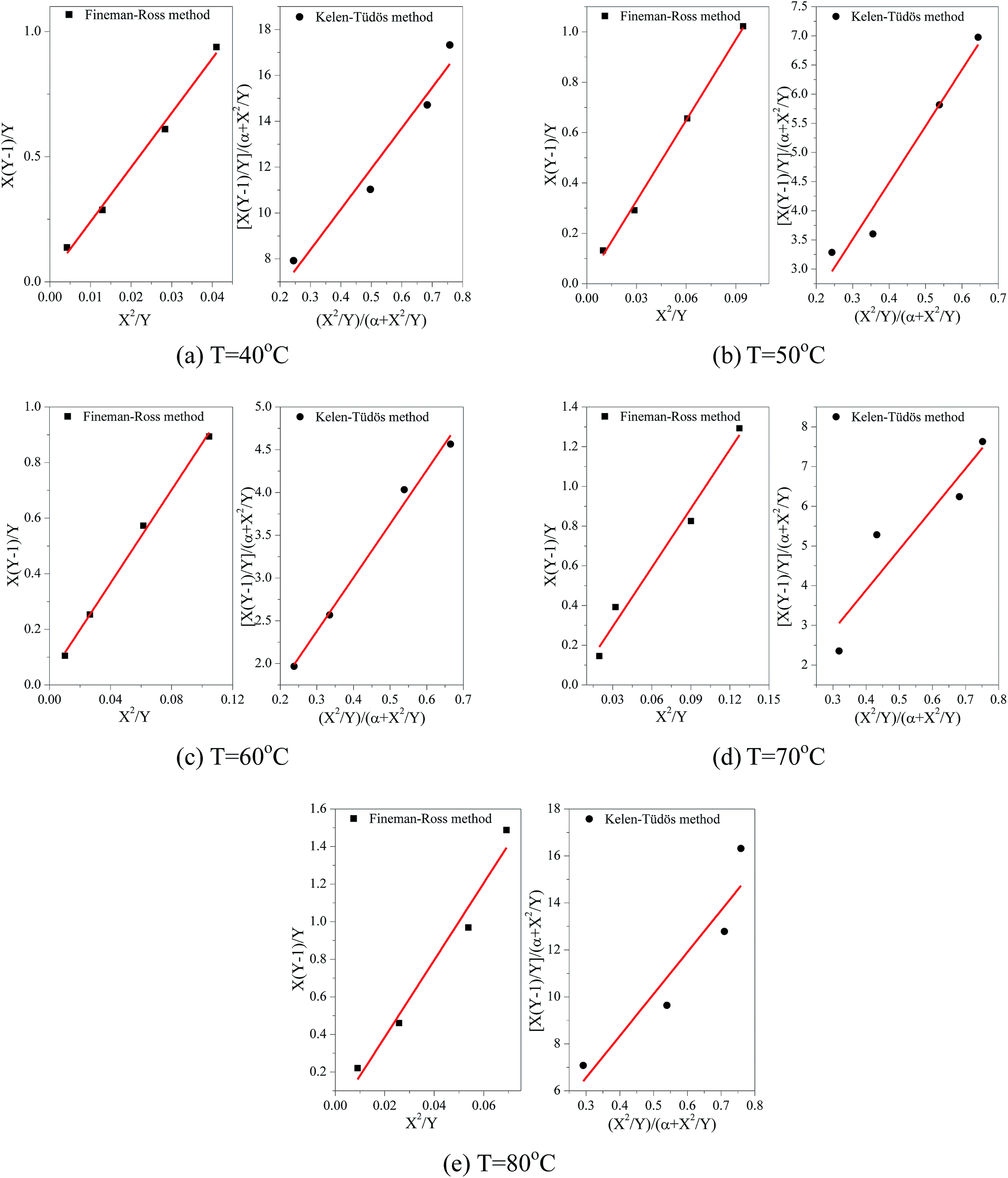 | ||
| Fig. 2 Comparison of both Fineman–Ross method and Kelen–Tüdös method at different temperature for E–P copolymerization. | ||
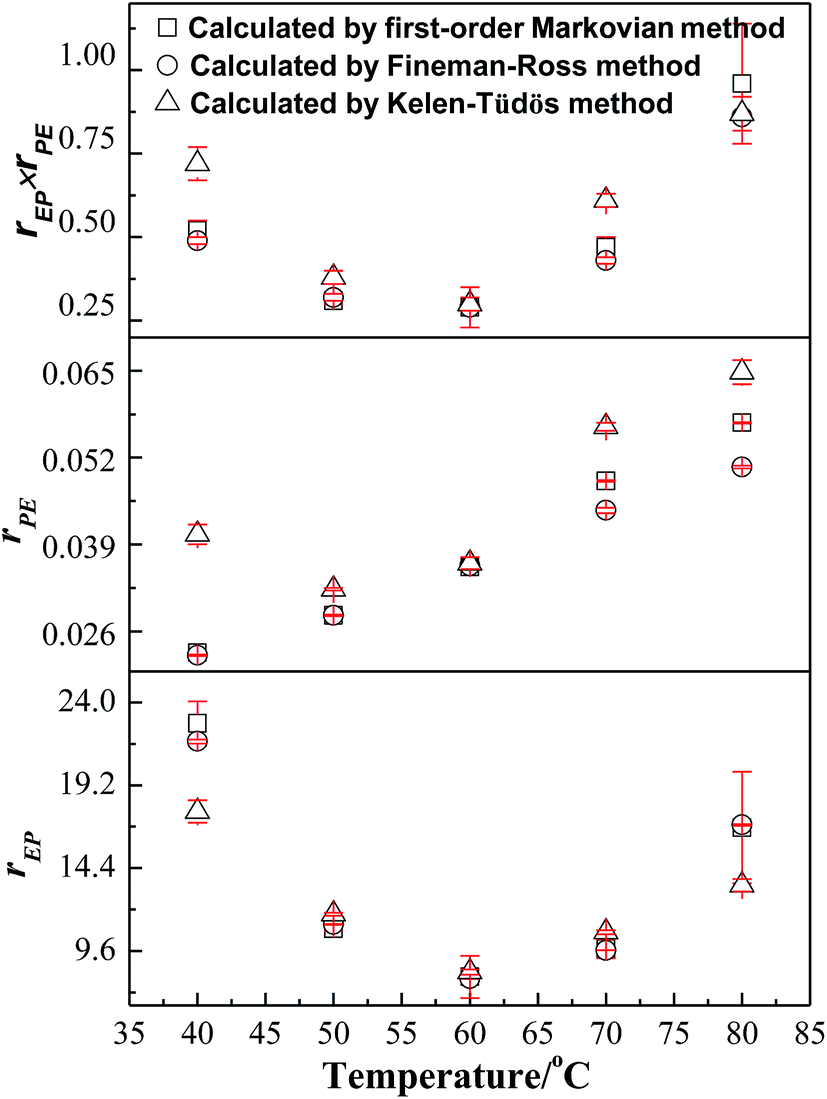 | ||
| Fig. 3 Reactivity ratios calculated by the various methods for E–P copolymerization. | ||
Generally, rEP > rPE, indicates that ethylene insertion is preferred when either ethylene or propylene is the last inserted unit. Here, rEP ≫ 1 and rPE ≪ 1, the higher the value of rEP/rPE, the longer the consecutive sequences of ethylene repeat units in the obtained copolymers. Moreover, the values of nE and nP in Table 1 shows that the resultant copolymers are essentially composed of long ethylene sequences with some isolated propylene units and less amount of PP diad which can be confirmed from the low values of PPE + EPP for all samples and the higher fraction for EPE triads. A raise in rEP with increasing polymerization temperature reveals that high temperature is helpful to propylene insertion. And a valley-shape variation of rEP with increasing temperature means there is a proper temperature (60 °C) to promoted copolymerization of ethylene and propylene. According to the product of reactivity ratios rEPrPE < 1, it is concluded that the [O-NS]TiCl3/MAO bring nearly random E–P copolymers. The product of reactivity ratios rEPrPE at 60 °C gets a minimum value 0.37 and reached a maximum value 0.96 at 80 °C.
Conclusions
In this work, the catalyst [O-NS]TiCl3 is used in ethylene and propylene copolymerization in toluene solution with MAO as the cocatalyst. The copolymerization behaviors dependent on reaction temperature, Al/Ti molar ratio and E/P feed ratios. It is revealed that the ethylene content in the obtained copolymers and activity of catalyst decrease with the increase of propylene addition. There is an optimum polymerization temperature and Al/Ti molar ratio for obtaining the highest yield and propylene content of copolymer. Tm, Tc and Xc of the samples increase with increasing the ethylene content due to the crystallization of EEE block segments. Tg of the final copolymers becomes visible when the propylene content is more than 22.3 mol%. A series of E–P copolymers with Mw = 20000–100000 are synthesized with a narrow MWD (1.7–2.6). The 13C-NMR results demonstrate that the copolymers are essentially composed of long ethylene sequences with some isolated propylene units and less amount of PP diad. Furthermore, the triad distribution data are elaborated in a statistical method to determine the reactivity ratios of the monomers, which are also obtained by the first-order direct fit method and the first-order Markovian method. The comparative results indicate that there is scarcely penultimate effect in this catalyst system.
Acknowledgements
This study was supported by the National High Technology Research and Development Program (863 Program) of China (2012AA040306). Here in 2017, it is dedicated to celebrate the 120th anniversary of the most prestigious Zhejiang University and 90th anniversary of the oldest chemical engineering in china.Notes and references
- G. Natta, G. Sartori, G. Mazzanti, D. Fiumani and A. Valvassori, J. Polym. Sci., 1961, 51(156), 411 CrossRef CAS.
- D. N. Matthews, Uniroyal Inc, US Pat. 3462399 A, 1969.
- H. Hagen, J. Boersma and G. van Koten, Chem. Soc. Rev., 2002, 31(6), 357–364 RSC.
- W. Kaminsky and M. Miri, J. Polym. Sci., Part A: Polym. Chem., 1985, 23(8), 2151–2164 CrossRef CAS.
- M. Arndt, W. Kaminsky, A. M. Schauwienold and U. Weingarten, Macromol. Chem. Phys., 1998, 199(6), 1135–1152 CAS.
- D. Arrowsmith, W. Kaminsky, A. M. Schauwienold and U. Weingarten, J. Mol. Catal. A: Chem., 2000, 160(1), 97–105 CrossRef CAS.
- W. Kaminsky, P. J. Lemstra, J. Loos, F. Muller, J. W. Niemantsverdriet, P. C. Thune and U. Weingarten, Isr. J. Chem., 2002, 42(4), 367–372 CrossRef CAS.
- B. Heuer and W. Kaminsky, Macromolecules, 2005, 38(8), 3054–3059 CrossRef CAS.
- C. Piel, F. G. Karssenberg, W. Kaminsky and V. B. F. Mathot, Macromolecules, 2005, 38(16), 6789–6795 CrossRef CAS.
- T. Seraidaris, W. Kaminsky, J. V. Seppala and B. Lofgren, Chem. Phys., 2005, 206(13), 1319–1325 CAS.
- W. Fan, M. K. Leclerc and R. M. Waymouth, J. Am. Chem. Soc., 2001, 123(39), 9555–9563 CrossRef CAS PubMed.
- S. Matsui, M. Mitani, J. Saito, Y. Tohi, H. Makio, N. Matsukawa, Y. Takagi, K. Tsuru, M. Nitabaru, T. Nakano, H. Tanaka, N. Kashiwa and T. Fujita, J. Am. Chem. Soc., 2001, 123(28), 6847–6856 CrossRef CAS.
- H. Makio, T. Ochiai, H. Tanaka and T. Fujita, Adv. Synth. Catal., 2010, 352(10), 1635–1640 CrossRef CAS.
- H. Terao, A. Iwashita, N. Matsukawa, S. Ishii, M. Mitani, H. Tanaka, T. Nakano and T. Fujita, ACS Catal., 2011, 1(4), 254–265 CrossRef CAS.
- S. Ishii, J. Saito, S. Matsuura, Y. Suzuki, R. Furuyama, M. Mitani, T. Nakano, N. Kashiwa and T. Fujita, Macromol. Rapid Commun., 2002, 23(12), 693–697 CrossRef CAS.
- C. Redshaw and Y. Tang, Chem. Soc. Rev., 2012, 41(12), 4484–4510 RSC.
- A. Tynys, T. Saarinen, K. Hakala, T. Helaja, T. Vanne, P. Lehmus and B. Lofgren, Macromol. Chem. Phys., 2005, 206, 1043 CrossRef CAS.
- W.-J. Wang and S. Zhu, Macromolecules, 2000, 33, 1157–1162 CrossRef CAS.
- M. Gao, C. Wang, X. Sun, C. T. Qian, Z. Ma, S. H. Bu, Y. Tang and Z. W. Xie, Macromol. Rapid Commun., 2007, 28(15), 1511–1516 CrossRef CAS.
- Y. V. Kissin, Polymers Properties and Applications, in Isospecific Polymerization of Olefins With Heterogeneous Ziegler-Natta Catalysts, ed. H. J. Cantow, H. J. Harwood, J. P. Kennedy, A. Ledwith, J. Meibner, S. Okamura and S. Olive, Springer-Verlag, New York, 1985, p. 3 Search PubMed.
- F. F. N. Escher, G. B. Galland and M. R. Ferreira, J. Polym. Sci., Part A: Polym. Chem., 2003, 41(16), 2531–2541 CrossRef CAS.
- R. Furayama, J. Saito, S. Ishii, M. Mitani, S. Matsui, Y. Tohi, H. Makio, N. Matsukawa, H. Tanaka and T. Fujita, J. Mol. Catal. A: Chem., 2003, 200(1–2), 31–42 CrossRef.
- A. Tynys, T. Saarinen, K. Hakala, T. Helaja, T. Vanne, P. Lehmus and B. Lofgren, Macromol. Chem. Phys., 2005, 206(10), 1043–1056 CrossRef CAS.
- J. C. Randall, J. Macromol. Sci., Rev. Macromol. Chem. Phys., 1989, 29(2–3), 201–317 CrossRef.
- C. J. Carman, R. A. Harrington and C. E. Wilkes, Macromolecules, 1977, 10(3), 536–544 CrossRef CAS.
- G. Odian in Principles of Polymerization, Wiley & Sons, New York, 1991, vol. 3, p. 455 Search PubMed.
- T. Kelen and F. Tüdős, J. Macromol. Sci., Part A: Pure Appl. Chem., 1975, 9(14), 1–27 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |