
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective synthesis of 2′-modified nucleosides by using ortho-alkynyl benzoate as a gold(I)-catalyzed removable neighboring participation group†
Haixin Ding‡
ab,
Chuang Li‡b,
Yirong Zhoub,
Sanguo Honga,
Ning Zhang*a and
Qiang Xiao *b
*b
aDepartment of Chemistry, Nanchang University, Nanchang, Jiangxi 330031, China
bJiangxi Key Laboratory of Organic Chemistry, Jiangxi Science & Technology Normal University, Nanchang, Jiangxi 330013, China. E-mail: xiaoqiang@tsinghua.org.cn
First published on 12th January 2017
Abstract
In the present paper, we report a novel strategy for highly efficient stereoselective synthesis of 2′-modified nucleosides by using ortho-alkynyl benzoate as neighboring participation group. Subsequently, ortho-alkynyl benzoate can be removed smoothly in the presence of 5 mol% Ph3PAuCl–AgOTf in dichloromethane with H2O (1 eq.) and ethanol (6 eq.) to afford 2′-OH nucleosides in high yields and selectivity.
Introduction
In the past decades, tremendous efforts have been devoted to the synthesis of novel nucleosides and the evaluation of their biological activities. Accordingly, a large number of nucleosides have been successfully developed as antiviral and antitumor drugs.1 Among these chemical entities, C-2′ substituted nucleosides have showed special importance.2–5 For instance, clofarabine, gemcitabine, nelarabine, clofrabine and most recently FDA approved sofosbuvir for chronic HCV treatment all contain the C-2′ substituted nucleoside core structure (Fig. 1).6–8 On the other hand, 2′-modified nucleosides have also been used as biochemical probes to investigate the structure and function of nucleic acid.9–11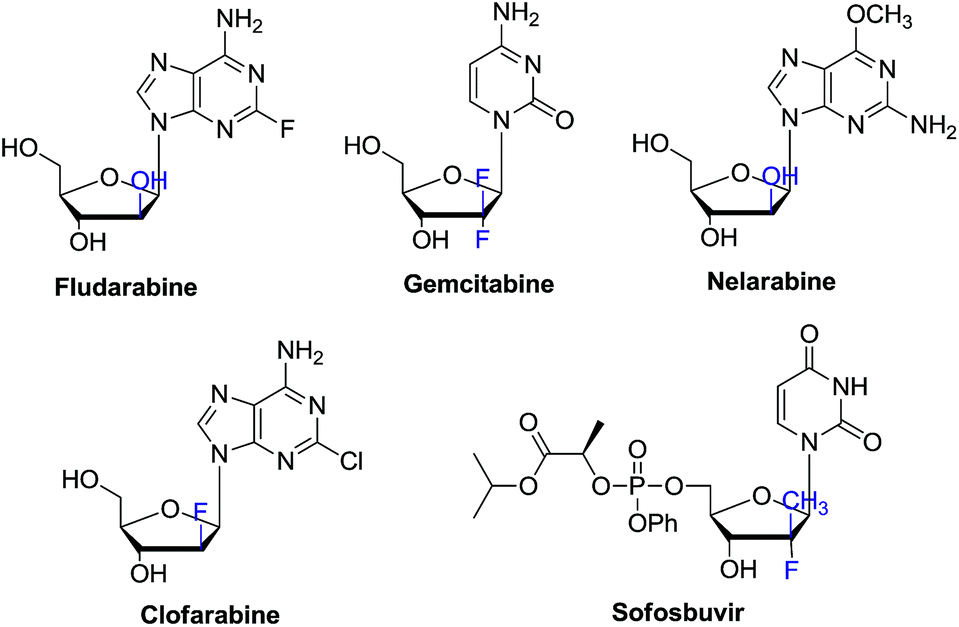 | ||
| Fig. 1 Examples of 2′-modified nucleoside drugs. | ||
Currently, there are generally two synthetic methodologies to access 2′-modified ribonucleosides, namely the convergent approach and the linear approach (Fig. 2). In the convergent approach, the nucleoside was produced by glycosylation of nucleobase with the corresponding sugar moiety. In the linear approach, the nucleoside was prepared by chemical modification of commercially available natural nucleosides or related compounds. From the synthetic point of view, the linear approach offers a relatively convenient way because of it could avoid the glycosylation step, which is often cumbersome. However, compared with the linear approach, the convergent approach is potentially more flexible. It could provide abundant structural diversity by using Vorbrüggen glycosylation of a variety of nucleobase with modified carbohydrate. But in absence of neighboring group participation (NGP), Vorbrüggen glycosylation always generated a mixture of α and β isomer in low selectivity and yield. Moreover, the separation is often time-consuming and labor intensive. Therefore, a general and efficient synthetic approach for C-2′ substituted nucleosides is highly desired.
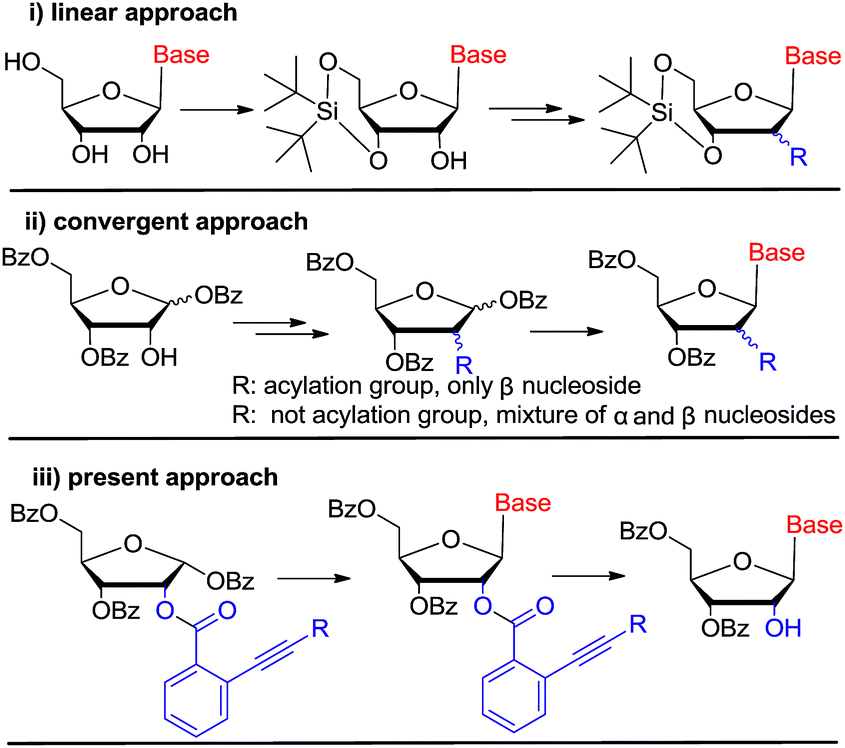 | ||
| Fig. 2 Synthetic methods of 2′-modified ribonucleosides. | ||
In the literature, some examples were reported using acyl group as neighboring participation group, which could be removed selectively by using NH2NH2, CH3NH2 or NH2CH2CH2NH2 etc.12 Nevertheless, these approches' reproducibilities were far from satisfied by our evaluation. At the meantime, acetyl transfer from 3′-OH to 2′-OH was inevitable, which will make the purification troublesome and sometimes impossible. So a general strategy is highly desired to solve this potential problem.
Gold(I) complexes are mild electrophiles and have wide usage in homogeneous catalysis. In recent years, the application of gold(I)-catalyzed electrophilic activation of alkynes has been extensively used for the construction of carbon–carbon or carbon–heteroatom bonds,13 especially in glycosylation for oligosaccharide synthesis by Yu's group.14 In 2008, Asao reported the first gold(I)-catalyzed transesterification of ortho-alkynylbenzoic acid esters with ethanol and its potential application as protecting group for alcohols and phenols.15 Afterwards, this approach was not further investigated. According to this preliminary report, we reasoned that ortho-alkynylbenzoic acid ester may act as neighboring participation group to prepare nucleosides stereoselectively by using Vorbrüggen glycosylation. Then, ortho-alkynylbenzoic acid ester could be removed regioselectively by gold(I) complexes to liberate 2′-OH (Fig. 2(iii)). If our assumption works, it could provide a practical alternative protocol for the synthesis of C-2′ substituted nucleosides.
Bearing the above considerations in mind, ortho-alkynyl benzoate 2 was firstly prepared starting from commercially available 1,3,5-tri-O-benzoyl-α-D-ribofuranose 1 in two steps with high overall yield. Then adenosine 3a was synthesized by using Vorbrüggen glycosylation. To our delight, in the presence of trimethylsilyl triflate (TMSOTf), ortho-alkynyl benzoate group of compound 2 acted as an excellent neighbouring participation group to form dioxolanylium with anomeric cation, which led to nucleophilic attack from β-face by silylated N6-benzoyl adenine to afford adenosine 3a stereoselectively in 82% yield (Scheme 1). Under the similar condition, a series of nucleosides 3(a–i) were successfully obtained in high yields and stereoselectivity.
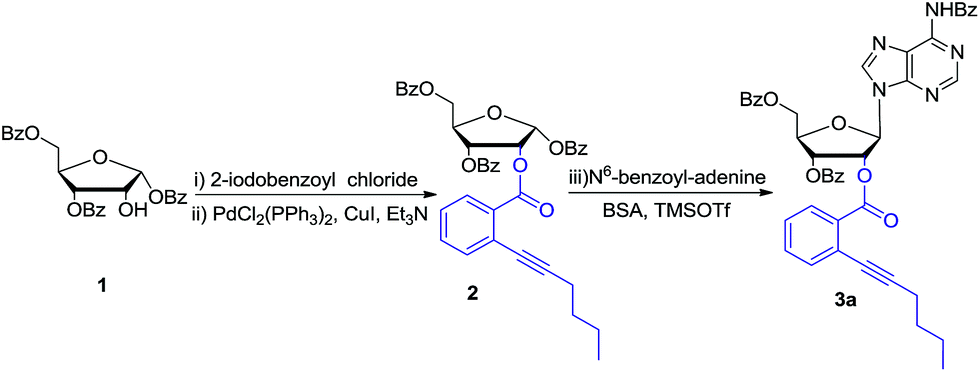 | ||
| Scheme 1 Synthesis of model substrate 3a: (i) 2-iodobenzoyl chloride, pyridine, r.t., 94%; (ii) PdCl2(PPh3)2, CuI, Et3N, THF, 50 °C, 72%; (iii) N6-benzyladenine, N,O-bis(trimethylsilyl) acetamide (BSA), trimethylsiyltriflate (TMSOTf), acetonitrile, 82%. | ||
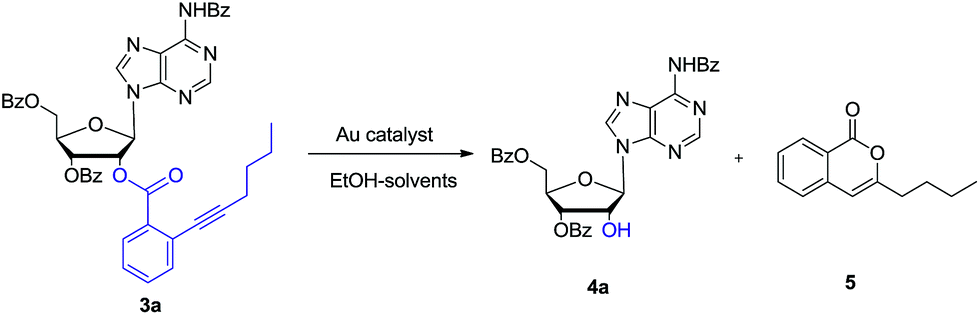
Then as a proof of concept, adenosine 3a was subjected to react with EtOH (6.0 eq.) in the presence of 5 mol% of AuCl in dichloromethane (DCM). After 5 hours, we were pleased to find that the desired 2′-OH nucleoside 4a can be obtained in 70% yield (entry 1). The ortho-alkynyl benzoate was released as isocoumarin 5. In Asao's preliminary report, gold-catalysed transesterification should give ortho-alkynylbenzoic acid ethyl ester as the main product together with a small amount isocoumarin. Because the reagent grade ethanol and DCM were directly used in our experiment, we reasoned that residue H2O in EtOH could have participated the reaction.
In order to further investigate the reaction, the freshly dried ethanol and 4 Å MS in DCM were used. It showed that the reaction became very sluggish. After 24 hours, the reaction still did not finish. In consistent with Asao's work, ortho-alkynylbenzoic acid ethyl ester was identified in the reaction mixture. Then H2O (1 eq.) was added to the reaction. After another 5 hours, the reaction proceeded smoothly. Both nucleoside 4a and isocoumarin 5 were obtained in high yields. So it was concluded that H2O (1 eq.) was essential for the reaction. In the absence of EtOH, the reaction was also sluggish. The reason might be that EtOH could improve the solubility of H2O in DCM.
Based on this promising result, a series of gold(I) catalysts were evaluated under H2O (1 eq.) and ethanol (6 eq.) (Table 1). Ph3PAuCl gave only trace amount of product (entry 2). Nearly quantitative yield of 4a was isolated by reaction with 5 mol% Ph3PAuCl–AgOTf (entry 6). But Ph3PAuCl–AgNTf afforded 4a in low yield (31%) (entry 7). When the catalysis loading of Ph3PAuCl–AgOTf was decreased to 2.5 mol% and 1 mol%, the corresponding yields were reduced to 81% and 49% respectively (entries 4–5).
|
||||
|---|---|---|---|---|
| Entrya | Catalyst (mol%) | Solvent | Time (h) | Yieldb (%) |
| a Each reaction was carried out with β-nucleoside 3a (0.20 mmol) with H2O (1 eq.) and ethanol (6 eq.) in the presence of catalyst at room temperature under N2 atmosphere for 5 hours.b Isolated yield. | ||||
| 1 | AuCl (5%) | DCM | 5 | 70% |
| 2 | Ph3PAuCl (5%) | DCM | 5 | Trace |
| 3 | AgOTf (10%) | DCM | 5 | NR |
| 4 | Ph3PAuCl–AgOTf (1.0%) | DCM | 5 | 49% |
| 5 | Ph3PAuCl–AgOTf (2.5%) | DCM | 5 | 81% |
| 6 | Ph3PAuCl–AgOTf (5.0%) | DCM | 5 | 96% |
| 7 | Ph3PAuCl–AgNTf (5.0%) | DCM | 5 | 31% |
| 8 | Ph3PAuCl–AgOTf (5.0%) | Toluene | 5 | Trace |
| 9 | Ph3PAuCl–AgOTf (5.0%) | DMF | 5 | Trace |
| 10 | Ph3PAuCl–AgOTf (5.0%) | CH3CN | 5 | 32 |
| 11 | Ph3PAuCl–AgOTf (5.0%) | THF | 5 | 26 |
| 12 | Ph3PAuCl–AgOTf (5.0%) | EtOH | 5 | 49 |
After 5 mol% Ph3PAuCl–AgOTf was indentified as the best catalyst loading, the solvents effect was also investigated by performing the reaction in a series of organic solvents under room temperature conditions (entries 8–12). It was observed that toluene and dimethylformamide (DMF) only gave a trace amount of product 4a (<5%). The reaction also proceeded less efficiently in THF, acetonitrile or ethanol. DCM was found to be the optimum solvent.
With the optimized condition in hand, the synthesized β-nucleoside substrates 3a–i were subjected to 5 mol% Ph3PAuCl–AgOTf, ethanol (6 eq.) and H2O (1 eq.) in DCM. The results are summarized in Scheme 2. Except for cytidine derivate 4c, the reaction of pyrimidine nucleosides (3b, 3d–e), purine nucleosides (3a, 3f–h), and 7-deazaguanine nucleoside (3i) all proceeded smoothly and the desired products (4a–b, 4d–i) were obtained in nearly quantitative yield. For cytidine derivate 4c, it was speculated that the unprotected N-4 primary amine group may chelate with gold(I) ion to deactivate its catalytic activity. The speculation was further proved that the reaction can afford corresponding nucleoside 4d in high yield after its N-4 amine was protected by benzoate. For nucleosides 3g and 3h, the electron-withdrawing substituents (F and Cl) in purine base might reduce the nucleophilicity of N-6 amine, but they did not interfere the gold(I) catalyst's activity. Therefore, nucleosides 4g and 4h could be obtained successfully. HPLC analysis of 4a–b and 4d–i showed that the no transesterification of 3′-benzoate to 2′-OH was noticed, which was crucial for following synthesis of 2′-modified ribonucleosides.
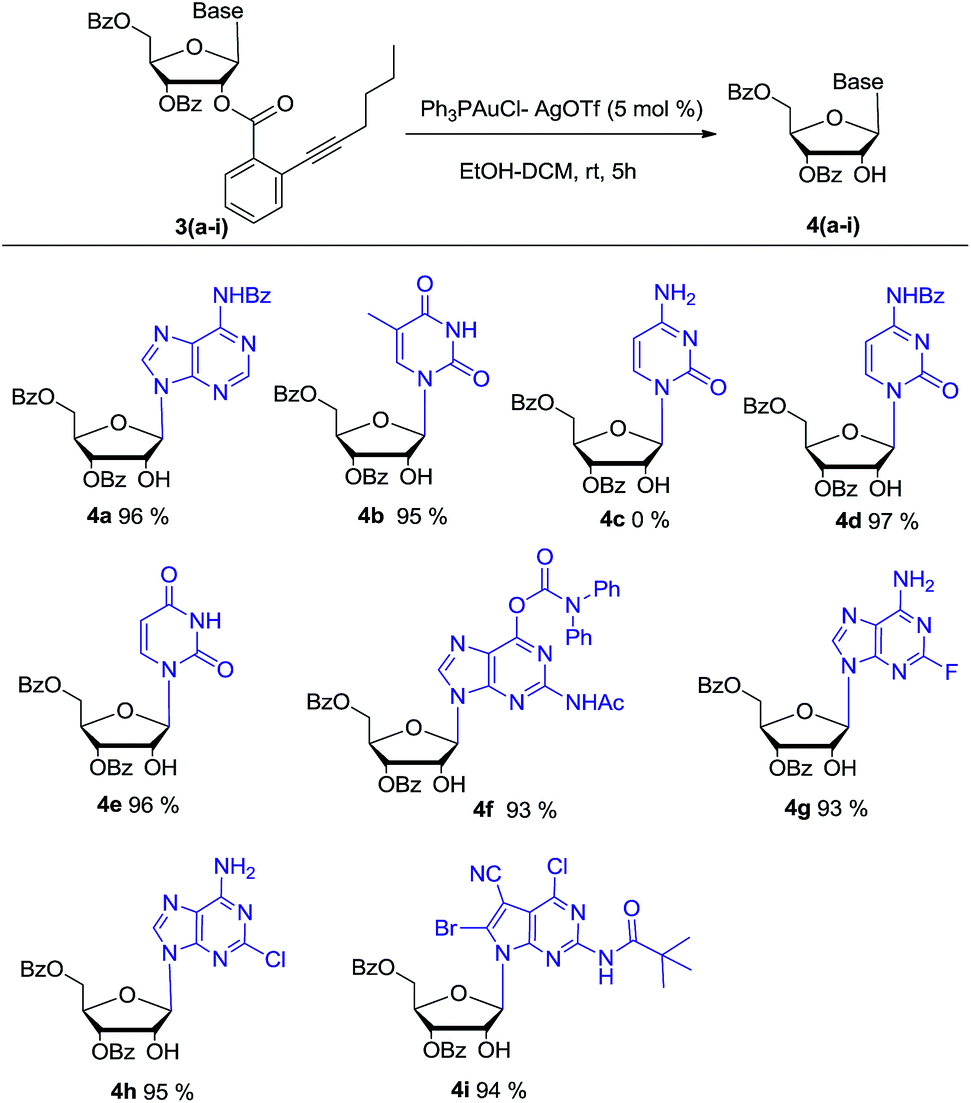 | ||
| Scheme 2 Gold(I)-catalyzed selective deprotection of β-nucleosides (4a–i). | ||
In order to further investigate the reaction mechanism, H2O18 was used in the control reaction. After reaction accomplished, O18-labeled isocoumarin G was obtained, which was confirmed by HRMS. According to the above evidences, a plausible catalytic cycle is proposed in Scheme 3. The coordination of the carbon–carbon triple bond of nucleoside A to the gold(I) catalyst improved the electrophilicity of alkyne (B). Subsequently, the carbonyl oxygen could attack the electron-deficient alkyne to form the intermediate C. While H2O addition to the formed onium ion would generate the intermediate D. After hydrogen atom transferred, nucleoside E would be released along with the generation of the intermediate F. Finally, isocoumarin G was yielded and the corresponding active gold(I) species were liberated to participate in the next catalytic cycle.
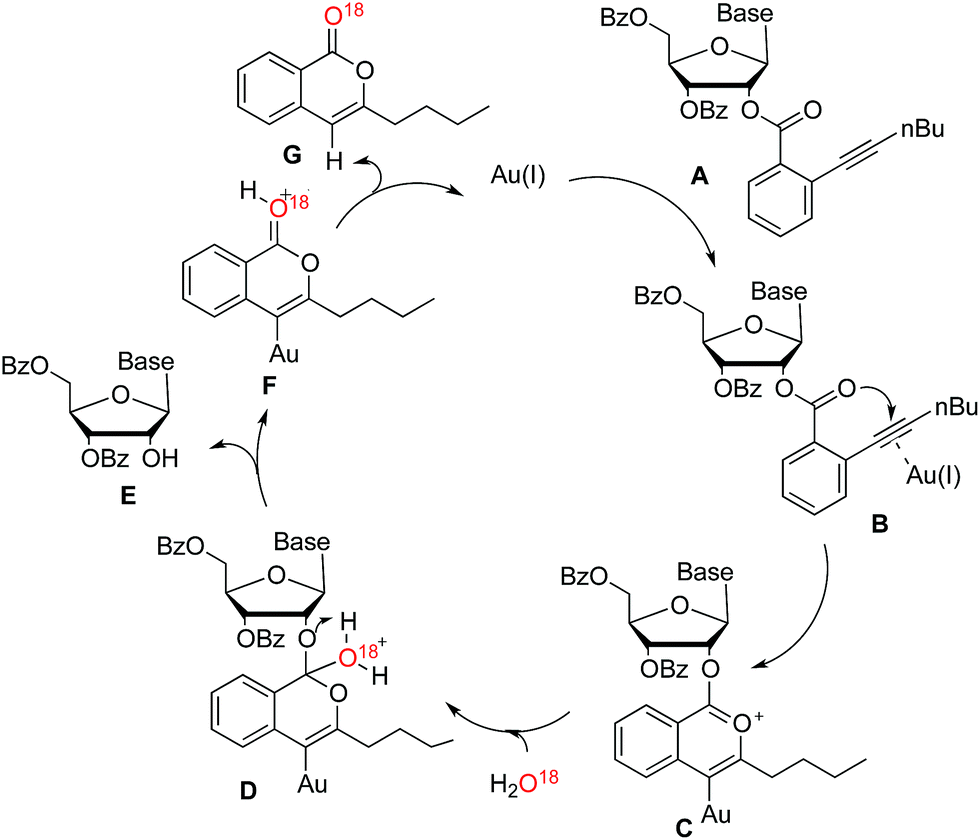 | ||
| Scheme 3 Proposed catalytic cycle for the gold(I)-catalysed removing ortho-alkynyl benzoate. | ||
In order to testify this strategy to prepare 2′-modified nucleosides, disaccharide nucleoside 6 (9-(2-O-β-D-ribofuranosyl-β-ribofuranosyl)-adenine) was synthesized. Disaccharide nucleoside is an important family of natural compounds, which is widely found in t-RNA, antibiotics, and other physiologically active compounds.16,17 Several synthetic approaches were reported. As shown in Scheme 4, glycosylation of nucleoside 4a with a little excess of 1-O-acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose gave the corresponding disaccharide nucleoside in 42% yield. After deprotection of all ester groups, nucleoside 6 (10 grams) was obtained in high purity. All the characterization spectra were identified with the reported data.
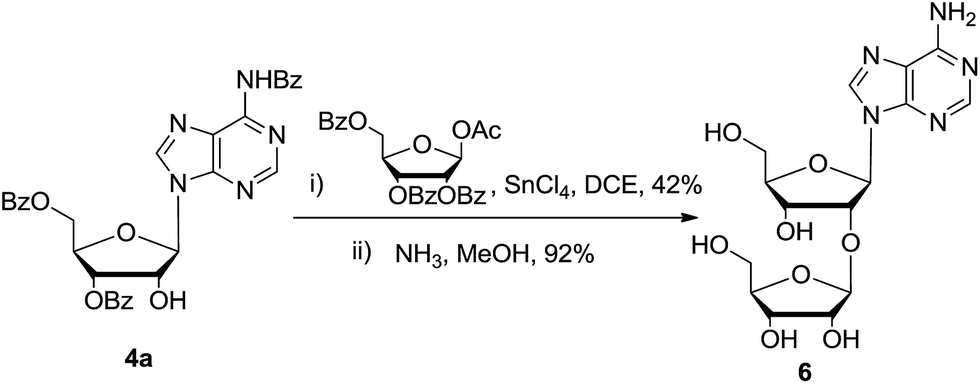 | ||
| Scheme 4 Synthesis of 2′-O-β-D-ribofuranosyladenosine: (i) 1-O-acetyl-2,3,5-tri-O-benzoyl-β-D-ribofuranose, SnCl4, 1,2-dichloroethane, −10 °C, 8 hours, 42%; (ii) NH3, MeOH, 60 °C, 5 hours, 92%. | ||
To further extend the application of this methodology, we also attempted to develop a new approach for synthesis of antitumor drug clofarabine 9.18,19 As presented in Scheme 5, the reaction of nucleoside 4h with trifluoromethanesulfonic anhydride (Tf2O) in pyridine gave corresponding trifluoromethanesulfonic ester of 2′-OH in almost quantitative yield. After fluorination with Et3N·HF, deacylation with ammonia in methanol gave clofarabine (16 grams) in 56% overall yield. HPLC analysis showed the purity is above 99% and coincidence with reference standard from Sigma Aldrich.
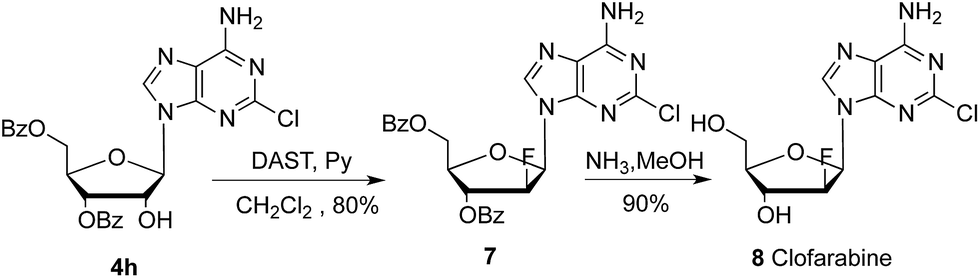 | ||
| Scheme 5 Synthesis of clofarabine 9: (i) Tf2O, pyridine, −20 °C, 5 hours, 95%; (ii) Et3N·3HF, ethyl acetate, 70 °C, 24 hours, 66%; (iii) NH3, MeOH, 60 °C, 5 hours, 90%. | ||
In summary, ortho-alkynyl benzoate was proved to be an efficient neighboring participation group in stereoselective synthesis of nucleosides by using Vorbrüggen glycosylation. It could be removed as isocoumarin 5 using gold(I)-catalysis to afford 2′-OH nucleosides in high yield and selectivity. The powerfulness of present strategy was further demonstrated by the synthesis of 9-(2-O-β-D-ribofuranosyl-β-ribofuranosyl)-adenine 6 and antitumor drug clofarabine 9 in high overall yields. This novel protocol could be used as a general alternative approach for the synthesis of 2′-modified nucleosides. Its application in carbohydrate synthesis is under way.
Acknowledgements
We thank National Science Foundation of China (No. 21462019 and No. 21676131), Bureau of Science & Technology of Jiangxi Province (20143ACB20012) for financial support.Notes and references
- (a) E. De Clercq, Med. Res. Rev., 2010, 31, 118–160 CrossRef PubMed; (b) E. De Clercq, Curr. Opin. Pharmacol., 2010, 10, 507–515 CrossRef CAS PubMed.
- (a) A. H. Cory, V. Samano, M. J. Robins and J. G. Cory, Biochem. Pharmacol., 1994, 47, 365–371 CrossRef CAS PubMed; (b) H. Awano, S. Shuto, M. Baba, T. Kira, S. Shigeta and A. Matsuda, Bioorg. Med. Chem. Lett., 1994, 4, 367–370 CrossRef CAS.
- S. Ganguly, V. V. Vithlani and K. A. Kesharwani, Acta Pharm., 2011, 61, 187–201 CrossRef CAS PubMed.
- J. Kim, J. Yu and V. Alexander, Eur. J. Med. Chem., 2014, 83, 208–225 CrossRef CAS PubMed.
- V. Alexander, J. Song and J. Yu, Arch. Pharmacal Res., 2015, 38, 966–972 CrossRef CAS PubMed.
- N. Goris, A. De Palma, J. F. Toussaint, I. Musch, J. Neyts and K. De Clercq, Antiviral Res., 2007, 73, 161–168 CrossRef CAS PubMed.
- J. L. Clark, J. C. Mason, L. Hollecker, L. J. Stuyver, P. M. Tharnish, T. R. McBrayer, M. J. Otto, P. A. Furman, R. F. Schinazi and K. A. Watanabe, Bioorg. Med. Chem. Lett., 2006, 16, 1712–1715 CrossRef CAS PubMed.
- (a) E. Lawitz, A. Mangia, D. Wyles and M. Rodriguez, et al., N. Engl. J. Med., 2013, 368, 1878–1887 CrossRef CAS PubMed; (b) M. Lagging, R. Wejstal, G. Norkrans, O. Karlstorm and F. Josephson, et al., Infect. Dis., 2016, 48, 251–261 CrossRef CAS PubMed; (c) P. Wang, B. K. Chun, S. Rachakonda, J. Du, N. Khan, J. Shi, W. Stec, D. Cleary, B. S. Ross and M. J. Sofia, J. Org. Chem., 2009, 74, 6819–6824 CrossRef CAS PubMed.
- J. Ye, N. Li, Q. Dai and J. A. Piccirilli, Angew. Chem., Int. Ed., 2007, 46, 3714–3717 CrossRef CAS PubMed.
- S. N. Mikhailov, M. Oivanen, P. Oksman and H. Lonnberg, J. Org. Chem., 1992, 57, 4122–4126 CrossRef CAS.
- M. Tanaka, R. Kozakai, Y. Saito and I. Saito, Bioorg. Med. Chem. Lett., 2011, 21, 7021–7024 CrossRef CAS PubMed.
- (a) Y. Ishido, N. Sakairi, K. Okazaki and N. Nakazaki, J. Chem. Soc., Perkin Trans. 1, 1980, 563–573 RSC; (b) S. Nishino, A. M. Rahman, H. Takamura and Y. Iahido, Tetrahedron, 1985, 41, 5503–5506 CrossRef CAS; (c) I. Nowak, C. T. Jones and M. J. Robins, J. Org. Chem., 2006, 71, 3077–3081 CrossRef CAS PubMed.
- (a) R. Dore and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef PubMed; (b) A. Corma, A. L. Perez and M. J. Sabater, Chem. Rev., 2011, 111, 1657–1712 CrossRef CAS PubMed.
- (a) M. Jia and M. Bandini, ACS Catal., 2015, 5, 1638–1652 CrossRef CAS; (b) Q. Zhang, J. Sun, Y. Zhu, F. Zhang and B. Yu, Angew. Chem., Int. Ed., 2011, 50, 4933–4936 CrossRef CAS PubMed; (c) Y. Zhu and B. Yu, Angew. Chem., Int. Ed., 2011, 50, 8329–8332 CrossRef CAS PubMed; (d) Y. Tang, J. Li, Y. Zhu, Y. Li and B. Yu, J. Am. Chem. Soc., 2013, 135, 18396–18405 CrossRef CAS PubMed; (e) B. Yu, J. Sun and X. Yang, Acc. Chem. Res., 2012, 45, 1227–1236 CrossRef CAS PubMed.
- (a) N. Asao, H. Aikawa, S. Tago and K. Umetsu, Org. Lett., 2007, 9, 4299–4302 CrossRef CAS PubMed; (b) K. Umetsu and N. Asao, Tetrahedron Lett., 2008, 49, 7046–7049 CrossRef CAS.
- S. N. Mikhailov, E. V. Efimtseva, A. A. Rodionov, G. V. Bobkov, I. V. Kulikova and P. Herdewijn, Curr. Protoc. Nucleic Acid Chem., 2007, 27, 1.14.1–1.14.19 Search PubMed.
- E. V. Efimtseva and S. N. Mikhailov, Russ. Chem. Rev., 2004, 73, 401–414 CrossRef CAS.
- P. L. Bonate, L. Arthaud, W. R. Cantrell, K. Stephenson, J. A. Secrist and S. Weitman, Nat. Rev. Drug Discovery, 2006, 5, 855–863 CrossRef CAS PubMed.
- R. Xia, Z. Guo, B. Qin, Z. Ji, M. Xie, G. Qu and H. Guo, Chin. J. Org. Chem., 2014, 34, 1154–1160 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedure, characterization data, 1H and 13C NMR spectra of compounds. See DOI: 10.1039/c6ra27790j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2017 |