
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoinduced structural changes of cationic azo dyes confined in a two dimensional nanospace by two different mechanisms†
Tomohiko Okada a,
Nozomi Nozakib,
Jangwon Seoc,
Ji Eon Kwonc,
Soo Young Parkc,
Hideo Hashizumed,
Takayoshi Sasakid and
Makoto Ogawa*be
a,
Nozomi Nozakib,
Jangwon Seoc,
Ji Eon Kwonc,
Soo Young Parkc,
Hideo Hashizumed,
Takayoshi Sasakid and
Makoto Ogawa*be
aDepartment of Chemistry and Materials Engineering, Shinshu University, 4-17-1 Wakasato, Nagano, Nagano 380-8553, Japan
bDepartment of Earth Sciences, Waseda University, 1-6-1 Nishiwaseda, Shinjuku, Tokyo 169-8050, Japan
cCenter for Supramolecular Optoelectronic Materials, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul 151-744, Korea
dNational Institute for Materials Science (NIMS), 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan
eInstitute of Energy Science and Engineering, Vidyasirimedhi Institute of Science and Technology (VISTEC), 555 Moo 1 Tumbol Payupnai, Amphoe Wangchan, Rayong, 21210, Thailand. E-mail: makoto.ogawa@vistec.ac.th
First published on 24th January 2017
Abstract
Photoresponsive change in the basal spacing of cationic azo dye-layered silicate intercalation compounds was investigated using X-ray diffraction under controlled humidity conditions to investigate the mechanism of the photoresponse triggered by trans–cis photoisomerization. The molecular design of azo dyes was conducted and two cationic azo dyes (phenylazobenzene and phenylazonaphthalene) were used to obtain intercalation compounds with different packing and hydration. Whereas the trans-phenylazobenzene-layered silicate hardly adsorbed water vapor, the trans-phenylazonaphthalene intercalation compound strongly interacted with water. Both azo dyes photoisomerized in the interlayer space reversibly. Under the relative humidity of 95%, the basal spacing of the phenylazobenzene–silicate increased upon UV irradiation, confirming that the change in the basal spacing was caused by the photoinduced hydration. Under the relative humidity of 6.8%, the basal spacing of the phenylazonaphthalene–silicate decreased upon UV irradiation, suggesting that the packing of the phenylazonaphthalene in the interlayer space was changed to be compacted by the photochemical conversion to the cis-form.
Introduction
Stimuli-responsive supramolecular assemblies have been designed for creating smart materials. Reversible trans-to-cis photoisomerization of azobenzene by light irradiation or by thermal treatment changes its dipole moment as well as molecular geometry. This well-known stimuli-responsive phenomenon has been utilized as a trigger for the construction of photoresponsive materials.1 Azobenzene-containing supramolecular systems were synthesized to achieve photoresponse of guest binding.2 Azobenzene monolayer3 and multilayer4 deposited on substrates have been used to control the alignment of liquid crystals and polymers on the surface by irradiation. Controls of plasmonic resonance5a and achromatic polarization5b are examples of applications of the command surfaces. The incorporation of azobenzene units into low-dimensional nanospaces (e.g., cylindrical pore of mesoporous silica6 and 2D interlayer space7–12) has been investigated as supramolecular photoresponsive materials. Reversible changes in the basal spacing (interlayer expansion) of layered inorganic solids have been achieved after the careful design of the nanostructures.13,14 This is a photomechanical change in response to the change of the molecular configuration to the packing of molecular assemblies,13,14 which cause the intercalation of additional molecular species.15 The molecular design of azo dyes13–16 and the molecular dynamics simulation17 indicated the importance of the functional groups attached to azobenzene in inducing the structural change.We have been interested in the photochemistry of organic dyes intercalated in the interlayer space of layered silicates.7–9,11–13,15,16,18 Among available layered silicates, Na–magadiite (the ideal formula of Na2Si14O29)19 possesses following unique and useful properties for organizing cationic azo dyes; (1) the density of the cation exchange sites on the layer surface is ca. 2.2 meq g−1, which is higher than that of smectite group of layered clay minerals; (2) it can conveniently be prepared in a laboratory by hydrothermal synthesis. We have reported the X-ray diffraction (XRD)-detectable change in the basal spacing upon irradiation, for a cationic phenylazobenzene (Scheme 1a: abbreviated as AZ+) intercalated magadiite.13b The observed structural change of the intercalation compound by the photoisomerization has been explained by the changes in the polarity, which accompany the adsorption of atmospheric water, and/or change in the packing of the intercalated AZ+. In order to understand the mechanisms of the photoinduced structural change as well as to apply the phenomena, more systematic investigation is worth conducting. In this study, a cationic phenylazonaphthalene (Scheme 1b: AzNaph+) was synthesized and intercalated in the interlayer space of Na–magadiite in the expectation of causing differences in the packing and the photoinduced hydration.
 | ||
| Scheme 1 Molecular structures of (a) AZ+ and (b) AzNaph+. | ||
Experimental section
Preparation of the cationic azo dyes
AZ+–Br−, p-[2-(2-hydroxyethyldimethylammonioethoxy)phenylazo]benzene bromide, was prepared by the reaction of phenylazophenol with dibromoethane and subsequent reaction with dimethylaminoethanol. The dye was used after recrystallization from ethanol and characterized by 1H NMR and CHN analysis (calculated for C18H24N3O2Br: C, 54.8; H, 6.13; N, 10.7; found C, 53.7; H, 5.97; N, 10.3 mass%) and mass spectrometry.13bAzNaph+–Br−, 1-[(4-trimethylammonioethoxy)phenylazo]naphthalene bromide, was synthesized by the reaction between phenylazonaphthalene and dibromoethane, followed by the quaternization reaction using trimethylamine. The product was used after recrystallization from ethanol and analyzed by 1H and 13C NMR analysis and mass spectrometry (the results are summarized in ESI†).
Intercalation of the cationic azo dyes into Na–magadiite
Na–magadiite was prepared by a hydrothermal reaction as reported previously.19a Intercalation of cationic azo dyes (AzNaph+ and AZ+) into Na–magadiite was conducted by the conventional ion-exchange reactions. Na–magadiite was mixed with an aqueous solution of the cationic azo dye (2.0 mM) at a molar ratio of Na![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :AzNaph+ = 1:1.7 or Na:AZ+ = 1:1.2. The mixture was allowed to react for 24 h at 343 K. After centrifugation, the resulting solid was washed with acetone, followed by being dried under a reduced pressure at room temperature. In order to record UV-vis absorption spectra, a supported film on a quartz substrate was prepared by embedding the mixture of the azo dye-magadiite with a chloroform solution of poly(methyl methacrylate) (PMMA).
:AzNaph+ = 1:1.7 or Na:AZ+ = 1:1.2. The mixture was allowed to react for 24 h at 343 K. After centrifugation, the resulting solid was washed with acetone, followed by being dried under a reduced pressure at room temperature. In order to record UV-vis absorption spectra, a supported film on a quartz substrate was prepared by embedding the mixture of the azo dye-magadiite with a chloroform solution of poly(methyl methacrylate) (PMMA).
Photochemical reactions
The photochemical reactions of the intercalated azo dyes were conducted using UV and visible light irradiation with a Hamamatsu Photonics 150 W high pressure Hg–Xe lamp. For the trans-to-cis reactions of AZ+ and AzNaph+, band-pass filters, HOYA U-340 and HOYA B390 (the transmittance centered at 350 and 405 nm), were used for isolating UV light, respectively. For the cis-to-trans backward reactions, sharp-cut filters, HOYA L42 (cut-off wavelength is 420 nm) and HOYA Y52 (cut-off wavelength is 520 nm) were used to irradiate visible light toward AZ+ and AzNaph+, respectively.Characterization
Adsorption–desorption isotherms of water from vapor phase were obtained using a BELSORP-max adsorption equipment (Microtrack BEL Co. Ltd.). Before the experiment, the AzNaph+–magadiite powder was dried at 333 K. XRD patterns were recorded on a RIGAKU 2100s humidity-controlled equipment using monochromatic Cu Kα radiation, operated at 20 mA and 40 kV. UV-visible absorption spectra were recorded on a Shimadzu UV 3100 PC spectrophotometer. The composition of the azo dye-intercalated magadiite was determined by CHN analysis (Perkin Elmer 2400 II instrument).Results and discussion
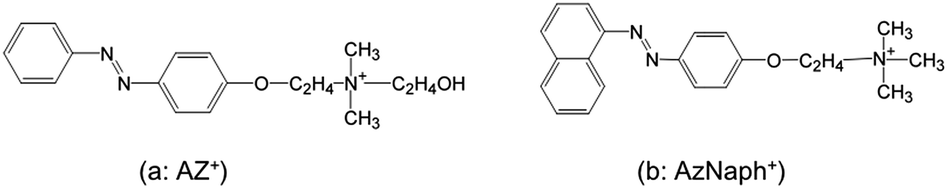
As shown in XRD patterns (Fig. 1a), the basal spacing of Na–magadiite (1.57 nm) increased to 3.01 and 2.74 nm for AzNaph+- and AZ+–magadiites, respectively, where the interlayer expansion (the gallery height) was derived by subtracting the thickness of the silicate layer of magadiite (1.12 nm)20 to be 1.89 and 1.62 nm. Considering the interlayer expansion and the molecular size (1.9 nm for the long axis of each azo cation), interdigitated monolayer or bilayer coverage is possible orientation of the interlayer azo dyes (Fig. 1b). The amount of AzNaph+ in the product was determined by CHN analysis (C, 16.0%; N, 2.2%) to be 0.85 mol/Si14O29, which was smaller than that (1.8 mol/Si14O29 from C, 25.7%; N, 3.6%)13b of AZ+. On the bases of the differences in the interlayer expansion, the adsorbed amount of azo dye, and the molecular size, there seems to be more space (lower packing density) to rotate naphthalene group in AzNaph+ in the interlayer space (calculation in detail has been described in ESI†).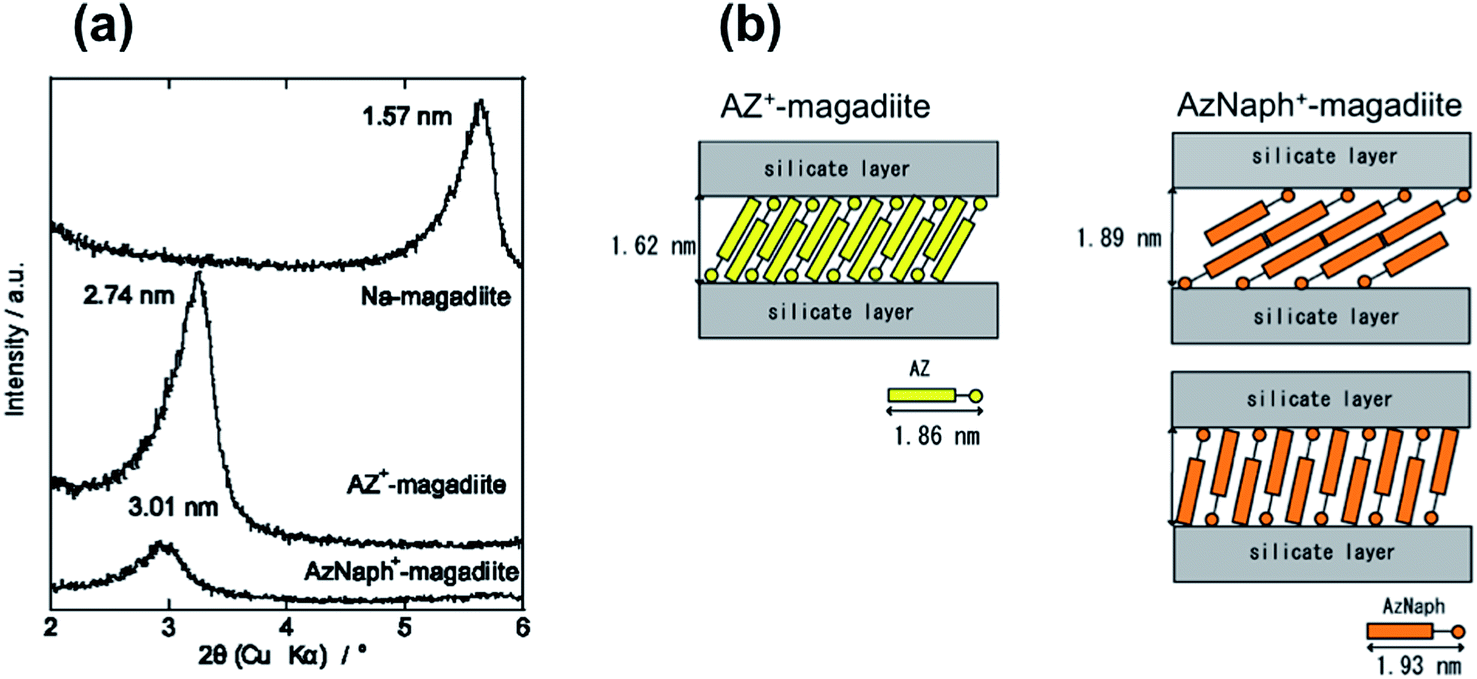 | ||
| Fig. 1 (a) XRD patterns of Na–magadiite, AZ+–magadiite, and AzNaph+–magadiite recorded under ambient atmosphere, (b) schematic drawing of the interlayer structures of AzNaph+- and AZ+–magadiite. | ||
The adsorption/desorption isotherms of water vapor (298 K) on the trans-forms are shown in Fig. 2. Whereas AZ+–magadiite hardly adsorbed water vapor, AzNaph+–magadiite was shown to have a larger water adsorption capacity. The basal spacing of AzNaph+–magadiite was recorded using a humidity-controlled XRD equipment. The value was 3.08 nm under the RH of 90%, which is larger than that (2.89 nm) observed for the sample under the RH of 10%, indicating that the interlayer space of AzNaph+–magadiite is expandable by the water adsorption (the XRD patterns are shown in ESI, Fig. S1†). The basal spacing of trans-AZ+–magadiite (2.74 nm) was not sensitive for the humidity (Fig. S1†). In AzNaph+–magadiite, residual Na ions (ca. 1.1 mol/Si14O29) and interlayer silanol groups are possible adsorption sites for water vapor. In addition, packing of the dyes in the interlayer space (loosely packed AzNaph+ and densely packed AZ+) is thought to be concerned for the adsorptive characteristics of the intercalation compounds.
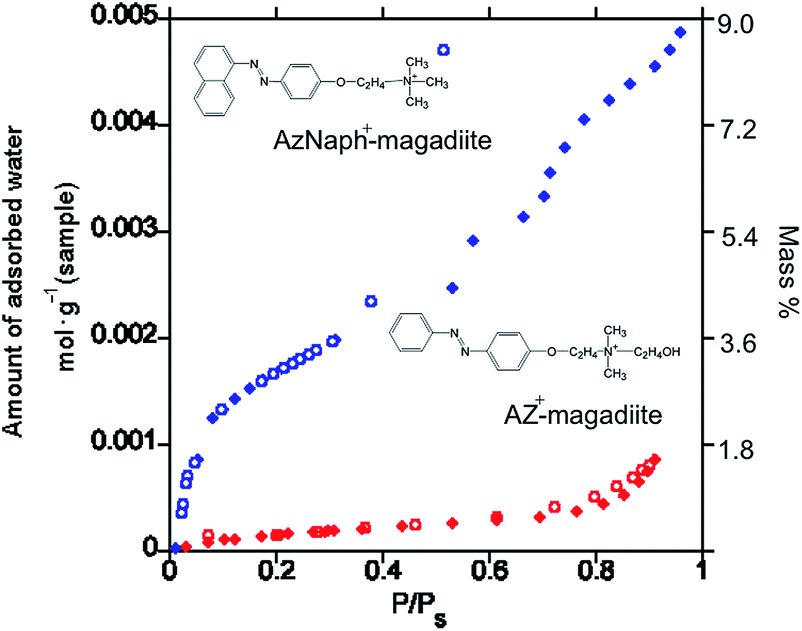 | ||
| Fig. 2 Adsorption (filled symbols)/desorption (open symbols) isotherms of water vapor (298 K) on AzNaph+- (red) and AZ+–magadiite (blue) intercalation compounds. | ||
UV irradiation led the change in the absorption spectrum of the AzNaph+–magadiite as shown in Fig. 3; absorbance of the band ascribed to the trans-isomer (at around 390 nm) decreased after the UV irradiation, indicating the trans-to-cis photoisomerization of the interlayer AzNaph+. The absorption spectrum was regenerated upon visible light irradiation, suggesting that the photochemically formed cis-AzNaph+ photoisomerized to trans-form. The absorbance of the cis-isomer (at around 500 nm) is quite small due to the n–π* forbidden transition. Based on the change in the absorbance of the trans-isomer, the fraction of the photochemically formed cis-isomer was estimated to be 14% at the photostationary state.21 Similar spectral change in responses to UV and visible light irradiations was observed in the AZ+–magadiite system, and the fraction of the cis-isomer was up-to 45%.13b
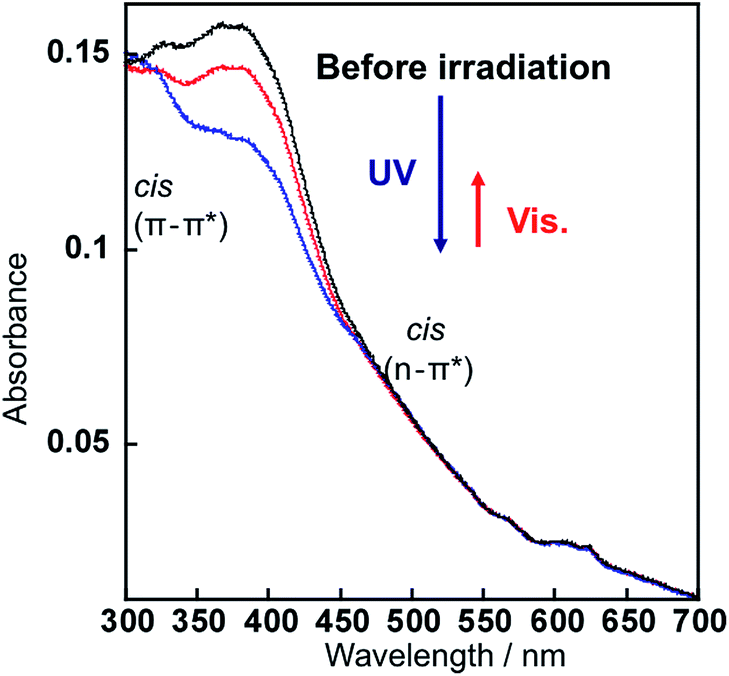 | ||
| Fig. 3 The absorption spectra of AzNaph+–magadiite embedded in PMMA: before (black), after UV irradiation (blue) for 10 min, and subsequent visible light irradiation (red) for 10 min. | ||
Photoinduced change in the basal spacing of AZ+–magadiite was observed when photoirradiation was conducted under a high humidity condition (RH = 95%) (Fig. 4); the basal spacing increased from 2.73 to 2.76 nm upon UV irradiation. It was explained that, due to the polar nature of cis-AZ+, the adsorption of atmospheric water to the photochemically formed cis-AZ+–magadiite occurred to cause the expansion of the interlayer space. In contrast, the basal spacing of AzNaph+–magadiite decreased from 2.89 to 2.79 nm when UV irradiation was conducted under a dry N2 atmosphere (RH = 6.8%) (Fig. 5). The cis-isomer co-existed with the trans-isomer in the same interlayer space as suggested by the single phase-XRD patterns before and during the irradiation. Geometrical change from trans to cis-AzNaph+ triggered the rearrangement of the overall guest packing in the interlayer space, which was thought to affect the decreased basal spacing upon UV irradiation. It is clear from a molecular simulation (Scheme 2) that a larger exclusion volume of the substituent bonded to azo group is required for AzNaph+ to photoisomerize. Once the basal spacing decreased, the value did not return to that before UV irradiation probably due to the slow recovery of a portion of the photochemically formed cis-form. Careful examination of the irradiation conditions will be required to understand the structural changes.
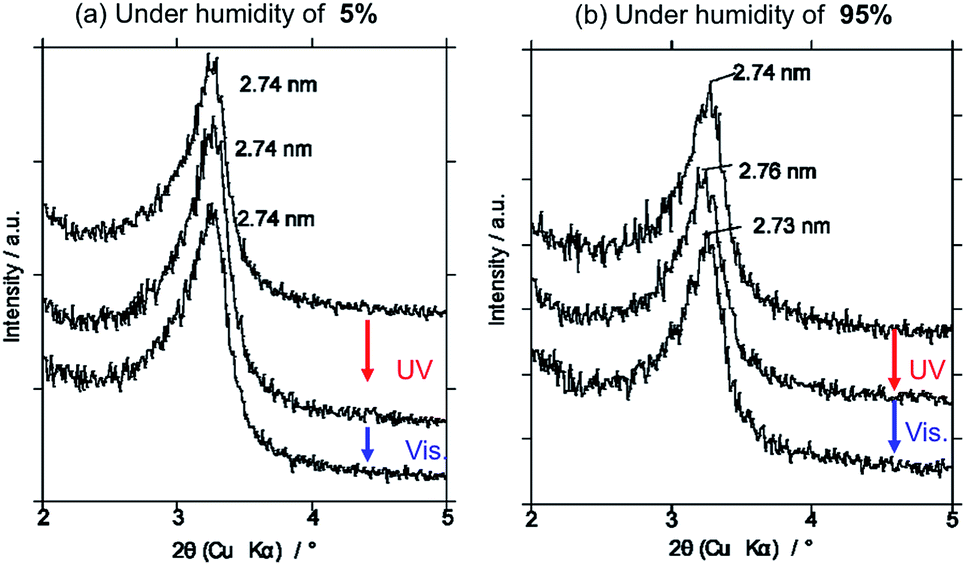 | ||
| Fig. 4 Change in the XRD pattern of trans-AZ+–magadiite upon UV and visible light irradiations at different humidity of (a) RH = 5 and (b) RH = 95 for 10 min each. | ||
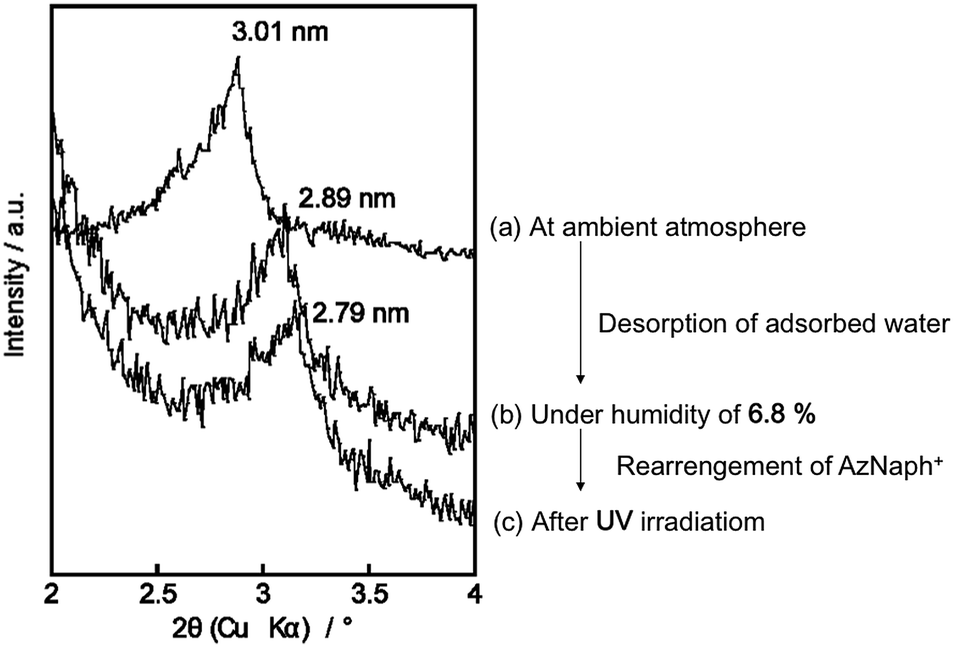 | ||
| Fig. 5 XRD patterns of AzNaph+–magadiite recorded under (a) ambient atmosphere, (b) RH of 6.8% at ground state and (c) UV irradiation for 10 min (RH = 6.8%). | ||
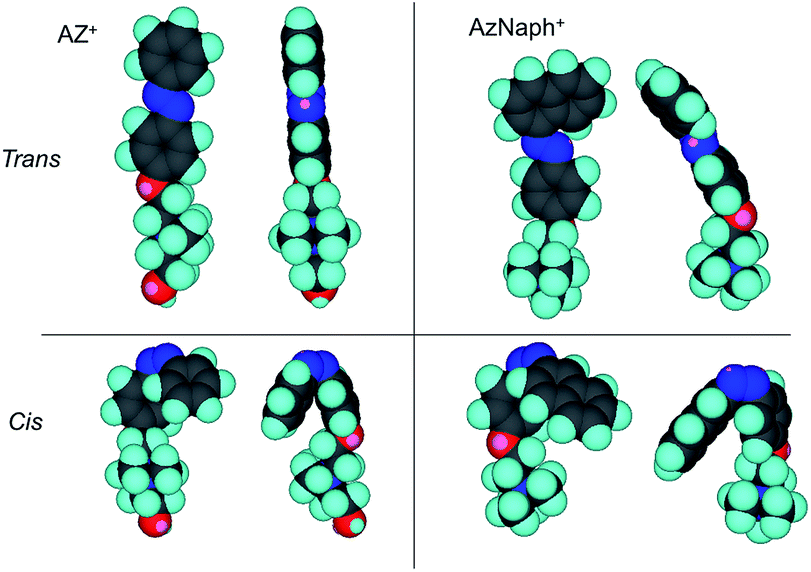 | ||
| Scheme 2 Molecular simulation structures of trans- and cis-forms for AZ+- and AzNaph+. | ||
We recorded the XRD patterns and the absorption spectra after reaching photo-stationary state (10 min). Time for structural transition (basal spacing change) was strongly affected whether accompanying intercalation of a molecule by light irradiation or not. When the photoinduced intercalation was not involved, the changes in absorption spectrum and basal spacing occurred simultaneously.14c On the other hand, a longer time than the spectral change was required to the transition because the intercalation of a molecule (phenol) was a rate determining step to reach the steady state.15b In the present AzNaph+–magadiite system, simultaneous transition with the trans-to-cis isomerization is plausible because of the effect of the water intercalation.
Conclusions
The structural changes of the cationic azo dye-magadiite intercalation compounds upon irradiation have been investigated under controlled humidity conditions. Two azo dyes (azobenzene and azonaphthalene) gave photoinduced structural changes from different mechanisms. Hydration of the photochemically formed cis-form of the cationic azobenzene–magadiite was the reason of the increase in the basal spacing by the UV irradiation. The change in the basal spacing of the cationic azonaphthalene–magadiite intercalation compound was observed by the irradiation under the dry condition (decrease by UV irradiation), which was thought to cause by the change in the packing of the azonaphthalene in the interlayer space of magadiite. The observed photoinduced structural changes based on different mechanisms reminded the importance of the mechanisms on the applications of the stimuli responsive supramolecular systems under different operating conditions.Notes and references
- Photochromism: Molecules and Systems, ed. H. Dürr and H. Bouas-Laurent, Elsevier, Amsterdam, 2003 Search PubMed.
- (a) G. S. Kumar and D. C. Neckers, Chem. Rev., 1989, 89, 1915 CrossRef CAS; (b) J. Anzai and T. Osa, Tetrahedron, 1994, 50, 4039 CrossRef CAS; (c) I. Wilier, Acc. Chem. Res., 1997, 30, 347 CrossRef; (d) K. Hoffmann, F. Marlow and J. Caro, Adv. Mater., 1997, 9, 567 CrossRef CAS; (e) J. Park, D. Yuan, K. T. Pham, J.-R. Li, A. Yakovenko and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 99 CrossRef CAS PubMed; (f) N. Yanai, T. Uemura, M. Inoue, R. Matsuda, T. Fukushima, M. Tsujimoto, S. Isoda and S. Kitagawa, J. Am. Chem. Soc., 2012, 134, 4501 CrossRef CAS PubMed; (g) A. Harada, Y. Takashima and M. Nakahata, Acc. Chem. Res., 2014, 47, 2128 CrossRef CAS PubMed; (h) New Frontiers in Photochromism, ed. M. Irie, Y. Yokoyama and T. Seki, Springer, Japan, 2013 Search PubMed; (i) T. H. Tran-Thi, R. Dagnelie, S. Crunaire and L. Nicole, Chem. Soc. Rev., 2011, 40, 621 RSC; (j) B. Lebeau and P. Innocenzi, Chem. Soc. Rev., 2011, 40, 886 RSC; (k) D.-H. Qu, Q.-C. Wang, Q.-W. Zhang, X. Ma and H. Tian, Chem. Rev., 2015, 115, 7543 CrossRef CAS PubMed.
- (a) K. Ichimura, Y. Suzuki, T. Seki, A. Hosoki and K. Aoki, Langmuir, 1988, 4, 1214 CrossRef CAS; (b) K. Ichimura, Chem. Rev., 2000, 100, 1847 CrossRef CAS PubMed.
- (a) T. Seki, S. Nagano and M. Hara, Polymer, 2013, 54, 6053 CrossRef CAS; (b) T. Nakai, D. Tanaka, M. Hara, S. Nagano and T. Seki, Langmuir, 2016, 32, 909 CrossRef CAS PubMed.
- (a) L. D. Sio, G. Kjein, S. Serak, N. Tabiryan, A. Cunningham, C. M. Tone, F. Ciuchi, T. Bürgi, C. Umeton and T. Bunning, J. Mater. Chem. C, 2013, 1, 7483 RSC; (b) C. Oh and M. J. Escuti, Opt. Lett., 2008, 33, 2287 CrossRef PubMed.
- (a) M. Ogawa, J. Photochem. Photobiol., C, 2002, 3, 129 CrossRef CAS; (b) M. Sohmiya, K. Saito and M. Ogawa, Sci. Technol. Adv. Mater., 2015, 16, 054201 CrossRef PubMed; (c) N. Mizoshita, T. Tani and S. Inagaki, Chem. Soc. Rev., 2011, 40, 789 RSC; (d) A. Stein, B. J. Melde and R. C. Schroden, Adv. Mater., 2000, 12, 1403 CrossRef CAS.
- T. Okada, M. Sohmiya and M. Ogawa, Photochromic Intercalation Compounds in Photofunctional Layered Materials, ed. D. Yan and M. Wei, Springer Int. Pub., Switzerland, 2015, pp. 177–211 Search PubMed.
- (a) M. Ogawa and K. Kuroda, Chem. Rev., 1995, 95, 399 CrossRef CAS; (b) Comprehensive Supramolecular Chemistry, ed. G. Alberti and T. Bein, Pergamon, Oxford, 1996, vol. 7 Search PubMed; (c) K. Takagi and T. Shichi, Photophysics and Photochemistry in Clay Materials in Solid State and Surface Photochemistry, ed. V. Ramamurthy and K. S. Schanze, Marcel Dekker, New York, 2000, pp. 31–110 Search PubMed; (d) S. Takagi, T. Shimada, Y. Ishida, T. Fujimura, D. Masui, H. Tachibana, M. Eguchi and H. Inoue, Langmuir, 2013, 29, 2108 CrossRef CAS PubMed.
- (a) M. Ogawa, H. Kimura, K. Kuroda and C. Kato, Clay Sci., 1996, 10, 57 CAS; (b) M. Ogawa, M. Hama and K. Kuroda, Clay Miner., 1999, 34, 213 CrossRef CAS; (c) M. Sasaki and T. Fukuhara, Photochem. Photobiol., 1997, 66, 716 CrossRef CAS PubMed; (d) H. Kandori, T. Ichioka and M. Sasaki, Chem. Phys. Lett., 2002, 354, 251 CrossRef CAS.
- W. Fujita and K. Awaga, J. Am. Chem. Soc., 1997, 119, 4563 CrossRef CAS.
- (a) T. Yamamoto, Y. Umemura, O. Sato and Y. Einaga, Chem. Mater., 2004, 16, 1195 CrossRef CAS; (b) T. Yamamoto, N. Saso, Y. Umemura and Y. Einaga, J. Am. Chem. Soc., 2009, 131, 13196 CrossRef CAS PubMed.
- T. Umemoto, Y. Ohtani, T. Tsukamoto, T. Shimada and S. Takagi, Chem. Commun., 2014, 50, 314 RSC.
- (a) M. Ogawa, K. Fujii, K. Kuroda and C. Kato, Mater. Res. Soc. Symp. Proc., 1991, 233, 89 CrossRef CAS; (b) M. Ogawa, T. Ishii, N. Miyamoto and K. Kuroda, Adv. Mater., 2001, 13, 1107 CrossRef CAS; (c) T. Fujita, N. Iyi and Z. Klapyta, Mater. Res. Bull., 1998, 33, 1693 CrossRef CAS; (d) N. Iyi, T. Fujita, C. V. Yelamaggad and F. L. Arbeloa, Appl. Clay Sci., 2001, 19, 47 CrossRef CAS; (e) S. Guo, K. Matsukawa, T. Miyata, T. Okubo, K. Kuroda and A. Shimojima, J. Am. Chem. Soc., 2015, 137, 15434 CrossRef CAS PubMed.
- (a) Z. W. Tong, S. Takagi, T. Shimada and H. Tachibana and Inoue, J. Am. Chem. Soc., 2006, 128, 684 CrossRef CAS PubMed; (b) Z. W. Tong, S. Sasamoto, T. Shimada, S. Takagi, H. Tachibana, X. B. Zhang, D. A. Tryk and H. Inoue, J. Mater. Chem., 2008, 18, 4641 RSC; (c) Y. Nabetani, H. Takamura, Y. Hayasaka, S. Sasamoto, Y. Tanamura, T. Shimada, D. Masui, S. Takagi, H. Tachibana, Z. Tong and H. Inoue, Nanoscale, 2013, 5, 3182 RSC; (d) Y. Nabetani, H. Takamura, Y. Hayasaka, T. Shimada, S. Takagi, H. Tachibana, D. Masui, Z. Tong and H. Inoue, J. Am. Chem. Soc., 2011, 133, 17130 CrossRef CAS PubMed; (e) J. Han, D. Yan, W. Shi, J. Ma, H. Yan, M. Wei, D. G. Evans and X. Duan, J. Phys. Chem. B, 2010, 114, 5678 CrossRef CAS PubMed.
- (a) T. Okada, Y. Watanabe and M. Ogawa, Chem. Commun., 2004, 320 RSC; (b) T. Okada, Y. Watanabe and M. Ogawa, J. Mater. Chem., 2005, 15, 987 RSC; (c) T. Okada, H. Sakai and M. Ogawa, Appl. Clay Sci., 2008, 40, 187 CrossRef CAS.
- (a) M. Ogawa, Chem. Mater., 1996, 8, 1347 CrossRef CAS; (b) M. Ogawa and A. Ishikawa, J. Mater. Chem., 1998, 8, 463 RSC; (c) M. Ogawa, J. Mater. Chem., 2002, 12, 3304 RSC; (d) J. Bujdak, N. Iyi and T. Fujita, J. Colloid Interface Sci., 2003, 262, 282 CrossRef CAS PubMed; (e) S. Guo, W. Chaikittisilp, T. Okubo and A. Shimojima, RSC Adv., 2014, 4, 25319 RSC.
- (a) H. Heinz, R. A. Vaia, H. Koerner and B. L. Farmer, Chem. Mater., 2008, 20, 6444 CrossRef CAS; (b) H. Heinz, Clay Miner., 2012, 47, 205 CrossRef CAS.
- M. Ogawa, M. Yamamoto and K. Kuroda, Clay Miner., 2001, 36, 263 CrossRef CAS.
- (a) K. Kosuge, A. Yamazaki, A. Tsunashima and R. Otsuka, J. Ceram. Soc. Jpn., 1992, 100, 326 CrossRef CAS; (b) G. Lagaly, Adv. Colloid Interface Sci., 1979, 11, 105 CrossRef CAS; (c) G. Lagaly, K. Beneke and A. Weiss, Am. Mineral., 1975, 60, 642 CAS; (d) G. W. Brindley, Am. Mineral., 1969, 54, 1583 CAS; (e) H. P. Eugster, Science, 1967, 157, 1177 CAS.
- J. M. Rojo, E. Ruiz-Hitzky and J. Sanz, Inorg. Chem., 1988, 27, 2785 CrossRef CAS.
- The absorption spectra of the film are different from that of solution, because of the light scattering by small particles of the azo dye-magadiite. In order to subtract the scattering effects, the baseline was normalized from the original spectrum before measuring the absorbance of the trans-isomer.
Footnote |
| † Electronic supplementary information (ESI) available: 1H, 13C NMR, and SPI-MS of AzNaph+; calculation of volume occupying azo dyes in the interlayer space and XRD patterns of trans-AZ+- and AzNaph+–magadiites recorded under different humidity (Fig. S1). See DOI: 10.1039/c6ra27749g |
| This journal is © The Royal Society of Chemistry 2017 |