
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn ultrafiltration membrane with enhanced photocatalytic performance from grafted N–TiO2/graphene oxide
Wei Chenab,
Ting Yeb,
Hang Xu*ab,
Taoyuan Chenb,
Nannan Gengb and
Xiaohong Gaob
aKey Laboratory of Integrated Regulation and Resource Development on Shallow Lakes of Ministry of Education, College of Environment, Hohai University, Nanjing 210098, China. E-mail: xuhang810826@163.com; Tel: +86 13770848137
bCollege of Environment, Hohai University, Nanjing 210098, China
First published on 3rd February 2017
Abstract
An enhanced photocatalytic ultrafiltration membrane was prepared by grafting with N–TiO2/graphene oxide. After N–TiO2/graphene oxide particles and a polysulfone membrane had been prepared, the N–TiO2/graphene oxide was distributed in deionized water, poured onto the membrane surface and grafted onto the membrane surface by using a pump filter. Scanning electron microscopy and atomic force microscopy were used to investigate the surface and morphological structure of the prepared membranes. Fourier transform infrared spectroscopy and X-ray photoelectron spectroscopy were used to analyze the elemental compositions and chemical bonds of the membranes. The hydrophilicity of the membrane surface was investigated by a sessile-drop method. The membrane water flux was tested in an ultrafiltration cup system. Methylene blue removal under darkness, ultraviolet light and sunlight were used to characterize the photocatalytic ability. The photocatalytic membrane exhibited an enhanced photocatalytic performance, especially in sunlight rather than in ultraviolet light. The synergistic effect of photocatalysis and filtration was tested; the photocatalytic membrane exhibited a better methylene-blue removal ability than a pure membrane as the methylene blue concentration in filtered water was lower. The recyclability of the photocatalytic membrane had a great improvement compared with the powder photocatalyst.
1. Introduction
In photocatalytic oxidation technology, which occurs under the effect of light, a substance absorbs light energy and is stimulated to cause molecular excitation. This results in hydroxyl free-radical generation with strong oxidizing properties on the photocatalyst surface and organic pollutant degradation.1 Titanium dioxide (TiO2) is a semiconductor and is termed a green environmental protection material in the twenty-first century because of its low cost, non-toxicity, excellent chemical stability, corrosion resistance and high photocatalytic activity.2–4 However, certain disadvantages limit its practical application. The energy gap of TiO2 between the valence and conduction bands is ∼3.2 eV, which means that it can only be triggered by ultraviolet (UV) light (λ ≤ 387.5 nm),5,6 which implies a utilization of only ∼4% of sunlight. Therefore, titanium-dioxide stimulation requires large amounts of energy that cannot be provided by the sun. Photocatalysis is most dependent on photogenerated hole oxidation and photogenerated electron reduction, but the photogenerated electron hole recombines easily,7,8 so the quantum efficiency of TiO2 is unsatisfactory. The TiO2 photocatalytic ability and its particle size are inversely proportional. Although the preparation of small TiO2 is advantageous, such a small particle size is difficult to recycle,4,9 TiO2 use in practice would be limited if there were no way to recycle the TiO2. To solve these problems, TiO2 modification is being researched worldwide.Current commonly used TiO2 modification methods include heavy-metal modification,10 metal modification,11 non-metal modification,12 dye-sensitized modification,13 semiconductor-compound modification14 and polyoxometalate modification.15 Nitrogen-doping of TiO2 is a non-metal modification that replaces oxygen vacancies with nitrogen12,16 or introduces nitrogen to TiO2 interstitially,3 is efficient in reducing the energy gap between valence and conduction bands and is considered a promising photocatalyst to increase photocatalytic efficiency. Graphene comprises a single sheet of sp2-hybridized carbon atoms in a hexagonal lattice and has received increased attention in various fields because of its high surface area and attractive electronic, thermal, mechanical and optical properties.1,11,16 Graphene oxide (GO) is a group of oxygen-containing functional groups (hydroxyl groups, carboxyl groups, epoxy groups and etc.) that is based on a graphene structure.17 The effect of GO on photocatalytic activity enhancement has been researched, in which GO acts as an electron acceptor1,18 to hinder photogenerated electron–hole pair recombination. GO behaves as an impurity11,19 to form Ti–O–C20 bonds, and therefore expands light absorption to sunlight.21
To solve difficulties in photocatalyst recycle, the embodiment of photocatalysts in some support materials (such as glass, ceramic, activated carbon, magnetic materials and membranes)4,9,22–25 has been researched. Photocatalytic membranes are considered a good support option. Photocatalyst immobilization on membranes prevents loss and improves its utilization.26 Membranes modified by photocatalysts may affect various membrane properties such as water permeability,27 hydrophilicity,28 contaminant rejection29 and fouling resistance.30 GO addition to photocatalytic membranes is advantageous; the hydroxyl, carbonyl and carboxylic groups of the GO make the membrane more hydrophilic and its mechanical properties may increase the membrane strength.31
Photocatalysts can be immobilized in the membrane by nesting them inside the membrane or grafting them on the surface.25,32 Nesting is achieved by photocatalyst addition into the casting membrane solution. During membrane formation, photocatalyst is distributed in the membrane. This type of photocatalytic membrane improves membrane properties (water permeability, hydrophilicity, fouling resistance). However, it is difficult to stimulate internal photocatalysts, and such photocatalytic membranes exhibit a weak photocatalytic performance.33 Photocatalysts are grafted on the membrane surface by physical or chemical binding. Such photocatalytic membranes exhibit excellent photocatalytic performance. It is difficult to deposit organics on the membrane surface because of the photocatalytic performance, and this results in a lower possibility of membrane fouling.25
Two types of study exist on this topic. N–TiO2/graphene oxide and modified TiO2 has been prepared as a powder and exhibits a superior photocatalytic performance,5,7 however, hardly can these photocatalysts be recycled. In other studies, the membrane surface has been modified with TiO2/graphene oxide,24,25,34 but its photocatalytic performance was better in UV light. To our best knowledge, the use of N–TiO2/graphene oxide to produce photocatalytic membranes has not been reported previously. In this study, a polysulfone-based ultrafiltration membrane was prepared by grafting N–TiO2/graphene oxide onto the membrane (NTG-M) and the membrane was characterized.
2. Materials and methods
2.1 Nanoparticles and chemicals
Tetrabutyl titanate (analytical reagent, AR, purity ≥ 98.5%), anhydrous ethanol (AR, purity ≥ 99.7%), urea (AR, purity ≥ 99.0%), glacial acetic acid (AR, purity ≥ 99.5%), hydrochloric acid (HCl, 6 M), polysulfone (AR, purity ≥ 99%, Mr = 1327.58), N-methylpyrrolidone (NMP, AR, purity ≥ 99%), polyethylene glycol (PEG 600, AR), polyvinylpyrrolidone (PVP, guarantee reagent, GR) and methylene blue (MB, purity ≥ 98.5%) were from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). Graphene oxide (GO, powder, diameter = 0.5–5 μm, thickness = 0.6–1 nm, purity ≥ 99.5%) was from Hengqiu Graphene Technology Co. Ltd. (SuZhou, China). Deionized water (DI) was made using a Milli-Q integral system.2.2 N–TiO2 (NT) preparation
NT nanoparticles were prepared by using the sol–gel approach. Tetrabutyl titanate (10.0 mL) was added slowly into anhydrous ethanol (40.0 mL) to form solution A. Urea was dissolved in a solution of anhydrous ethanol (10.0 mL), DI (4.0 mL) and glacial acetic acid (2.0 mL) at pH 2.0 (adjusted using HCl) with stirring to form solution B. Solution B was added dropwise into solution A with vigorous stirring. The solution was stirred for a further 0.5 h before allowing it to settle for ∼3 h to form a gel. The solution was dried at 100 °C for ∼24 h to form yellow crystals. The solids were ground to a white powder and calcined at 500 °C for 2 h, and NT was formed. The TiO2 preparation was the same as for the NT but without urea addition.2.3 N–TiO2/GO (NTG) preparation
NTG was prepared by a hydrothermal method. GO (0.025 g) was dispersed in 80 mL of DI with ultrasonic treatment for 2 h, and then TiO2 (0.475 g) was added into the suspension for an additional 1 h of ultrasonic treatment. Soon afterwards, the suspension was moved to a 100 mL Teflon-lined stainless autoclave with the pH being adjusted to 3–4 (using HCl). After the reactor had been kept at 180 °C for 15 h, the mixture was cooled to room temperature naturally. NTG was obtained by freeze drying. TiO2/GO (TG) was prepared by the same process except for the difference in starting materials where TiO2 was used to prepare the TG and NT was used for the NTG.2.4 Polysulfone membrane preparation
Polysulfone (18.0 g) was dried and added into NMP (80.0 g) solution with continuous stirring for 5 h at 50 °C. PEG (1.6 g) and PVP (0.4 g) were added to the solution with further stirring for 0.5 h. The solution was maintained in a drying atmosphere for ∼24 h to release bubbles. The solution was poured on a spotless glass plate, and using a 100 μm casting knife and with a constant casting rate, a cast film was formed. The cast film was immediately transferred into a non-solvent coagulation bath (DI at 25 °C). The membrane were washed with DI to remove residual solvent and preserved in DI for at least 24 h before it was used.2.5 NTG-M preparation
NTG (0.05 g) was dispersed in 100 mL DI by ultrasonic treatment to form 0.5 g L−1 NTG solution. NTG solution (20 mL) was poured onto the membrane surface using a pump filter. N–TiO2/graphene oxide particles were grafted on the membrane surface while the DI was filtered. The NTG-M was dried at 50 °C, washed with DI and preserved in DI at least 24 h before use. Membranes grafted using TiO2 (T-M), NT (NT-M) and TG (TG-M) were prepared by the same process except for differences in starting materials.2.6 Characterization
The surface and morphology of the prepared membranes were studied by scanning electron microscopy (SEM, Zeiss Ultra 55, Oberkochen, Germany) and atomic force microscopy (AFM, Multimode 8, Bruker, Santa Barbara, CA, USA). The elemental compositions and chemical bonds of the membranes were analyzed by using Fourier transform infrared spectroscopy (FT-IR, Nicolet 6700, Thermo Scientific, Waltham, MA, USA) and X-ray photoelectron spectroscopy (XPS, K-Alpha, Thermo Scientific, West Palm Beach, FL, USA). The membrane surface hydrophilicity was studied by the sessile-drop method (G10, Kruss, Hamburg, Germany).The membrane water flux was tested in an ultrafiltration cup system as illustrated in Fig. 1(a). The system consisted of a nitrogen gas cylinder to provide pressure, a feed tank to avoid backflow of liquid, an ultrafiltration cup as the core part of the system and an electronic balance to measure filtered water. The synergistic effect of photocatalysis and filtration of the NTG-M was measured by the removal of MB in the equipment shown in Fig. 1(b), with attachments that were similar to the ultrafiltration cup system but differed at the core. The core contained a light source, a sealed quartz glass container with a water inlet and a sampling mouth, and a collet to support the membrane.
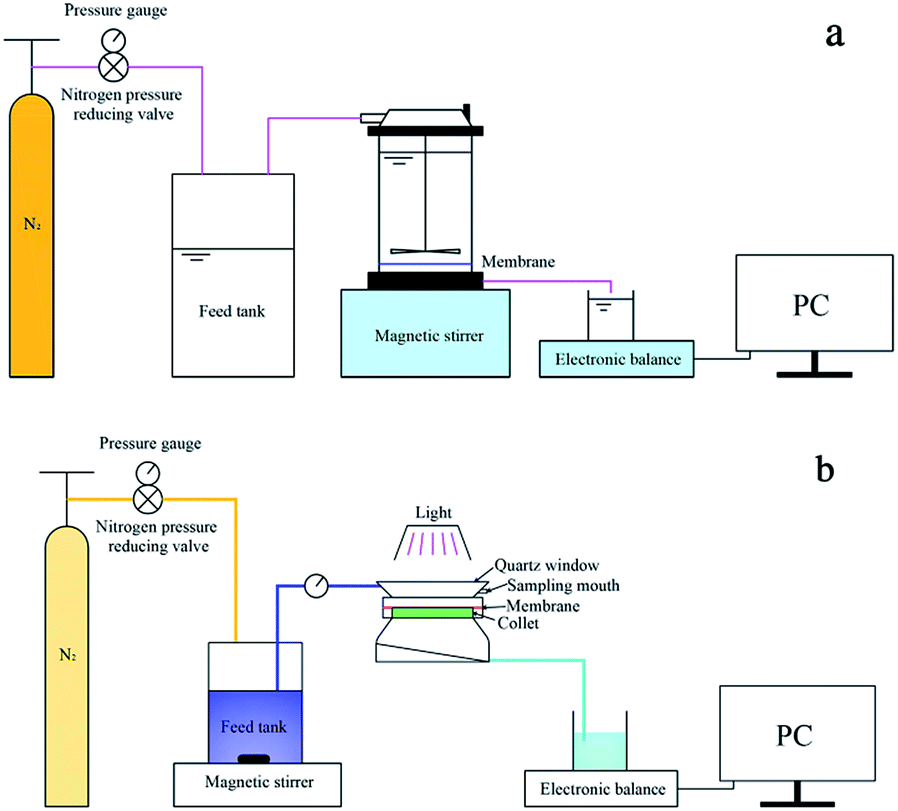 | ||
| Fig. 1 Schematic diagram of (a) ultrafiltration cup system and (b) equipment to measure the removal of MB. | ||
3. Results and discussion
3.1 Membrane characterization
SEM (Fig. 2) was used to assess the membrane surface modification of NTG-M and other prepared membranes (M, T-M, NT-M, TG-M). Fig. 2(a) shows a pure membrane with nothing grafted on its surface. The top surface of T-M and NT-M are shown in Fig. 2(b) and (c), TiO2 and NT are distributed evenly on the membrane surface but some TiO2 (NT) recombines. The morphologies of TG-M and NTG-M are shown in Fig. 2(d) and (e), the arrangement of the TG (NTG) platelets leads a good TiO2 (NT) distribution on the GO sheets, which increases the TiO2 (NT) surface area and the photocatalytic performance of the corresponding photocatalytic membranes. Fig. 2(f) shows the SEM image of NTG, the mean particle size of NT is about 30–50 nm. The weigh difference between membranes before and after grafting NTG is ∼9 mg, which means the loading amount of NTG in NTG-M is ∼3 g m−2. | ||
| Fig. 2 Top surface SEM images of (a) M, (b) T-M, (c) NT-M, (d) TG-M, (e) NTG-M and (f) SEM image of NTG. | ||
Fig. 3 shows the three-dimensional surface AFM images of M, T-M, NT-M, TG-M and NTG-M at a scan size of 5 μm × 5 μm. The brightest regions represent the highest peaks of the membrane surface, the dark areas on the membrane are the pores or lowest valley. The surface-roughness parameters (mean roughness Sa, root mean square of Z data Sq, height difference between the highest peak and the lowest valley Sy) of bare and grafted membranes are listed in Table 1. These surface-roughness parameters are connected with the ability to absorb and desorb pollutant on the membrane surface. Because photocatalysis takes place in the surface of photocatalyst, photocatalyst with higher surface area is expected to have a higher photocatalytic efficiency. NTG-M (TG-M) has a higher surface roughness than M, T-M and NT-M, we predict that NTG-M (TG-M) can absorb more pollutant and then speed up photocatalytic rate.
 | ||
| Fig. 3 Three-dimensional AFM images with a scan area of 5 μm × 5 μm for (a) M, (b) T-M, (c) NT-M, (d) TG-M and (e) NTG-M. | ||
| Membrane | Roughness parameters | ||
|---|---|---|---|
| Sa (nm) | Sq (nm) | Sy (nm) | |
| M | 9.450 | 11.942 | 42.200 + 58.529 = 100.729 |
| T-M | 13.137 | 17.167 | 73.397 + 62.532 = 135.929 |
| NT-M | 16.505 | 20.081 | 53.325 + 70.392 = 123.717 |
| TG-M | 16.959 | 21.332 | 111.675 + 164.387 = 276.062 |
| NTG-M | 21.361 | 27.270 | 165.907 + 132.087 = 297.994 |
Fig. 4 shows the surface FT-IR spectra for M, T-M, NT-M, TG-M and NTG-M. Peaks at 1732 cm−1 and 3000–3600 cm−1 can be assigned to the carbonyl groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and hydroxyl groups (OH) 16 that appear in the spectrum of TG-M and NTG-M. Peaks at 1401 cm−1, 1226 cm−1 and 1050 cm−1 can be attributed to the skeletal vibration of unoxidized graphitic, epoxy groups C–O and the hydroxyl group stretching mode C–O,35,36 which are stronger in the spectra of TG-M and NTG-M over those of M, T-M and NT-M. The increase in oxygen-containing functional groups shows that membrane hydrophilicity is enhanced. These oxygen-containing functional groups (hydroxyl groups, carboxyl groups and epoxy groups) can only be derived from GO. A broad band at wavenumbers between 1000 and 450 cm−1 is linked with Ti–O–Ti and Ti–O–C.37
O) and hydroxyl groups (OH) 16 that appear in the spectrum of TG-M and NTG-M. Peaks at 1401 cm−1, 1226 cm−1 and 1050 cm−1 can be attributed to the skeletal vibration of unoxidized graphitic, epoxy groups C–O and the hydroxyl group stretching mode C–O,35,36 which are stronger in the spectra of TG-M and NTG-M over those of M, T-M and NT-M. The increase in oxygen-containing functional groups shows that membrane hydrophilicity is enhanced. These oxygen-containing functional groups (hydroxyl groups, carboxyl groups and epoxy groups) can only be derived from GO. A broad band at wavenumbers between 1000 and 450 cm−1 is linked with Ti–O–Ti and Ti–O–C.37
 | ||
| Fig. 4 FT-IR spectra of M, T-M, NT-M, TG-M and NTG-M. | ||
M and NTG-M were investigated by XPS spectra to confirm the nitrogen doping. Fig. 5 shows that peaks at 161, 284, 399, 458 and 530 eV can be assigned to S 2p, C 1s, N 1s, Ti 2p and O 1s, respectively. The atomic percentages of S 2p, C 1s, N 1s, Ti 2p and O 1s in NTG-M are 6.62 at%, 74.54 at%, 0.55 at%, 2.47 at% and 15.82 at%, respectively, whereas their atomic percentages in M are 7.51 at%, 77.49 at%, 0.22 at%, 0.00 at% and 14.78 at%. The small amount of N 1s that is contained in M may originate from membrane fabrication because polysulfone was dissolved in NMP that contains N. The percentage of N in NTG-M is higher than in pure membranes, and can originate only from NTG, which indicates that nitrogen was doped on TiO2. With the grafting of NTG on the membrane, the percentages of C and S were reduced, whereas Ti and O increased because the main ingredient in NTG is TiO2.
 | ||
| Fig. 5 XPS spectra of M and NTG-M. | ||
Contact angle measurements are shown in Fig. 6 (M and NTG-M) and Table 2 (M, T-M, NT-M, TG-M and NTG-M). In Fig. 6(a) and (b), once a bare membrane had been grafted by NTG, the apparent pure water-contact angle decreased from 81° to 64°, which represented a much higher affinity between the membrane surface and water because of the hydrophilicity of TiO2 and oxygen-containing functional groups in GO. The contact angle of NTG-M improved slightly compared with TG-M, and was ∼65°, which implies that nitrogen doping increases the TiO2 hydrophilicity and then increases the hydrophilicity of the grafted membranes.
 | ||
| Fig. 6 Water contact angle of (a) M and (b) NTG-M. | ||
| Membrane | Contact angle (°) | Water flux (L m−2 h−1) |
|---|---|---|
| M | 81 ± 1.5 | 108 ± 4.2 |
| T-M | 78 ± 1.4 | 78 ± 3.3 |
| NT-M | 76 ± 1.9 | 82 ± 3.0 |
| TG-M | 65 ± 2.2 | 66 ± 3.9 |
| NTG-M | 64 ± 1.8 | 70 ± 2.5 |
The pure water flux of membranes was tested in the system as illustrated in Fig. 1(a) at 0.1 MPa and at room temperature, and the results are shown in Table 2. The pure water flux for a pure membrane is 108 L m−2 h−1, and this value decreases after grafting by TiO2, NT, TG and NTG, respectively. T-M exhibits a decrease in pure water flux by approximately 28% relative to the water flux of M because of the obstruction of TiO2, which recombined partly on the surface. TG-M has an additional 15% membrane resistance to water flux compared with T-M, possibly because of the additional film thickness from the GO nanosheets. NTG-M promotes the pure water flux slightly compared with TG-M, possibly because nitrogen doping increases the TiO2 hydrophilicity and thus the pure water flux.
3.2 Photocatalytic performance
Without filtration performance, the photocatalytic performance of M, T-M, NT-M, TG-M and NTG-M were tested in a container with a membrane and MB solution. The initial MB solution concentration was 50 mg L−1. Subsequent MB concentrations were calculated using a standard curve that was measured by UV-vis spectrophotometry. The absorbances of pure water, 1 mg L−1, 3 mg L−1, 5 mg L−1, 7 mg L−1 and 9 mg L−1 MB solutions at 665 nm were measured and a concentration–absorbance standard curve was plotted.The photocatalytic performance of all membranes was tested for different irradiations (darkness, UV and sunlight) as shown in Fig. 7. Fig. 7(a) shows that in darkness, the MB absorption capability of all membranes is almost the same, whereas TG-M and NTG-M have a slightly higher absorption capability because of the participation of GO. The GO structure provides a high specific surface area that results in good adsorption performance. However, the membrane or GO absorption is too low to have a significant effect on the MB removal.
 | ||
| Fig. 7 Residual MB concentration treated by M, T-M, NT-M, TG-M and NTG-M in (a) darkness, (b) UV and (c) sunlight. (d) MB degradation kinetics for NTG-M in darkness, UV and sunlight. C0 is the initial MB concentration (50 mg L−1), C is MB concentration at any time t. | ||
A comparison of NTG-M and NT-M (Fig. 7(b) and (c)) shows that NTG grafting improves photocatalytic performance in UV light and sunlight significantly, just as reported by Gao,25 the presences of GO is efficient in improving photocatalytic performance, because GO has an excellent electrical conduction ability and forms an impurity level in TiO2. The MB concentration in a pure membrane is reduced continuously in UV light and in sunlight. It is expected that MB exhibits a degradation performance in light without catalyst. NTG-M and TG-M have almost the same photocatalytic performance in UV, whereas NTG-M performs better than TG-M in sunlight, which indicates that nitrogen doping did not have a significant impact in UV light but it did in sunlight. Nitrogen doping reduces the TiO2 forbidden band width, and thus TiO2 has a longer response range for light.
The kinetics of MB photodegradation was calculated using a first-order reaction kinetics equation:
| −ln(C/C0) = kt |
3.3 Synergistic effect of photocatalysis and filtration
The synergistic effect of photocatalysis and filtration was tested in the equipment shown in Fig. 1(b). The membrane was placed on the collet with a light source from above to provide energy to power the electron transition. Under nitrogen pressure, MB solution in the feed tank was transported continuously into the photocatalytic membrane reactor. Because of the synergistic effect of photocatalysis and filtration, filtered water flowed out the reactor, whereas MB was intercepted and photodegraded simultaneously.We used a photocatalytic filtration concentration (Cpf, mg L−1) as a numerical representative for the MB photodegradation ability:

Fig. 8 shows the synergistic effect of photocatalysis and filtration of NTG-M and M in sunlight. Fig. 8(a) shows that all of the NTG-M concentration curves exhibit an increasing trend, whereas the concentration curve in the reactor exhibits the largest increase. At 0.5 h, the photocatalytic filtration concentration is ∼23.3 mg L−1, which means that 1 L water is filtered and 22.3 mg MB will be photodegraded. Because the total MB concentration (35.8 mg L−1) of the photocatalytic filtration concentration and filtered water (12.5 mg L−1) is lower than the initial MB concentration (50 mg L−1), the MB concentration in the reactor will increase. Because the MB concentration in the reactor increases, the photocatalytic rate will increase according to first-order reaction kinetics, and thus, the photocatalytic filtration concentration will increase. Fig. 8(b) shows the concentration curves of M; the trend in basic concentration curves in the reactor and filtered water are the same as NTG-M. Because the photocatalytic performance of NT-M is lower than NTG-M, the photocatalytic filtration concentration is lower than NTG-M, thus the MB concentration in the reactor increase faster than NTG-M. Fig. 8(c) shows the concentration curves of M; the trend in basic concentration curves in the reactor and filtered water are also the same as NTG-M, but its degree of change is larger. Without a photocatalyst, the photodegradation of MB can almost be ignored as the photocatalytic filtration concentration of M is less than 5 mg L−1. NTG-M exhibits a higher removal of MB because its MB concentration of filtered water is lower than M because of its photocatalytic performance. Because the MB concentration of NTG-M in the reactor is lower than M, the possibility of membrane pollution is reduced significantly. Thus, under the synergistic effect of photocatalysis and filtration, the entire MB removal ability of NTG-M exhibits a great improvement.
 | ||
| Fig. 8 MB concentration in the reactor, filtered water MB concentration and photocatalytic filtration concentration for (a) NTG-M, (b) NT-M and (c) M in sunlight. The initial MB concentration in the reactor was 50 mg L−1. | ||
3.4 Recycle performance
To evaluate the recycle performance of NTG-M, we measured the pure water flux of NTG-M continuously and tested the photocatalytic performance of NTG-M repeatedly. The pure water flux of NTG-M was tested in the system as illustrated in Fig. 1(a) at 0.1 MPa and at room temperature. Similarly, the photocatalytic performance of NTG-M were tested in a container with a membrane and MB solution in sunlight, the initial MB solution concentration was 50 mg L−1 and adjusted to 50 mg L−1 every 3 h.As shown in Fig. 9, the residual MB concentration is ∼25 mg L−1 after being photodegraded by NTG-M for the first 3 h in sunlight, and NTG-M shows the similar photocatalytic performance in the second 3 h while the residual MB concentration is raised gradually in the following tests. At the end of the eighth 3 h, the residual MB concentration is ∼33.5 mg L−1, its photodegradation ability decreases ∼34% compare with which in the first 3 h. This is possibly caused by the loss of photocatalyst or MB deposition in the photocatalyst. The pure water flux of NTG-M is also shown in Fig. 9, its changing curve exhibits a weak increase as the increasement of pure water flux is ∼9% after 24 h. Because the photocatalyst on the membrane surface has an obstructive performance, we guess that a small amount of photocatalyst has lost and thus pure water flux raise. Considering above two kinds of test results, we think that a small part of the photocatalyst on NTG-M is losing during use, but the recyclability of photocatalyst has a great improvement compare with powder photocatalyst.
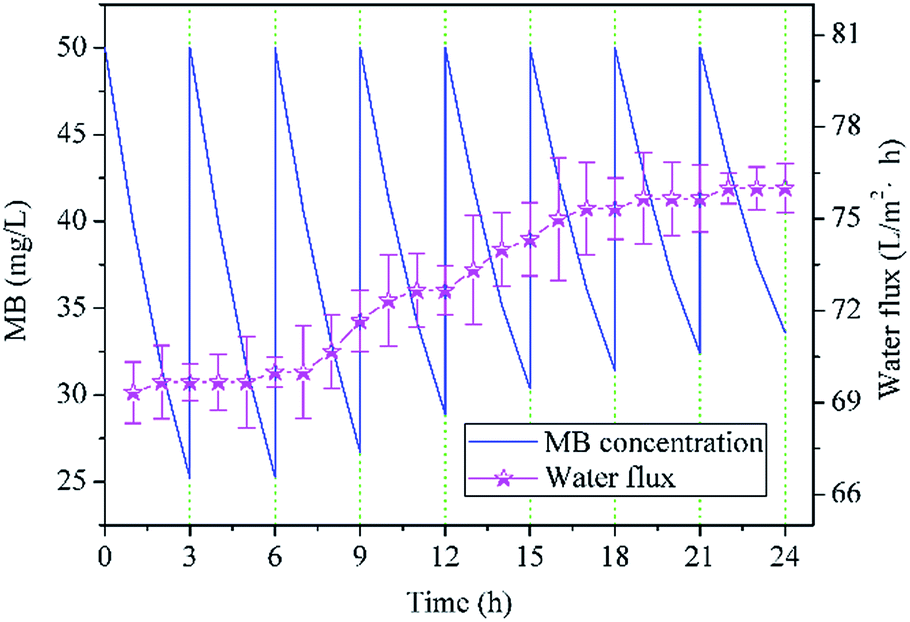 | ||
| Fig. 9 Residual MB concentration treated by NTG-M in sunlight (the initial MB concentration was always 50 mg L−1) and pure water flux of NTG-M at 0.1 MPa at room temperature. | ||
4. Conclusions
An ultrafiltration membrane grafted by N–TiO2/graphene oxide (NTG-M) was prepared. SEM images show membrane surfaces that are covered with flakes. FT-IR signals of oxygen-containing functional groups show a graphene oxide success load on the membrane surface. XPS signals of the peaks and elemental composition of the nitrogen and titanium reveal successful N-doping on TiO2 and NT loading on the membrane surface. The surface roughness as tested by AFM shows that NTG-M exhibits a rougher surface, which is advantageous to its photodegradation. Contact-angle measurements show that NTG-M has a lower water-contact angle, which implies better hydrophilicity. However, a lower pure water flux of NTG-M was tested by using the ultrafiltration cup system because of obstructive performance of the NTG, indicating membrane grafted by N–TiO2/graphene oxide is not all advantages. By testing its photocatalytic performance, we believe that N-doping and graphene oxide have a positive effect on the TiO2 photocatalytic performance, and NTG-M is used better in sunlight than in UV light. By testing the synergistic effect of photocatalysis and filtration, we conclude that NTG-M has a better MB removal ability and antifouling ability because of its photocatalytic performance. Furthermore, the recyclability of the NTG-M has a great improvement compare with powder photocatalyst.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 51678213 and No. 51578209), and A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions.References
- G. Moon, D. Kim, H. Kim, A. D. Bokare and W. Choi, Environ. Sci. Technol. Lett., 2014, 1, 185–190 CrossRef CAS.
- M. N. Chong, B. Jin, C. W. K. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- W. Qian, P. A. Greaney, S. Fowler, S. Chiu, A. M. Goforth and J. Jiao, ACS Sustainable Chem. Eng., 2014, 2, 1802–1810 CrossRef CAS.
- D. Kanakaraju, J. Kockler, C. A. Motti, B. D. Glass and M. Oelgemöller, Appl. Catal., B, 2015, 166–167, 45–55 CrossRef CAS.
- F. Pei, Y. Liu, S. Xu, J. Lü, C. Wang and S. Cao, Int. J. Hydrogen Energy, 2013, 38, 2670–2677 CrossRef CAS.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne, K. O'Shea, M. H. Entezari and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS.
- R. Wang, Q. Wu, Y. Lu, H. Liu, Y. Xia, J. Liu, D. Yang, Z. Huo and X. Yao, ACS Appl. Mater. Interfaces, 2014, 6, 2118–2124 CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- W. Sangkhun, L. Laokiat, V. Tanboonchuy, P. Khamdahsag and N. Grisdanurak, Superlattices Microstruct., 2012, 52, 632–642 CrossRef CAS.
- H. Zhang, G. Wang, D. Chen, X. Lv and J. Li, Chem. Mater., 2008, 20, 6543–6549 CrossRef CAS.
- H. Zhang, L. Guo, D. Wang, L. Zhao and B. Wan, ACS Appl. Mater. Interfaces, 2015, 7, 1816–1823 CAS.
- H. Li, Y. Hao, H. Lu, L. Liang, Y. Wang, J. Qiu, X. Shi, Y. Wang and J. Yao, Appl. Surf. Sci., 2015, 344, 112–118 CrossRef CAS.
- G. K. Mor, K. Shankar, M. Paulose, O. K. Varghese and C. A. Grimes, Nano Lett., 2006, 6, 215–218 CrossRef CAS PubMed.
- W. Sun, Y. Yu, H. Pan, X. Gao, Q. Chen and L. Peng, J. Am. Chem. Soc., 2008, 130, 1124–1125 CrossRef CAS PubMed.
- C. Gu and C. Shannon, J. Mol. Catal. A: Chem., 2007, 262, 185–189 CrossRef CAS.
- Y. Liu, S. Wang, S. Xu and S. Cao, Mater. Res. Bull., 2015, 65, 27–35 CrossRef CAS.
- T. Nguyen-Phan, V. H. Pham, E. W. Shin, H. Pham, S. Kim, J. S. Chung, E. J. Kim and S. H. Hur, Chem. Eng. J., 2011, 170, 226–232 CrossRef CAS.
- W. Wang, Y. Xie, C. Xia, H. Du and F. Tian, Microchim. Acta, 2014, 181, 1325–1331 CrossRef CAS.
- D. Zhao, G. Sheng, C. Chen and X. Wang, Appl. Catal., B, 2012, 111–112, 303–308 CrossRef CAS.
- X. Pan, M. Yang, Z. Tang and Y. Xu, J. Phys. Chem. C, 2014, 118, 27325–27335 CAS.
- S. Liu, H. Sun, S. Liu and S. Wang, Chem. Eng. J., 2013, 214, 298–303 CrossRef CAS.
- W. Yang, C. Li, L. Wang, S. Sun and X. Yan, Appl. Surf. Sci., 2015, 353, 307–316 CrossRef CAS.
- G. Xiao, X. Zhang, W. Zhang, S. Zhang, H. Su and T. Tan, Appl. Catal., B, 2015, 170–171, 255–262 CrossRef CAS.
- L. M. Pastrana-Martínez, S. Morales-Torres, J. L. Figueiredo, J. L. Faria and A. M. T. Silva, Water Res., 2015, 77, 179–190 CrossRef PubMed.
- Y. Gao, M. Hu and B. Mi, J. Membr. Sci., 2014, 455, 349–356 CrossRef CAS.
- A. Alem, H. Sarpoolaky and M. Keshmiri, J. Eur. Ceram. Soc., 2009, 29, 629–635 CrossRef CAS.
- Y. Wei, H. Chu, B. Dong, X. Li, S. Xia and Z. Qiang, Desalination, 2011, 272, 90–97 CrossRef CAS.
- J. Xu, C. Chang, J. Hou and C. Gao, Chem. Eng. J., 2013, 223, 722–728 CrossRef CAS.
- X. Li, X. Fang, R. Pang, J. Li, X. Sun, J. Shen, W. Han and L. Wang, J. Membr. Sci., 2014, 467, 226–235 CrossRef CAS.
- X. Huang, K. L. Marsh, B. T. McVerry, E. M. V. Hoek and R. B. Kaner, ACS Appl. Mater. Interfaces, 2016, 8, 14334–14338 Search PubMed.
- H. M. Hegab and L. Zou, J. Membr. Sci., 2015, 484, 95–106 CrossRef CAS.
- M. Kumar, Z. Gholamvand, A. Morrissey, K. Nolan, M. Ulbricht and J. Lawler, J. Membr. Sci., 2016, 506, 38–49 CrossRef CAS.
- F. Shi, Y. Ma, J. Ma, P. Wang and W. Sun, J. Membr. Sci., 2012, 389, 522–531 CrossRef CAS.
- M. Safarpour, A. Khataee and V. Vatanpour, J. Membr. Sci., 2015, 489, 43–54 CrossRef CAS.
- O. C. Compton, B. Jain, D. A. Dikin, A. Abouimrane, K. Amine and S. T. Nguyen, ACS Nano, 2011, 5, 4380–4391 CrossRef CAS PubMed.
- N. A. Almeida, P. M. Martins, S. Teixeira, J. A. Lopes Da Silva, V. Sencadas, K. Kühn, G. Cuniberti, S. Lanceros-Mendez and P. A. A. P. Marques, J. Mater. Sci., 2016, 51, 6974–6986 CrossRef CAS.
- M. Safarpour, A. Khataee and V. Vatanpour, Ind. Eng. Chem. Res., 2014, 22211943 Search PubMed.
- M. Nie, Y. Yang, Z. Zhang, C. Yan, X. Wang, H. Li and W. Dong, Chem. Eng. J., 2014, 246, 373–382 CrossRef CAS.
- A. E. Cassano and O. M. Alfano, Catal. Today, 2000, 58, 167–197 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |