
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dicarabrol A, dicarabrone C and dipulchellin A, unique sesquiterpene lactone dimers from Carpesium abrotanoides†
Jie-Wei Wuab,
Chunping Tanga,
Chang-Qiang Kea,
Sheng Yaoa,
Hong-Chun Liua,
Li-Gen Lin *c and
Yang Ye*ad
*c and
Yang Ye*ad
aState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, P. R. China. E-mail: yye@mail.shcnc.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cState Key Laboratory of Quality Research in Chinese Medicine, Institute of Chinese Medical Sciences, University of Macau, Macao 999078, P. R. China. E-mail: ligenl@umac.mo
dSchool of Life Science and Technology, Shanghai Tech University, Shanghai 201203, P. R. China
First published on 16th January 2017
Abstract
Three rare sesquiterpene lactone dimers, dicarabrol A (1), dicarabrone C (2) and dipulchellin A (3), were isolated from the whole plants of Carpesium abrotanoides. Their structures were elucidated by comprehensive analyses of NMR and MS spectroscopic data. The structure of dipulchellin A was further confirmed by single-crystal X-ray crystallography. Compounds 1 and 2 possessed an unusual carbon skeleton with two carabranolide moieties linking through a spirotetrahydrofuran ring, which was presumably formed by a [4 + 2] cycloaddition. Compound 3 was a [3 + 2] cycloaddition product of a guaianolide moiety and a carabranolide moiety linking through a cyclopentane ring. Their plausible biosynthetic pathways were also proposed. Compounds 1–3 showed moderate cytotoxicity against HL-60 cells with IC50 values of 8.7 ± 0.3, 8.2 ± 0.3 and 8.9 ± 0.4 μM, respectively.
Introduction
In the past decade, a series of sesquiterpenoid dimers with unusual carbon skeletons have been reported.1 The complex linkage patterns, along with their multiple chiral centres, make the sesquiterpenoid dimers exhibit more “biologically friendly” and “drug-like” molecular features than their monomers.1,2 Sesquiterpenoid dimers have been considered as promising drug candidates, and have gathered considerable research interest recently.The genus Carpesium (Asteraceae) consists approximately of 21 species, which are mainly distributed in Asia, especially in Southwest China.3 The fruits of Carpesium abrotanoides L., known as “Nan-He-Shi” in Chinese, are used as insecticides in Northern China.4 Phytochemical investigations on this species have led to the isolation of a number of sesquiterpenoid monomers and dimers.5–8 C. abrotanoides was also discovered with diverse biological activities such as anti-inflammatory,9 antitumor,7 antiplasmodial,10,11 and bactericidal effects.12
In our previous studies, dicarabrones A and B, dimeric sesquiterpene lactones connecting by a cyclopentane ring, were isolated from the whole plants of C. abrotanoides.7 As a continuing search for cytotoxic sesquiterpenoids, the whole plants of C. abrotanoides was thoroughly isolated. Three new sesquiterpene lactone dimers, dicarabrol A (1), dicarabrone C (2) and dipulchellin A (3), were identified. Compounds 1 and 2 possessed an unusual carbon skeleton with two carabranolide moieties linking through a spiro-tetrahydrofuran ring, which was presumably formed by a [4 + 2] cycloaddition. Compound 3 was a [3 + 2] cycloaddition product of a guaianolide moiety and a carabranolide moiety linking through a cyclopentane ring. Herein, the isolation, structure characterization, proposed biosynthetic pathways, and cytotoxicity of these three compounds are presented.
Results and discussion
The air-dried whole plant of C. abrotanoides was extracted with 95% ethanol at room temperature (3 times, each 3 days) to afford a crude extract. After partitioned between CH2Cl2 and H2O, a CH2Cl2 fraction was obtained. The CH2Cl2 fraction was subjected to column chromatography (CC) over MCI gel eluted with EtOH/H2O (50% to 95%) to yield fractions A–C. Fraction B was then subjected to CC over silica gel eluted with CH2Cl2/MeOH (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 10:1) to give seven subfractions (B1–B7). Subsequently, subfractions B3–B5 were further purified by CC over Sephadex LH-20 (MeOH) and then preparative HPLC (CH3CN/H2O from 30% to 60%), yielding 1 (32 mg), 2 (38 mg) and 3 (88 mg) (Fig. 1).
:1 to 10:1) to give seven subfractions (B1–B7). Subsequently, subfractions B3–B5 were further purified by CC over Sephadex LH-20 (MeOH) and then preparative HPLC (CH3CN/H2O from 30% to 60%), yielding 1 (32 mg), 2 (38 mg) and 3 (88 mg) (Fig. 1).
 | ||
| Fig. 1 The structures of dicarabrol A (1), dicarabrone C (2) and dipulchellin A (3). | ||
Dicarabrol A (1) was obtained as a yellow amorphous powder, and its molecular formula was assigned to be C30H44O7 from the positive ion peak at m/z 539.2974 ([M + Na]+, calcd 539.2985) in the HRESIMS, requiring 9 degrees of unsaturation. The 13C NMR and DEPT spectra of 1 displayed 30 carbon resonances including 4 methyls, 10 methylenes, 10 methines, and 6 quaternary carbons. The IR absorption at 1772 cm−1, along with two carbonyl carbons at δC 176.8 and 175.4, clearly suggested the existence of two lactone rings in the molecule of 1. The IR spectrum also suggested the existence of hydroxyl groups (3432 cm−1). The 1H NMR and HSQC spectra of 1 revealed the presence of four oxygenated methines at δH 4.96 (m), 4.82 (m) and 3.80 (m, 2H), one oxygenated methylene at δH 3.97 (m, 2H), two singlet methyls at δH 1.09 and 1.07, and two doublet methyls at δH 1.20 (J = 6.2 Hz) and 1.19 (J = 6.2 Hz) (Table 1). Additionally, two typical methine protons at δH 0.46 (m) and 0.38 (m) (Table 1) were indicative of the presence of two cyclopropane fragments. These characteristic signals, together with the MS data, suggested that 1 might be a sesquiterpene lactone dimer.
| No. | 1 | 2 | 3 | |||
|---|---|---|---|---|---|---|
| δH (mult, J Hz) | δC | δH (mult, J Hz) | δC | δH (mult, J Hz) | δC | |
| a Signals overlapped. | ||||||
| 1 | 0.46, m | 35.1 | 0.43a, m | 33.8 | 1.48a, m | 48.2 |
| 2 | 1.35a, m | 25.6 | 1.55a, m | 23.4 | α 1.76a, m | 25.5 |
| β 1.40, m | ||||||
| 3 | 1.52a, m | 39.3 | 2.53a, m | 43.6 | 1.97, m | 29.2 |
| 4 | 3.80a, m | 68.0 | 208.9 | 3.72, m | 83.8 | |
| 5 | 0.38, m | 23.7 | 0.43a, m | 23.8 | 45.1 | |
| 6α | 1.99, m | 25.3 | 1.97m m | 25.2 | 1.87, m | 36.2 |
| 6β | 0.70a, m | 0.72a, m | 1.70a, m | |||
| 7 | 2.31, m | 44.2 | 2.30, m | 44.1 | 2.30, m | 53.5 |
| 8 | 4.96, m | 76.9 | 4.95, m | 76.8 | 4.24, dt, 4.3, 10.9 | 79.1 |
| 9α | 2.44a, m | 37.5 | 2.44a, m | 37.0 | 2.31, dt, 3.5, 13.3 | 45.2 |
| 9β | 0.96, m | 1.04, m | 1.19a, m | |||
| 10 | 15.9 | 16.1 | 0.91, d, 6.5 | 20.7 | ||
| 11 | 85.3 | 85.3 | 57.4 | |||
| 12 | 175.4 | 175.2 | 178.5 | |||
| 13a | 2.71, d, 13.2 | 40.7 | 2.71, d, 13.1 | 40.7 | 2.11, m | 40.8 |
| 13b | 2.15, m | 2.15a, m | 1.74a, m | |||
| 14 | 1.09, s | 19.1 | 1.08, s | 19.2 | 0.91, d, 6.5 | 20.7 |
| 15 | 1.19, d, 6.2 | 23.8 | 2.16, s | 30.3 | 0.88, s | 17.0 |
| 1′ | 0.43, m | 34.6 | 0.43a, m | 34.4 | 2.06, m | 20.6 |
| 2′ | 1.35a, m | 25.5 | 1.55a, m | 23.5 | 1.65, m | 30.7 |
| 1.36a, m | ||||||
| 3′ | 1.52a, m | 39.3 | 2.53a, m | 43.7 | 1.48a, m | 38.6 |
| 1.35a, m | ||||||
| 4′ | 3.80a, m | 68.1 | 208.9 | 3.77, m | 68.8 | |
| 5′ | 0.30, m | 23.5 | 0.34, m | 23.6 | 1.79, m | 50.9 |
| 6′α | 2.19a, m | 24.7 | 2.15a, m | 24.6 | 1.55, m | 26.3 |
| 6′β | 0.73a, m | 0.67a, m | 1.10, m | |||
| 7′ | 2.19a, m | 44.0 | 2.15a, m | 44.0 | 2.99, m | 36.4 |
| 8′ | 4.82, m | 75.7 | 4.82, m | 75.6 | 4.69, dt, 7.2, 10.3 | 75.4 |
| 9′α | 2.44a, m | 37.2 | 2.44a, m | 37.4 | 1.85, m | 30.4 |
| 9′β | 1.03, m | 0.94, m | 1.48a, m | |||
| 10′ | 16.0 | 16.0 | 43.5 | |||
| 11′ | 52.8 | 52.8 | 139.0 | |||
| 12′ | 176.8 | 176.7 | 170.1 | |||
| 13′ | 3.97, m | 71.4 | 3.97, m | 71.3 | 6.23, d, 1.9 | 123.3 |
| 5.60, d, 1.9 | ||||||
| 14′ | 1.07, s | 19.2 | 1.10, s | 19.0 | 1.15, s | 26.4 |
| 15′ | 1.20, d, 6.2 | 23.8 | 2.15, s | 30.4 | 1.19, d, 6.1 | 24.1 |
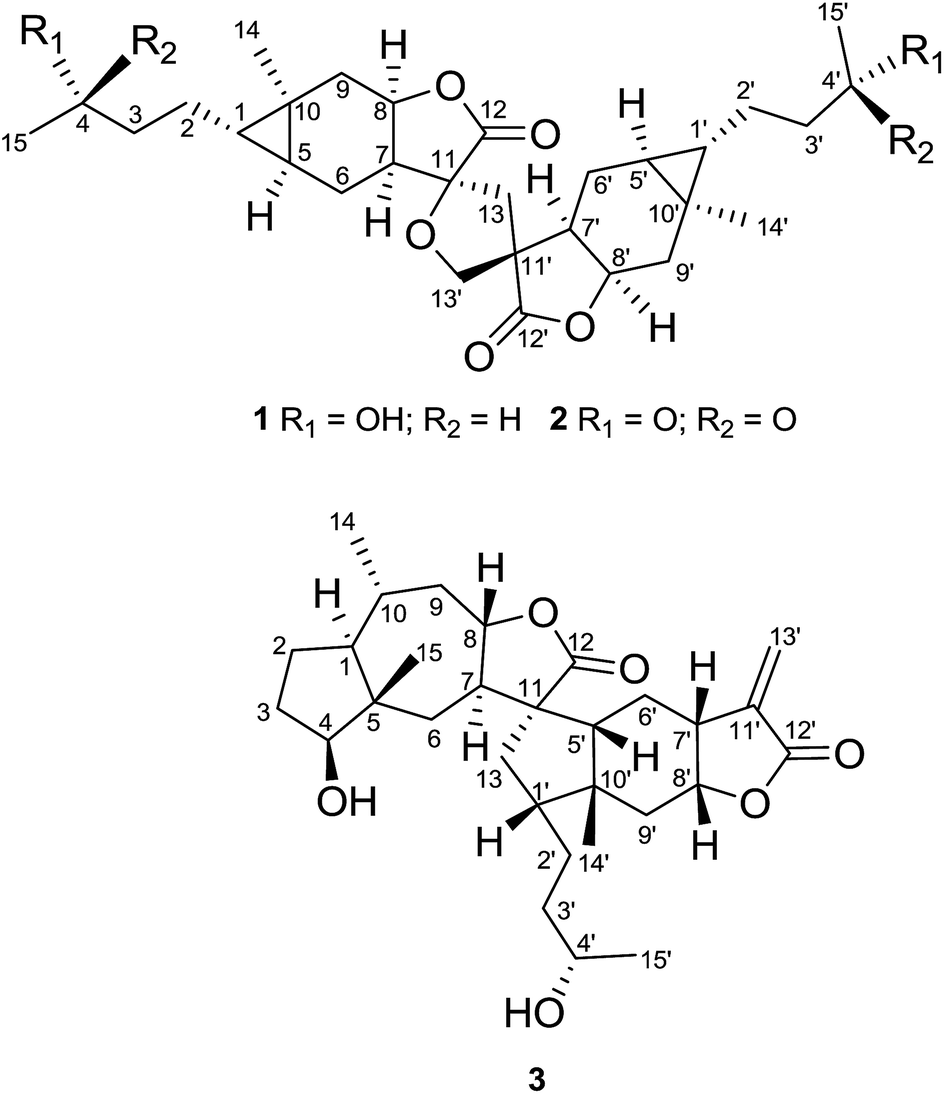
On the basis of analysis of the 1H–1H COSY and HMBC spectra of 1, two carabranolide moieties were constructed (Fig. 2), whose 1H and 13C NMR data showed great similarity to those of carabrol.13 The major difference was an oxygen-bearing quaternary carbon (δC 85.3) and a methylene (δH 2.71, d, J = 13.2 Hz; 2.15, m/δC 40.7) in the moiety 1a, and a quaternary carbon (δC 52.8) and an oxygen-bearing methylene (δH 3.97, m, 2H/δC 71.4) in the moiety 1b, were observed instead of the exocyclic double bond between C-11 and C-13 in carabrol. The linkage between the two moieties was established by the HMBC correlations (Fig. 2). The HMBC cross-peaks between H-13 (δH 2.71; 2.15) and C-12 (δC 175.4), C-7′ (δC 44.0), and C-11′ (δC 52.8) indicated a C–C bond between C-13 and C-11′. And the HMBC correlations from H-13′ (δH 3.97) to C-11 (δC 85.3) and C-13 (δC 40.7) suggested a C–O–C linage between C-11 and C-13′. Thus, a unique spiro-tetrahydrofuran ring was constructed connecting the two carabranolide moieties.
 | ||
| Fig. 2 Key 1H–1H COSY (–) and HMBC correlations (H → C) of 1. | ||
The relative configuration of 1 was inferred from the ROESY experiment (Fig. 3). The cross-peaks of H-5/H3-14, H-7/H-8, H-5′/H3-14′, and H-7′/H-8′ suggested cis-diaxial like structure of two carabranolide units. Additionally, the ROESY cross-peaks of H-5/H-6α, H-6α/Hb-13, H2-13′/H-6′β, H3-14′/H-6′α, suggested that H-5, H-6α, and Hb-13 are on the same face, while H-6′β, and H2-13′ are on the other face. Considering the biogenetic relationship between 1 and carabrol, the absolute configuration of 1 was determined as 1S, 4S, 5S, 7S, 8R, 10R, 11S, 1′S, 4′S, 5′S, 7′R, 8′R, 10′R, 11′R. Taken together the structure of 1 was determined.
 | ||
| Fig. 3 Key ROESY correlations of 1. | ||
Dicarabrone C (2) was obtained as a yellow amorphous powder. The HRESIMS indicated a molecule formula of C30H40O7 (m/z 535.2680, [M + Na]+, calcd for 535.2672). Comparison of the 1H and 13C NMR data of 2 with those of 1 revealed a high degree of similarity. The only difference was two hydroxy-methines in 1 were replaced by two carbonyl groups (δC 208.9, C-4/208.9, C-4′) in 2. Further analysis of the 1H–1H COSY and HMBC data revealed that 2 was a dimer of two carabrone moieties with the same skeleton as 1. The NOESY correlations of H2-13′/H-6′β and H-6α/Hb-13 indicated the spiro-tetrahydrofuran ring possessed the same configuration as that of 1. Similarly, the absolute configuration of 2 was determined as 1S, 5S, 7S, 8R, 10R, 11S, 1′S, 5′S, 7′R, 8′R, 10′R, 11′R, based on its biogenetic relationship with carabrone. Thus, the structure of 2 was determined.
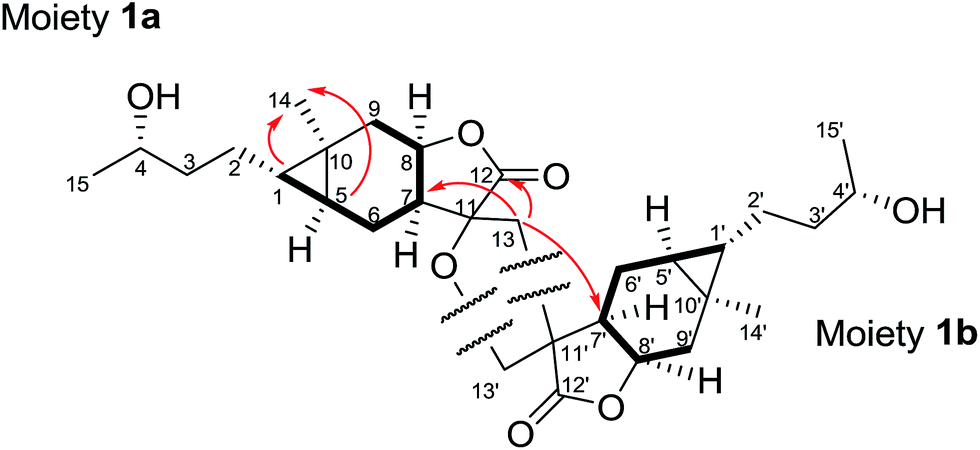
Dipulchellin A (3) was obtained as white lamellar crystals. Its molecular formula C30H44O6 was determined by the positive ion peak at m/z 523.3020 ([M + Na]+, calcd 523.3030) in the HRESIMS, indicating 9 degrees of unsaturation. The IR spectrum indicated the presences of hydroxyl groups (3444 cm−1) and ester carbonyl groups (1755 cm−1). The 13C and DEPT NMR spectra of 3 showed 30 carbon resonances due to 4 methyls, 10 methylenes, 10 methines, and 6 quaternary carbons (Table 1). The 1H NMR spectrum of 3 showed the signals of four oxygenated methines at δH 3.72 (m), 3.77 (m), 4.24 (dt, J = 4.3, 10.9 Hz), and 4.69 (dt, J = 7.2, 10.3 Hz), two singlet methyls at δH 0.88 and 1.15, and two doublet methyls at δH 0.91 (J = 6.5 Hz) and 1.19 (J = 6.1 Hz) (Table 1). From these resonances, one α-methylene-γ-lactone moiety (δH 5.60, d, J = 1.9 Hz; 6.23, d, J = 1.9 Hz; δC 170.1, 139.0 and 123.3) and a lactone carbonyl carbon (δC 178.5) could be identified. The NMR and MS data suggested that 3 might be a sesquiterpene lactone dimer. Extensive analysis of the 1D and 2D NMR spectra of 3 led to the identification of two moieties (Fig. 4). The moiety 3a was assembled based on the 1H–1H COSY correlations (H2-2/H2-3/H-4 and H2-6/H-7/H-8/H2-9/H-10/H3-14) and the key HMBC correlations (H3-14/C-1, C-9, H3-15/C-4, C-6, and H-8/C-11, C-12). The 1H and 13C NMR data of the moiety 3a were very similar to those of 2-desoxy-4-epi-pulchellin6 except a methylene (δH 1.74, m; 2.11, m/δC 40.8) and a quaternary carbon (δC 57.4) in the moiety 3a instead of the exocyclic double bond between C-11 and C-13 in 2-desoxy-4-epi-pulchellin. The remaining signals indicated a 1,5-seco carabrol unit for moiety 3b.
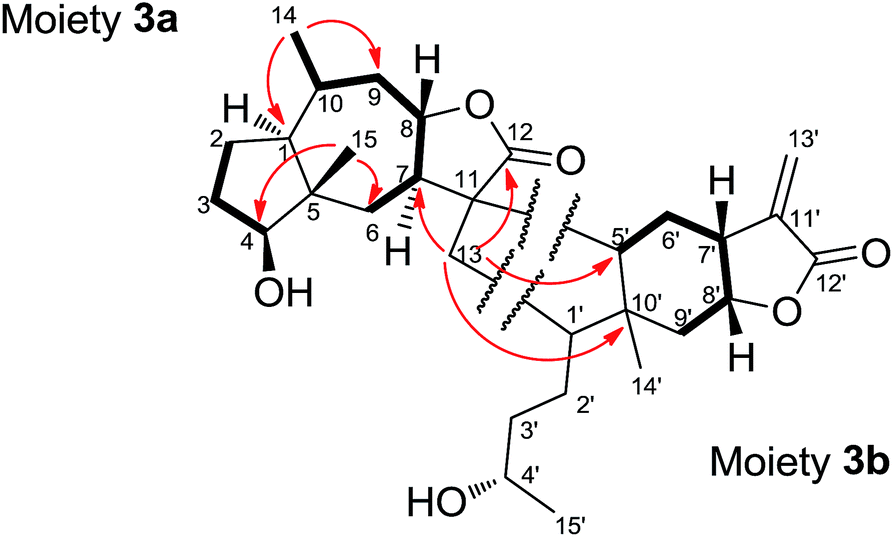 | ||
| Fig. 4 Key 1H–1H COSY (–) and HMBC correlations (H → C) of 3. | ||
The connection between the moieties 3a and 3b was first indicated by the 1H–1H COSY correlation between H2-13 and H-1′. Further evidence from the HMBC correlations between H2-13 and C-7 (δC 53.5), C-12 (δC 178.5), C-5′ (δC 50.9), and C-10′ (δC 43.5) confirmed the C–C bond between C-13 and C-1′. Additionally, the HMBC correlations from H-5′ (δH 1.79, m) to C-7, and H-7 (δH 2.30, m) to C-5′ indicated another C–C bond between C-11 and C-5′. Thus, a cyclopentane ring was constructed to connect the 2-desoxy-4-epi-pulchellin and carabrol moieties. Due to the overlapping of the key ROESY correlations, the configuration of the cyclopentane moiety could not be fully determined. The X-ray crystallographic analysis established unambiguously the absolute configuration of 3 as 1S, 4S, 5S, 7R, 8S, 10R, 11S, 1′R, 4′S, 5′R, 7′R, 8′R, and 10′R (Fig. 5).
 | ||
| Fig. 5 X-ray crystallographic structure of 3. | ||
Dicarabrol A (1) and dicarabrone C (2) possess two carabranolide units linking through a spiro-tetrahydrofuran ring, which are rarely discovered in nature and, thus, of particular significance from the perspective of biosynthesis. To the best of our knowledge, only one example of sesquiterpenoid dimer, featured with the spiro-tetrahydrofuran ring, was reported in the previous study.14 Accordingly, a plausible biosynthetic pathway of dicarabrol A (1) and dicarabrone C (2) was proposed in Scheme 1A. The key step of this biosynthetic pathway might be the [4 + 2] Diels–Alder cycloaddition. Carabrol and carabrone were previously reported from this species, which could be considered as the parent compounds. First, the double bond (C-11′![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2-13′) of one carabrol/carabrone might undergo [4 + 2] Diels–Alder cycloaddition with the α,β-unsaturated carbonyl (CH2-13C-11–C-12O) of another carabrol/carabrone, to form the key intermediate molecule I (KIM I) with a dihydropyran ring. Then the double bond of the dihydropyran could be enzymatically oxygenated to yield an oxirane fragment in KIM II. Last, two C–O bonds could be displaced each other to generate the tetrahydrofuran ring in dicarabrol A/dicarabrone C.
CH2-13′) of one carabrol/carabrone might undergo [4 + 2] Diels–Alder cycloaddition with the α,β-unsaturated carbonyl (CH2-13C-11–C-12O) of another carabrol/carabrone, to form the key intermediate molecule I (KIM I) with a dihydropyran ring. Then the double bond of the dihydropyran could be enzymatically oxygenated to yield an oxirane fragment in KIM II. Last, two C–O bonds could be displaced each other to generate the tetrahydrofuran ring in dicarabrol A/dicarabrone C.
 | ||
| Scheme 1 Proposed biosynthetic pathway for dicarabrol A (1), dicarabrone C (2) and dipulchellin A (3). | ||
The skeleton of dipulchellin A (3) features a cyclopentane ring linking a guaianolide unit and a carabranolide unit. The formation of the cyclopentane ring is quite unique. So far a number of structurally complicated sesquiterpenoid dimers have been isolated, and most of them were proposed to be biosynthesized through a [4 + 2] or [2 + 2] cycloaddition of two monomers.1,2 In the current case, the carabrone monomer possesses a cyclopropane ring and the 2-desoxy-4-epi-pulchellin monomer possesses a double bond; therefore, the cyclopentane ring might be envisioned to arise from a [3 + 2] cycloaddition starting from the cyclopropane ring in one monomer and the double bond in the other (Scheme 1B), as proposed in our previous study.7 In the proposed biosynthetic pathway, the reaction might take place via radical intermediates. The bond between C-1′ and C-5′ could be cleaved to form a radical intermediate, and the radical at C-5′ might be then trapped with the double bond. Due to steric hindrance in the structure, C-1′ could form a new bond with C-13 first, and subsequently C-5′ could form the other new bond with C-11. Therefore, a cyclopentane ring might be introduced to fuse two monomers.
In order to identify anti-tumour substances, compounds 1–3 were evaluated for their cytotoxic effects on A549 and HL-60 cancer cells. Compounds 1–3 exhibited selective cytotoxicity against HL-60 cells with IC50 values of 8.7 ± 0.3, 8.2 ± 0.3 and 8.9 ± 0.4 μM, respectively, but no activity on A549 cells (IC50 > 10 μM).
Conclusions
In conclusion, three novel sesquiterpoen lactone dimers were identified from the whole plants of C. abrotanoides. Till now, most sesquiterpenoid dimers were proposed to be biosynthesized from two monomeric sesquiterpenoids through a cyclohexene or 3,4-dihydro-2H-pyran ring via regular or hetero Diels–Alder cycloadditions.1,2 To the best of our knowledge, compounds 1 and 2 are the first examples of carabranolide dimers linking through a spiro-tetrahydrofuran ring; and compound 3 is the first dimer of a guaianolide unit and a carabranolide unit linking through a cyclopentane ring. Moreover, dipulchellin A (3) represents the second example of sesquiterpenoid dimer formed via a [3 + 2] cycloaddition. Our finding enriches the structure and biosynthesis diversity of sesquiterpenoid dimers. Furthermore, these compounds showed potential cytotoxic effect on HL-60 cells, which might be developed as anti-tumour agents.Experimental section
General experimental procedures
Melting points were obtained on a SGM X-4 apparatus (Shanghai Precision & Scientific Instrument Co., Ltd., P. R. China). Optical rotations were measured on a Perkin-Elmer 341 polarimeter. IR spectra were recorded on a Nicolet Magna FT-IR 750 spectrophotometer using KBr disks. NMR spectra were recorded on a Varian Mercury-500 spectrometer. The chemical shift (δ) values are given in ppm with TMS as internal standard, and coupling constants (J) are in Hz. ESI-MS and ESI-HRMS data were recorded on Waters 2695-3100 LC-MS and Waters Q-TOF Ultima Global mass spectrometers, respectively. Silica gel (Qingdao Marine Chemical Industrials) was used for flash chromatography. MCI gel CHP20P (75–150 μm, Mitsubishi Chemical Industries, Japan) and Sephadex LH-20 (Pharmacia Biotech AB, Uppsala, Sweden) were used for column chromatography (CC). TLC was carried out on precoated silica gel GF254 plates (Yantai Chemical Industrials) and the TLC spots were viewed at 254 nm and visualized with 5% H2SO4 in EtOH containing 10 mg mL−1 vanillin. Analytical HPLC was performed on a Waters 2690 instrument with a 996 PAD (photodiode array detector) and coupled with an Alltech ELSD 2000 detector. All solvents used for CC were of at least analytical grade (Shanghai Chemical Reagents Co., Ltd., Shanghai, P. R. China), and solvents used for HPLC were of HPLC grade (J & K Scientific Ltd., Shanghai, P. R. China).Plant material
The whole plants of C. abrotanoides L. were collected in Henan Province, P. R. China in 2012, and identified by Professor Jin-Gui Shen from the Shanghai Institute of Materia Medica. A voucher (20120901) was deposited at the Herbarium of the Shanghai Institute of Materia Medica, Chinese Academy of Sciences.Extraction and isolation
The air-dried whole plants of C. abrotanoides (50 kg) were extracted three times with 95% ethanol at room temperature to afford a crude extract (3.29 kg). The extract was further partitioned between CH2Cl2 and H2O, giving a CH2Cl2-soluble fraction (1.66 kg). The CH2Cl2-soluble fraction was fractionated over MCI gel (EtOH/H2O, from 50% to 95%) to yield fractions A–C. Fraction B (300 g) was then subjected to column chromatography (CC) over silica gel eluted with CH2Cl2/MeOH (50:1 to 10:1) in a stepwise manner to give seven subfractions (B1–B7). Subsequently, subfractions B3–B5 were selected for purification by CC over Sephadex LH-20 (MeOH) and then preparative HPLC (CH3CN/H2O from 30% to 60%), yielding 1 (32 mg), 2 (38 mg) and 3 (88 mg).
X-ray crystallographic analysis
Dipulchellin A (3) was crystallized from acetone at room temperature. The X-ray crystallographic data was obtained on a Bruker SMART CCD detector employing graphite monochromated Cu-Kα radiation (λ = 1.54178 Å) (operated in the φ–ω scan mode). The structure was solved by direct method using SHELXS-97 program and refined with full-matrix least-squares calculations on F2 using SHELXL-97. Crystallographic data for 3 (key parameters see Table S1†) have been deposited at the Cambridge Crystallographic Data Centre (Deposition No. CCDC 1479445).Cytotoxicity bioassays
The anti-proliferative activities of compounds 1–3 against HL-60 (human leukemia) and A-549 (human lung adenocarcinoma) tumor cell lines were evaluated. Doxorubicin was used as a positive control. Both the HL-60 and A-549 cells were obtained from ATCC.The anti-proliferative activity of the compounds against HL-60 cells was evaluated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.15 Exponentially growing HL-60 cells were seeded into 96-well plates. Upon approximately 70–80%, the cells were treated with series concentrations of different compounds for 48 h. DMSO was used as the vehicle control. Then, 1 mg mL−1 MTT solution was added to each well and the 96-well plates were further incubated for 4 h at 37 °C. 100 μL DMSO was added to each well to dissolve the formazan crystals. Absorbance at 570 nm was measured by a microplate reader (SpectraMax, Molecular Devices, USA). The inhibition rate was calculated as (1 − A570 treated/A570 vehicle control) × 100%.
The anti-proliferative activity of the compounds against A-549 cells was evaluated using the sulforhodamine B (SRB) assay.16 A-549 cells (4 × 103 cell per well) were seeded into 96-well plates and grown for 24 h. Cells were then treated with increasing concentrations of compounds and grown for further 72 h. DMSO was used as the vehicle control. At the end of exposure time, 100 μL ice-cold 10% trichloroacetic acid (TCA) was added to each well, left at 4 °C for 1 h, and washed five times with distilled water. The TCA-fixed cells were stained for 15 min with 100 μL 4 mg mL−1 SRB in 1% HOAc. The plates were washed five times with 1% HOAc and air-dried overnight. After air-drying, protein-bound dye was dissolved in 150 μL 10 mM Tris base for 5 min and was measured at 515 nm using multiwall spectrophotometer (SpectraMax, Molecular Devices, USA). The inhibition rate was calculated as (1 − A515 treated/A515 control) × 100%.
Acknowledgements
We thank the financial support of the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program” (No. 2011ZX09307-002-03). Our thanks are also given to the National Natural Science Funds for Distinguished Young Scholar (No. 30925043), grants from the Ministry of Science and Technology (2007DFC31030, 2010DFA30980), the Shanghai Commission of Science and Technology (10DZ1972700, 11DZ1970700, 12JC1410300) and the Science and Technology Development Fund of Macau (FCDT 120/2013/A3).Notes and references
- Z.-J. Zhan, Y.-M. Ying, L.-F. Ma and W.-G. Shan, Nat. Prod. Rep., 2011, 28, 594–629 RSC.
- G.-W. Wang, J.-J. Qin, X.-R. Cheng, Y.-H. Shen, L. Shan, H.-Z. Jin and W.-D. Zhang, Expert Opin. Invest. Drugs, 2014, 23, 317–345 CrossRef CAS PubMed.
- Editor committee, Flora of China, Science Press, Beijing, China, 1979, vol. 75, p. 293 Search PubMed.
- Editor committee, Flora of China, Science Press, Beijing, China, 1979, vol. 75, p. 313 Search PubMed.
- J. S. Lee, B. S. Min, S. M. Lee, M. K. Na, B. M. Kwon, C. O. Lee, Y. H. Kim and K. H. Bae, Planta Med., 2002, 68, 745–747 CrossRef CAS PubMed.
- F. Wang, K. Yang, F.-C. Ren and J.-K. Liu, Fitoterapia, 2009, 80, 21–24 CrossRef CAS PubMed.
- J.-W. Wu, C.-P. Tang, L. Chen, Y. Qiao, M.-Y. Geng and Y. Ye, Org. Lett., 2015, 17, 1656–1659 CrossRef CAS PubMed.
- J.-P. Zhang, G.-W. Wang, X.-H. Tian, Y.-X. Yang, Q.-X. Liu, L.-P. Chen, H.-L. Li and W.-D. Zhang, J. Ethnopharmacol., 2015, 163, 173–191 CrossRef CAS PubMed.
- H. J. Lee, H. J. Lim, D. Y. Lee, H. Jung, M. R. Kim, D. C. Moon, K. I. Kim, M. S. Lee and J. H. Ryu, Biochem. Biophys. Res. Commun., 2010, 391, 1400–1404 CrossRef CAS PubMed.
- E. J. Kim, H. K. Jin, Y. K. Kim, H. Y. Lee, S. Y. Lee, K. R. Lee, O. P. Zee, J. W. Han and H. W. Lee, Biochem. Pharmacol., 2001, 61, 903–910 CrossRef CAS PubMed.
- I. M. Chung and H. I. Moon, J. Enzyme Inhib. Med. Chem., 2009, 24, 131–135 CrossRef CAS PubMed.
- C. Yang, Y.-P. Shi and Z.-J. Jia, Planta Med., 2002, 68, 626–630 CrossRef CAS PubMed.
- F. Bohlmann, P. K. Mahanta, J. Jakupovic, R. C. Rastogi and A. A. Natu, Phytochemistry, 1978, 17, 1165–1172 CrossRef CAS.
- H.-L. Jiang, J. Chen, X.-J. Jin, J.-L. Yang, Y. Li, X.-J. Yao and Q.-X. Wu, Tetrahedron, 2011, 67, 9193–9198 CrossRef CAS.
- J. Heilmann, M.-R. Wasescha and T.-J. Schmidt, Bioorg. Med. Chem., 2001, 9, 2189–2194 CrossRef CAS PubMed.
- P.-A. Skehan, R. Storeng, A. Monks, J. McMahon, D. Vistica, J.-T. Warren, H. Bokesch, S. Kenney and M.-R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107–1112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed description of the experimental procedures, a listing of NMR, IR and HRESIMS spectra of compounds 1–3, as well as crystallographic data of 3 in CIF (CCDC 1479445). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra27626a |
| This journal is © The Royal Society of Chemistry 2017 |