DOI:
10.1039/C6RA27536B
(Paper)
RSC Adv., 2017,
7, 23265-23271
Synthesis and biological evaluation of 3-amino-3-hydroxymethyloxindoles as potential anti-cancer agents†
Received
29th November 2016
, Accepted 15th April 2017
First published on 27th April 2017
Abstract
A series of substituted 3-amino-3-hydroxymethyloxindoles (4a–4i) synthesized through a multi-component approach showed anticancer potency via in vitro cytotoxicity screening. Further, to develop the efficiency, derivatives (5a–5m) were synthesized and evaluated for their anti-proliferation activity. The most potent compound 5m showed cytotoxic effect toward SJSA-1 cells (IC50 = 3.14 μM) as well as inhibiting the growth of other cancer cell lines (HCT-116, Jurkat, KB and Bel7402). Further investigation revealed that 5m induced a significant G2/M cell cycle arrest and time- and dose-dependent cellular apoptosis in SJSA-1 cells. These results suggested that new compound 5m has anti-proliferating and pro-apoptotic effects, which might be a candidate for cancer therapies.
Introduction
Although tremendous efforts and resources have been invested for many years in the development of novel anti-cancer therapies, cancer remains the second leading cause of mortality in the world. It has been reported that a total of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 658370 new cancer cases and 589430 cancer deaths are projected to occur in the United States in 2015.1 New approaches to treat cancers are urgently needed. The paradigm of novel anticancer drug discovery has been shifted to molecularly-targeted therapeutics, but the conventional approach to drug discovery based on identification of cytotoxic agents on multiple human tumor cell lines, one kind of phenotypic screening, remains the single most successful route.2
658370 new cancer cases and 589430 cancer deaths are projected to occur in the United States in 2015.1 New approaches to treat cancers are urgently needed. The paradigm of novel anticancer drug discovery has been shifted to molecularly-targeted therapeutics, but the conventional approach to drug discovery based on identification of cytotoxic agents on multiple human tumor cell lines, one kind of phenotypic screening, remains the single most successful route.2
Compared to target-based screening, phenotypic screening without particular target in drug discovery highly depends on the quality of new molecule entities with molecular complexity and diversity.3,4 Fortunately, such screening library of complex and diverse compounds simultaneously with new scaffolds can be quickly and easily built through multi-component reactions (MCRs).5 Our group has developed a series of ylides or zwitterionic intermediates “trapping” MCRs which have been verified as an efficient approach to form heterocycle.6–10 Rh(II) catalyzed MCR of 3-diazooxindoles, anilines and formalin synthesized 3-amino-3-hydroxymethyloxindoles type scaffolds.11 Further, screening of synthesized compounds has revealed their anticancer activity. Inspired by the primary results, we further synthesized a series of oxindole derivatives to evaluate their anti-proliferation activity for advance study. The compounds with the best activity were assessed for wide-spectrum anticancer studies and analyzed for its effects on cell cycle and apoptosis.
Results and discussion
Chemistry
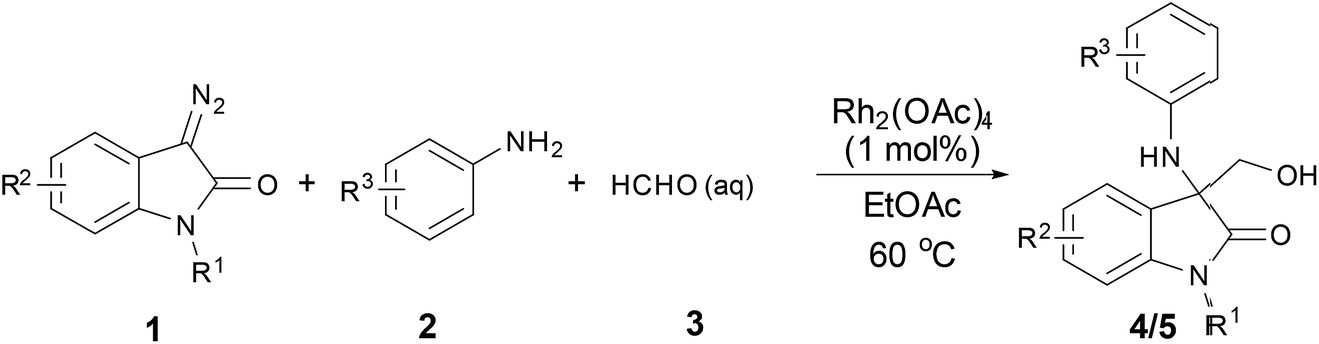
Herein, oxindole derivatives (4a–4i, 5a–5m) were synthesized by three-component “trapping” reaction (Scheme 1) as following: in ethyl acetate (EA) solution of catalyst Rh2(OAc)4, (1 mol%), aniline 2 and formalin 3 was added sequentially. The suspension was stirred at 60 °C for several minutes. Afterwards, 3-diazooxindoles 1 in EA was added to suspension over 1 hour via a syringe pump at the same temperature. Upon completion of the addition, the reaction mixture was left on stirring for an additional hour until no further transformation was monitored by TLC test. Solvent was evaporated and the residue was purified by flash chromatography on silica gel to afford the products in moderate yield (53–89%).
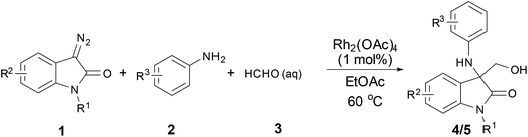 |
| | Scheme 1 Synthesis of 3-amino-3-hydroxymethyloxindoles 4 & 5. | |
As marked in the Scheme 2, the benzyl, methyl and phenyl group which is represented by R1, R2 and R3 in Table 1 were chemically modified. According to primary biological test results mentioned in Table 1, we realized that the benzyl group at R1 position and the methyl group at R2 position were more efficient at respective positions for the antitumor activity of these compound analogues. These results diverted our attention modification at R3 position. In order to compare the effect on bioactivity of different abundant groups at R3 position, we have prepared a series of analogues having groups such as –F, –Cl, –Br, –I and –NO2, on the phenyl ring at R3 position and studied subsequently.
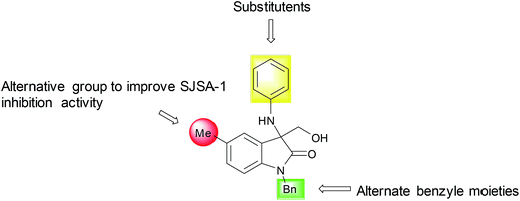 |
| | Scheme 2 Library optimization strategy for 3-amino-3-hydroxymethyloxindoles to improve SJSA-1 inhibition activity. | |
Table 1 Inhibitory activity of compounds 4a–4i and 5a–5m on SJSA-1 cell proliferation present as inhibition (%) and IC50 (μM)a
| Compound |
R1 |
R2 |
R3 |
Inhibition (%) |
IC50 (μM) |
| Inhibition of SJSA cell proliferation produced by the tested compounds at 10 μM. The IC50 of compounds were not determined since the inhibition rate at 10 μM was lower than 50%. |
| 4a |
Me |
H |
2,6-Di-Cl |
16.14 |
—b |
| 4b |
Bn |
H |
2,6-Di-Cl |
32.69 |
— |
| 4c |
Bn |
5-Br |
2,6-Di-Cl |
90.78 |
5.74 |
| 4d |
Bn |
H |
2,4-Di-Cl |
−1.73 |
— |
| 4e |
Bn |
H |
2-I |
41.59 |
— |
| 4f |
Bn |
H |
4-Cl |
89.23 |
3.5 |
| 4g |
Bn |
H |
4-Br |
41.46 |
— |
| 4h |
Bn |
H |
4-I |
69.46 |
2.94 |
| 4i |
Bn |
H |
4-NO2 |
97.12 |
10.81 |
| 5a |
Bn |
H |
2-F |
7.51 |
— |
| 5b |
Bn |
H |
3,5-Di-CF3 |
37.90 |
— |
| 5c |
Bn |
H |
3,4-Di-F |
23.66 |
— |
| 5d |
Bn |
H |
2-Cl-4-F |
25.83 |
— |
| 5e |
Bn |
H |
2,6-Di-F |
−4.67 |
— |
| 5f |
Me |
H |
2,6-Di-F |
7.29 |
— |
| 5g |
Me |
5-F |
2,6-Di-F |
19.86 |
— |
| 5h |
Bn |
5-Me |
2,6-Di-F |
38.56 |
— |
| 5i |
4-Me–Bn |
H |
2,6-Di-F |
90.08 |
6.99 |
| 5j |
4-Me–Bn |
H |
2,4-Di-F |
81.03 |
6.19 |
| 5k |
Bn |
H |
2,4-Di-F |
25.31 |
— |
| 5l |
Bn |
5-F |
2,4-Di-F |
48.41 |
— |
| 5m |
Bn |
5-Me |
2,4-Di-F |
93.65 |
3.14 |
Biological activities
Anti-proliferative activity evaluation. Our random screening had found some of 3-amino-3-hydroxymethyloxindoles had antitumor activity. To obtain new compounds with anticancer activity, CCK-8 assay was used to measure the cytotoxicity of all the known and newly-synthesized substituted 3-amino-3-hydroxymethyloxindoles in SJSA-1 cells. The results are summarized in Table 1. A total of seven compounds had good growth inhibitory activity when used at 10 μM. These compounds were further measured their IC50 values of cytotoxic effects. The IC50 value ranged from 2.94 to 10.81 μM. Among them, compounds 4h and 5m showed the most potent inhibitory effects with IC50 values of about 3 μM.Regarding the structure–activity relationship, several correlations can be made from these data. Compound 4c with 5-Br substituent on oxindole ring has shown more inhibition activity than 4b. Through the evaluation of R1 group, with benzyl (4b) moiety replaced by methyl group (4a), a previous suppression on the inhibition percentage has been observed. As the affection of R2 group was concerned, we attempted to refine the hydrogen (5e, 5f) with fluorine atom (5g, 5l) and methyl group (5h, 5m). We were pleased to find a considering increase on the cytotoxic activity when methyl group (5h, 5m) appears on R1 position. So, with this optimal condition in hand, we introduced a series of substitute group on R3 phenyl group. After comparison of the bioactivity data, the 2,4-difluorine-phenyl group was considered competent in the R3 position. Another noteworthy point is 4-Cl (4f) and 4-I (4h) were expelled from preferable list due to its high IC50 value which failed to satisfy our basic requirement even though they show off significantly valuable activity on SJSA-1 inhibition percentage test.
To ensure if these compounds had broad-spectrum anti-cancer activity and preference of killing cancer cells over normal cells, the IC50 of proliferation inhibition was also measured in other four cancer cell lines and two normal cell lines. Compound 5m also showed some extent of anti-proliferation activities in all the four cells lines (IC50 = 5.1 μM, 5.38 μM, 18.35 μM, 25.04 μM for HCT116, Jurkat, KB, Bel7402 cells, respectively, Table 2). The IC50 of cytotoxicity in HL7702 and HEK293 cells is 39.19 μM and 11.83 μM, respectively. Therefore, 5m showed better effects of growth inhibition in SJSA-1, HCT116 and Jurkat cells than other cancer and normal cells.
Table 2 In vitro cytotoxicity of compound 5m on five human cancer cell lines and two normal cell lines
| Cell |
SJSA-1 |
HCT116 |
Jurkat |
KB |
Bel7402 |
HEK293 |
HL7702 |
| IC50 (μM) |
3.14 |
5.1 |
5.38 |
18.35 |
25.04 |
11.83 |
39.19 |
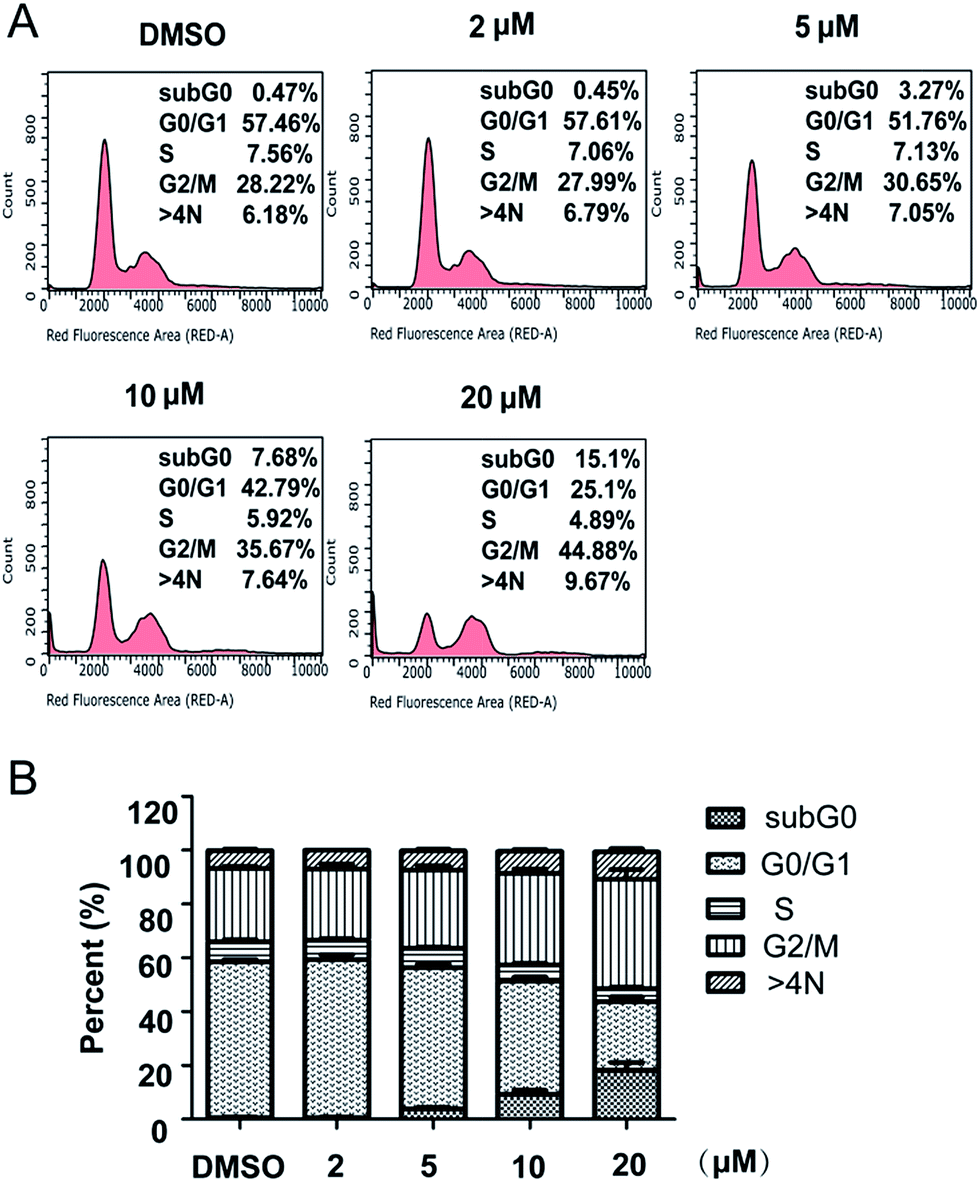
Cell cycle analysis. It is well known that inhibitors of cell proliferation work by interfering with cell cycle or inducing cell death. In order to identify whether compound 5m has any effects on cell cycle, SJSA-1 cells were treated with compound 5m at different concentration (0 μM, 2 μM, 5 μM, 10 μM or 20 μM) for 48 h, the cells were collected, stained with PI, and analyzed their cell cycle distribution by flow cytometry. The results showed that 57.46% of cells were in the G0/G1 phrase, 7.56% in the S phrase, and 28.22% in the G2/M phrase in the control group (Fig. 1A and B). On the contrast, a significant decreased percentage of cells were in G0/G1 phrase 48 h following compound 5m treatment at higher concentration (51.76%, 42.79% and 25.1% for 5 μM, 10 μM and 20 μM, respectively). Meanwhile, the proportion of cells in G2/M phrase were significantly increased (30.65%, 35.67% and 44.88% for 5 μM, 10 μM and 20 μM, respectively) (Fig. 1A and B). These results suggested that compound 5m treatment induced cell cycle arrest at G2/M in a dose-dependent manner, which might be one of the primary mechanisms responsible for the anti-cancer activity of the compound.
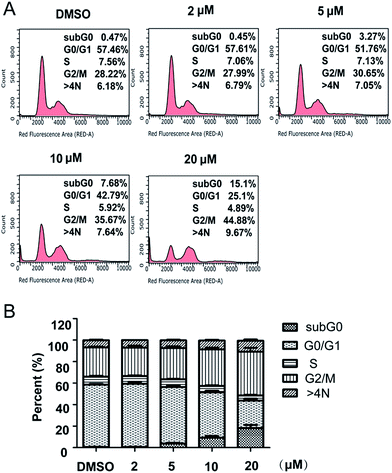 |
| | Fig. 1 Effect of compound 5m on cell cycle progression in SJSA-1 cells. Cells were treated with compound 5m at 0, 2, 5, 10 or 20 μM for 48 h, stained with PI. Then cell cycle was analyzed by flow cytometry. (A) Representative photographs from three independent experiments were displayed. (B) Cell cycle proportion in (A) was quantitated. Data presented are the mean ± SD of three independent experiments. | |
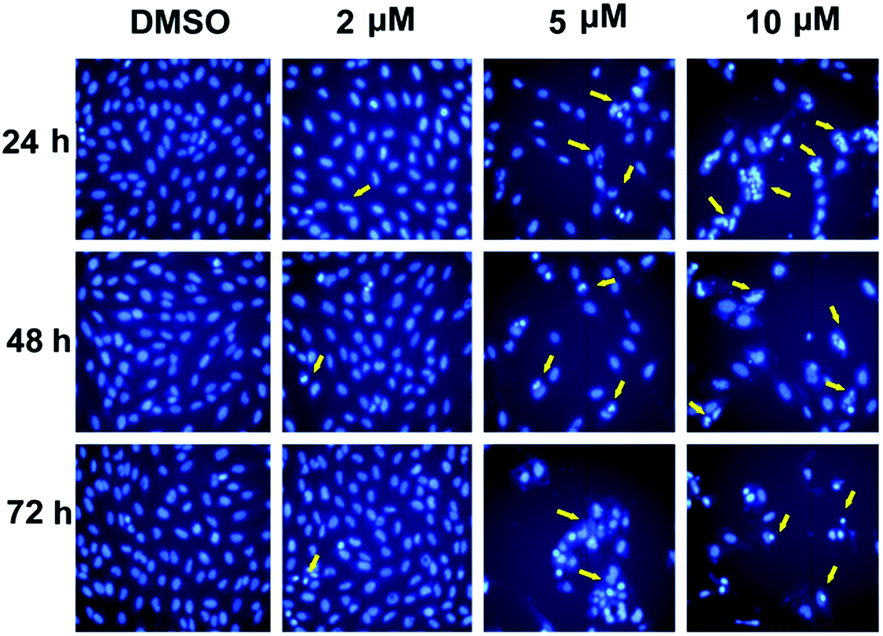
Apoptosis induction. In the experiment of cell cycle analysis by flow cytometry, we observed that more and more compound-treated cells entered subG0 phrase (3.27%, 7.68% and 15.1% for 5 μM, 10 μM and 20 μM, respectively; compared with control, 0.47%) (Fig. 1), a phrase related to cell apoptosis, which suggested that compound 5m could also induced cell apoptosis. In order to further confirm the effect, SJSA-1 cells were treated with 5m at different concentrations (0 μM, 2 μM, 5 μM or 10 μM) and Hoechst 33342 staining using microscopy were carried out 24 h, 48 h or 72 h after treatment. In the control cells (treated with only the vehicle DMSO), the cells kept growing without visible apoptosis. On the contrast, shrunken nucleus and peripherally clumped and fragmented chromatin were observed following compound treatment at higher concentration (5 μM and 10 μM), a typical characteristic of cells undergoing apoptosis (Fig. 2).
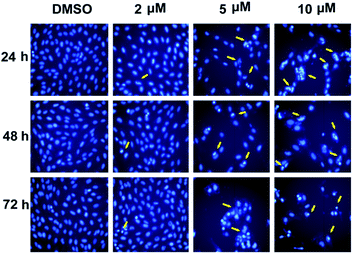 |
| | Fig. 2 Compound 5m induced apparent cell apoptosis. Cells were treated with compound 5m at 0, 2, 5, or 10 μM for 24 h, 48 h or 72 h and nuclear morphology was checked by Hoechst 33342 staining by microscopy at 200× and representative photographs were captured. The yellow arrows stand for nucleus of apoptotic cells. | |
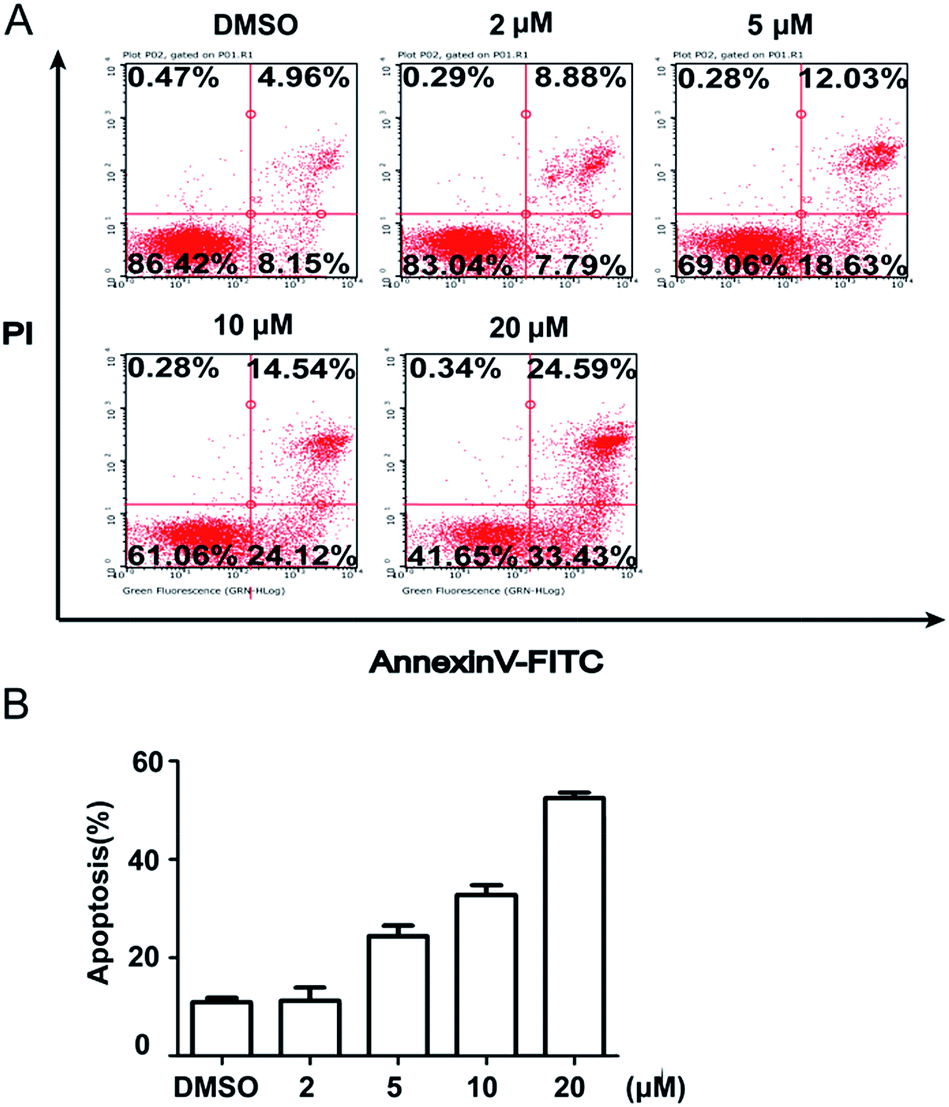
To further quantify the effects of 5m-induced apoptosis, Annexin V–FITC and PI dual staining were performed using flow cytometry. The analysis of stained cells distinguished between four groups: (1) viable cells (Annexin V−, PI−); (2) early apoptotic cells (Annexin V+, PI−); (3) late apoptotic cells (Annexin V+, PI+); (4) necrotic cells (Annexin V−, PI+). Firstly, we detected the time-dependent apoptosis. SJSA-1 cells were treated by 5m at 10 μM or 20 μM for 24 h, 48 h or 72 h and then were subject to apoptosis evaluation. The results showed that the percentage of apoptotic cells (including early apoptotic and late apoptotic population) at both concentrations was elevated as the treatment time was extended (Fig. 3). In another experiment, 5m at different concentration were added into SJSA-1 cells and its effects were analyzed after 48 h. Similarly, 5m at high concentration induced more apoptotic cells than lower concentration (2 μM, 16.67%; 5 μM, 30.66%; 10 μM, 38.66%; 20 μM, 58.02%) (Fig. 4), suggesting a dose-dependent effect. Therefore, compound 5m induced apoptosis of SJSA-1 cells in a time- and dose-dependent manner. Moreover, western blotting experiments were performed to study the underlying mechanism of 5m-induced apoptosis. Our preliminary data (refer ESI†) showed that 5m caused the ratio of Bax/Bcl-2, two apoptosis-related proteins, to be increased, suggesting that the intrinsic mitochondrial apoptosis pathway, rather than the extrinsic pathway, was involved in 5m-induced apoptosis. Further investigations are essential to illuminate the accurate mechanism.
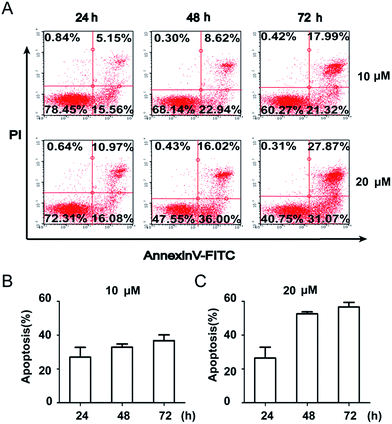 |
| | Fig. 3 Time-dependent cellular apoptosis. Cells were treated with compound 5m at 10 or 20 μM for 24 h, 48 h or 72 h and apoptosis was evaluated using Annexin V–FITC/PI dual staining kit by flow cytometry. (A) Representative photographs from three independent experiments were displayed. (B) Cell apoptosis rates were calculated and plotted in response to different time. | |
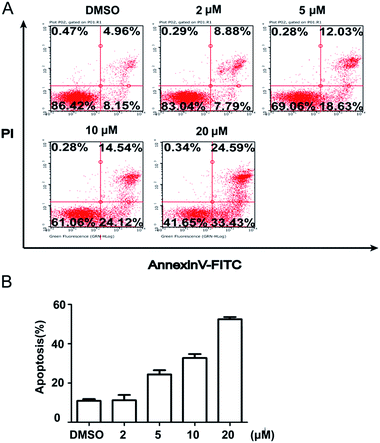 |
| | Fig. 4 Concentration-dependent cellular apoptosis. Cells were treated with compound 5m at 0, 2, 5, 10 or 20 μM for 48 h and apoptosis was evaluated using Annexin V–FITC/PI dual staining kit by flow cytometry. (A) Representative photographs from three independent experiments were displayed. (B) Cell apoptosis rates were calculated and plotted in response to different concentrations. | |
Conclusion
In this study, we synthesized a potent series of oxindole compounds (4a–4i, 5a–5m) using MCR approach.11 These compounds were screened for anticancer activity using the CCK-8 assay. Among them, compound 4h and 5m displayed the most potent anti-proliferating effect against SJSA-1 cells (IC50 ≈ 3.0 μM). Besides, 5m showed broad-spectrum anti-cancer activity because it can inhibit the proliferation of other cancer cells. The flow cytometry results showed that 5m inhibited the cell growth and caused the cell cycle to be arrested at the G2/M phase. Meanwhile, 5m induced apoptosis of SJSA-1 cells in a dose- and time-dependent manner. These data proved the potent cell proliferation inhibitory abilities and pro-apoptotic activities of compound 5m in SJSA-1 cells. To sum up, we regarded compound 5m as a promising, potent anti-cancer candidate among the 3-amino-3-hydroxymethyloxindoles studied. Further study on the in vivo anticancer effects of 5m compound and its underlying mechanism is in progress.
Materials and methods
Chemicals
Diazocompound 1 was prepared according to the literature methods.12–14 All isolated compounds were characterized on the basis of 1H NMR and 13C NMR spectroscopic data and HRMS (TOF-Q) data. 1H NMR and 13C NMR chemical shifts are reported in ppm using tetramethylsilane (TMS) as an internal standard.
General procedure for the synthesis of 3-amino-3-hydroxymethyloxindoles (4a–4i and 5a–5m)
To a mixture of Rh2(OAc)4 (0.001 mmol), aniline 2 (0.10 mmol), formalin 3 (37% aqueous solution of formaldehyde, 0.60 mmol) in EtOAc (1.0 mL) at 60 °C and diazo compound 1 (0.1 mmol) in EtOAc (1.0 mL) were added via a syringe pump for 1 h. After the completion of the addition, the reaction mixture was stirred for additional 1 h. Solvents were removed under reduced pressure to give the crude products. The crude products were purified by flash chromatography on silica gel (ethyl acetate/petroleum ether = 1:10 to 1:1) to give pure products. The compounds (4a–4i) were prepared according to literature methods.11
Cell culture
Five human cancer cell lines and two human normal cells were used in the study, including SJSA-1 (osteosarcoma), HCT116 (colon cancer), Bel7402 (liver cancer), KB (Oral epithelial carcinoma), Jurkat (human T lymphocyte leukaemia), HL7702 (normal liver) and HEK293 cells (embryonic kidney). They were purchased from Cell Bank of China Science Academy (Shanghai, China), and cultured aseptically using the corresponding media supplemented with 10% (v/v) fetal bovine serum and 100 units per mL each of penicillin and streptomycin, pH 7.2 and 5% CO2 humidified atmosphere at 37 °C.
Cell viability assay (cck-8 assay)
In vitro cytotoxicity of the compounds was evaluated by the CCK-8 assay. Briefly, SJSA-1 cells were respectively trypsinized and seeded in 96-well plates at a proper density the day before compound administration. Each tested compound was dissolved in DMSO (10 mM) and diluted in media. Then the compound was added to the cells at 10 μM. The control cells were treated with the vehicle DMSO. After 72 h incubation, 10 μL of CCK8 solution (5 g L−1; Yeasen) in the media was added to each well and incubated for additional 4 h. Finally, the optical density (OD) was measured at 450 nm using a microplate reader (spectraMax M5/M5e, Sunnyvale, CA, USA) and a reference wavelength at 620 nm. If one compound inhibits cell proliferation with percentage inhibition higher than 50%, the IC50 value was determined by testing the inhibitory effects of the compound with 10 gradient-dilution concentrations with at least three replicates per concentration. As for the compound with the best growth-inhibitory activity, its IC50 value in other six cell lines (HCT116, Bel7402, KB, Jurkat, HL7702 and HEK293) were further determined.
Cell cycle analysis
Cell cycle analysis was performed to check the distribution of SJSA-1 cells in different phases caused by compound 5m using propidium iodide (PI) staining method. SJSA-1 cells were plated in 6-well plates (1 × 105 cells per well), and allowed to grow to about 70% to 80% confluence and then the cells were exposed to compound 5m at a concentration of 0, 2, 5, 10 or 20 μM, respectively. After 48 h, cells were trypsinized, washed with cold PBS, and permeabilized with 70% ethanol overnight. On the next day, the cells were stained with PI (Beyotime) according to the manufacturer's instructions. Then cells were measured using an easyCyte 6HT-2L (Millipore) and analyzed by FlowJo software. All experiments were conducted in triplicate, and for each measurement, at least 20000 cells were counted.
Hoechst staining
Nuclear morphology was detected by staining nuclei with Hoechst 33342. SJSA-1 cells (1 × 104) were seeded in twenty-four-well plates for 24 h. Thereafter, cells were treated with compound 5m at different concentration for 24 h, 48 h or 72 h. Cells were washed with PBS and fixed with 4% paraformaldehyde. Cells were stained with Hoechst 33342 and images were captured using fluorescence microscope (Leica).
Annexin V–FITC/PI dual staining
To determine effect of compound 5m on apoptosis in SJSA-1 cells, Annexin V–FITC/PI dual staining was carried out by flow cytometry. SJSA-1 cells (1 × 105 cells per well) were seeded in six-well plates and allowed to grow to 70 to 80% confluence. Then the cells were treated with compound 5m at concentrations of 0, 2, 5, 10 or 20 μM for 48 h, respectively (dose–response experiment) or 10 μM and 20 μM for 24 h, 48 h or 72 h, respectively (time–response experiment). At the end of the incubation, the cells were harvested by trypsinization and washed twice with cold PBS. Then the cells were resuspended in 400 μL of 1× binding buffer and stained with Annexin V–FITC and PI using the apoptosis detection kit from Beyotime. Cells were measured using an easyCyte 6HT-2L (Millipore) and analyzed by FlowJo software. The percentage of cells for the lower and upper right quadrant was used for statistical analysis using Graphpad Prism 5. All experiments were conducted in triplicate, and for each measurement, at least 1 × 104 cells were counted.
1-Benzyl-3-((2-fluorophenyl)amino)-3-(hydroxymethyl)indolin-2-one (5a). A white solid (73% yield).1H NMR (400 MHz, CDCl3) δ 7.30 (qd, J = 6.6, 3.3 Hz, 7H), 7.08 (t, J = 7.5 Hz, 1H), 6.96 (ddd, J = 11.6, 8.0, 1.4 Hz, 1H), 6.88 (d, J = 7.8 Hz, 1H), 6.65–6.58 (m, 1H), 6.53 (t, J = 7.6 Hz, 1H), 5.78–5.71 (m, 1H), 5.47 (d, J = 3.2 Hz, 1H), 5.12 (d, J = 15.4 Hz, 1H), 4.80 (d, J = 15.4 Hz, 1H), 3.97 (t, J = 11.4 Hz, 1H), 3.73 (dd, J = 11.5, 2.5 Hz, 1H), 2.91 (dd, J = 11.3, 2.6 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 177.58, 153.55, 151.17, 141.86, 135.32, 134.03 (d, J = 11.2 Hz), 129.77, 128.87, 127.98, 127.72, 127.25, 124.23 (d, J = 3.7 Hz), 124.03, 123.64, 118.96 (d, J = 7.2 Hz), 114.91, 114.73, 114.30 (d, J = 2.4 Hz), 110.11, 99.99, 68.07, 64.38, 44.18. HRMS (ESI) calcd for C22H19FN2O2 [M + Na]+: 385.1328, found 385.1320.
1-Benzyl-3-((3,5-bis(trifluoromethyl)phenyl)amino)-3-(hydroxymethyl)indolin-2-one (5b). A white solid (85% yield). 1H NMR (400 MHz, CDCl3) δ 7.38–7.26 (m, 7H), 7.12 (dd, J = 14.3, 6.4 Hz, 2H), 6.91 (d, J = 7.8 Hz, 1H), 6.61 (s, 2H), 5.63 (s, 1H), 5.06–4.88 (m, 2H), 3.95 (t, J = 11.6 Hz, 1H), 3.75 (dd, J = 11.6, 1.8 Hz, 1H), 2.90 (dd, J = 11.1, 1.8 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 176.90, 146.36, 141.92, 134.95, 132.34 (d, J = 32.9 Hz), 130.40, 129.06, 128.03, 127.15, 125.60, 124.52, 123.94 (d, J = 8.6 Hz), 121.81, 114.03 (q, J = 3.8, 2.3 Hz), 112.26, 110.51, 67.95, 64.27, 44.36. HRMS (ESI) calcd for C24H18F6N2O2 [M + Na]+: 503.1170, found 503.1163.
1-Benzyl-3-((3,4-difluorophenyl)amino)-3-(hydroxymethyl)indolin-2-one (5c). A white solid (67% yield). 1H NMR (400 MHz, CDCl3) δ 7.40–7.27 (m, 5H), 7.21 (d, J = 7.0 Hz, 2H), 7.10 (t, J = 7.4 Hz, 1H), 6.86 (d, J = 7.8 Hz, 1H), 6.74 (dd, J = 18.6, 9.2 Hz, 1H), 6.13–5.93 (m, 2H), 5.12 (d, J = 15.2 Hz, 2H), 4.75 (d, J = 15.4 Hz, 1H), 3.91 (t, J = 11.4 Hz, 1H), 3.69 (d, J = 11.3 Hz, 1H), 2.94 (d, J = 11.1 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 177.51, 142.46 (dd, J = 8.2, 2.0 Hz), 141.90, 135.12, 129.97, 128.93, 128.02, 127.45, 126.87, 123.86 (d, J = 24.2 Hz), 117.35 (d, J = 18.5 Hz), 111.31 (dd, J = 5.6, 3.3 Hz), 110.30, 104.74 (d, J = 20.6 Hz), 67.88, 65.13, 44.13. HRMS (ESI) calcd for C22H18F2N2O2 [M + Na]+: 403.1234, found 403.1233.
1-Benzyl-3-((2-chloro-4-fluorophenyl)amino)-3-(hydroxymethyl)indolin-2-one (5d). A white solid (53% yield). 1H NMR (400 MHz, CDCl3) δ 7.37–7.27 (m, 7H), 7.09 (t, J = 7.5 Hz, 1H), 7.03 (dd, J = 8.2, 2.5 Hz, 1H), 6.91 (d, J = 7.7 Hz, 1H), 6.37 (td, J = 8.6, 2.6 Hz, 1H), 5.72 (s, 1H), 5.64 (dd, J = 8.9, 5.1 Hz, 1H), 5.13 (d, J = 15.3 Hz, 1H), 4.77 (d, J = 15.3 Hz, 1H), 3.98 (t, J = 11.4 Hz, 1H), 3.73 (dd, J = 11.5, 1.7 Hz, 1H), 2.88 (dd, J = 11.3, 1.8 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 177.27, 141.82, 138.47 (d, J = 2.5 Hz), 135.29, 129.90, 128.89, 128.10, 127.80, 126.94, 123.98, 123.75, 116.76 (d, J = 25.7 Hz), 113.93 (dd, J = 20.8, 14.9 Hz), 110.16, 68.05, 64.78, 44.20. HRMS (ESI) calcd for C22H18ClFN2O2 [M + Na]+ = 419.0939, found 419.0919.
1-Benzyl-3-((2,6-difluorophenyl)amino)-3-(hydroxymethyl)indolin-2-one (5e). A white solid (77% yield). 1H NMR (400 MHz, CDCl3) δ 7.30 (dd, J = 12.0, 4.8 Hz, 3H), 7.25–7.12 (m, 4H), 6.97 (t, J = 7.5 Hz, 1H), 6.82–6.57 (m, 4H), 5.02–4.82 (m, 3H), 3.95–3.82 (m, 2H), 2.82 (dd, J = 9.5, 4.0 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 177.34, 154.81 (dd, J = 242.9, 6.7 Hz), 142.62, 135.56, 129.36, 128.77, 127.70, 127.59, 127.31, 123.74, 122.75, 120.58 (t, J = 9.5 Hz), 111.28 (dd, J = 17.2, 7.0 Hz), 109.58, 68.35, 65.92, 44.04. HRMS (ESI) calcd for C22H18F2N2O2 [M + Na]+ = 403.1234, found 403.1224.
3-((2,6-Difluorophenyl)amino)-3-(hydroxymethyl)-1-methylindolin-2-one (5f). A white solid (89% yield). 1H NMR (400 MHz, CDCl3) δ 7.41 (t, J = 7.7 Hz, 1H), 7.33 (d, J = 7.3 Hz, 1H), 7.14 (t, J = 7.5 Hz, 1H), 6.97 (d, J = 7.8 Hz, 1H), 6.75 (ddd, J = 11.2, 8.5, 2.8 Hz, 1H), 6.43–6.33 (m, 1H), 5.73 (td, J = 9.3, 5.4 Hz, 1H), 5.30 (s, 1H), 5.27 (d, J = 1.8 Hz, 1H), 3.92 (t, J = 11.3 Hz, 1H), 3.68 (dd, J = 11.5, 2.5 Hz, 1H), 3.28 (s, 3H), 2.82 (dd, J = 11.2, 2.5 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 177.31, 156.80, 142.77, 130.57, 130.00, 127.12, 124.00, 123.70, 114.29 (dd, J = 8.9, 3.6 Hz), 110.49 (dd, J = 21.8, 3.9 Hz), 109.08, 103.76 (dd, J = 26.6, 23.3 Hz), 67.84, 64.65, 53.50, 26.46. HRMS (ESI) calcd for C16H14F2N2O2 [M + Na]+ = 327.0921, found 327.0934.
3-((2,6-Difluorophenyl)amino)-5-fluoro-3-(hydroxymethyl)-1-methylindolin-2-one (5g). A white solid (69% yield). 1H NMR (400 MHz, CDCl3) δ 7.00 (td, J = 8.9, 2.6 Hz, 1H), 6.92 (dd, J = 7.5, 2.6 Hz, 1H), 6.79 (dd, J = 8.5, 4.0 Hz, 1H), 6.72–6.62 (m, 3H), 4.95 (s, 1H), 3.88–3.75 (m, 2H), 3.22 (s, 3H), 2.93 (dd, J = 8.6, 4.7 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 177.13 (d, J = 0.9 Hz), 160.38, 157.98, 155.48 (d, J = 6.8 Hz), 153.07 (d, J = 6.6 Hz), 139.34, 129.85–129.57 (m), 122.06 (t, J = 15.0 Hz), 120.25 (t, J = 9.6 Hz), 115.71, 115.47, 112.06, 111.81, 111.45, 111.38, 111.28, 111.20 (d, J = 1.7 Hz), 109.06 (d, J = 8.0 Hz), 68.02, 66.01, 26.55. HRMS (ESI) calcd for C16H13F3N2O2 [M + Na]+ = 345.0827, found 345.0817.
1-Benzyl-3-((2,6-difluorophenyl)amino)-3-(hydroxymethyl)-5-methylindolin-2-one (5h). A white solid (75% yield). 1H NMR (400 MHz, CDCl3) δ 7.32–7.27 (m, 2H), 7.26–7.19 (m, 3H), 7.05–6.94 (m, 2H), 6.80–6.64 (m, 3H), 6.62 (d, J = 7.9 Hz, 1H), 5.01–4.80 (m, 3H), 3.93–3.79 (m, 2H), 2.86 (dd, J = 9.9, 3.7 Hz, 1H), 2.24 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 177.23, 155.91 (d, J = 6.7 Hz), 153.50 (d, J = 6.7 Hz), 140.14, 135.66, 132.44, 129.59, 128.73, 127.79, 127.64, 127.30, 124.46, 122.40 (t, J = 15.3 Hz), 120.39 (t, J = 9.5 Hz), 111.30 (dd, J = 17.1, 7.0 Hz), 109.32, 68.40, 65.88, 44.04, 21.01. HRMS (ESI) calcd for C23H20F2N2O2 [M + Na]+ = 417.1391, found 417.1376.
3-((2,6-Difluorophenyl)amino)-3-(hydroxymethyl)-1-(4-methylbenzyl)indolin-2-one (5i). A white solid (67% yield). 1H NMR (400 MHz, CDCl3) δ 7.32 (dd, J = 15.1, 7.6 Hz, 2H), 7.18–7.04 (m, 5H), 6.88 (d, J = 7.8 Hz, 1H), 6.77 (ddd, J = 11.1, 8.6, 2.7 Hz, 1H), 6.28 (t, J = 8.5 Hz, 1H), 5.70 (td, J = 9.3, 5.5 Hz, 1H), 5.32 (d, J = 2.8 Hz, 1H), 5.09 (d, J = 15.3 Hz, 1H), 4.70 (d, J = 15.3 Hz, 1H), 3.96 (t, J = 11.5 Hz, 1H), 3.72 (dd, J = 11.5, 2.2 Hz, 1H), 2.90 (dd, J = 11.4, 2.2 Hz, 1H), 2.33 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 177.47, 141.92, 137.82, 132.17, 130.46 (dd, J = 11.5, 2.6 Hz), 129.88, 129.52, 127.67, 127.13, 124.05, 123.64, 115.10 (dd, J = 8.8, 3.5 Hz), 110.41 (dd, J = 21.9, 3.8 Hz), 110.23, 103.80 (dd, J = 26.5, 23.5 Hz), 67.86, 64.80, 43.89, 21.14. HRMS (ESI) calcd for C22H20F2N2O2 [M + Na]+ = 417.1391, found 417.1385.
3-((2,4-Difluorophenyl)amino)-3-(hydroxymethyl)-1-(4-methylbenzyl)indolin-2-one (5j). A white solid (83% yield). 1H NMR (400 MHz, CDCl3) δ 7.31 (dd, J = 15.6, 7.2 Hz, 2H), 7.21–6.98 (m, 5H), 6.88 (d, J = 7.8 Hz, 1H), 6.76 (ddd, J = 11.2, 8.5, 2.8 Hz, 1H), 6.28 (ddd, J = 10.6, 3.2, 1.9 Hz, 1H), 5.71 (td, J = 9.3, 5.5 Hz, 1H), 5.31 (d, J = 2.1 Hz, 1H), 5.08 (d, J = 15.3 Hz, 1H), 4.70 (d, J = 15.3 Hz, 1H), 3.96 (t, J = 11.4 Hz, 1H), 3.72 (dd, J = 11.5, 2.4 Hz, 1H), 2.89 (dd, J = 11.3, 2.4 Hz, 1H), 2.33 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 177.45, 141.97, 137.81, 132.18, 130.46 (dd, J = 11.3, 4.1 Hz), 129.96, 129.81, 129.64, 129.39, 127.78, 127.53, 127.14, 124.11, 123.97, 123.75, 123.51, 110.22 (d, J = 32.0 Hz), 64.77, 43.90 (dd, J = 20.6, 13.2 Hz), 21.12 (d, J = 18.8 Hz). HRMS (ESI) calcd for C23H20F2N2O2 [M + Na]+ = 417.1391, found 417.1382.
1-Benzyl-3-((2,4-difluorophenyl)amino)-3-(hydroxymethyl)indolin-2-one (5k). A white solid (53% yield). 1H NMR (400 MHz, CDCl3) δ 7.39–7.27 (m, 5H), 7.24–7.17 (m, 2H), 7.10 (t, J = 7.5 Hz, 1H), 6.87 (d, J = 7.8 Hz, 1H), 6.82–6.70 (m, 1H), 6.27 (t, J = 8.4 Hz, 1H), 5.72 (td, J = 9.2, 5.8 Hz, 1H), 5.31 (s, 1H), 5.11 (d, J = 15.4 Hz, 1H), 4.75 (d, J = 15.4 Hz, 1H), 3.97 (t, J = 11.4 Hz, 1H), 3.74 (d, J = 11.5 Hz, 1H), 2.89 (d, J = 11.1 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 177.48, 141.95, 135.24, 130.42 (dd, J = 11.6, 3.2 Hz), 129.89, 128.86, 128.03, 127.66, 127.18, 124.11, 123.68, 115.31 (dd, J = 8.8, 3.5 Hz), 110.41 (dd, J = 21.6, 3.9 Hz), 110.14, 103.80 (dd, J = 26.5, 23.4 Hz), 67.91, 64.87, 44.12. HRMS (ESI) calcd for C22H18F2N2O2 [M + Na]+ = 403.1234, found 403.1222.
1-Benzyl-3-((2,4-difluorophenyl)amino)-5-fluoro-3-(hydroxymethyl)indolin-2-one (5l). A white solid (86% yield). 1H NMR (400 MHz, CDCl3) δ 7.35–7.27 (m, 3H), 7.24–7.16 (m, 2H), 7.10 (dd, J = 7.4, 2.5 Hz, 1H), 6.98 (td, J = 8.8, 2.6 Hz, 1H), 6.85–6.71 (m, 2H), 6.36–6.26 (m, 1H), 5.72 (td, J = 9.2, 5.4 Hz, 1H), 5.28 (s, 1H), 5.11 (d, J = 15.4 Hz, 1H), 4.73 (d, J = 15.4 Hz, 1H), 3.94 (t, J = 11.3 Hz, 1H), 3.75 (dd, J = 11.4, 2.4 Hz, 1H), 2.88 (dd, J = 11.1, 2.4 Hz, 1H). 13C NMR (100 MHz, CDCl3) δ 177.20, 137.77, 134.91, 129.09, 129.02 (d, J = 1.0 Hz), 128.94 (d, J = 3.0 Hz), 128.88, 128.26, 127.75, 127.54, 115.39 (d, J = 3.5 Hz), 115.18, 112.59 (d, J = 6.7 Hz), 112.34, 112.06, 111.07, 110.72 (dd, J = 5.0, 1.9 Hz), 104.14 (d, J = 2.0 Hz), 65.15 (d, J = 1.8 Hz), 44.31, 17.99. HRMS (ESI) calcd for C22H17F3N2O2 [M + Na]+ = 421.1140, found 421.1142.
1-Benzyl-3-((2,4-difluorophenyl)amino)-3-(hydroxymethyl)-5-methylindolin-2-one (5m). A white solid (75% yield). 1H NMR (400 MHz, CDCl3) δ 7.35–7.26 (m, 3H), 7.22 (dd, J = 7.0, 2.3 Hz, 2H), 7.15 (s, 1H), 7.08 (d, J = 8.0 Hz, 1H), 6.83–6.68 (m, 2H), 6.38–6.19 (m, 1H), 5.72 (td, J = 9.3, 5.5 Hz, 1H), 5.30 (d, J = 6.6 Hz, 1H), 5.09 (d, J = 15.4 Hz, 1H), 4.72 (d, J = 15.4 Hz, 1H), 4.72 (d, J = 15.4 Hz, 1H), 3.95 (t, J = 11.4 Hz, 1H), 3.71 (dd, J = 11.5, 1.8 Hz, 1H), 2.96 (d, J = 11.3 Hz, 1H), 2.29 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 177.39, 156.78 (d, J = 11.1 Hz), 154.40 (d, J = 11.0 Hz), 153.31 (d, J = 11.6 Hz), 150.89 (d, J = 11.7 Hz), 139.47, 135.36, 133.48, 130.54 (dd, J = 11.5, 3.2 Hz), 130.19, 128.82, 127.98, 127.70, 127.13, 115.08 (dd, J = 8.8, 3.6 Hz), 110.41 (dd, J = 21.7, 3.9 Hz), 109.94, 103.77 (dd, J = 26.6, 23.4 Hz), 67.93, 64.76, 44.14, 21.05. HRMS (ESI) calcd for C23H20F2N2O2 [M + Na]+ = 417.1391, found 417.1389.
Acknowledgements
We wish to thank the Natural Science Foundation of Shanghai (16ZR1410500) for financial support.
Notes and references
- R. L. Siegel, K. D. Miller and A. Jemal, Ca-Cancer J. Clin., 2015, 65, 5–29 CrossRef PubMed.
- A. S. Narang and D. D. Desai, in Pharmaceutical Perspectives of Cancer Therapeutics, ed, Y. Lu and R. I. Mahato, 2009, pp. 49–92 Search PubMed.
- S. L. Schreiber, Science, 2000, 287, 1964–1969 CrossRef CAS PubMed.
- E. Ruijter, R. Scheffelaar and R. V. Orru, Angew. Chem., Int. Ed., 2011, 50, 6234–6246 CrossRef CAS PubMed.
- Y. Xia, Y. Zhang and J. Wang, ACS Catal., 2013, 3, 2586–2598 CrossRef CAS.
- X. Guo and W. Hu, Acc. Chem. Res., 2013, 46, 2427–2440 CrossRef CAS PubMed.
- D. Xing and W. Hu, Tetrahedron Lett., 2014, 55, 777–783 CrossRef CAS.
- W. Hu, X. Xu, J. Zhou, W. J. Liu, H. Huang, J. Hu, L. Yang and L. Z. Gong, J. Am. Chem. Soc., 2008, 130, 7782–7783 CrossRef CAS PubMed.
- J. Jiang, H. D. Xu, J. B. Xi, B. Y. Ren, F. P. Lv, X. Guo, L. Q. Jiang, Z. Y. Zhang and W. H. Hu, J. Am. Chem. Soc., 2011, 133, 8428–8431 CrossRef CAS PubMed.
- H. Qiu, M. Li, L. Q. Jiang, F. P. Lv, L. Zan, C. W. Zhai, M. P. Doyle and W. H. Hu, Nat. Chem., 2012, 4, 733–738 CrossRef CAS PubMed.
- C. Wang, D. Xing, D. Wang, X. Wu and W. Hu, J. Org. Chem., 2014, 79, 3908–3916 CrossRef CAS PubMed.
- C. Marti and E. M. Carreira, J. Am. Chem. Soc., 2005, 127, 11505–11515 CrossRef CAS PubMed.
- S. Muthusamy, C. Gunanathan and M. Nethaji, J. Org. Chem., 2004, 69, 5631–5637 CrossRef CAS PubMed.
- M. P. Cava and D. R. Napier, J. Am. Chem. Soc., 1957, 79, 1701–1705 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27536b |
| ‡ These authors contribute to this paper equally. |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*