
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bio-inspired enantioselective full transamination using readily available cyclodextrin†‡
Shiqi Zhanga,
Guangxun Lib,
Hongxin Liub,
Yingwei Wanga,
Yuan Caoa,
Gang Zhao*a and
Zhuo Tang *b
*b
aSchool of Chemical Engineering, Sichuan University, Sichuan 610065, China. E-mail: gzhao@scu.edu.cn
bNatural Products Research Center, Chengdu Institution of Biology, Chinese Academy of Science, Chengdu, Sichuan 610041, China. E-mail: tangzhuo@cib.ac.cn
First published on 16th January 2017
Abstract
The mimics of vitamin B6-dependent enzymes that catalyzed an enantioselective full transamination in the pure aqueous phase have been realized for the first time through the establishment of a new “pyridoxal 5′-phosphate (PLP) catalyzed non-covalent cyclodextrin (CD)-keto acid inclusion complexes” system, and various optically active amino acids have been obtained.
Amino acids play important roles in almost all biological processes, and most of them are synthesized through transamination in the cell, which is promoted by transaminase (also known as amino acid aminotransferase).1 Transaminase is a pyridoxal-5′-phosphate-dependent enzyme that catalyzes aminotransferation from an amino acid (1) to a prochiral α-keto acid (3) with the assistance of its unique structure generating the corresponding new chiral amino acid (4), as shown in Fig. 1.2,3 Moreover, the original amino acid (1) is converted into the corresponding α-keto acid (2).
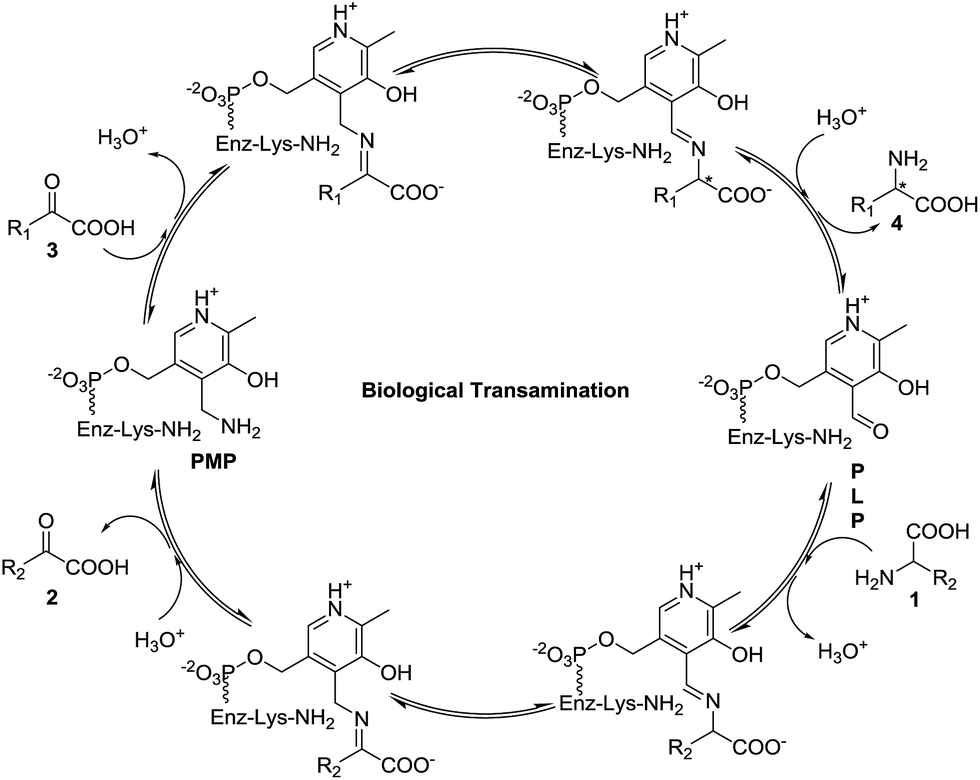 | ||
| Fig. 1 Biological transamination of α-keto acids. | ||
Inspired by biological transamination, extensive studies have been conducted to mimic this process for the purpose of obtaining optically active amino acids in an environmentally friendly method since the 1970s.4–8 Various strategies have been designed, which mainly focused on the enantioselective half-transamination,8–14 whereas rarely on the full transamination.
In prior research studies, addition of different Lewis acids (including copper, iron, aluminum, silver, indium, and zinc salts)8–14 to form chiral metal complexes with chiral ligands (L*)11–14 (Fig. 2A, M–L*) or chiral pyridoxamine (PM) derivatives5,8,15,16 (Fig. 2A, 5b), and application of various chiral pyridoxamine analogues through bounding with chiral basic side chains (Fig. 2A, 5c) or β-cyclodextrin (β-CD) (Fig. 2A, 5d) without metal ions were two most common strategies in the half-transamination reaction. Since the 1980s, various cyclodextrin-pyridoxamine artificial enzymes have been designed by covalently linking pyridoxamine with β-cyclodextrin at primary17 or secondary7 edges, and moderate enantioselectivity was obtained in half-transamination, attributed to the binding of the phenyl group in the β-CD cavity.18,19 Well-designed bifunctionalized cyclodextrins5,20,21 consisting of two cooperating units, a modified B6 coenzyme grouping, and an amino apoenzyme grouping (Fig. 2A, 5d), showed remarkable enantioselectivity in half-transamination, generating phenylalanine (Phe) with up to 96% ee's in MeOH. Employing disubstituted glycine as a sacrificial amine source instead of a monosubstituted α-amino acid, record turnover numbers (up to 100 turnovers) were obtained for the oxidative decarboxylation process with polyethylenimine (PEI) derivatives as enzyme mimics. Breslow and coworkers22 for the first time realized the non-enzymatic biomimic of the full transamination (Fig. 2B). Taking advantage of the abovementioned findings, the first chiral-pyridoxal-catalyzed asymmetric biomimetic transamination was achieved by the Zhao's group23 last year, where the chiral pyridoxal-diarylprolinol derivatives (Fig. 2B, 5f) were applied as catalysts in a mixed system of THF and H2O (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) (Fig. 2B). However, to date, enantioselective full transamination in the pure aqueous phase has not been realized.
:3) (Fig. 2B). However, to date, enantioselective full transamination in the pure aqueous phase has not been realized.
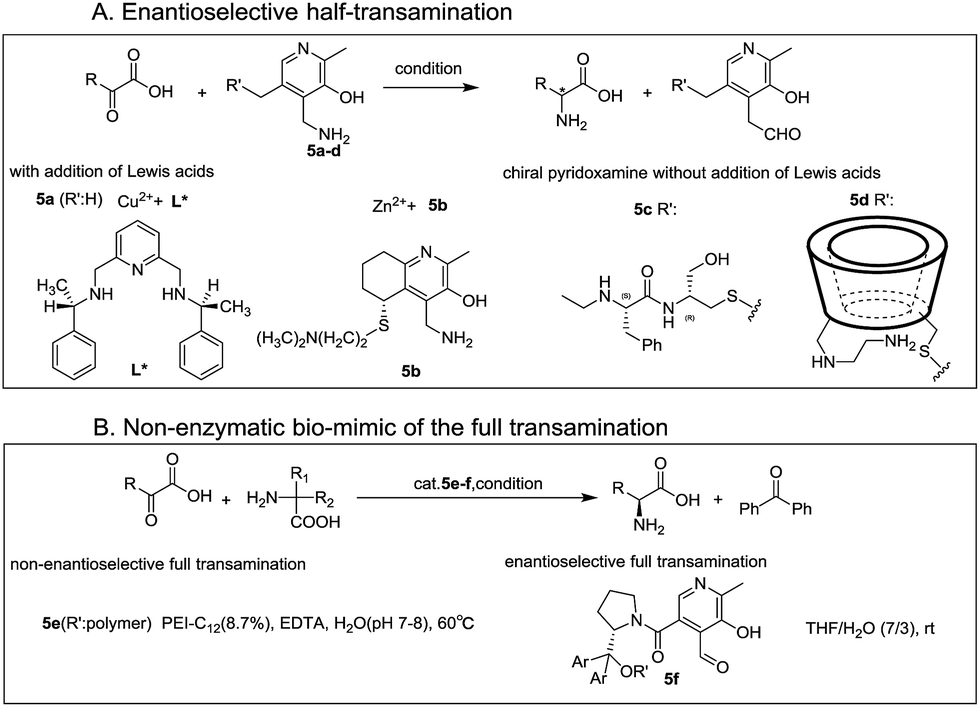 | ||
| Fig. 2 Vitamin B6-based biomimetic asymmetric transamination. | ||
Based on previous research, the bottlenecks of mimicking the enantioselective full transamination in the pure aqueous phase could be summarized as follows: (1) the creation of a local hydrophobic region to mimic the hydrophobic interaction between transaminase and substrates. Moreover, the whole process of transamination should take place in a less than a fully aqueous environment,24,25 (2) the selection of a suitable amine source to improve the relatively low turnovers of the entire cycle, which is composed of two reversible half-transamination processes, and (3) the introduction of an appropriate chiral source to realize the enantioselectivity of the products, and the crux of this is how to ensure the matching degree of a chiral source and the substrates. As shown, there are still plenty of unsolved problems. As a result, mimicking the enantioselective full transamination in the pure aqueous phase is extremely challenging and meaningful; therefore, we conducted a series of studies.
Inspired by Breslow's studies about employing a modified vitamin B6-β-CD (5d) as the artificial pyridoxamine enzyme for the asymmetry half-transamination20 (Fig. 2A) and designing a non-covalent PEI/PM system22 (Fig. 2B), we considered using well-defined commercially available β-CD instead of PEIs, whose structures are poorly defined to develop a non-bond system for a better transaminase mimic, due to the chirality and inclusion property of CD. Furthermore, 2,2-disubstituted glycines22,26 were applied as a sacrificial amine source instead of monosubstituted α-amino acids to improve the turnovers of the process by oxidative decarboxylation (Table 1). To realize the mimic of vitamin B6-dependent enzymes to catalyze the enantioselective full transamination in the pure aqueous phase, we developed a new non-bond system, which is easy to obtain with inexpensive raw materials.
|
|||||
|---|---|---|---|---|---|
| Entry | 7 | Buffer | CD | Yieldb (%) | eec (%) |
| a All reactions were carried out with phenylpyruvic acid (3a) (0.05 mmol), diphenylglycine (7) (0.05 mmol), CD (0.06 mmol), EDTA (0.01 mmol), and PLP (0.01 mmol) in 300 mM Tris buffer (4.0 mL), pH 8.0, at 50 °C for 36 h.b The yield was determined by chiral HPLC through the standard addition method27,28 (Table S1).c The ee's were determined by chiral HPLC (Chiralpak AD-H column) after the amines were converted into their N-Bz derivatives.29 The absolute configuration of 4a was assigned as S by comparison with the standard product.d 150 mM Tris buffer, pH 8.0.e 300 mM Tris buffer, pH 7.0.f 300 mM Tris buffer, pH 9.0. | |||||
| 1 | 7a (R2: Ph, M: H) | Tris buffer | β-CD | 9 | 20 |
| 2 | 7a | Carbonic acid buffer | β-CD | 4 | 16 |
| 3 | 7a | Phosphate buffer | β-CD | 5 | 9 |
| 4 | 7a | Tris bufferd | β-CD | 6 | 17 |
| 5 | 7a | Tris buffere | β-CD | 1 | 8 |
| 6 | 7a | Tris bufferf | β-CD | 3 | 24 |
| 7 | 7a | Tris buffer | α-CD | 2 | 5 |
| 8 | 7a | Tris buffer | γ-CD | 2 | 0 |
| 9 | 7b (R2: Me, M: H) | Tris buffer | β-CD | 11 | 19 |
| 10 | 7c (R2: Ph, M: Na+) | Tris buffer | β-CD | 27 | 20 |
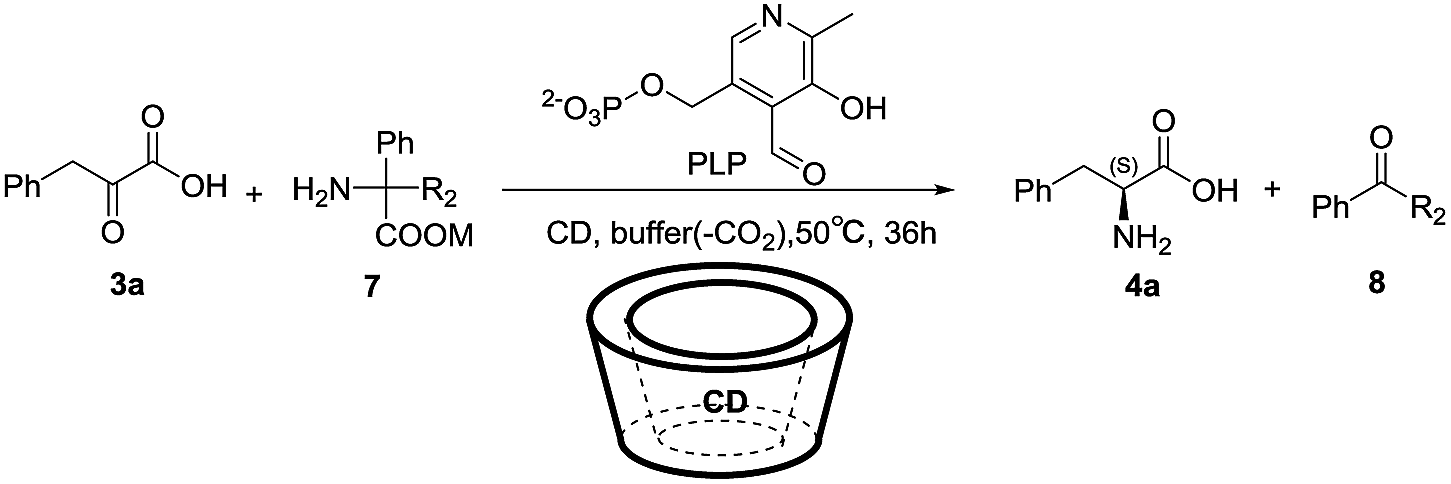
Our initial studies started with phenylpyruvic acid (3a) as a substrate, diphenylglycine (7a) as an amine donor, 1.2 equiv. of β-CD for chiral induction, and PLP as a catalyst at a concentration of 2.5 mM in 300 mM various weak basic buffer solutions. When the reaction mixture was stirred at 50 °C for 24 hours, a new spot with a strong fluorescence intensity could be detected by TLC, which had been identified by NMR to be benzophenone (as the oxidative decarboxylation byproduct) (Fig. S1‡), and phenylalanine was obtained in 7% yields with 12% ee's (Table S1‡). When EDTA was added to the model system to further block the effect of any possible metal ion contaminants, the enantioselectivity was dramatically enhanced with a slower reaction rate (Table 1, entry 1), which could be explained by the addition of EDTA that eliminated the possible acceleration of the transamination by trace metals in the buffer.12–14 Thus, EDTA was added to the system as an additive in the following reactions. To obtain a higher yield, prolonging the reaction time was considered; however, after 36 hours, no significant increase in yield was found with a slight decrease of ee probably due to the partial racemization of the product, for details see the ESI (Table S1).‡ Then, we studied the influence of temperature by varying the reaction temperature in controlled trials. The results showed that temperatures that were too high or too low disfavored the reaction (Table S2‡), so 50 °C was chosen as the final temperature.
Next, we studied the effects of the buffer on this transamination reaction (Table 1, entries 1–6 and Table S3‡). Examination of various buffers (Table 1, entries 1–3 and Table S3‡) indicated that the transamination worked better in nitrogen-based buffers, such as Tris, MOPS, HEPES, and EDTA, than others and Tris provided the best yield and enantioselectivity, which could be possibly explained by general acid-base catalysis mimicking of the transaminase's lysine amino group (Fig. 1).8,13,30 Tris buffer is also well-known for the ability of absorbing CO2, as the byproduct of oxidative decarboxylation; so, a higher concentration resulted in better promotion of the full-transamination (Table 1, entries 1, 4 and Table S3‡). The behavior of the system at different pH-values was also evaluated, which showed an obvious pH-dependence. When the pH value exceeded 8, the enantioselectivity was improved at the expense of yield (Table 1, entries 1, 5–6 and Table S3‡). Integrating both ee and yield, 300 mM pH 8.0 Tris buffer was selected as the optimum condition.
Subsequently, we studied the relationship between the reaction results and the dosage of PLP. The ee and yield showed the opposite changes. Upon the addition of increasing amounts of the catalyst, the yield increased, whereas the ee decreased (Table S4‡). After this, the impact of the types of CD's on the transamination was also investigated by varying the chiral pocket size of the CD from six D-glucopyranose units to eight or enhancement of its water solubility (Table 1, entries 1, 7–8 and Table S5‡). α-CD consisting of six glucose units yielded chiral phenylalanine with 20% ee's, smaller α-CD with only 5% ee's and extremely low yield (Table 1, entry 7), bigger γ-CD and better water solubility HP-β-CD yielded a racemic product that is same as in the reaction without chiral induction (β-CD) (Table 1, entry 8 and Table S5‡), which indirectly proved that a β-CD inclusion complex might be formed, leading to the enantioselectivity results. When we studied the different dosage of β-CD, 0.06 mmol (1.2 equiv.) was still chosen as the appropriate amount of β-CD considering both the enantioselectivity and yield. A lower yield was obtained despite a slight increase in ee when the dosage exceeded 0.06 mmol (Table S5‡).
Based on the status of the low yield, the poor solubility of the sacrificial amine source was considered as one of the possible reasons; thus, various amine sources with different amounts were examined in this system (Table 1, entries 1, 9–10 and Table S6‡). The result of (±) methylphenylglycine (Table 1, entry 9) was almost the same as diphenylglycine, both of which underwent oxidative decarboxylation in the transamination. When diphenylglycine was made into its alkali metal salts including lithium (Table S6‡), sodium (Table 1, entry 10), and potassium (Table S6‡), higher yields without loss of ee were obtained. However, the reaction was not affected by the amount of sodium diphenylglycine, and the same results were achieved when 4 equiv. or 1 equiv. of amine source were used (Table S6‡), which indicated that the second half-transamination was the rate-determining step. Therefore, the optimal reaction conditions were phenylpyruvic acid (1) (0.05 mmol), sodium diphenylglycine (7c) (0.05 mmol), β-CD (0.06 mmol), EDTA (0.01 mmol), and PLP (0.01 mmol) in 300 mM Tris buffer (4.0 mL), pH 8.0, at 50 °C stirred for 36 h. Phenylpyruvic acid was obtained with 20% ee's and in 27% yield.
Under the optimal conditions, the substrate scope was then examined with a series of pyruvic acid derivatives, and the results are shown in Table 2. A series of analogs of pyruvic acids were synthesized following a modified method reported in literature,31 in which the corresponding aldehydes were condensed with hydantoin with ethanolamine as catalyst, and were then hydrolyzed under alkaline conditions (Fig. S1‡). Results showed that the electronic and steric effects of the phenyl substituent of phenylpyruvic acids had significant impacts on the reactivity and enantioselectivity of the transamination. In general, 4-phenylpyruvic acid with electron-donating groups (methoxy (4b) and methyl (4e)) was more effective than those with electron-withdrawing groups (4f). para-Substituted substrates including methoxy (4b) and methyl (4e) all exhibited better enantioselectivity than that of the phenylpyruvic acid, yet the highly electron-deficient system (4f) was ineffective with less than 3% product obtained even in an increased temperature and with a prolonged reaction time. Halogens (4i, 4j, 4k) also exhibited good enantioselectivity and activity as a special case for the common effect of both the negative inductive effect and the electron donating conjugative effect. We attributed the three different results to the combined effects of the inductive effect, conjugation, and the size of the three halogens, which could affect the matching degree of the guest (the α-keto acid) host (the β-CD cavity) molecules, which further influenced the yield and ee. When the substituted group was replaced by a hydroxyl group (4h), tyrosine was obtained with 16% ee's. This relatively poor result could be explained by the hydrogen bonding between the keto acid and hydroxyl group of β-CD. In addition, it was easy to find that the position of the substituent had a major impact on the reaction, where the para-substitution showed the highest selectivity, meta second, and ortho gave the racemic product (4b–4d). The disubstituted substrate (4g) gave the corresponding α-amino acid with no ee probably because it was too sterically bulky for this system. When the phenyl ring was replaced by thiophene (4l), 14% ee's were obtained. However, with the shortening of carbon chain (4m), almost no enantioselectivity was observed, which is the same as that for pyruvic acid (4n), but a low ee could be achieved with the increase of the aliphatic chain (4o), likely due to the matching between the substrates and the cyclodextrin cavity during chirality induction.
| a All reactions were carried out with α-keto acid (3) (0.05 mmol), sodium diphenylglycine (7c) (0.05 mmol), β-CD (0.06 mmol), EDTA (0.01 mmol), and PLP (0.01 mmol) in 300 mM Tris buffer (4.0 mL), pH 8.0, at 50 °C for 36 h or 72 h. The ee's were determined by chiral HPLC analysis after the α-amino acids were converted into the corresponding N-benzoyl derivatives29 for 4a, 4d, 4e, 4g, 4i, 4k, 4l, and 4o or N-Fmoc derivatives32 for the rest of them. The absolute configurations of 4b–4o were tentatively proposed by the analogue. The yield was determined by chiral HPLC through the standard addition method.27,28 |
|---|
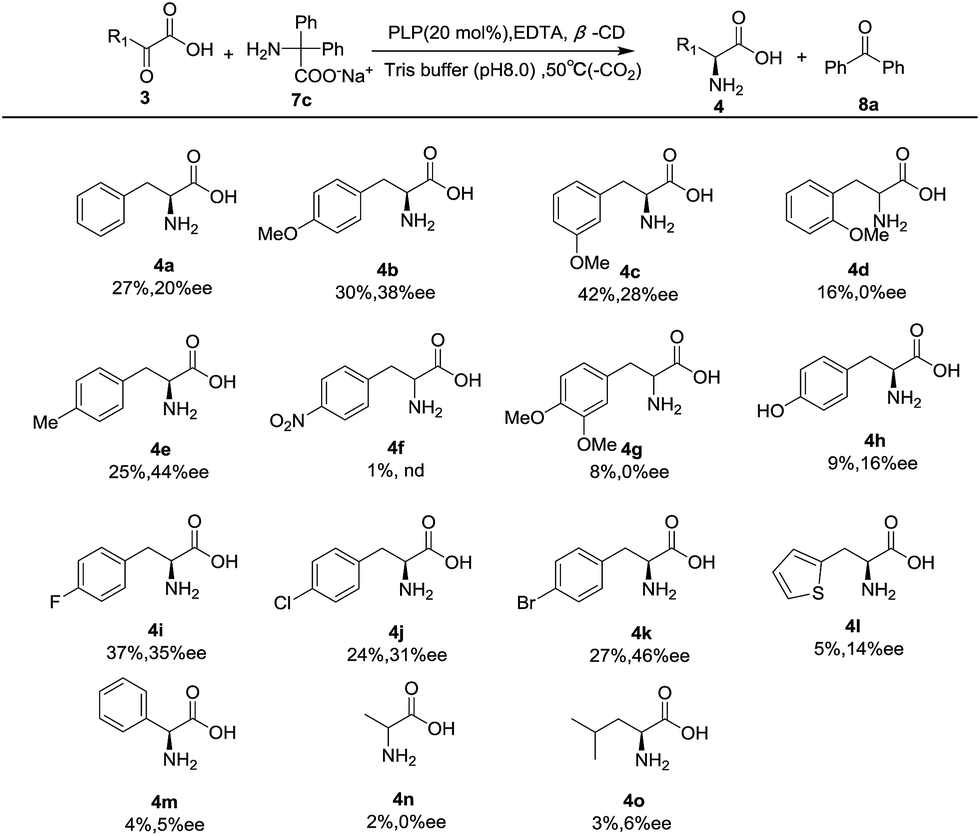 |
It appeared that the enantioselectivity was primarily influenced by the α-keto acid moiety, very little by the amine sources, and the type of CD also greatly influenced the results. To further understand the mechanism of transamination and how β-CD works as the only chiral source in the whole process, several control experiments were designed, and 4-fluorophenylpyruvic acid (4i) was chosen as the substrate for the studies because of its good performance in the reaction process. The binding experiments were designed as follows: 4-fluorophenylpyruric acid (4i) and sodium diphenylglycine (7c) were respectively added to the β-CD Tris buffer solution (D2O was applied to replace H2O) and the resulting mixtures were stirred overnight at 50 °C. Nuclear magnetic resonance (NMR) analysis showed that only 4-fluorophenylpyruvic acid could form the β-CD inclusion complexes, resulting in an upfield shift of H3 and downfield shift of H5 on the 1H NMR spectrum33 (Fig. 3B), since the magnetic screening environment of aromatic molecule34. To gain more information about the conformational geometry in the complex, a NOESY experiment was employed to study the inclusion complex.35 The contour map expansion for a 1:1 molar ratio of 4-fluorophenylpyruric acid/β-CD is presented in Fig. 3C. As shown in Fig. 3C (top), cross peak correlation between the aromatic hydrogen atoms of 4-fluorophenylpyruric acid (K-Aro H) and the internal (H3 and H5) protons of the β-CD molecule was observed, which is in agreement with the 1H NMR results. In Fig. 3C (bottom), cross-peak correlation between the benzyl hydrogen in 4-fluorophenylpyruric acid (K–H3′) and protons of β-CD showed that the benzyl hydrogens were correlated with H5 and not with H3, suggesting that the chain of 4-fluorophenylpyruric acid was in close proximity to the secondary rim of the cyclodextrin (the wider side, Fig. 3A).
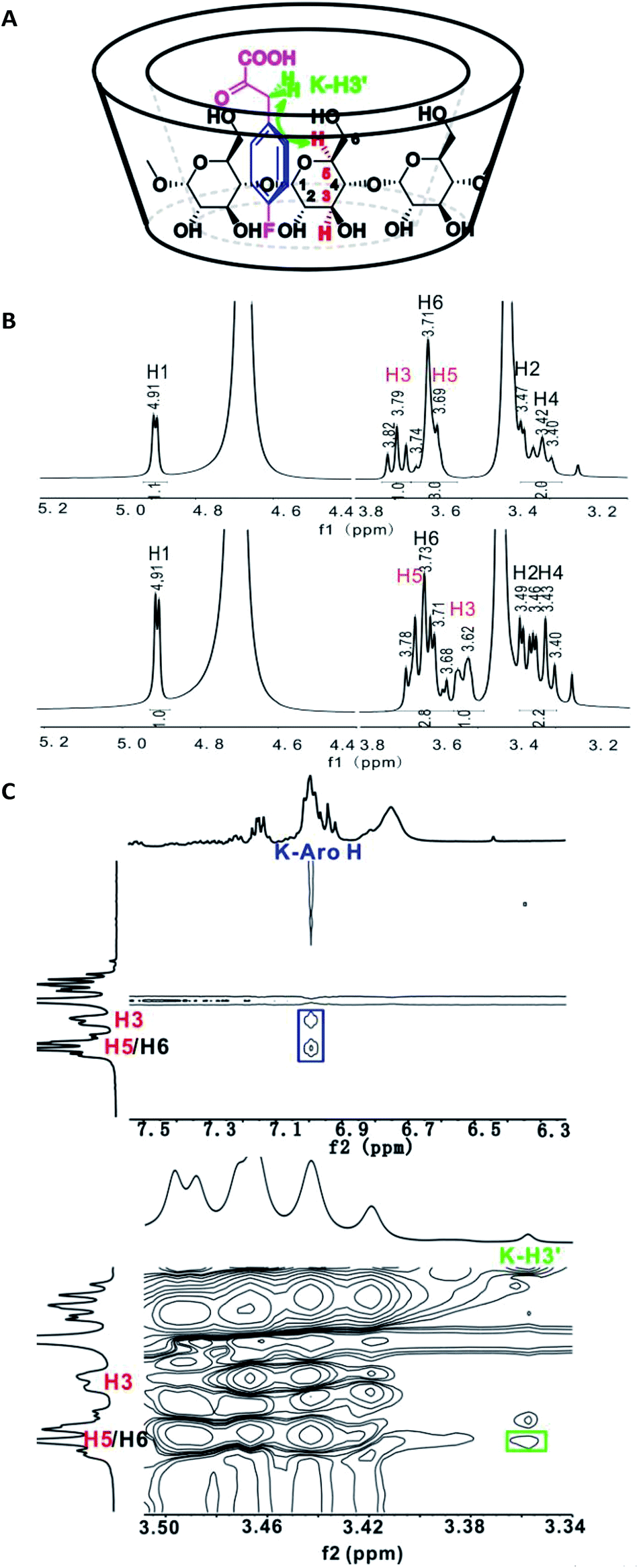 | ||
| Fig. 3 NMR study of the inclusion of 4-fluorophenylpyruric acid in β-CD. (A) Possible conformational geometry of 4-fluorophenylpyruric acid-β-CD inclusion complex. (B) 1H NMR solution spectra of β-CD solution (top), complex of 4-fluorophenylpyruric acid with β-CD solution (bottom), (0.02 M) in Tris buffer (pH = 8.0, D2O). (C) Partial contour plot of the 400 MHz NOESY spectrum for the binding of 4-fluorophenylpyruric acid (50 mM) to β-CD (50 mM) in Tris buffer (pH = 8.0, D2O). | ||
The same conclusion was drawn from the control experiments (Table S7‡) when the catalyst and the amine source were added to the reaction system containing the well prepared 4-fluorophenylpyruric acid-β-CD inclusion complex. About a 9% higher ee could been obtained than the one-pot method obtaining 4-fluorophenylalanine with 44% ee's, which indirectly indicated that the self-assembly of phenylpyruvic acid derivatives and β-CD resulted in the enantioselectivity of the transamination. As for the flat enantioselectivity of the reaction, which could be explained as the strong competition between the enclosed keto acid and the free one with the amine source, the latter one reacted out of the cavity, leading to the racemic products. Under the improved conditions, increasing the amount of β-CD could obtain up to 50% ee's due to the improvement of the inclusive rate (Table S7‡).
All of the abovementioned evidence pointed to the conclusion that the enantioselectivity of the product was induced by the chirality of β-CD, and the inclusion complex was formed by the self-assembly of keto acid and β-CD. This was in good agreement with the phenomenon, as shown in Table 2, that the different enantioselectivity and activity of the keto acid was positively correlated with the difficulty and stability of the formation of the inclusion complexes.36 Also, the previous assumptions about the substituent position (4b–4d) and the substrate size (4a, 4g, 4l, 4n, and 4o) were verified.
Therefore, a plausible catalytic cycle of this enantioselective full transamination could be considered as the composition of two half-transaminations, and β-CD only played a role in the second part for chiral induction. The first half-transamination started with the condensation of PLP with 7a to form aldimine 9, followed by decarboxylation and a 1,3 hydrogen transfer to generate a ketimine 10, and a subsequent hydrolization afforded PMP and the byproduct benzophenone 8. Then, PMP condenses with α-keto acid 3i, which self-assembled with β-CD, as shown in Fig. 4, to form ketimine 11. The product 11 isomerized to optically active aldimine 12 via an asymmetric 1,3 hydrogen transfer in the presence of β-CD. Hydrolysis of 12 gave the chiral amino acid 4i and regenerated PLP, finishing a whole catalytic cycle of the transamination. The key step for the whole process is regarded as the stereoselective protonation of ketimine 11 at the α-C of the carboxyl group affording the optically active aldimine 12. Therefore, the formation of the inclusion complexes is particularly important.
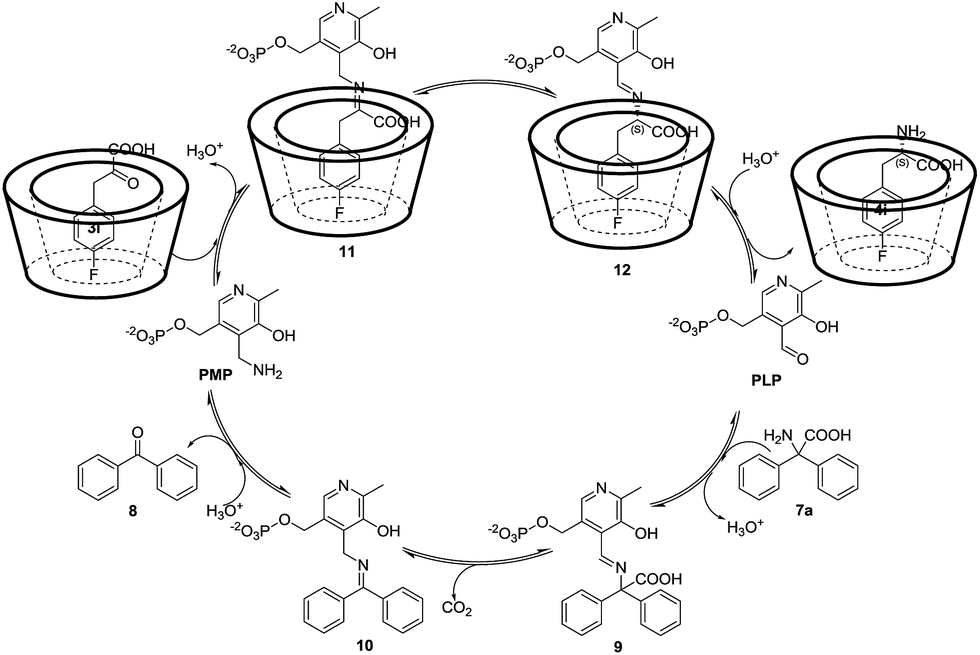 | ||
| Fig. 4 A proposed mechanism for the asymmetric transamination of α-keto acids with the assistance of β-CD. | ||
To summarize, mimicking the enantioselective biological full transamination in the pure aqueous phase has been realized for the first time by establishing a new “PLP catalyzed CD-keto acid inclusion complexes” system. Readily available β-CD was added to the system as a well-defined macromolecular transaminase model, successfully self-assembly with the α-keto acid forming the inclusion complex, which closely mimicked the real transaminase as a non-covalent complex formed from the pyridoxamine cofactor and the enzyme protein. Through a series of optimization experiments, various optically active α-amino acids were obtained up to 42% yields with up to 50% ee's under mild conditions. The 1H NMR study showed that the possible mechanism of this enantioselective transamination could be explained by the α-keto acid reversibly binding into the chiral β-cyclodextrin. While a precise understanding of the origin of the enantioselectivity still awaited further study, the ee and yield of the product remained to be improved probably by application of different modified cyclodextrins. However, note that the successful mimicking of this enantioselective transamination process using β-CD encourage us to make more attempts to utilize various chiral cavity macromolecules for enantioselective biological processes.
Notes and references
- M. Fuchs, K. Tauber, J. Sattler, H. Lechner, J. Pfeffer, W. Kroutil and K. Faber, RSC Adv., 2012, 2, 6262 RSC.
- A. Bujacz, M. Rutkiewicz-Krotewicz, K. Nowakowska-Sapota and M. Turkiewicz, Acta Crystallogr., Sect. D: Struct. Biol., 2015, 71, 632 CrossRef CAS PubMed.
- J. Ward and R. Wohlgemuth, Curr. Org. Chem., 2010, 14, 1914 CrossRef CAS.
- H. J. Pan, Y. Xie, M. Liu and Y. Shi, RSC Adv., 2014, 4, 2389 RSC.
- H. Kuzuhara, T. Komatsu and S. Emoto, Tetrahedron Lett., 1978, 3563 CrossRef CAS.
- M. Ando and H. Kuzuhara, Bull. Chem. Soc. Jpn., 1990, 63, 1925 CrossRef CAS.
- S. C. Zimmerman, A. W. Czarnik and R. Breslow, J. Am. Chem. Soc., 1983, 105, 1694 CrossRef CAS.
- S. C. Zimmerman and R. Breslow, J. Am. Chem. Soc., 1984, 106, 1490 CrossRef CAS.
- D. E. Metzler and E. E. Snell, J. Am. Chem. Soc., 1952, 74, 979 CrossRef CAS.
- D. E. Metzler, J. Olivard and E. E. Snell, J. Am. Chem. Soc., 1954, 76, 644 CrossRef CAS.
- K. Bernauer, R. Deschenaux and T. Taura, Helv. Chim. Acta, 1983, 66, 2049 CrossRef CAS.
- R. Deschenaux and K. Bernauer, Helv. Chim. Acta, 1984, 67, 373 CrossRef CAS.
- K. R. Knudsen, S. Bachmann and K. A. Jorgensen, Chem. Commun., 2003, 2602 RSC.
- S. Bachmann, K. R. Knudsen and K. A. Jorgensen, Org. Biomol. Chem., 2004, 2, 2044 CAS.
- J. T. Koh, L. Delaude and R. Breslow, J. Am. Chem. Soc., 1994, 116, 11234 CrossRef CAS.
- R. Breslow, S. Wei and C. Kenesky, Tetrahedron, 2007, 63, 6317 CrossRef CAS PubMed.
- R. Breslow, M. Hammond and M. Lauer, J. Am. Chem. Soc., 1980, 102, 421 CrossRef CAS.
- R. Kataky and E. Morgan, Biosens. Bioelectron., 2003, 18, 1407 CrossRef CAS PubMed.
- W. Weiner, J. Winkler, S. C. Zimmerman, A. W. Czarnik and R. Breslow, J. Am. Chem. Soc., 1985, 107, 4093 CrossRef CAS.
- I. Tabushi, Y. Kuroda, M. Yamada, H. Higashimura and R. Breslow, J. Am. Chem. Soc., 1985, 107, 5545 CrossRef CAS.
- E. Fasella, S. D. Dong and R. Breslow, Bioorg. Med. Chem., 1999, 7, 709 CrossRef CAS PubMed.
- L. Liu, W. J. Zhou, J. Chruma and R. Breslow, J. Am. Chem. Soc., 2004, 126, 8136 CrossRef CAS PubMed.
- L. M. Shi, C. A. Tao, Q. Yang, Y. E. Liu, J. Chen, J. F. Chen, J. X. Tian, F. Liu, B. Li, Y. L. Du and B. G. Zhao, Org. Lett., 2015, 17, 5784 CrossRef CAS PubMed.
- K. Toth, T. L. Amyes, J. P. Richard, J. P. G. Malthouse and M. E. NiBeilliu, J. Am. Chem. Soc., 2004, 126, 10538 CrossRef CAS PubMed.
- L. Liu and R. Breslow, Bioorg. Med. Chem., 2004, 12, 3277 CrossRef CAS PubMed.
- J. J. Chruma, L. Liu, W. J. Zhou and R. Breslow, Bioorg. Med. Chem., 2005, 13, 5873 CrossRef CAS PubMed.
- G. Schumann, R. Klauke and J. Buttner, Fresenius' J. Anal. Chem., 1992, 343, 89 CrossRef.
- A. V. Sakkiadi, C. A. Georgiou and S. A. Haroutounian, Molecules, 2007, 12, 2259 CrossRef CAS PubMed.
- M. E. Gunay and C. J. Richards, Organometallics, 2009, 28, 5833 CrossRef CAS.
- Y. Morino and S. Tanase, J. Biol. Chem., 1978, 253, 252 CAS.
- A. Schouteeten and Y. Christidis, US Patent 4518800, 21 May 1985.
- Y. Tang, J. Zukowski and D. W. Armstrong, J. Chromatogr. A, 1996, 743, 261 CrossRef CAS PubMed.
- D. J. Wood, F. E. Hruska and W. Saenger, J. Am. Chem. Soc., 1977, 99, 1735 CrossRef CAS.
- J. W. Apsimon, W. G. Craig, P. V. Demarco, D. W. Mathieso and W. B. Whalley, Chem. Commun., 1966, 361 RSC.
- W. K. Chan, W. Y. Yu, C. M. Che and M. K. Wong, J. Org. Chem., 2003, 68, 6576 CrossRef CAS PubMed.
- H. J. Schneider, T. Blatter and S. Simova, J. Am. Chem. Soc., 1991, 113, 1996 CrossRef CAS.
Footnotes |
| † This paper is dedicated to Prof. Thomas C. W. Mak on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27525g |
| This journal is © The Royal Society of Chemistry 2017 |