
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSyntheses, structures, luminescent and photocatalytic properties of various polymers based on a “V”-shaped dicarboxylic acid†
Xiang-min Menga,
Xia Zhanga,
Peng-fei Qia,
Zi-ao Zonga,
Fan Jin*b and
Yu-hua Fan *a
*a
aKey Laboratory of Marine Chemistry Theory and Technology, Ministry of Education, College of Chemistry and Chemical Engineering, Ocean University of China, Qingdao, Shandong 266100, P. R. China. E-mail: fanyuhua304@163.com
bMax Planck Institute for Terrestrial Microbiology & LOEWE Center for Synthetic Microbiology(SYNMIKRO), Marburg, Germany. E-mail: pkujinfan@gmail.com
First published on 17th January 2017
Abstract
Solvothermal reactions of “V”-shaped 3,5-di(4′-carboxyl-phenyl)benzene acid (H2dpb), and Cd(II)/Zn(II) salts in the presence of four ancillary bridging imidazole linkers afford four novel luminescent coordination polymers (LCPs), namely, {[Cd(dpb)2(bimb)2]·0.5H2O}n (1), [Cd(dpb)(tib)]n (2), [Zn(Hdpb)2(4,4′-bibp)]n (3) and [Cd(dpb)(1,3-bitl)]n (4), (bimb = 1,4-bis(imidazol-1-ylmethyl)benzene, tib = 1,3,5-tris(1-imidazolyl) benzene, 4,4′-bibp = 4,4′-bis(imidazol-1-yl)biphenyl and 1,3-bitl = 1,3-bis(1-imidazoly)toluene). Then complexes 1–4 are characterized by elemental analysis, IR spectroscopy, powder X-ray diffraction (PXRD), thermogravimetric (TG) analysis and single-crystal X-ray diffraction. Single-crystal X-ray diffraction analysis shows that complex 1 shows an unprecedented 3-nodal (2,2,4)-connected 2D + 2D → 3D two-fold interpenetration framework with the Schläfli symbol (125·16)(12)2. Complexes 2 and 3 show a 2D layer structure and 1D zigzag chain, which are further assembled into a 3D supramolecular structure by π⋯π interactions and H-bonds, respectively. While complex 4 displays a 3-nodal (2,2,8)-connected network with a –ABAB– fashion. The luminescent recognition properties of polymers 1–4 to metal cations and anions have also been explored systematically. The results demonstrate that 2 and 3 display highly sensitive and selective luminescence sensing towards Fe3+ ions, and 4 displays highly sensitive and selective luminescence sensing towards Cu2+ ions. 1–4 can work as highly sensitive sensors to Cr2O72− by luminescence quenching. Moreover, the photocatalytic properties of four Cd(II)/Zn(II) compounds (complex 1–4) have also been investigated.
Introduction
During the past decades, the construction of luminescent coordination polymers (LCPs) has attracted considerable attention not only for their potential applications in luminescence,1 catalysis,2 magnetism,3 gas storage,4 separation,5 proton conduction,6 nonlinear optics7 and so on, but also for their aesthetically pleasing structures and diversities in topology.8 In particular, based on their great physical and chemical characteristics, their capability of detecting heavy metal ions plays a significant role in public health,9 environmental protection10 and medicinal science.11 For example, Wang's group has reported that Cd-based LCPs were assembled using aromatic hexa-carboxylate which shows high sensitivity to Fe3+ ions in waste-water.12 The Eu-MOF reported by Zhu's group can detect Cu2+ ions.13 Liu synthesized a NH2–Zn-MOF that can detect Cr(III) and Cr(IV) which could cause water pollution.14 Meanwhile, the photocatalytic capability of the Zn/Cd-based CPs which are constructed using V-shaped ligands, has received wide attention from the viewpoint of chemists.15 However, the LCPs sensors for detecting metal ions which assemble using V-shaped ligands are still quite few.As we known, the aromatic multicarboxylate ligands play an important role in tuning the coordination framework structures due to their abundant coordination modes. Furthermore, due to their bent backbones and versatile bridging fashions, V-shaped aromatic multicarboxylate ligands are excellent candidates for building highly connected, interpenetrating, or helical coordination frameworks.16,17 Compared with some other reported V-shaped ligands (4-bpah,15a 4-bpab15b and 2,4′-bpdc17b), the V-shaped H2dpb we selected could show more advantages: (1) the H2dpb ligand with high solubility is easy to synthesize; (2) the suitable size makes it a reasonable candidate to generate LCPs with open frameworks; (3) the reports on LCPs assembled by V-shaped aromatic multicarboxylate ligands are rare; (4) the free rotation of two carboxylic benzene ring can promote the flexibility of the H2dpb ligand to meet the requirements of coordination geometries of metal ions; (5) the carboxylic group could show more coordination modes while coordinating to metal ions. Meanwhile, to further investigate the influence of auxiliary N-donor ligands on the formation of final architectures, a series of semi-rigid/rigid imidazole ligands with different conformations were into the system (Scheme 1).
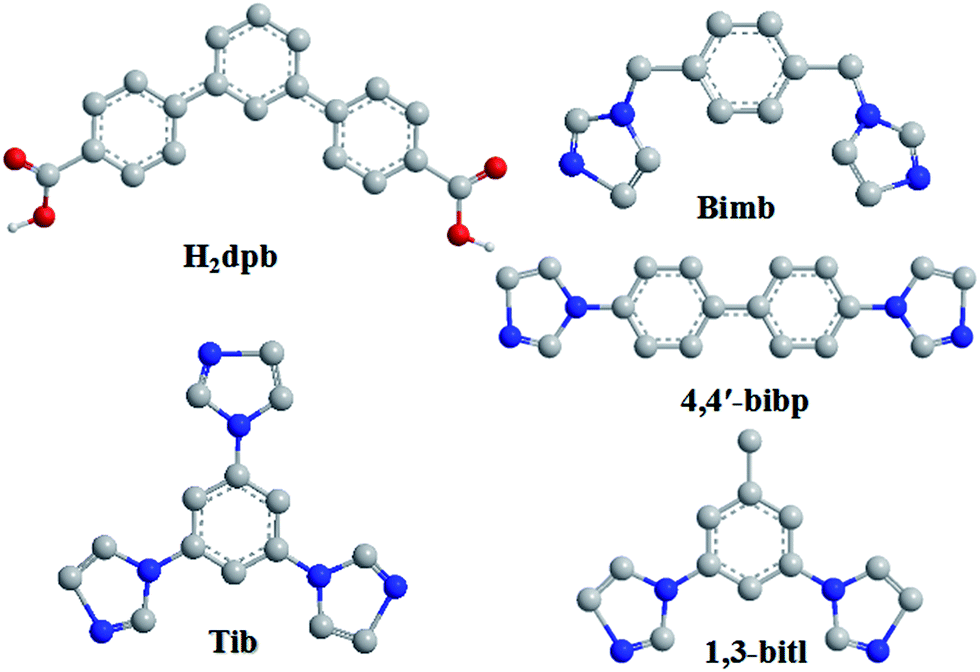 | ||
| Scheme 1 Structures of H2dpb and four auxiliary N-donor bridging linkers. | ||
Herein, four mixed-ligand LCPs have been successfully synthesized under solvothermal conditions, namely, {[Cd(dpb)2(bimb)2]·0.5H2O}n (1), [Cd(dpb)(tib)]n (2), [Zn(Hdpb)2(4,4′-bibp)]n (3) and [Cd(dpb)(1,3-bitl)]n (4), which show a systematic variation of architectures from 1D chain based on supramolecular to 3D interpenetrated framework (Scheme 2). The packing structures of these four complexes show that the nature of the bridging N-donor linkers has a significant effect on the H2dpb coordination modes. Moreover, the luminescent recognition properties of metal cations/anions and photocatalytic activities of the four Cd(II)/Zn(II) compounds were also investigated.
 | ||
| Scheme 2 Various polymeric structures of complexes 1–4. | ||
Experimental section
Materials and physical measurements
All reagents and solvents were purchased from Jinan Henghua Sci. & Tec. Co. Ltd. and were used without further purification. Infrared spectrum was recorded as KBr pellets on the Nicolet 170SX spectrometer in the 4000–400 cm−1 region. Elemental analysis (C, H, and N) was performed in a model 2400 Perkin-Elmer analyzer. Thermogravimetric analyses (TGA) were measured on a Perkin-Elmer TGA-7 thermogravimetric analyzer under air conditions from room temperature to 800 °C with a heating rate of 10 °C min−1. The X-ray powder diffractions (XRPD) were collected on an Enraf-Nonius CAD-4 X-ray single-crystal diffractometer with Cu-Kα radiation. Topological analysis were performed and confirmed by the Topos program and the Systre software.18,19Synthesis of {[Cd(dpb)2(bimb)2]·0.5H2O}n (1)
A mixture of H2dpb (0.20 mmol, 0.064 g), bimb (0.20 mmol, 0.048 g), Cd(NO3)2·4H2O (0.20 mmol, 0.062 g) and 8 mL of DMF–H2O (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were placed in a Teflon-lined stainless steel vessel, heated to 120 °C for 3 days, and then cooled (a descent rate of 5 °C h−1) to room temperature. Colourless block crystals of 1 were obtained. Yield of 51% (based on Cd). Anal. (%) calcd. for C34H26CdN4O4.5: C, 60.41; H, 4.03; N, 8.29. Found: C, 60.31; H, 3.91; N, 8.31. IR (KBr pellet, cm−1): 3588 (m), 3170 (m), 1625 (s), 1550 (m), 1370 (vs), 1100 (m), 855 (m), 791 (m) 659 (w).
:1) were placed in a Teflon-lined stainless steel vessel, heated to 120 °C for 3 days, and then cooled (a descent rate of 5 °C h−1) to room temperature. Colourless block crystals of 1 were obtained. Yield of 51% (based on Cd). Anal. (%) calcd. for C34H26CdN4O4.5: C, 60.41; H, 4.03; N, 8.29. Found: C, 60.31; H, 3.91; N, 8.31. IR (KBr pellet, cm−1): 3588 (m), 3170 (m), 1625 (s), 1550 (m), 1370 (vs), 1100 (m), 855 (m), 791 (m) 659 (w).
Synthesis of [Cd(dpb)(tib)]n (2)
A mixture of H2dpb (0.20 mmol, 0.064 g), tib (0.20 mmol, 0.056 g), Cd(NO3)2·4H2O (0.20 mmol, 0.062 g), NaOH (0.40 mmol, 0.016 g) and 8 mL of water was placed in a Teflon-lined stainless steel vessel, heated to 150 °C for 3 days, and then cooled (a descent rate of 5 °C h−1) to room temperature. Colourless block crystals of 2 were obtained. Yield of 60% (based on Cd). Anal. (%) calcd. for C35H24CdN6O4: C, 59.63; H, 3.43; N, 11.92. Found: C, 59.51; H, 3.32; N, 11.64. IR (KBr pellet, cm−1): 3126 (s), 1653 (m), 1506 (s), 1401 (vs), 1244 (m), 775 (m), 669 (m).Synthesis of [Zn(Hdpb)2(4,4′-bibp)]n (3)
Complex 3 was synthesized following the same synthetic procedure as that for complex 2 except that 4,4′-bibp was used instead of tib and Cd(NO3)2·4H2O was instead by Zn(NO3)2·6H2O. Pink block crystals of 3 were obtained. Yield of 51% (based on Zn). Anal. (%) calcd. for C58H40ZnN4O8: C, 70.62; H, 4.09; N, 5.68. Found: C, 70.46; H, 4.02; N, 5.56. IR (KBr pellet, cm−1): 3146 (m), 1721 (s), 1608 (s), 1554 (s), 1405 (vs), 1310 (m), 824 (m), 774 (s).Synthesis of [Cd(dpb)(1,3-bitl)]n (4)
Complex 4 was synthesized following the same synthetic procedure as that for complex 1 except that 1,3-bitl was used instead of bimb. Colourless block crystals of 4 were obtained. Yield of 48% (based on Cd). Anal. (%) calcd. for C33H24CdN4O4: C, 60.70; H, 3.70; N, 8.58. Found: C, 60.56; H, 3.62; N, 8.55. IR (KBr pellet, cm−1): 3125 (s), 1590 (m), 1506 (s), 1398 (vs), 1247 (w), 859 (m), 771 (m).X-ray crystallography
X-ray crystallography data of complexes 1–4 were collected on a Bruker Apex Smart CCD diffractometer at 293(2) K with graphite-monochromatized Mo-Kα radiation (λ = 0.71073 Å) by using the ω–2θ scan mode. The structure was solved by direct methods using SHELXS-97.20 The non-hydrogen atoms were defined by the Fourier synthesis method. Positional and thermal parameters were refined by the full matrix least-squares method (on F2) to convergence.21 Crystallographic data for complexes 1–4 are given in Table 1. Selected bond lengths and angles for 1–4 are listed in Table S1.† CCDC numbers for complexes of 1–4 are 1430347 for 1, 1402587 for 2, 1061113 for 3 and 1479858 for 4.| a R1 = ∑||Fo| − |Fc||/∑|Fo|, wR2 = [∑w(Fo2 − Fc2)2]/[∑w(Fo2)2]1/2. | ||||
|---|---|---|---|---|
| Compound | 1 | 2 | 3 | 4 |
| Empirical formula | C34H27CdN4O4.5 | C35H24CdN6O4 | C58H40N4O8Zn | C33H24CdN4O4 |
| Formula weight | 676.00 | 705.00 | 986.31 | 652.96 |
| Crystal system | Orthorhombic | Monoclinic | Monoclinic | Monoclinic |
| Space group | I212121 | P21/c | C2/c | P2/c |
| a (Å) | 14.0719(13) | 17.3405(15) | 32.919(3) | 17.7764(15) |
| b (Å) | 21.6271(19) | 13.6821(11) | 6.8673(5) | 13.6685(12) |
| c (Å) | 9.9750(9) | 25.213(2) | 21.6901(19) | 15.2008(13) |
| α (°) | 90 | 90 | 90 | 90 |
| β (°) | 90 | 109.982(2) | 114.515(2) | 102.863(2) |
| γ (°) | 90 | 90 | 90 | 90 |
| V (Å3) | 3035.7(5) | 5621.9(8) | 4461.4(6) | 3600.8(5) |
| Z | 4 | 4 | 4 | 4 |
| Dcalcd (Mg m−3) | 1.479 | 0.833 | 1.468 | 1.204 |
| μ (mm−1) | 0.766 | 0.416 | 0.618 | 0.642 |
| Reflections collected | 7612 | 27885 |
10537 |
17884 |
| Data/parameters | 2697/201 | 9891/597 | 3913/321 | 6336/507 |
| F(000) | 1372 | 1424 | 2040 | 1320 |
| T (K) | 298(2) | 298(2) | 298(2) | 298(2) |
| Rint | 0.0750 | 0.0634 | 0.0460 | 0.0674 |
| Final R indices [I > 2σ(I)] | R1 = 0.0470 | R1 = 0.0550 | R1 = 0.0438 | R1 = 0.0764 |
| wR2 = 0.0929 | wR2 = 0.1251 | wR2 = 0.0952 | wR2 = 0.2214 | |
| R indices (all data) | R1 = 0.0742 | R1 = 0.1063 | R1 = 0.0819 | R1 = 0.1115 |
| wR2 = 0.1013 | wR2 = 0.1338 | wR2 = 0.1080 | wR2 = 0.2346 | |
| Gof | 1.062 | 1.059 | 1.026 | 1.096 |
Results and discussion
Synthesis and characterization
The crystals of complexes 1–4 is dependent on the experimental method and the nitrate salts of Cd(NO3)2·4H2O and Zn(NO3)2·6H2O. Then suitable crystal were characterized by single-crystal X-ray diffraction after cooling to room temperature. Those complexes 1–4 are stable in the solid state even after extended exposure to air. Meanwhile, all of the complexes 1–4 have poor solubility in water and common organic solvent, but can be slightly soluble in very high polarity solvents.The IR spectra of complexes 1–4 display characteristic absorption bands of the coordinated carboxylate groups appear at 1590–1653 cm−1 for asymmetric stretching and 1320–1401 cm−1 for the symmetric.22 Meanwhile, a adsorption bands are observed in ca. 1721 cm−1,23 suggesting the partially deprotonation of carboxylic groups in 3, which is consistent with the results of the X-ray diffraction analysis.
Structure description of {[Cd(dpb)2(bimb)2]·0.5H2O}n (1)
Single-crystal X-ray diffraction analysis reveals that 1 crystallizes in the orthorhombic system with space group I2(1)2(1)2(1). As can be seen from Fig. 1a, the asymmetric unit consists of one Cd(II) ion, two dpb2− carboxylate ligands and two bimb N-donor ligands. The Cd(II) is six-coordinated by four O atoms from two dpb2− ligands [Cd(1)–O(1)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Cd(1)–O(1)#1 = 2.248(5) Å, Cd(1)–O(2)Cd(1)–O(2)#1 = 2.545(6) Å] and two N atoms from two bimb bridging linkers [Cd(1)–N(2)Cd(1)–N(2)#1 = 2.283(5) Å], which adopts a distorted octahedral {CdN2O4} coordination geometry.
Cd(1)–O(1)#1 = 2.248(5) Å, Cd(1)–O(2)Cd(1)–O(2)#1 = 2.545(6) Å] and two N atoms from two bimb bridging linkers [Cd(1)–N(2)Cd(1)–N(2)#1 = 2.283(5) Å], which adopts a distorted octahedral {CdN2O4} coordination geometry.
 | ||
| Fig. 1 (a) Coordination environment of the Cd(II) ion in complex 1 (symmetry codes: (A) −x + 3/2, y + 0, −z + 1; (B) −x + 1/2, y + 0, −z + 2; (C) x, −y + 2, −z + 1/2). (b) View of the 1D [Cd-dpb]n linear chain, 1D [Cd-bimb]n helical chain and 2D network of 1 (blue spheres: bimb ligands; red spheres: dpb2− ligands). (c) The 2D + 2D → 3D two-fold interpenetration parallel networks viewed along the a axis. (d) The 2D + 2D → 3D interpenetrated (2,2,4)-connected topology (125·16)(12)2 sheets in 1. | ||
As shown in Scheme 3, each “V”-shaped dpb2− ligand coordinates with two adjacent Cd(II) ion by adopting a μ2-(κ1-κ1)-(κ1-κ1) coordination mode (Scheme 3, Mode I) forming an infinite 1D [Cd-dpb]n linear chain, in which the two carboxyl groups show same μ1-(κ1-κ1) coordination. Interestingly, the μ2-bridging bimb ligands exhibits a “U” shape conformation to connect the adjacent Cd(II) ions to result in a single-strand helix chain [Cd-bimb]n with the Cd⋯Cd separation of 12.86 Å. The two types of 1D chains ([Cd-dpb]n and [Cd-bimb]n) are alternatively connected with each other, constructing a 2D network (Fig. 1b). The two adjacent 2D networks are further interact with each other through helix chain ([Cd-bimb]n) twining round the “V”-shaped H2dpb ligands, finally resulting in a rarely 2D + 2D → 3D two-fold interpenetration parallel framework (Fig. 1c). The effective free volume of 1 was 10.5% of the crystal volume (318.7 out of the 3035.7 Å3 unit cell volumes), calculated by PLATON analysis.24
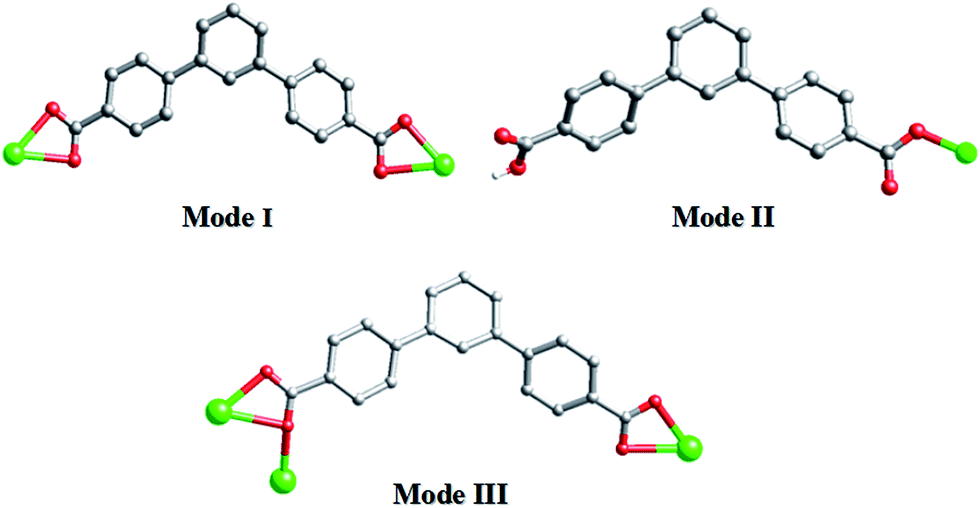 | ||
| Scheme 3 Diverse coordination modes of H2dpb in complexes 1–4. | ||
From the viewpoint of structural topology, the Cd(II) ions are coordinated by two dpb2− ligands and two bimb ligands, which can be viewed as a 4-connected node and the dpb2− ligand as a “V”-shaped spacer, then an unprecedented 3-nodal (2,2,4)-connected 3D interpenetrated framework is created with (125·16)(12)2 topology (Fig. 1d).
Structure description of [Cd(dpb)(tib)]n (2)
The crystal structure determined by single-crystal X-ray diffraction showed that complex 2 crystallizes in the monoclinic system, space group P21/c. The asymmetric unit of 2 contains one Cd(II) cation, one dpb2− ligand and one tib ligand. As shown in Fig. 2a, each Cd(II) cation with an unusual {CdN3O4} distorted decahedral coordination geometry is seven-coordinated by three imidazolyl nitrogen atoms from three tib ligands [Cd–O = 2.320(3)–2.580(4) Å], and four oxygen atoms from two dpb2− anions [Cd(1)–N(2) = 2.322(4) Å, Cd(1)–N(4)#1 = 2.306(4) Å and Cd(1)–N(5) = 2.326(4) Å]. Cd to O/N distances and bond angles are within the normal range.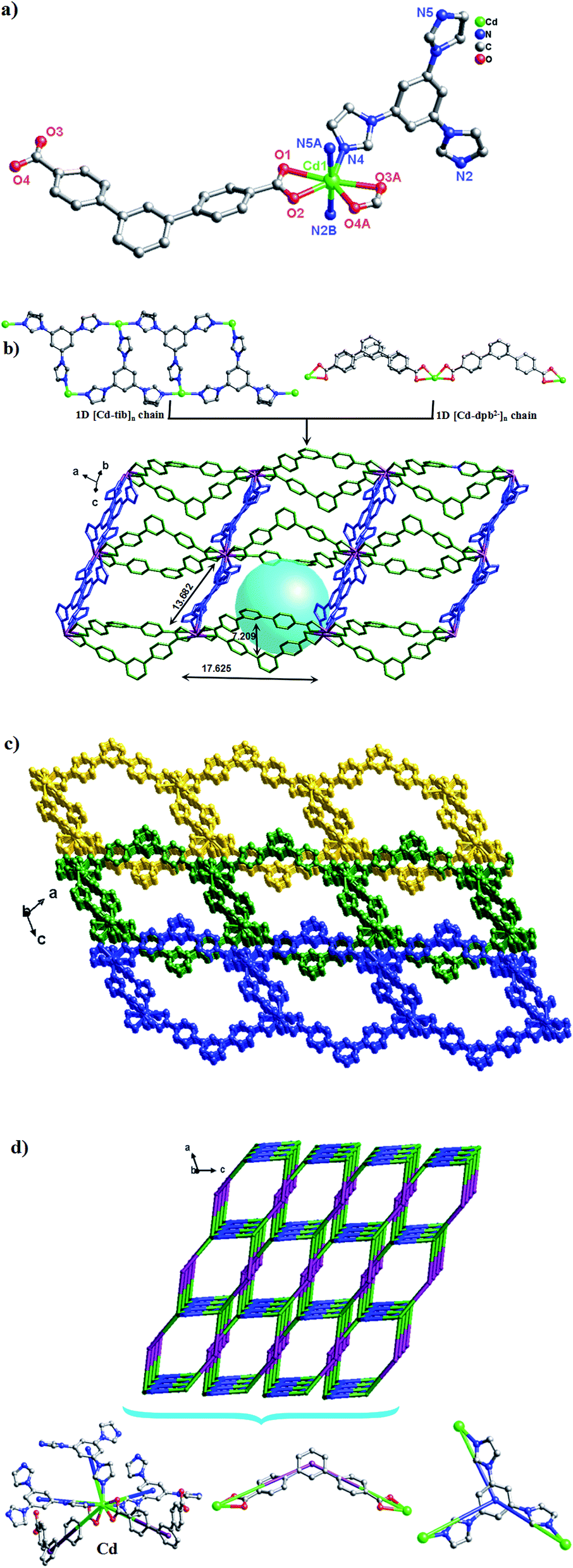 | ||
| Fig. 2 (a) Coordination environment of the Cd(II) ion in complex 2 (symmetry codes: (A) −x + 1, −y + 1, −z + 2; (B) x, y − 1, z; (C) x, y + 1, z). Note: the dangling benzyl rings are omitted for clarity. (b) View of the 2D network of 2 (blue spheres: tib ligands; green spheres: dpb2− ligands). (c) The 2D → 3D supramolecular networks by C–H⋯C and π–π interactions. (d) Views of the (2,3,5)-connected 3D supramolecular frameworks with unprecedented (42·6·86·12)(42·6)(8) topology. | ||
In complex 2, the completely deprotonated dpb2− ligand exhibits a μ2-(κ1-κ1)-(κ1-κ1) coordination mode (Scheme 3, Mode I). Two neighbour Cd(II) cation were connected by “V”-shaped dpb2− ligand form a 1D [Cd-dpb2−]n chain with the Cd⋯Cd separation is 17.625 Å, and tib ligand form a 1D [Cd-tib]n ladder shaped chain with different Cd⋯Cd separation (10.573, 11.411 and 13.682 Å), respectively. Each ladder-like chain contains two types of binuclear ({Cd2N8C10} 20-membered ring and {Cd2N8C12} 22-membered ring) loops. The adjacent 1D chains are further intersected with each other by sharing the same Cd(II) cation, finally giving a 2D network (Fig. 2b). The final 3D supramolecular framework of complex 2 is through the C–H⋯C between the carbon atoms from phenyl ring and imidazolyl [C(35′)–H(35′)⋯C(6) = 2.915 Å] (Fig. 2c and S1†). Meanwhile, π⋯π stacking interactions make a more stable 3D supramolecular structure. Besides, the void volume of 2 is 44.3% of the crystal volume (2489.9 out of the 5621.9 Å3 unit cell volume), calculated by PLATON.
From the viewpoint of topology, the “V”-liked dpb2− anions can be defined as 2-connected nodes, while the tib ligands can be considered as 3-connected nodes. Thus the 3D supramolecular structure can be simplified as an unprecedented 3-nodal (2,3,5)-connected framework with the point Schläfli symbol (42·6·86·12)(42·6)(8) (Fig. 2d).
Structure description of [Zn(Hdpb)2(4,4′-bibp)]n (3)
A similar reaction environment compared with complex 1 was used except for the H2O–CH3CN system being replaced with the H2O, results in a 3D supramolecular structure. Complex 3 crystallizes in monoclinic space group C2/c. The asymmetric unit of 3 contains one Zn(II) ion, two Hdpb− carboxylate ligands, and one 4,4′-bibp N-donor ligand as shown in Fig. 3a. The Zn(1) center is a four-coordinated by two O atoms from two Hdpb− ligands and two N atoms from two 4,4′-bibp ligands, displaying a distorted triangle cone geometry. The bond length of Zn–O is 1.940(2) Å, and the Zn–N distance is 2.019(2) Å, respectively. Both Zn–O and Zn–N bond length also well-matched to similar complexes.25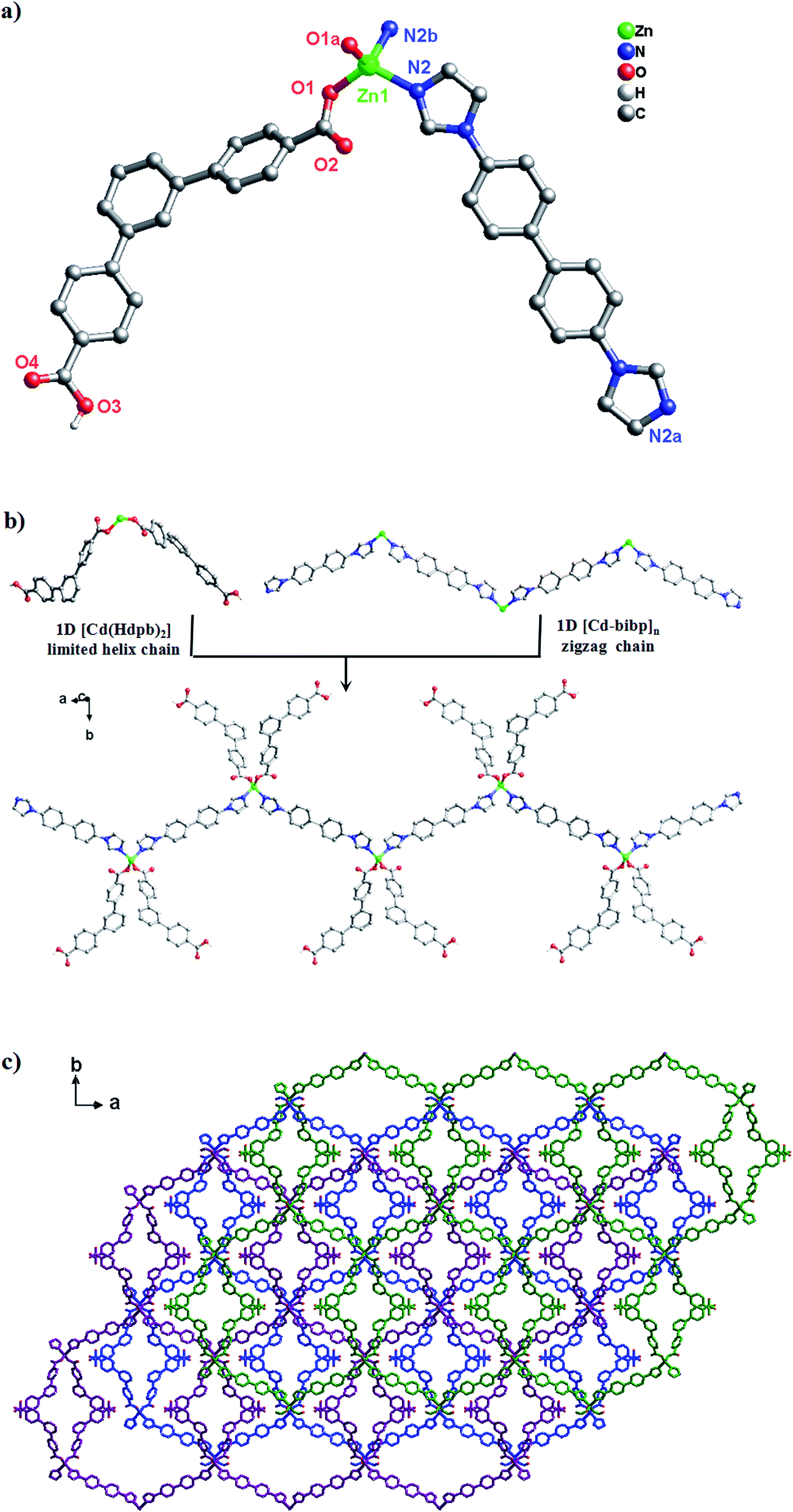 | ||
| Fig. 3 (a) Coordination environment of the Zn(II) ion in complex 3 (symmetry codes: (A) −x + 1, y, −z + 3/2; (B) −x + 3/2, −y + 5/2, −z + 2). (b) The simplified 1D zigzag chain structure of along the a axis in complex 3. (c) From 1D chain to 3D supramolecular structure of complex 3, which are linked by O–H⋯O hydrogen bonds along [1,1,0] orientation. | ||
The two partially deprotonated carboxyl groups of “V”-liked Hdpb− ligands exhibit a μ1-(κ1-κ0) coordination mode (Scheme 3, Mode II). Two adjacent “V”-liked Hdpb− ligands linked the same Zn(II) ions hold a limited helix chain, and the N-donor ligands linking two adjacent Zn(II) ions along the axis to form a 1D zigzag chain [Zn(bibp)]n (Fig. 3b). Such chains are linked through O–H⋯O hydrogen bonds along [1,1,0] orientation to form 3D cone supramolecular structure (Fig. 3c and S2†). The dihedral angle between the three phenyl rings in Hdpb− are 18.78(4)°, 34.31(3)° and 47.91(4)° respectively.
Structure description of [Cd(dpb)(1,3-bitl)]n (4)
A similar reaction environment compared with complex 3 was used except for the bimb N-donor ligand being replaced by 1,3-bitl, results in another 3D supramolecular ladder structure. Compound 4 crystallizes in the monoclinic space group P2/c. In the asymmetric unit, there exist one crystallographically unique Cd(II) ions, one dpb2− carboxylate ligands and one coordinated 1,3-bitl N-donor ligand. As shown in Fig. 4a, each Cd(II) cation also show an unusual seven-coordinated by five O atoms from three individual dpb2− carboxylate ligands (Cd–O = 2.316(6)–2.622(6) Å) and two N atoms from two different 1,3-bitl N-donor ligand (Cd–N(2) = 2.231(7) and Cd–N(3) = 2.314(8) Å) with a {CdN2O5} coordination environment, displaying a slightly distorted decahedral geometry. Both Cd–O and Cd–N bond length also well-matched to similar complexes.26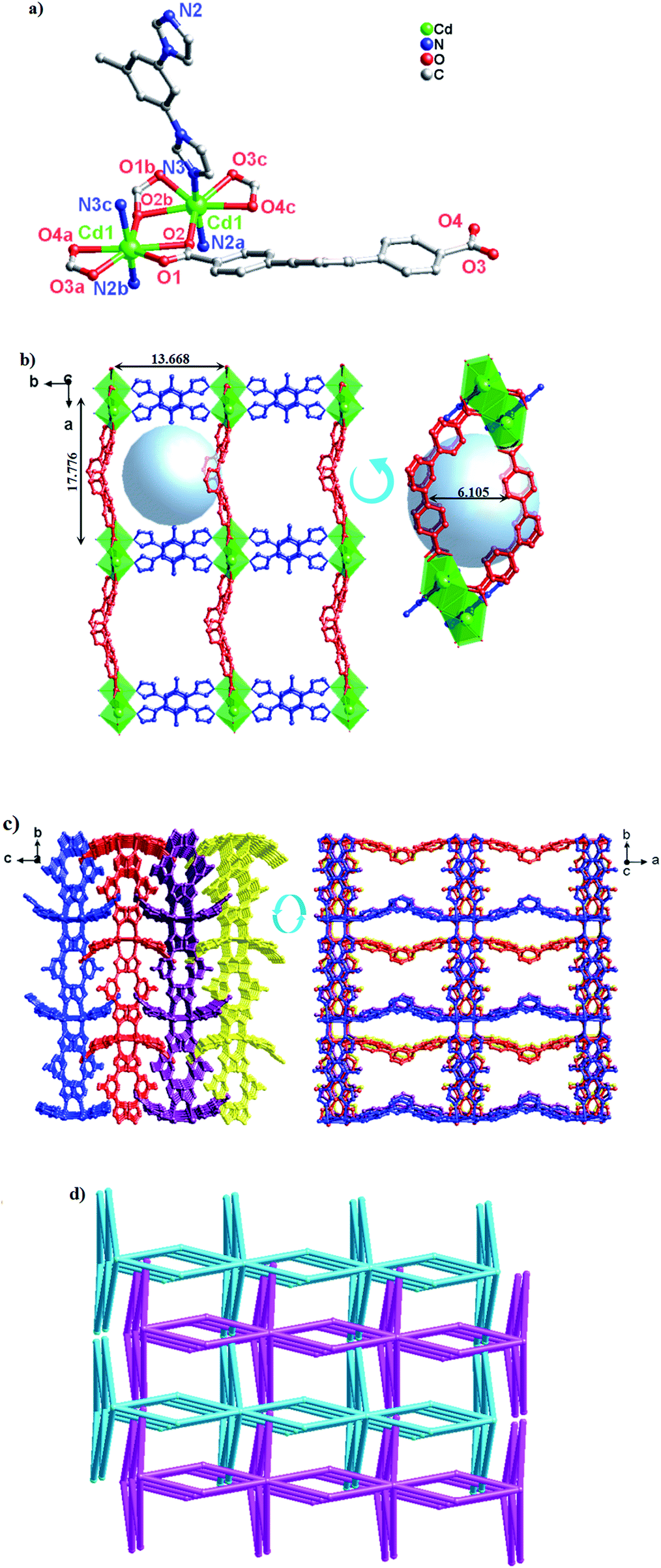 | ||
| Fig. 4 (a) Coordination environment of the Cd(II) ion in complex 4 (symmetry codes: (A) −x + 1, y, −z + 3/2; (B) x, y + 1, z; (C) x + 1, y, z; (D) x − 1, y, z). Note: the dangling benzyl rings are omitted for clarity. (b) View of the 2D network of 4 (blue spheres: 1,3-bitl ligands; red spheres: dpb2− ligands). Note: the dangling benzyl rings are omitted for clarity. (c) From 2D network to 3D framework of complex 4, which are linked by C–H⋯O bonds along [0,1,1] orientation. Note: the dangling benzyl rings are omitted for clarity. (d) Views of the (2,2,8)-connected 2D → 3D network with a –ABAB– fashion. | ||
In complex 4, the completely deprotonated dpb2− ligand exhibits a μ3-(κ1-κ2)-(κ1-κ1) coordination mode (Scheme 3, Mode III). The dihedral angles between the three phenyl rings in dpb2− are 35.78(2), 27.96(3) and 19.18(3)°, respectively. In the structure, two Cd(1) ions are connected by the μ2-(κ1-κ2) carboxyl group, forming {Cd2(COO)2} SBUs, which can be viewed as a shared nodal. Meanwhile, the two adjacent {Cd2(COO)2} SBUs are connected by two “V”-shaped dpb2− ligands forming a bigger {Cd4(dpb2−)2} (28-membered) ring and two bitl ligands forming a {Cd4(bitl)2(COO)2} (26-membered) ring. Then the two different loops intersected with each other forming a stable 2D network with a 17.776(15) × 13.668(12) × 6.1052(28) Å3 quadrilateral opening (Fig. 4b). The adjacent 2D network are alternatively connected with each other through C–H⋯O bonds (Table S2,† C(28)–H(28)⋯O(3) = 2.411 and C(21)–H(21)⋯O(3) = 2.431 Å), constructing a stable 3D network (Fig. 4c and S3†). The effective free volume of 4 was 22.9% of the crystal volume (823.0 out of the 3600.8 Å3 unit cell volumes), calculated by PLATON analysis.
From the topologically, the {Cd2(COO)2} SBUs can be regarded as eight-connected nodes and “V”-shaped dpb2− ligands as two-connected nodes; thus, this layer displays a 3-nodal (2,2,8)-connected network (Fig. 4d).
Structural comparison and discussion
The structures of LCPs have been found to be greatly influenced by N-donor ligands based on previous literature. As shown in Scheme 3 and Table S3,† H2dpb exhibits versatile coordination modes, resulting in different new structures (1D–3D). In complexes 1 and 2, the “V”-shaped dpb2− ligands chelate the metal atoms (chelate both in 1 and 2) through their two carboxylate groups, to further stabilize the 1D chain. After that, those chains further interacted with bimb (1D [Cd-bimb]n helix chain)/tib (1D [Cd-tib]n ladder chain) chain to form the final 2D + 2D → 3D interpenetrated (2,2,4)-connected unprecedented and (2,3,5)-connected unprecedented 3D/2D topologies. For complex 3, only one carboxylate group of the H2dpb ligand coordinated with the metal atoms (monodentate in 3), and the other carboxylate group interacted with the adjacent [Zn(Hdpb)2(4,4′-bibp)]n chain to form the 3D supramolecular structure through H-bonding interactions. For 4, one carboxylate group of the dpb2− adopt in μ2-(κ1-κ2)-chelate fashion and the other carboxylate group show μ2-(κ1-κ1)-chelate coordination modes. And each carboxylate group connecting neighboring Cd(II) atoms and 1,3-bitl N-donor ligand to form the 2D network, which is further interacted by C–H⋯O to form a 3D supramolecular structure. Moreover, H2dpb is completely deprotonated in 1, 2 and 4, and partly deprotonated (Hdpb−) in 3; their asymmetric units contain two H2dpb ligands in 1 and 3, and one H2dpb ligand in 2 and 4. Meanwhile, the “V”-shaped ligand play important roles in the construction of bigger open spacer framework in 1, 2 and 4.For N-donor ligands, the differences in their lengths and configurations greatly influence the final structure of the targeted CPs. Except 1 is a semi-flexible-auxiliary ligand, the N-donor ligands can be seen as rigid-auxiliary ligand in complexes 2–4. Thus the complex 1 show a 2D + 2D → 3D two-fold interpenetrated framework, and 3D supramolecular framework in 2–4, respectively. Due to the different configurations of the N-donor ligands, complex 2 shows a higher effective free volume (44.3%) than complexes 1 (10.5%) and 4 (22.9%). Hence, complex 2 may be a good candidate for application in gas/dye absorption.
Thermal analyses
To understand the thermal stabilities of compounds 1–4, their thermal behaviors were investigated by TGA (Fig. S4†). The experiments were performed on samples consisting of numerous single crystals of 1–4 under a N2 atmosphere with a heating rate of 10 °C min−1. For complex 1, the first weight loss in the temperature range of 80–150 °C is consistent with the removal of the lattice H2O (obsd: 1.1%, calcd: 1.3%). The second weight loss corresponds to the loss of the organic ligands at ca. 350 °C. The remaining weight corresponds to the formation of CdO (obsd: 23.7%, calcd: 19.1%). For complex 2, the decomposition of the compound occurs at ca. 330 °C, and the remaining weight corresponds to the formation of CdO (obsd: 33.1%, calcd: 18.2%). For complex 3, the whole structure starts to collapse at ca. 350 °C with a result of thermal decomposition and the remaining residue of ZnO (obsd: 30.2%, calcd: 8.2%). For complex 4, the destruction of the framework occurs at ca. 230 °C, finally leading to the formation of the stoichiometric amount of CdO as a residue (obsd: 20.2%, calcd: 19.8%).Optical band gaps
Because of the energy band gap (Eg) of CPs is closely related to its photocatalytic ability. Thus investigate the conductivity potentials of titled compounds are necessarily. And the band gap energy of CPs can be evaluated by Kubelka–Munk transformation (F):27 F = (1 − R)/2R, where transformed from the recorded diffuse reflectance data and R is the reflectance of an infinitely thick layer at a given wavelength.28 As shown in Fig. 5, the Eg values assessed from the steep absorption edge for complexes 1–4 are 2.70, 2.74, 2.28 and 2.59 eV, respectively, which indicate that complexes 1–4 are potential semi-conductive materials.29,30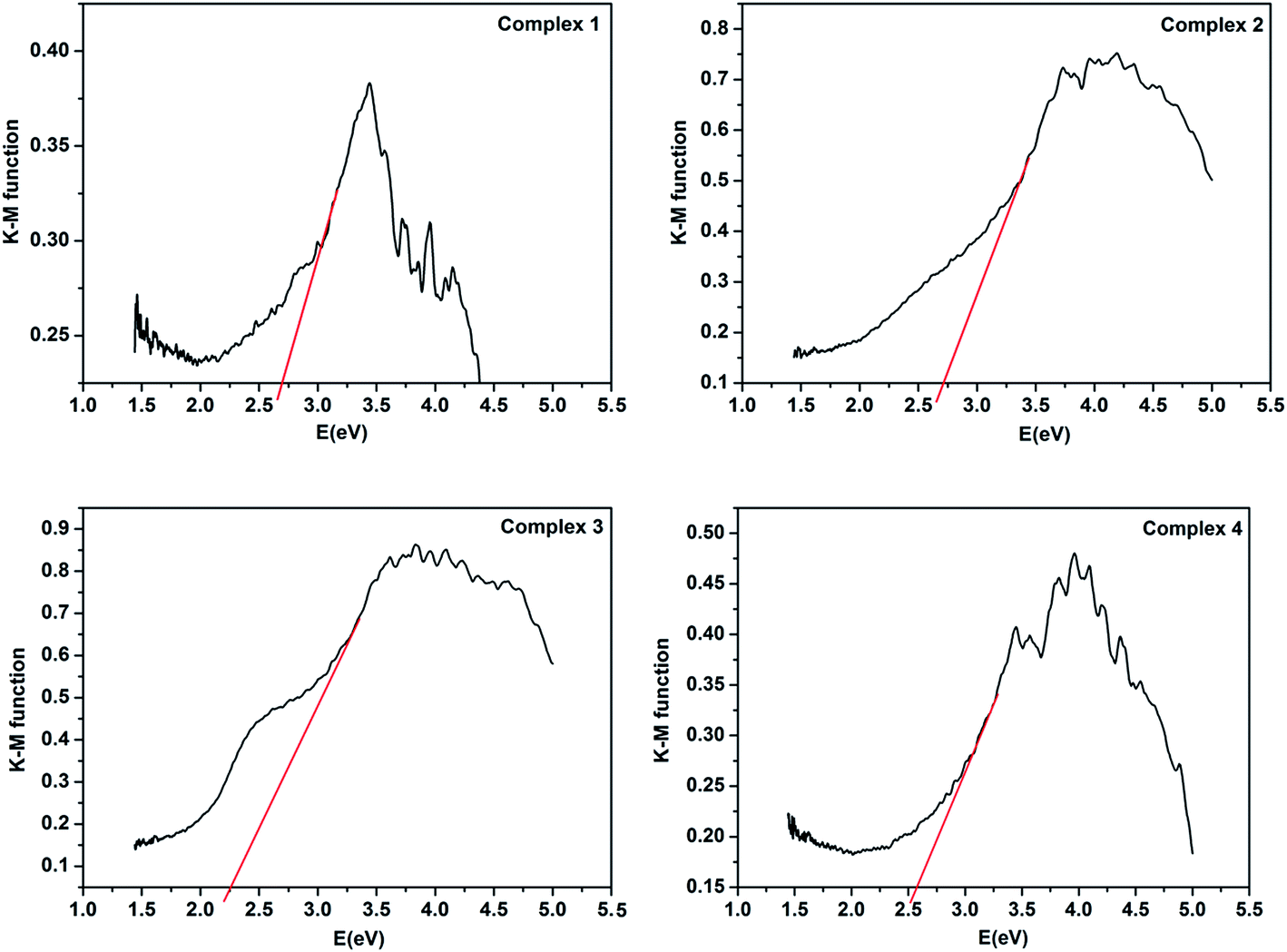 | ||
| Fig. 5 Kubelka–Munk-transformed diffuse reflectance spectra of complexes 1–4. | ||
Luminescence properties
The luminescence properties of complexes 1–4 together with the free H2dpb, bimb, tib, 4,4′-bibp and 1,3-bitl ligands, were investigated in the solid state at ambient temperature (Table S4†). The free ligands emit strong fluorescence centered at 405 nm (λex = 275 nm) for H2dpb, 380 nm (λex = 220 nm) for bimb, 371 nm (λex = 275 nm) for tib, 378 nm (λex = 270 nm) for 4,4′-bibp and 385 nm (λex = 280 nm) for 1,3-bitl respectively. After metallization of these ligands with Cd(II)/Zn(II) atom, the main emission peaks occur at 375 nm (λex = 275 nm) for 1, 369 nm (λex = 270 nm) for 2, 376 nm (λex = 265 nm) for 3, and 383 nm (λex = 255 nm) for 4. In comparison to the free ligands, the emission peaks of 1–4 are close to imidazol ligands, so the emission bands of these complexes can probably be attributed to the N-donor ligand fluorescence emission. Compared to the free N-donor ligand, their peaks are blue-shifted by 5 nm, 2 nm, 2 nm and 2 nm, respectively. These emissions are neither metal-to-ligand charge transfer (MLCT) nor ligand-to-metal charge transfer (LMCT) in nature, since Cd(II) and Zn(II) ions are difficult to oxidize or reduce due to their d10 configuration.31 The photoluminescent of 1–4 may originate from the intraligand π*–π or π*–n transition since similar emissions were also observed for the ligands themselves. The emission discrepancy of these compounds is probably due to the differences of organic ligands and coordination environments of central metal ions, which have a close relationship to the photoluminescence behavior.32 As shown in Fig. 6a, with the maximum emission peaks of 1–4 are mainly centered at 375, 369, 376 and 383 nm, respectively, and bright blue, blue, blue and blue fluorescence are found by visual observation at the optimum excitation. Their chromaticity coordinates (1–4) are (0.1585, 0.0221), (0.1441, 0.0486), (0.1428, 0.0518) and (0.1492, 0.0386) (Fig. 6b). | ||
| Fig. 6 (a) Photoluminescence of CPs 1–4 at room temperature in the solid state. The inset shows the relative fluorescence images. (b) CIE chromaticity diagrams of 1–4. | ||
As we know that the luminescence characteristic of PLCCs is closely related to their structures.33 The size of the metal, the structure of the secondary building units (SBUs) and the orientation of the linkers all affect the emission properties of the material.34 Compounds 1–4 contain the same linker (H2dpb) in different coordination modes and geometries, allowing comparative study of their photoluminescence diversity. Here we observed that compounds 2 (blue), 3 (blue) and 4 (blue) display similar emission character, which is different from that of 1 (bright blue). The different visual fluorescence maybe attributed to the various structures of four complexes (1 (3D), 2 (2D), 3 (1D) and 4 (2D)).
Fe3+/Cu2+-sensing properties
Currently, the luminescent MOFs have great advantages in the ion detection and separation.35 Among the metal cations, the Fe3+/Cu2+ cations are the most necessary elements involved in biochemical processes in humans.36 Based on the strong luminescence and good water stabilities of 1–4, the sensing properties of complexes on metal cations were investigated. In order to examine their sensing abilities, the luminescence spectra of 1–4 (3 mg) dispersed in water solutions (3 mL) of 0.01 M (mol L−1) AgNO3 or MClx (M = Na+, K+, Ca2+, Mg2+, Cd2+, Mn2+, Co2+, Zn2+, Ni2+, Cu2+, Fe3+) were studied. Each suspension solution was sonicated in the dark for 30 min before fluorescence testing. As can be seen in Fig. 7, complexes 2 and 3 exhibit the obvious luminescence quenching when Fe3+ ions were loaded, whereas other metal ions exhibit no quenching effects. Meanwhile, complex 1 also show luminescence quenching when Ag+, Cu2+, Fe3+, Ni2+ and other ions were added. Interestingly, the Cu2+ ions can bring pronounced complete quenching in 4, and the Fe3+ ions do not exhibit the obvious luminescence quenching. Those results demonstrate that compounds 2 and 3 could be highly effective and selective luminescent sensors for Fe3+ ions, and complex 4 could be highly effective and selective luminescent sensors for Cu2+ ions. It is generally accepted that the N atoms in the complexes can donate their lone-pair of electrons to the Fe3+/Cu2+ cation, which can form electron-deficient regions and act as acceptors. When under light, the luminophore in the complex can become electron donors. The energy migrations occur in case of the electrons transferred from luminophores to acceptors (Fe3+/Cu2+), which leading to the fluorescence quenching.37,38 | ||
| Fig. 7 Emission spectra and intensities for 1 (a), 2 (b), 3 (c) and 4 (d) in solutions of different metal cations. | ||
For further examining the sensitivity of luminescence quenching, the concentration-dependent studies were carried out in the presence of Fe3+ (in 2 and 3)/Cu2+ (in 4) with different concentrations. As shown in Fig. 8, the emission intensities 2–4 decreased gradually with increasing of Fe3+ (in 2 and 3)/Cu2+ (in 4) from 0 to 5 × 10−3 M.39 The Stern–Volmer equation (I0/I − 1 = KsvC[M]) can be used to calculated the corresponding quenching coefficient, in which the values I0 and I are the luminescent intensities of 2–4 without and with addition of Fe3+/Cu2+, respectively, Ksv is the quenching constant, and C[M] is the concentration of metal ions.40 Based on the luminescent data, the Ksv were 1.212 × 104 for 2, 9.383 × 103 for 3 and 4.894 × 104 for 4, respectively. The Ksv value can be used to evaluate the metal ions selective and sensitive sensing of complexes. The higher Ksv of 2 demonstrates that the luminescent quenching effect for Fe3+ ions of 2 are more sensitive than 3. Meanwhile, the luminescent quenching effect for Cu2+ ions of 4 are more sensitive than 2 and 3.
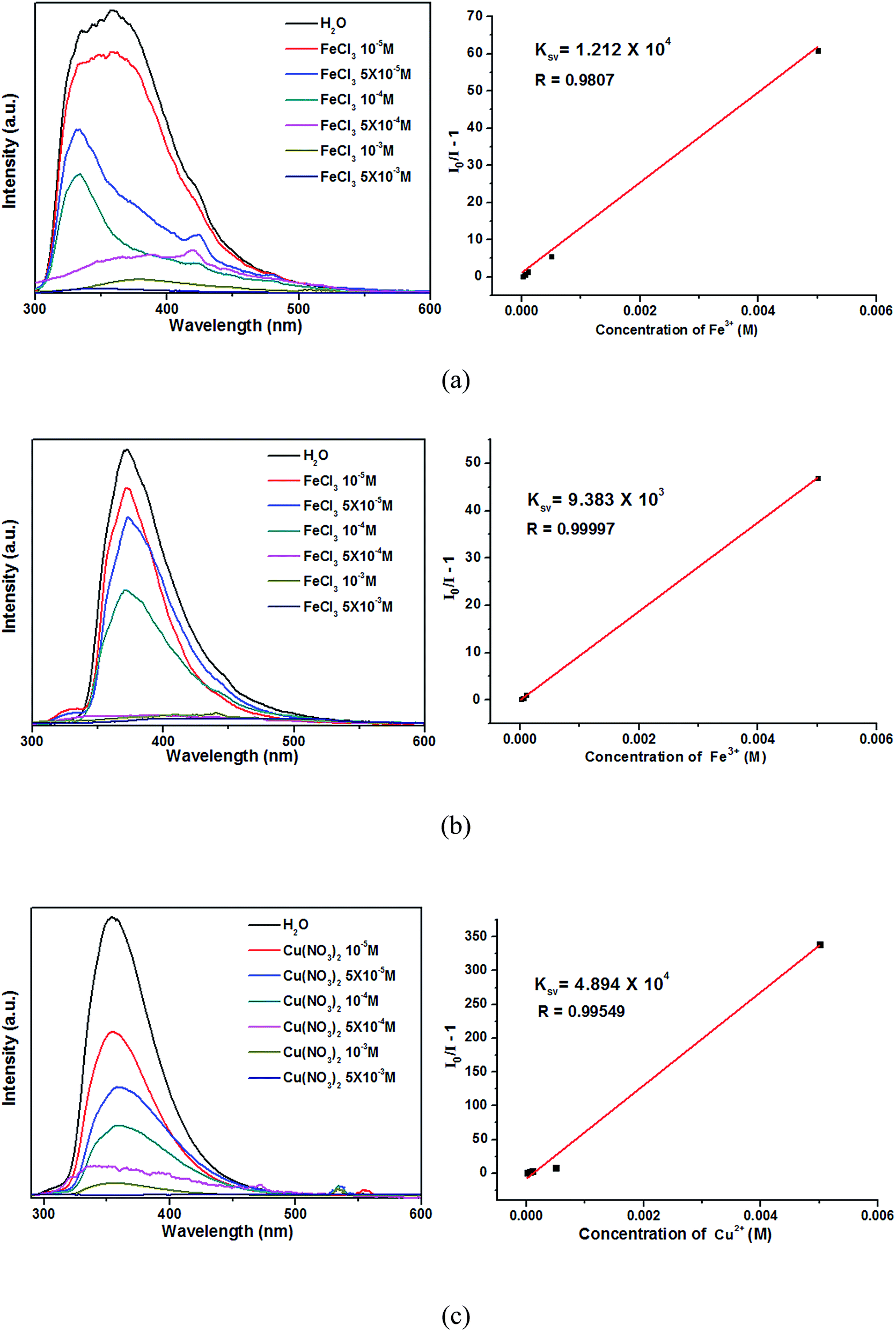 | ||
| Fig. 8 Emission spectra and linear relationships for 2 (a), 3 (b) and 4 (c) in aqueous solutions of different Fe3+/Cu2+ concentrations. | ||
The above luminescent studies indicate that 2 and 3 could selectively sense the exoteric Fe3+ through the luminescent quenching, and 4 could be highly effective and selective luminescent sensors for Cu2+ ions. The PXRD patterns of the Fe3+/Cu2+ loaded 2–4 were nearly corresponded with the original samples (Fig. S5†). The results show that their basic frameworks are stable after loaded metal ions.
Cr2O72−-sensing properties
Encourage by the metal ions' high selective and sensitive of 1–4, their sensing of trace anions in aqueous were investigated through the way that polymers 1–4 (3 mg) were soaked in an aqueous solution (3 mL) of 0.01 M K2CO3, K2Cr2O7, K2S2O8, K2SO4, KSCN, CH3COOK, KCl, KClO3, KIO3, KNO3, KOH and KH2PO4. After the polymer-anion suspensions were sonicated in the dark for 30 min, the photoluminescent spectra were investigated. As shown in Fig. 9, the four titled complexes with other anions do not exhibit the obvious luminescence quenching except Cr2O72−, which exhibited an obviously quenching effect. The absorption bands of the Cr2O72− (two wide absorption bands from 230 to 413 nm)41 almost cover the whole ranges of absorption bands that arise from complexes 1–4 (320–450 nm). Therefore, the luminescence quenching may be caused by the competition of excitation energy between the complexes and Cr2O72−.38 The PXRD results reveal that four titled compounds were stable after sensing Cr2O72− (Fig. S5†). | ||
| Fig. 9 Emission spectra and intensities for 1 (a), 2 (b), 3 (c) and 4 (d) in solutions of different anions. | ||
As shown in Fig. 10, the luminescence intensities of 1–4 are gradually decreased with increasing Cr2O72− concentrations, and the luminescence intensities are almost completely quenched at the concentrations of 5 × 10−4 M. The Ksv values calculated by Stern–Volmer equation are 6.254 × 103, 4.894 × 104, 4.357 × 104 and 1.045 × 104, respectively. Thus, the complexes 2–4 are much more sensitive than 1 in detecting Cr2O72−. Moreover, complexes 2–4 show high sensitivities in sensing Cr2O72− noxious anion compared with other reported CPs.41–43 The high sensitivity indicates that complexes 2–4 may be a good candidate for the sensing of Cr2O72− anion in industry.
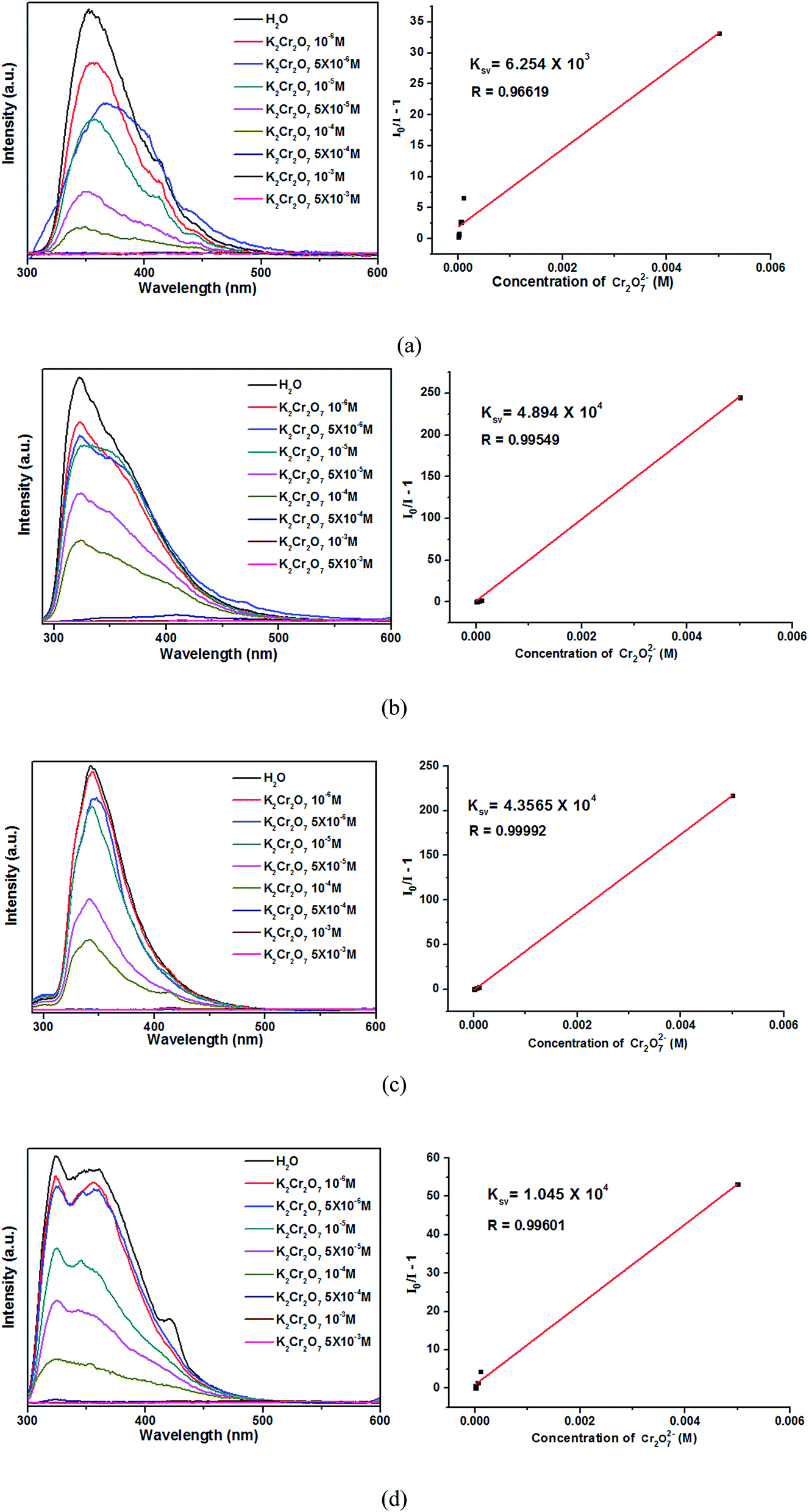 | ||
| Fig. 10 Emission spectra and linear relationships for 1 (a), 2 (b), 3 (c) and 4 (d) in aqueous solutions of different Cr2O72− concentrations. | ||
Photocatalysis properties
Photocatalysts have attracted much attention due to their potential applications in purifying water by thoroughly decomposing organic compounds.44 It is well known that rhodamine B (RhB) and methylene blue (MB) are the most common organic dyes in waste water, and some MOFs show photocatalytic activity in the degradation of organic dyes under UV irradiation by oxidation of organic materials.45 Here, we investigated the photocatalytic ability of the title complexes (1–4) towards the degradation of RhB and MB under UV irradiation. The photocatalytic experiments were performed by the following process:46 20 mg of 1, 2, 3 and 4 was dispersed in 100 mL aqueous solution of RhB (6 mg L−1) or MB (6 mg L−1), respectively. For the adsorption–desorption equilibrium, the mixture was stirred in the dark for 30 min before turning on the Hg lamp (125 W). After centrifugation of the sample, the transparent solution was tested under UV measurement.47The photocatalytic activities of 1–4 in RhB solution are shown in Fig. 11a and S6.† From the Fig. 11a and S6,† we can found that the absorbance peaks of RhB decreased obviously with different photocatalytic efficiency after the compounds were added. Moreover, the concentration ratios of RhB (c/c0) against irradiation time (min) in the presence of the complexes 1–4 were plotted, with c0 representing the initial concentration of RhB after magnetically stirring in the dark for 30 min (Fig. 11b). For complexes 1–4, the RhB solution has been taken out every 15 min, but the photocatalytic efficiency is different. The degradation ratios of RhB are 90.2% for 1, 82.7% for 2 and 95.1% for 4 after 135 min of UV irradiation except complex 3 (97.1% and 98.7% after 75 and 90 min), and the remarkable photocatalytic activity is 3 > 4 > 1 > 2. Meanwhile, all the RhB degradation ratios of control experiments did not surpass 30% and nearly cease after 150 min (91.8% for 1, 85.3% for 2, 98.7% for 3 and 95.1% for 4) under UV irradiation.
 | ||
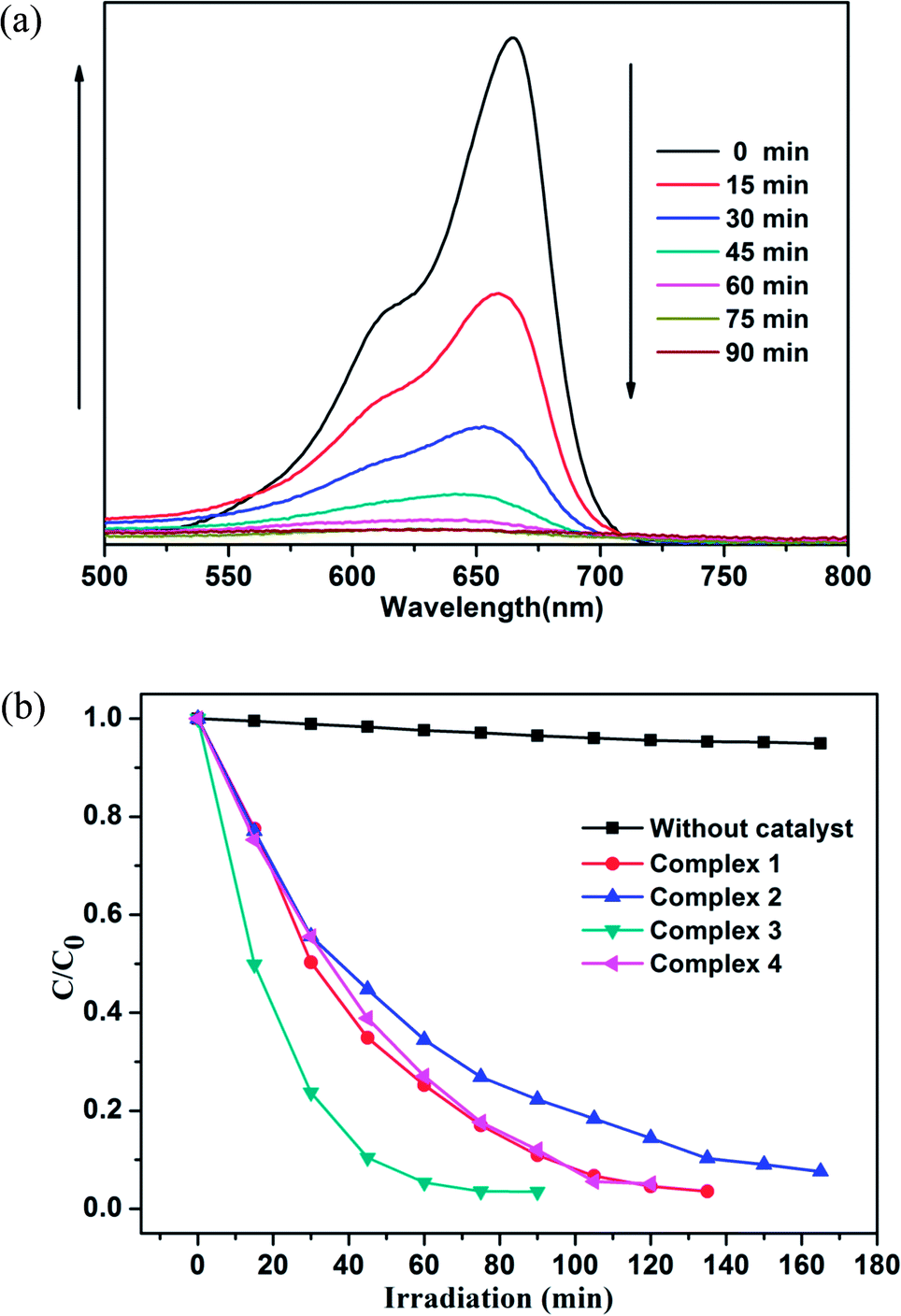
| Fig. 11 (a) Absorption spectra of the RhB solution in the presence of complex 3. (b) Plots of concentration ratios of RhB (c/c0) against irradiation time (min) in the presence of complexes 1–4 and without any catalyst during the decomposition reaction under UV irradiation. | ||
As shown in Fig. 12 and S7,† the degradation ratios of MB are 93.2% for 1, 85.6% for 2 and 94.9% for 4 after 120 min of UV irradiation except complex 3 (96.5% after 75 min). The MB degradation ratios of control experiments did not surpass 30% and nearly cease after 150 min under UV irradiation. From photocatalytic degradation results, we found that the complex 3 much better than the complex 1, 2 and 4 in RhB and MB. Moreover, the photocatalytic activity of the complex 1, 2, 3 and 4 is better in MB solution compared with RhB solution in 120 min (Fig. 13).
 | ||
| Fig. 12 (a) Absorption spectra of the MB solution in the presence of complex 3. (b) Plots of concentration ratios of MB (c/c0) against irradiation time (min) in the presence of complexes 1–4 and without any catalyst during the decomposition reaction under UV irradiation. | ||
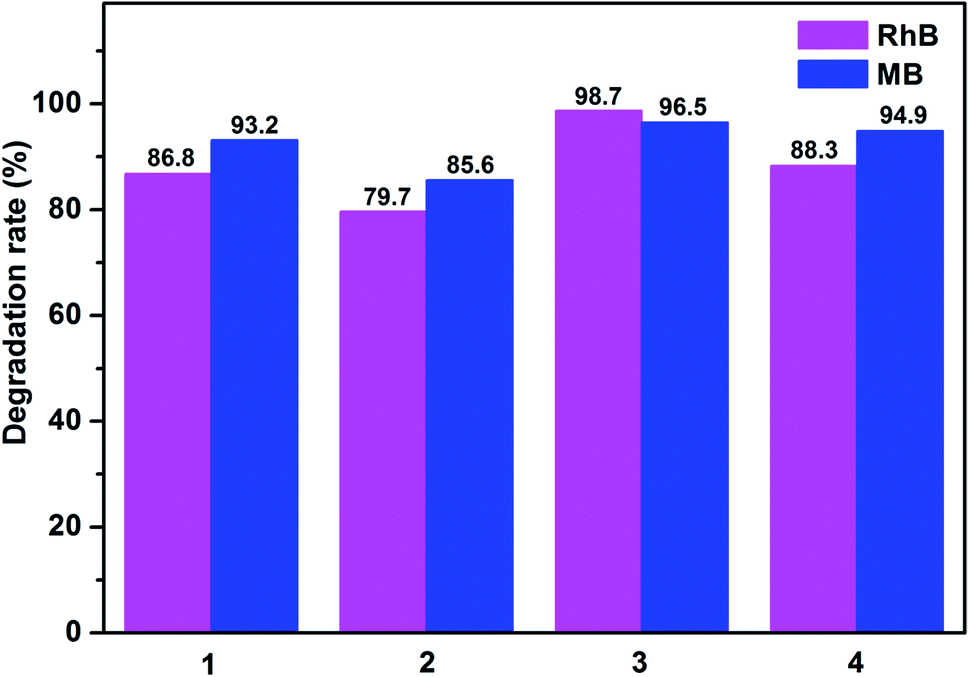 | ||
| Fig. 13 Degradation rates of the RhB and MB solutions in the presence of complexes 1–4. | ||
Fig. 11b and 12b show that the titled complexes have good photocatalytic activities for the photodegradation of RhB and MB under UV irradiation, and the degradation activity 3 > 4 > 1 > 2, which corresponding with the optical band gaps (Eg values: 2.70 eV for 1, 2.74 eV for 2, 2.28 eV for 3 and 2.59 eV for 4). As we know, for complex structures, the numbers of the coordinated water molecules, the coordination environments of the central metals, the extent of the conjugation48 and optical band gap could influence the photocatalytic activities. Therefore, it can be surmised that the low optical band gap and coordination environments of Zn(II) (four-coordination) of 3 aided in the transport of excited holes/electrons to the surface to initiate the photocatalytic decomposition reaction. Moreover, all of the titled d10 compounds (1–4) show a better photocatalytic activity compared with reported similar Cd(II)/Zn(II) CPs28,49 in the degradation of MB under the same condition (Table S5†). Thus, complex 3 may be candidates for photocatalytic activity in the degradation of some organic dyes.
When the complex is exposed under UV light, the 2p bonding orbital of the oxygen and/or nitrogen atom which in metal complex could transfer an electron from the highest occupied molecular to the lowest unoccupied molecular orbital (an empty metal orbital). Then the water molecule would captured one electron for the stable of highest occupied molecular orbital. Meanwhile, the water molecule was oxygenated into the ˙OH radical. The ˙OH active species could decompose RhB and MB effectively to complete the photocatalytic process.50 Meanwhile, the PXRD results revealed that four Cd(II)/Zn(II) compounds were stable in degradation process, as shown in Fig. S8.†
Conclusions
In summary, four LCPs were synthesized based on “V”-shaped 3,5-di(4′-carboxyl-phenyl)benzene acid (H2dpb) and four different imidazole bridging linkers (bimb, tib, 4,4′-bibp and 1,3-bitl) under hydrothermal conditions, with the final packing structures exhibiting a systematic variation of architectures from 1D chains based supramolecular to 3D interpenetrated frameworks. These results reveal that nature of the bridging N-donor linkers have significant effects on the H2dpb coordination modes and the final packing structures. Moreover, the luminescent sensing of metal cations and anions through fluorescence quenching were investigated systematically and quantitatively. The results demonstrate that 2 and 3 display high sensitive and selective luminescent sensing towards Fe3+ ion, and 4 display high sensitive and selective luminescent sensing towards Cu2+ ion. Both 1–4 can work as high sensitive sensors to Cr2O72− by luminescent quenching. And the photocatalytic studies indicate that complex 3 is good candidates for the photocatalytic degradation of rhodamine B (RhB) and methylene blue (MB).Acknowledgements
This research was supported by the National Natural Science Foundation of China to C. F. Bi (No. 21371161), the Specialized Research Fund for the Doctoral Program of Higher Education of China (No. 20120132110015).References
- (a) Y. J. Cui, Y. F. Yue, G. D. Qian and B. L. Chen, Chem. Rev., 2012, 2, 703 Search PubMed; (b) H. Yang, F. Wang, Y. X. Tan, T. H. Li and J. Zhang, Chem.–Asian J., 2012, 7, 1069 CrossRef CAS PubMed; (c) M. J. Sie, Y. J. Chang, P. W. Cheng, P. T. Kuo, C. W. Yeh, C. F. Cheng, J. D. Chen and J. C. Wang, CrystEngComm, 2012, 14, 5505 RSC; (d) G. M. Sun, Y. M. Song, Y. Liu, X. Z. Tian, H. X. Huang, Y. Zhu, Z. J. Yuan, X. F. Feng, M. B. Luo, S. J. Liu, W. Y. Xu and F. Luo, CrystEngComm, 2012, 14, 5714 RSC; (e) Y. W. Li, H. Ma, Y. Q. Chen, K. H. He, Z. X. Li and X. H. Bu, Cryst. Growth Des., 2012, 12, 189 CrossRef CAS; (f) Z. Y. Du, H. B. Xu and J. G. Mao, Inorg. Chem., 2006, 45, 9780 CrossRef CAS PubMed; (g) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330 RSC.
- (a) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196 CrossRef CAS PubMed; (b) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450 RSC; (c) M. B. Lalonde, O. K. Farha, K. A. Scheidt and J. T. Hupp, ACS Catal., 2012, 2, 1550 CrossRef CAS; (d) K. S. Jeong, Y. B. Go, S. M. Shin, S. J. Lee, J. Kim, O. M. Yaghi and N. Jeong, Chem. Sci., 2011, 2, 877 RSC.
- (a) J. Li, J. Tao, R. B. Huang and L. S. Zheng, Inorg. Chem., 2012, 51, 5988 CrossRef CAS PubMed; (b) H. L. Wang, D. P. Zhang, D. F. Sun, Y. T. Chen, K. Wang, Z. H. Ni, L. J. Tian and J. Z. Jiang, CrystEngComm, 2010, 12, 1096 RSC; (c) M. Kurmoo, Chem. Soc. Rev., 2009, 38, 1353 RSC; (d) L. H. Jia, R. Y. Li, Z. M. Duan, S. D. Jiang, B. W. Wang, Z. M. Wang and S. Gao, Inorg. Chem., 2011, 50, 144 CrossRef CAS PubMed; (e) S. Y. Qian, H. Zhou, A. H. Yuan and Y. Song, Cryst. Growth Des., 2011, 11, 5676 CrossRef CAS.
- (a) L. J. Murray, M. Dinca and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC; (b) H. Uehara, S. Diring, S. Furukawa, Z. Kalay, M. Tsotsalas, M. Nakahama, K. Hirai, M. Kondo, O. Sakata and S. Kitagawa, J. Am. Chem. Soc., 2011, 133, 11932 CrossRef CAS PubMed; (c) Y. Kang, F. Wang, J. Zhang and X. Bu, J. Am. Chem. Soc., 2012, 134, 117881 Search PubMed; (d) J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC; (e) C. T. He, J. Y. Tian, S. Y. Liu, G. F. Ouyang, J. P. Zhang and X. M. Chen, Chem. Sci., 2013, 4, 351 RSC.
- (a) G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC; (b) H. X. Zhang, F. Wang, Y. X. Tian, Y. Kang and J. Zhang, J. Mater. Chem., 2012, 22, 16288 RSC; (c) J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869 CrossRef CAS PubMed; (d) H. L. Jiang, Y. Tatsu, Z. H. Lu and Q. Xu, J. Am. Chem. Soc., 2010, 132, 5586 CrossRef CAS PubMed.
- (a) M. Yoon, K. Suh, S. Natarajan and K. Kim, Angew. Chem., Int. Ed., 2013, 52, 2688 CrossRef CAS PubMed; (b) P. Ramaswamy, N. E. Wong and G. K. H. Shimizu, Chem. Soc. Rev., 2014, 43, 5913 RSC; (c) S. Horike, D. Umeyama and S. Kitagawa, Acc. Chem. Res., 2013, 46, 2376 CrossRef CAS PubMed.
- (a) F. E. Kuhn, J. L. Zuo, F. F. Biani, A. M. Santos, Y. M. Zhang, J. Zhao, A. Sandulache and E. Herdtweck, New J. Chem., 2004, 28, 43 RSC; (b) W. Lu, N. Y. Zhu and C. M. Che, J. Am. Chem. Soc., 2003, 125, 16081 CrossRef CAS PubMed; (c) A. Albinati, P. Leoni, L. Marchetti and S. Rizzato, Angew. Chem., Int. Ed., 2003, 42, 5990 CrossRef CAS PubMed.
- L. Carlucci, G. Ciani and D. M. Proserpio, Coord. Chem. Rev., 2003, 246, 247 CrossRef CAS.
- L. Y. Pang, G. P. Yang, J. C. Jin, M. Kang, A. Y. Fu and Y. Y. Wang, Cryst. Growth Des., 2014, 14, 2954 CAS.
- C. J. Watras, R. C. Back, S. Halvorsen, R. J. M. Hudson, K. A. Morrison and S. P. Wente, Sci. Total Environ., 1998, 219, 183 CrossRef CAS PubMed.
- J. Kong, N. R. Franklin, C. Zhou, M. G. Chapline, S. Peng, K. Cho and H. Dai, Science, 2000, 287, 622 CrossRef CAS PubMed.
- J. C. Jin, L. Y. Pang, G. P. Yang, L. Hou and Y. Y. Wang, Dalton Trans., 2015, 44, 17222 RSC.
- X. Wang, T. Qin, S. S. Bao, Y. C. Zhang, X. Shen, L. M. Zheng and D. R. Zhu, J. Mater. Chem. A, 2016, 4, 16484 CAS.
- R. Lv, J. Y. Wang, Y. P. Zhang, H. Li, L. Y. Yang, S. Y. Liao, W. Gu and X. Liu, J. Mater. Chem. A, 2016, 4, 15494 CAS.
- (a) X. L. Wang, J. Luan, H. Y. Lin, C. Xu, G. C. Liu, J. W. Zhang and A. X. Tian, CrystEngComm, 2013, 15, 9995 RSC; (b) X. L. Wang, J. J. Huang, L. L. Liu, G. C. Liu, H. Y. Lin, J. W. Zhang, N. L. Chen and Y. Qu, CrystEngComm, 2013, 15, 1960 RSC.
- (a) X. Lin, I. Telepeni, A. J. Blake, A. Dailly, C. M. Brown, J. M. Simmons, M. Zoppi, G. S. Walker, K. M. Thomas, T. J. Mays, P. Hubberstey, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2009, 131, 2159 CrossRef CAS PubMed; (b) H. S. Choi and M. P. Suh, Angew. Chem., Int. Ed., 2009, 48, 6865 CrossRef CAS PubMed; (c) B. L. Chen, N. W. Ockwig, A. R. Millward, D. S. Contreras and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4745 CrossRef CAS PubMed; (d) J. Jia, X. Lin, C. Wilson, A. J. Blake, N. R. Champness, P. Hubberstey, G. Walker, E. J. Cussen and M. Schroder, Chem. Commun., 2007, 840 RSC.
- (a) J. M. Lim, P. Kim, M. C. Yoon, J. Sung, V. Dehm, Z. J. Chen, F. Wurthner and D. Kim, Chem. Sci., 2013, 4, 388 RSC; (b) F. Guo, F. Wang, H. Yang, X. L. Zhang and J. Zhang, Inorg. Chem., 2012, 51, 9677 CrossRef CAS PubMed; (c) D. R. Xiao, Y. G. Li, E. B. Wang, L. L. Fan, H. Y. An, Z. M. Su and L. Xu, Inorg. Chem., 2007, 46, 4158 CrossRef CAS PubMed; (d) S. N. Wang, R. R. Yun, Y. Q. Peng, Q. F. Zhang, J. Lu, J. M. Dou, J. F. Bai, D. C. Li and D. Q. Wang, Cryst. Growth Des., 2012, 12, 79 CrossRef CAS; (e) X. M. Meng, C. B. Fan, C. F. Bi, Z. A. Zong, X. Zhang and Y. H. Fan, CrystEngComm, 2016, 18, 2901 RSC.
- (a) V. A. Blatov, IUCr CompComm Newsletter, 2006, 7, 4 Search PubMed; (b) V. A. Blatov, A. P. Shevchenko and V. N. Serezhkin, J. Appl. Crystallogr., 2000, 33, 1193 CrossRef CAS; (c) V. A. Blatov, M. O' Keeffe and D. M. Proserpio, CrystEngComm, 2010, 12, 44 RSC.
- O. D. Friedrichs, Program SYSTRE 1.14 beta, 2007, http://www.gavrog.sourceforge.net/ Search PubMed.
- Bruker, SMART, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 1998 Search PubMed.
- G. M. Sheldrick, SHELXTL NT, version 5.1, Program for Solution and Refinement of Crystal Structures, University of GÖttingen, GÖttingen, Germany, 1997 Search PubMed.
- (a) K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley & Sons, New York, 5th edn, 1997 Search PubMed; (b) G. Socrates, Infrared Characteristic Group Frequencies, Wiley, New York, 1980 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination complexes, John Wiley & Sons, New York, 1986 Search PubMed.
- (a) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS; (b) A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 2002 Search PubMed.
- S. S. Chen, J. Fan, T. Okamura, M. S. Chen, Z. Su, W. Y. Sun and N. Ueyama, Cryst. Growth Des., 2010, 10, 812 CAS.
- Z. H. Yan, X. W. Zhang, H. D. Pang, Y. H. Zhang, D. F. Sun and L. Wang, RSC Adv., 2014, 4, 53608 RSC.
- (a) L. Zhang, Y. Wei, C. Wang, H. Guo and P. J. Wang, Solid State Chem., 2004, 177, 3433 CrossRef CAS; (b) J. H. Liao, J. S. Juang and Y. C. Lai, Cryst. Growth Des., 2006, 6, 354 CAS; (c) Y. Xia, P. F. Wu, Y. G. Wei, Y. Wang and H. Y. Guo, Cryst. Growth Des., 2006, 6, 253 CrossRef CAS.
- W. Q. Kan, B. Liu, J. Yang, Y. Y. Liu and J. F. Ma, Cryst. Growth Des., 2012, 12, 2288 CAS.
- H. S. Liu, Y. Q. Lan and S. L. Li, Cryst. Growth Des., 2010, 10, 5221 CAS.
- J. Guo, J. F. Ma, B. Liu, W. Q. Kan and J. Yang, Cryst. Growth Des., 2011, 11, 3609 CAS.
- (a) H. Y. Liu, H. Wu, J. F. Ma, Y. Y. Liu, B. Liu and J. Yang, Cryst. Growth Des., 2010, 10, 4795 CrossRef CAS; (b) W. Q. Kan, Y. Y. Liu, J. Yang, Y. Y. Liu and J. F. Ma, CrystEngComm, 2011, 13, 4256 RSC; (c) H. Y. Bai, J. Yang, B. Liu, J. F. Ma, W. Q. Kan and Y. Y. Liu, CrystEngComm, 2011, 13, 5877 RSC.
- (a) J. Yang, Q. Yue, G.-D. Li, J.-J. Cao, G.-H. Li and J.-S. Chen, Inorg. Chem., 2006, 45, 2857 CrossRef CAS PubMed; (b) X.-H. Jin, C.-X. Ren, J.-K. Sun, X.-J. Zhou, L.-X. Cai and J. Zhang, Chem. Commun., 2012, 48, 10422 RSC.
- Y. Xiao, S. H. Wang, Y. P. Zhao, F. K. Zheng and G. C. Guo, CrystEngComm, 2016, 18, 2524 RSC.
- (a) C. A. Bauer, T. V. Timofeeva, T. B. Settersten, B. D. Patterson, V. H. Liu, B. A. Simmons and M. D. Allendorf, J. Am. Chem. Soc., 2007, 129, 7136 CrossRef CAS PubMed; (b) F. P. Doty, C. A. Bauer, A. J. Skulan, P. G. Grant and M. D. Allendorf, Adv. Mater., 2009, 21, 95 CrossRef CAS.
- X. Zhao, X. H. Bu, T. Wu, S. T. Zheng, L. Wang and P. Y. Feng, Nat. Commun., 2013, 4, 2344 Search PubMed.
- (a) J. F. Wang, Y. B. Li, N. G. Patel, G. Zhang, D. Zhou and Y. Pang, Chem. Commun., 2014, 50, 1225 Search PubMed; (b) K. G. Qu, J. S. Wang, J. S. Ren and X. G. Qu, Chem.–Eur. J., 2013, 19, 7243 CrossRef CAS PubMed.
- Z. H. Xiang, C. Q. Fang, S. H. Leng and D. P. Cao, J. Mater. Chem. A, 2014, 2, 7662 CAS.
- M. Shi, J. Yang, Y. Y. Liu and J. F. Ma, Dyes Pigm., 2016, 129, 106 CrossRef.
- S. T. Zhang, J. Yang, H. Wang, Y. Y. Liu and J. F. Ma, Chem.–Eur. J., 2015, 21, 15806 CrossRef CAS PubMed.
- S. W. Thomas, G. D. Joly and T. M. Swager, Chem. Rev., 2007, 107, 1339 CrossRef CAS PubMed.
- F. Y. Yi, J. P. Li, D. Wu and Z. M. Sun, Chem.–Eur. J., 2015, 21, 11475 CrossRef CAS PubMed.
- X. Li, H. Xu, F. Kong and R. Wang, Angew. Chem., Int. Ed., 2013, 52, 13769 CrossRef CAS PubMed.
- P. F. Shi, B. Zhao, G. Xiong, Y. L. Hou and P. Cheng, Chem. Commun., 2012, 48, 8231 RSC.
- (a) B. Liu, Z. T. Yu, J. Yang, H. Wu, Y. Y. Liu and J. F. Ma, Inorg. Chem., 2011, 50, 8967 CrossRef CAS PubMed; (b) H. X. Li, X. Y. Zhang, Y. N. Huo and J. Zhu, Environ. Sci. Technol., 2007, 41, 4410 CrossRef CAS PubMed.
- (a) X. Zhang, L. Fan, W. Zhang, Y. Ding, W. Fan and X. Zhao, Dalton Trans., 2013, 42, 16562 RSC; (b) X. J. Liang, X. D. Chen and J. C. Zhao, Chem. Soc. Rev., 2014, 43, 473 RSC; (c) H. Fu, Y. G. Li, Y. Liu, W. L. Chen, Q. Wu, J. X. Meng, X. L. Wang, Z. M. Zhang and E. B. Wang, Cryst. Growth Des., 2011, 11, 458 CrossRef CAS; (d) W. Wang, J. Yang, W. Q. Kan and J. F. Ma, CrystEngComm, 2013, 15, 5844 RSC.
- J. Lü, J. X. Lin, X. L. Zhao and R. Cao, Chem. Commun., 2012, 48, 669 RSC.
- X. L. Wang, C. H. Gong, J. W. Zhang, G. C. Liu, X. M. Kan and N. Xu, CrystEngComm, 2015, 17, 4179 RSC.
- A. K. Paul, R. Karthik and S. Natarajan, Cryst. Growth Des., 2011, 11, 5741 CAS.
- X. L. Wang, J. Luan, H. Y. Lin, C. Xu, G. C. Liu, J. W. Zhang and A. X. Tian, CrystEngComm, 2013, 15, 9995 RSC.
- (a) Y. Q. Chen, S. J. Liu, Y. W. Li, G. R. Li, K. H. He, Y. K. Qu, T. L. Hu and X. H. Bu, Cryst. Growth Des., 2012, 12, 5426 CrossRef CAS; (b) J. X. Meng, Y. Lu, Y. G. Li, H. Fu and E. B. Wang, CrystEngComm, 2011, 13, 2479 RSC; (c) J. Guo, J. Yang, Y. Y. Liu and J. F. Ma, CrystEngComm, 2012, 14, 6609 RSC; (d) H. X. Yang, T. F. Liu, M. N. Cao, H. F. Li, S. Y. Gao and R. Cao, Chem. Commun., 2010, 46, 2429 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1061113, 1402587, 1430347 and 1479858. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra27509e |
| This journal is © The Royal Society of Chemistry 2017 |