DOI:
10.1039/C6RA27496J
(Paper)
RSC Adv., 2017,
7, 4960-4974
Isocyano compounds newly recognized in photochemical reaction of thiazole: matrix-isolation FT-IR and theoretical studies†
Received
29th November 2016
, Accepted 27th December 2016
First published on 17th January 2017
Abstract
UV-induced photoreactions of thiazole isolated in low-temperature argon matrices have been investigated by a joint use of infrared spectroscopy and density-functional-theory calculations. Photoproducts have been identified by comparison of the observed infrared spectra with the corresponding calculated spectral patterns, leading to the conclusion that undetected open-chain molecules, syn-2-isocyanoethenethiol (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N–CH
N–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–SH) and 2-isocyanothiirane, are initially produced by cleavage of the CS–CN bond with hydrogen-atom migration, when the matrix samples are exposed to UV radiation coming from a super high-pressure mercury lamp for 3 min. In the secondary photolysis, syn-2-isocyanoethenethiol and 2-isocyanothiirane change to another unknown molecule, 2-isocyanoethanethial (CN–CH2–CHS), by hydrogen-atom migration with generation of the CS double bond. These photoreaction pathways are supported by kinetic analysis of the absorbance changes of IR bands against irradiation time. We have also found that HCN and the ˙CHCH–S˙ biradical are photodecomposed from thiazole by cleavage of the CN–CC bond following the cleavage of the CS–CN bond, where the hydrogen atom on the center carbon atom of ˙CHCH–S˙ immediately migrates to the end carbon atom to form CH2CS or to the sulfur atom to form HCC–SH. In addition, weak bands of the species of interest in astrophysics and astrochemistry such as HCCH, NC–SH, HNCS, HCNS, and the ˙CN radical are detected, but the photoconversion from thiazole to isothiazole or Dewar thiazole is not found. The ring-opening photoreaction, photoisomerization and photodecomposition pathways of thiazole isolated in low-temperature argon matrices are discussed comprehensively.
CH–SH) and 2-isocyanothiirane, are initially produced by cleavage of the CS–CN bond with hydrogen-atom migration, when the matrix samples are exposed to UV radiation coming from a super high-pressure mercury lamp for 3 min. In the secondary photolysis, syn-2-isocyanoethenethiol and 2-isocyanothiirane change to another unknown molecule, 2-isocyanoethanethial (CN–CH2–CHS), by hydrogen-atom migration with generation of the CS double bond. These photoreaction pathways are supported by kinetic analysis of the absorbance changes of IR bands against irradiation time. We have also found that HCN and the ˙CHCH–S˙ biradical are photodecomposed from thiazole by cleavage of the CN–CC bond following the cleavage of the CS–CN bond, where the hydrogen atom on the center carbon atom of ˙CHCH–S˙ immediately migrates to the end carbon atom to form CH2CS or to the sulfur atom to form HCC–SH. In addition, weak bands of the species of interest in astrophysics and astrochemistry such as HCCH, NC–SH, HNCS, HCNS, and the ˙CN radical are detected, but the photoconversion from thiazole to isothiazole or Dewar thiazole is not found. The ring-opening photoreaction, photoisomerization and photodecomposition pathways of thiazole isolated in low-temperature argon matrices are discussed comprehensively.
1. Introduction
Thiazole (1) and isothiazole (1′) are fundamental heterocyclic compounds with a five-membered ring including one nitrogen and one sulfur atom (Fig. 1). The thiazole ring is one of the components of natural products such as thiamine (Vitamin B1) and D-firefly luciferin, and structurally diverse alkaloids containing thiazole subunits are widely distributed in terrestrial and marine organisms and microorganisms.1,2 Thiazole derivatives in various natural and synthetic products, for example Bleomycin, with a wide range of pharmacological activities1–3 are known to be useful for human health.
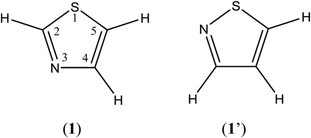 |
| | Fig. 1 Structures of thiazole (1) and isothiazole (1′), and numbering of atoms. | |
In astrochemical and astrophysical studies, UV photochemistry of heterocyclic molecules, which have yet to be detected in the interstellar medium (ISM), gives important information on small photofragments composed of H, C, N, O, and S because they are exposed to the strong UV radiation. In addition, the heterocyclic compounds are interested in astrobiology as the origins of S and N involved in biological materials.4 Indeed, 2-aminothiazole5 and 2-aminooxazole6 have been recently considered as important nucleotide precursors, implying that azole derivatives have potentials of prebiotic molecules. Since they have attractive electronically excited states due to calcogens (O, S, and Se) and N, a various photochemical reaction pathways are possible, implying that the photochemical reaction mechanism of azoles derivatives is more complicated than that of aromatic hydrocarbons. Although photochemical behavior of some five-membered ring containing two hetero atoms has been investigated,7 the whole reaction pathways have not been completed yet. In the present study, we focus our attention on the photoreaction pathways of thiazole.
Several research groups have investigated the photochemistry of thiazole (1) and isothiazole (1′) experimentally and theoretically. For example, Cateau et al. reported that the photoconversion from (1′) to (1) proceeds but not the reverse photoconversion from (1) to (1′) in any solvents.8 By contrast, the final photodecomposition products of thiazole (1) in the gas phase were found to be cyano radical (˙CN) and thiocyanato radical (˙NCS) using a flash photolysis technique,9 although the photodecomposition pathways have not been elucidated yet.
The low-temperature rare-gas matrix-isolation experiment is one of the most powerful techniques to study photoreaction pathways, because unstable or noble species are immediately frozen and trapped in low-temperature solid, which is chemically inert and no absorption in UV and visible-light regions, and stabilized in the environment without interactions between species and solvents. For example, the structure of a thiazole/carbon suboxide (C3O2) complex was investigated in argon matrices,10 while the phosphorescence of thiazole in Ne, Ar, Xe, SF6, and CH4 matrices at 4 K was measured.11 Most recently, the conformational changes of a thiazole derivative (thiazole-2-carboxylic acid) isolated in Ar and N2 matrices have been reported by Halasa et al., using narrowband excitation with near-infrared (IR) and UV light.12 However, the photoreaction pathways of matrix-isolated thiazole, the parent molecule with no substitution groups, have never been reported yet because of their complication, even though thiazole is one of the most fundamental heterocyclic compounds having two hetero atoms.
In the present study, the photochemical reactivity and stability of thiazole isolated in solid argon matrices are investigated by a joint use of IR spectroscopy and density-functional-theory (DFT) calculations. Our purposes are to identify intermediates and final products in the photolysis of thiazole as many as possible, to examine absorbance changes of IR bands against irradiation time, to elucidate when and where and why ring-opening reaction, photoisomerization, and photodecomposition occur, and finally to propose the whole UV-induced photoreaction pathways of thiazole.
2. Experimental
The sample of thiazole (Tokyo Chemical Industry Co., Ltd., more than 98.0% purity) was degassed and purified by freeze–thaw cycles at 77 K and room temperature before using. Argon gas (Taiyo Nippon Sanso, 99.9999% purity) was used without further purification. The sample vapor was premixed with argon gas in a vacuum line at room temperature. The experimental setup was reported in previous papers.13,14 The mixed gas was condensed on a cold substrate (CsI) cooled to 20 K by a closed-cycle helium refrigerator (Iwatani, Cryo Mini M310) through a stainless pipe of 1/8-inch o.d. over a period of 40 min so as to show good signal-to-noise spectra. After the deposition, the matrix sample was cooled down and kept at 10 K for recording IR spectral changes before and after UV irradiation.
IR spectra were measured with an FT-IR spectrometer (JEOL, JIR-WINSPEC50) equipped with an MCT detector cooled by liquid N2. All the spectra were measured at 0.5 cm−1 resolution and averaged over 64 scans. A super high-pressure mercury lamp (SHPML) (USHIO, BA-H500, λ > 200 nm) was used to induce photoreactions. The IR region in the light source was filtered out using a water filter, the wavelength of the light source was controlled with/without optical glass filters.
DFT calculations were performed to obtain the optimized geometry, relative energies and IR spectral patterns of reactants, intermediates, and products using the GAUSSIAN03 program15 at the UB3LYP/aug-cc-pVTZ level among some calculation levels, where their electronic ground state is singlet. Other calculation conditions are default values in the program. Scaling factors of 0.96, 0.97 and 0.98 were applied to the regions over 2800 cm−1, between 2800 and 1900 cm−1, and below 1900 cm−1, respectively, in the present study so as to reproduce the observed wavenumbers for bands of the reactant, thiazole. TD-DFT calculations were performed to obtain the vertical transition energy and oscillator strength at the same level.
3. Results and discussion
3.1 IR spectra of thiazole in argon matrices
IR spectra of thiazole in the vapor, liquid, and crystalline states were reported in the ref. 16 and those in the low-temperature inert-gas matrices were also reported in the ref. 12 The IR spectrum of thiazole (1) in an argon matrix at 10 K, shown in Fig. 2a, is consistent with the reported IR spectra. To confirm the assignment of the observed bands, we decided to calculate the spectral pattern using a basis set of UB3LYP/aug-cc-pVTZ among some calculation levels, because this basis set is applicable to species containing sulfur atoms. Indeed, this basis set worked well for the assignments of photoisomerized products of thiophenol in argon matrices17 and the fundamental bands of thiirane in the gas phase.18 The calculated spectral pattern for thiazole (1) is shown in Fig. 2b, and the observed and calculated wavenumbers and the IR intensities are summarized in Table S1 of the ESI† with the assignments of vibrational modes. It is found that the observed IR spectrum in Fig. 2a is in good agreement with the calculated spectral pattern in Fig. 2b.
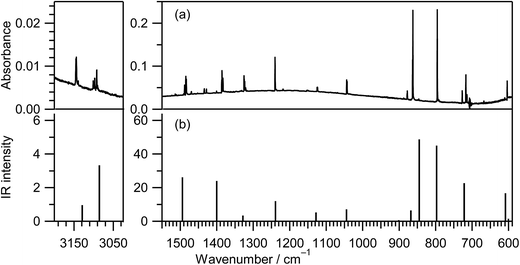 |
| | Fig. 2 (a) Observed IR spectrum of thiazole (1) isolated in a solid argon matrix at 10 K. (b) Calculated spectral pattern of thiazole at the DFT/UB3LYP/aug-cc-pVTZ level. Scaling factors of 0.96 and 0.98 are applied to the regions over 2800 cm−1 and below 1900 cm−1, respectively. | |
3.2 Ring-opening reactions by cleavage of the S1–C2 bond with hydrogen-atom migration
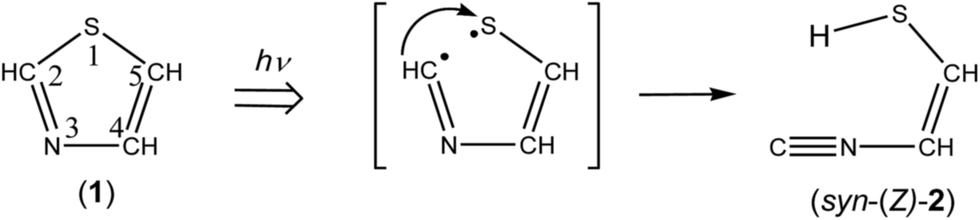
3.2.1. Identification of 2-isocyanoethenethiol (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) N–CH
N–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CH–SH). In order to investigate the photochemical stability and reactivity of thiazole (1), we exposed the matrix sample to UV radiation coming from a super high-pressure mercury lamp (SHPML) through various UV short-cutoff glass filters. When the light in the region with shorter wavelength than 270 nm was cut, no spectral changes were observed. This finding is consistent with the report on UV-visible absorption spectra of thiazole in six kinds of solvents,19 where thiazole in solution has absorption around 235 and 205 nm due to π → π* transition. Therefore, we conclude that the photochemistry of thiazole (1) in the present study is promoted by radiation between 200 and 270 nm through the π → π* transition.
CH–SH). In order to investigate the photochemical stability and reactivity of thiazole (1), we exposed the matrix sample to UV radiation coming from a super high-pressure mercury lamp (SHPML) through various UV short-cutoff glass filters. When the light in the region with shorter wavelength than 270 nm was cut, no spectral changes were observed. This finding is consistent with the report on UV-visible absorption spectra of thiazole in six kinds of solvents,19 where thiazole in solution has absorption around 235 and 205 nm due to π → π* transition. Therefore, we conclude that the photochemistry of thiazole (1) in the present study is promoted by radiation between 200 and 270 nm through the π → π* transition.A difference IR spectrum between the spectra measured before and after irradiation for only 3 min is shown in Fig. 3a, where the negative and positive bands indicate the reactant and the product, respectively. For example, the intensity of the 1484 and 1043 cm−1 bands of thiazole (1) decreased, and new bands appeared at 2113, 1591, and 1013 cm−1. The intense 2113 cm−1 band is characteristic of species containing an isocyano (CN–) or cyano (NC–) group.20 There are a few candidates of C3H3NS species including the triple bond, which have the same chemical formula as the parent molecule, thiazole (1). 2-Isocyanoethenethiol (CN–CHCH–SH) (2) is one of the candidates for the photoproducts of thiazole (1), which is produced by cleavage of the S1–C2 bond with the hydrogen-atom migration from C2 to S1 (see Scheme 1). The existence of (2) was predicted previously by the DFT calculation (B3LYP/6-31G*),21 but neither spectral parameters nor experimental evidences have yet been reported. Thus we have performed the DFT calculation for the geometrical optimization and the vibrational analysis of (2) to assign the bands newly appearing in Fig. 3.
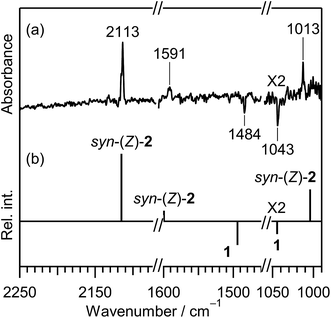 |
| | Fig. 3 (a) Difference IR spectrum of thiazole (1) isolated in a solid argon matrix between the spectra measured before and after UV irradiation for 3 min. (b) Calculated spectral patterns of the reactant, thiazole (1), (negative) and the product, syn-(Z)-2-isocyaonoethenethiol (syn-(Z)-2), (positive) at the DFT/UB3LYP/aug-cc-pVTZ level. Scaling factors of 0.97 and 0.98 are applied to the regions between 2800 to 1900 cm−1 and below 1900 cm−1, respectively. | |
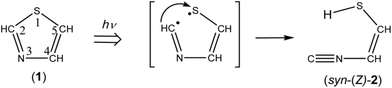 |
| | Scheme 1 Photoproduction pathway for syn-2-isocyanoethenethiol (syn-(Z)-2). | |
Two conformations are possible around both the C–S single bond and the CC double bond of (2), resulting in the four planar conformers shown in Fig. 4. We call anti and syn for the former conformation and (E) and (Z) for the latter conformation. The optimized geometrical parameters and the relative energies are shown in Fig. 4. Syn-(Z)-2 is the most stable one among the four conformers, and anti-(Z)-2 is less stable than syn-(Z)-2 by 4.6 kJ mol−1, while both syn-(E)-2 and anti-(E)-2 are less stable than syn-(Z)-2 by ∼8 kJ mol−1. The calculated wavenumbers and IR intensities of the bands for the four conformers are listed in Table S2 of the ESI.†
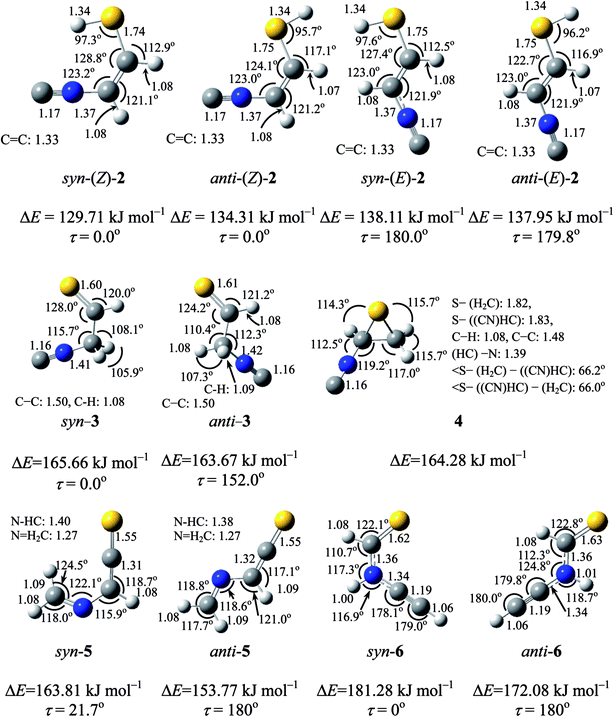 |
| | Fig. 4 Optimized structures and relative energies (ΔE in kJ mol−1 based on thiazole (1)) of open-chain species calculated at the UB3LYP/aug-cc-pVTZ level after ZPE correction. The numbers in figures represent bond lengths (in angstrom) and bonded angles (in degrees). The dihedral angle τ is defined as CN–HCHC–SH (2), CN–H2C–CHS (3), CH2N–CHC (5), and C–NH–CHS (6). | |
The calculated spectral pattern of the most stable syn-(Z)-2 is shown in Fig. 3b to compare with the observed spectrum of the photoproduct. The most intense band at 2113 cm−1 is assignable to the CN– stretching mode of syn-(Z)-2, while the 1591 and 1013 cm−1 bands are assignable to the CC stretching and S–H bending modes, respectively. Since the wavenumbers predicted by the DFT calculation are consistent with the corresponding observed values within ∼10 cm−1, as compared in Table 1, we conclude that syn-(Z)-2 is the initial photoproduct of thiazole (1) in argon matrices.
Table 1 Observed and calculated wavenumbers, and relative intensities of 2-isocyanoethenethiol (2), 2-isocyanoethanethial (3), and 2-isocyanothiirane (4) in solid argon matrices
| Molecules |
Observed |
Calculateda |
Assignment |
| ν/cm−1 |
Intensityb |
ν/cm−1 |
Intensityb |
| Calculated at the DFT/UB3LYP/aug-cc-pVTZ level. Scaling factors of 0.96, 0.97, and 0.98 are applied to the regions over 2800 cm−1, between 2800 to 1900 cm−1, and below 1900 cm−1, respectively. Relative intensity is normalized to the most intense band. Overlapped with a band of (4). Overlapped with a band of ethenethione. Overlapped with a band of syn-3. |
| syn-(Z)-2-Isocyaonoethenethiol(syn-(Z)-2) |
1013 |
29.4 |
1003.99 |
23.70 |
S–H bending |
| 1591 |
7.7 |
1599.32 |
15.69 |
CC stretching |
| 2113 |
100.0 |
2115.16 |
100.00 |
CN– stretching |
| syn-2-Isocyanoethanethial(syn-3) |
627c |
|
624.94 |
6.47 |
C–C–N bending |
| 700d |
|
704.46 |
3.93 |
CH2 rocking + C–H bending |
| 804 |
2.4 |
782.45 |
2.95 |
C–C stretching |
| 970 |
1.4 |
971.92 |
6.89 |
C–H bending |
| 998 |
9.4 |
992.26 |
5.77 |
C–N stretching |
| 1139 |
8.1 |
1134.31 |
19.49 |
CS stretching |
| 1140 |
5.4 |
| 1302 |
3.2 |
1297.50 |
11.01 |
C–H bending |
| 1303 |
11.0 |
| 1304 |
7.2 |
| 1368 |
12.6 |
1375.91 |
18.99 |
C–H bending |
| 1425 |
4.1 |
1425.35 |
11.45 |
CH2 scissoring |
| 2157 |
100.0 |
2163.30 |
100.00 |
CN– stretching |
| 2915 |
3.7 |
2889.28 |
5.20 |
C–H stretching |
| 2971 |
2.2 |
2952.58 |
12.37 |
C–H stretching |
| 2972 |
2.7 |
| anti-2-Isocyanoethanethial(anti-3) |
700d |
|
704.37 |
4.95 |
CH2 rocking + C–H bending |
| 936 |
4.8 |
942.31 |
11.72 |
C–N stretching |
| 954 |
24.8 |
947.40 |
7.12 |
C–H bending |
| 1135 |
10.2 |
1117.97 |
22.74 |
CS stretching |
| 1285 |
10.6 |
1287.05 |
26.39 |
C–H bending |
| 2151 |
100.0 |
2155.71 |
100.00 |
CN– stretching |
| 2975 |
1.3 |
2976.14 |
2.64 |
C–H stretching |
| 2-Isocyanothiirane (4) |
627e |
|
625.79 |
17.72 |
Ring stretching |
| 855 |
5.4 |
840.54 |
4.49 |
CH2 rocking |
| 987 |
18.2 |
977.62 |
8.72 |
C–H bending |
| 1348 |
16.3 |
1347.44 |
12.73 |
C–H bending |
| 2132 |
100.0 |
2138.63 |
100.00 |
CN– stretching |
| 2133 |
68.9 |
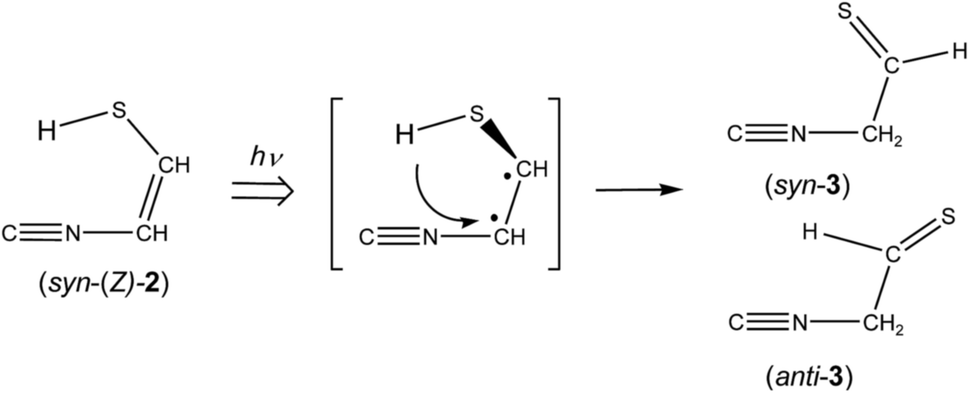
3.2.2. Identification of 2-isocyanoethanethial (CN–CH2–CHS). When the matrix sample was exposed to UV light for 10 min, new bands appeared at 2157, 2151, and 2132 cm−1 near the CN– stretching band at 2113 cm−1 of syn-(Z)-2, as shown in Fig. 5a. The intensities of the 2157 and 2151 cm−1 bands attractively increased when the irradiation time increased to 45 min, as shown in Fig. 5b. These new bands could be due to the CN– stretching mode of the photoproducts like the 2113 cm−1 band of syn-(Z)-2. We assume that the 2157 and 2151 cm−1 bands are assigned to 2-isocyanoethanethial (CN–CH2–CHS) (3), which has two conformations around the C–C single bond, syn and anti. The optimized geometrical parameters and the relative energies of syn-3 and anti-3 are shown in Fig. 4, where the relative energy of anti-3 is nearly equal to that of syn-3. All the calculated wavenumbers and IR intensities of the bands for syn-3 and anti-3 are summarized in Table S3 of the ESI.† It is found that the bands observed at 2157 and 2151 cm−1 in Fig. 5b correspond with the calculated IR bands due to the CN– stretching modes of syn-3 and anti-3, 2163 and 2155 cm−1 in Fig. 5c, respectively.
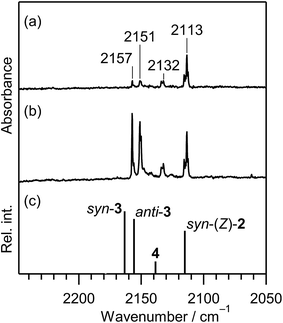 |
| | Fig. 5 Difference IR spectra of thiazole (1) isolated in a solid argon matrix at 10 K (2250–2050 cm−1) between the spectra measured before and after UV-irradiation. Irradiation time is (a) 10 min and (b) 45 min. (c) Calculated spectral patterns of 2-isocyanoethenethiol (syn-(Z)-2), 2-isocyanoethanethial (syn-3 and anti-3), and 2-isocyanothiirane (4) at the DFT/UB3LYP/aug-cc-pVTZ level. A scaling factor of 0.97 is applied to this region. | |
IR bands of syn-3 and anti-3 in other spectral regions are very weak after UV irradiation for 45 min. Thus we tried to find the bands of syn-3 and anti-3 in the difference spectrum between the spectra measured after 180 min minus after 45 min UV irradiation (see Fig. 6). It is noted that the bands due to syn-(Z)-2 initially produced from thiazole (1) are negligible in this difference spectrum because their intensities are almost unchanged during this prolonged irradiation period, as described in Section 3.2.4. Several weak bands of syn-3 and anti-3 are detectable in Fig. 6 and marked with “syn-3” and “anti-3”, respectively. The observed wavenumbers and relative intensities of the bands of syn-3 (12 vibrational modes) and anti-3 (7 vibrational modes) are consistent with the corresponding calculated values, as shown in Table 1. Therefore, we conclude that both syn-3 and anti-3 are produced from thiazole (1) upon prolonged UV irradiation. The photoproduction pathways for syn-3 and anti-3 are discussed in Section 3.2.5 using the kinetic analysis of the IR absorbance changes against the irradiation time.
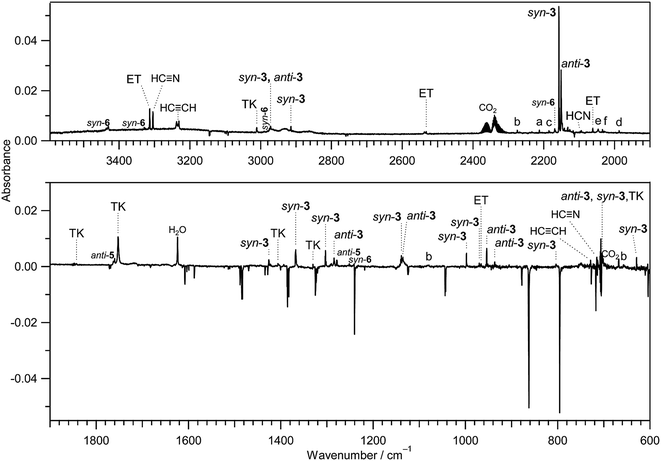 |
| | Fig. 6 Difference IR spectra of thiazole (1) isolated in a solid argon matrix at 10 K between the spectra measured after 180 min minus after 45 min UV irradiation. TK and ET denote thioketene (CH2CS) and ethynethiol (HCC–SH), respectively. Bands marked with “a”, “b”, “c”, “d”, “e”, and “f” are tentatively assigned to 2-cyanoethenethiol (NC–CHCH–SH), 2-cyanothiirane, NC–SH, HNCS, ˙CN radical, and HCNS, respectively. | |
3.2.3. Identification of 2-isocyanothiirane. A weak band is detected at 2132 cm−1 in Fig. 5a and b. This band appeared even in the photolysis of thiazole (1) for only 3 min (see Fig. 3), which is assignable to the CN– stretching mode like the bands of (Z)-2, syn-3 and anti-3 appearing at 2113, 2157, and 2151 cm−1, respectively. Since species including a three-membered ring are usually produced in the photolysis of five-membered heterocyclic compounds such as furan,22 thiophene,22 and isoxazole,23 we assume that this band is due to 2-isocyanothiirane (4), which is produced by cleavage of the S1–C2 bond, hydrogen-atom migration from C2 to C5, and then electron migration to make the S1–C4 bond (see Scheme 2). The optimized geometrical parameters and the relative energy of (4) are shown in Fig. 4; this species is less stable than that of syn-3 by only 0.61 kJ mol−1. All the calculated wavenumbers and IR intensities of (4) are listed in Table S3 of the ESI.† The wavenumbers and the relative intensities of the observed bands for (4) are compared with the corresponding calculated values in Table 1.
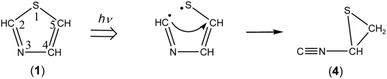 |
| | Scheme 2 Photoproduction pathway for 2-isocyanothiirane (4). | |
Initially, we considered that 2-isocyanothiirane (4) was produced from thiazole (1) via Dewar thiazole, because Dewar structures of five-membered ring molecules such as furan,22 thiophene,22 and cyclopentadiene24 were previously detected as intermediates in photolysis using the matrix-isolation technique. In addition, Dewar thiazole derivatives were theoretically proposed as candidates of the photochemical intermediates for 2-phenylthiazole and 2-acetylthiazole.25 However, no IR bands of Dewar thiazole are found in our IR spectra around the wavenumbers predicted by our DFT calculations (see Table S4, ESI†). For example, the most intense band due to the C–H in-plane bending mode is predicted at 1262.75 cm−1, but undetectable. The relative energy of Dewar thiazole is estimated to be higher than those of 2-isocyanothiirane (4) and thiazole (1) by 94.5 and 258.8 kJ mol−1, respectively, implying that Dewar thiazole is so unstable under our experimental condition to be detected. The photoproduction pathway shown in Scheme 2, where (4) is directly produced from thiazole (1), is supported by the kinetic analysis of the absorbance change against the irradiation time, as explained in the following section.
The photochemical behavior of thiazole derivatives such as methylthiazole and phenylthiazole in solvents were investigated theoretically25,26 and experimentally.27,28 It is reported that the photoconversion from the thiazole derivatives to the corresponding isothiazole derivatives occurs, where Dewar structures play a role of intermediates. If the photoconversion from thiazole (1) to isothiazole (1′) is accelerated in low-temperature argon matrices, we could detect the IR bands of isothiazole (1′). However, no corresponding bands near the 726, 819, 1239, and 1391 cm−1 bands reported in the gas phase29 were detected in the difference spectrum shown in Fig. 6. This fact also supports our conclusion that Dewar thiazole is not produced from thiazole (1) under our experimental condition.
3.2.4. Absorbance changes of IR bands for 1, 2, 3, and 4. To the best of our knowledge, the isocyano compounds, (2), (3), and (4), have been experimentally identified for the first time in the present study. To support our assignments and understand the photoreaction pathways of thiazole (1), we examined the absorbance changes of the 862 cm−1 (thiazole (1)), 2113 cm−1 (syn-(Z)-2), 2157 cm−1 (syn-3), 2151 cm−1 (anti-3), and 2132 cm−1 (4) bands against the irradiation time in Fig. 7. The absorbance of the band for thiazole (1) decreases, while that of the bands for syn-(Z)-2 and (4) increases immediately after starting UV irradiation (see the inserted panel of Fig. 7). Since syn-(Z)-2 and (4) reveal no induction periods at the early irradiation stage, these species could be directly photoproduced from thiazole (1).
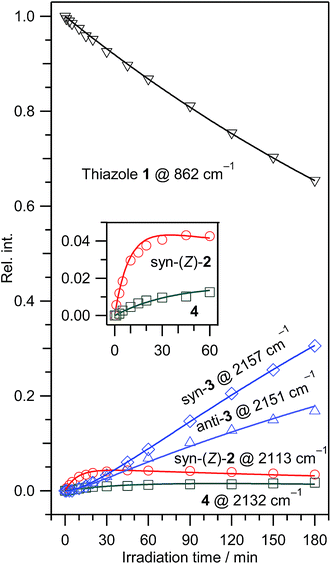 |
| | Fig. 7 Absorbance changes against irradiation time for thiazole (1), 2-isocyanoethenethiol (syn-(Z)-2), 2-isocyanoethanethial (syn-3 and anti-3), and 2-isocyanothiirane (4). The absorbance is normalized using the absorbance of the band at 862 cm−1 for thiazole (1) before UV irradiation. The solid lines represent calculated values obtained by least-squares fitting (see the text). The panel in the graph shows the enlarged absorbance changes of syn-(Z)-2 and (4) at the early irradiation stage (0–60 min). | |
The absorbance of syn-(Z)-2 slightly decreased upon prolonged irradiation over 45 min, as shown in Fig. 7, indicating that syn-(Z)-2 changes to other species by the secondary photo-induced reactions. On the other hand, the absorbance of syn-3 and anti-3 increases more slowly than that of syn-(Z)-2 and clearly reveals induction periods at the early irradiation stage. Thus we assume that syn-3 and anti-3 are photoproduced from syn-(Z)-2 by hydrogen-atom migration from S1 to C4 with generation of the CS double bond (see Scheme 3). If the SH group in the photoexcited state of syn-(Z)-2 is perpendicular to the other part (CN–CH), it is reasonable to assume that both syn-3 and anti-3 are produced from syn-(Z)-2.
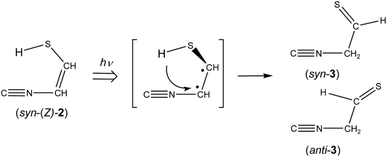 |
| | Scheme 3 Photoisomerization from 2-isocyanoethenethiol (2) to 2-isocyanoethanethial (3). | |
In contrast to the fact that the absorbance of thiazole (1) continues to decrease upon prolonged irradiation over 45 min, the absorbance of (4) is nearly constant as shown in Fig. 7, indicating that a partial amount of (4) changes to other species by the secondary photo-induced reactions like syn-(Z)-2. It is noted that the absorbance of the band for syn-3 increases more strongly over 45 min irradiation than that for anti-3. Thus we assume that syn-3 is also photoproduced from (4) by hydrogen-atom migration from C5 to C4 with generation of the CS double bond (see Scheme 4). To explain this selective photoproduction of syn-3 but not anti-3, we assume that the isocyano group in (4) moves to the opposite side against the hydrogen atom migrating from the methylene group. In this case, the attraction between the sulfur atom and the nitrogen atom in the photoexcited state of (4) may play an important role for the selectivity, which is an interesting problem especially for theoretical chemists. We support the selective photoreaction pathway by the kinetic analysis of the absorbance changes described in the following section.
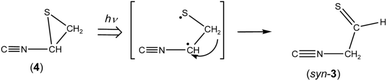 |
| | Scheme 4 Photoisomerization from 2-isocyanothiirane (4) to syn-2-isocyanoethanethial (syn-3). | |
3.2.5. Kinetic analysis. In order to confirm Schemes 1–4 proposed above, we have performed kinetic analysis of the absorbance changes of the IR bands for (1), syn-(Z)-2, syn-3, anti-3, and (4) against the irradiation time shown in Fig. 7, where the rate constants for the photoreaction pathways are defined as follows:| | |
(1) → (syn-(Z)-2) + (4), rate constant: k1
| (1) |
| | |
(syn-(Z)-2) → syn-3 + anti-3, rate constant: k2
| (2) |
| | |
(4) → syn-3, rate constant: k3
| (3) |
Five rate equations are derived from the above reaction pathways of (1)–(3) as follows,
| | |
d[(1)]/dt = −k1[(1)],
| (4) |
| | |
d[syn-(Z)-2]/dt = k1[(1)] − k2[syn-(Z)-2],
| (5) |
| | |
d[(4)]/dt = k1[(1)] − k3[(4)],
| (6) |
| | |
d[syn-3]/dt = k2[syn-(Z)-2] + k3[(4)],
| (7) |
| | |
d[anti-3]/dt = k2[(syn-(Z)-2)],
| (8) |
where [
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
] represents the number of each species. These differential equations are solved easily in terms of absorbance
A and absorption coefficient
ε, resulting in the following equations,
| | |
A(1) = A0exp(–k1t),
| (9) |
| | |
A(syn-(Z)-2) = (ε(syn-(Z)-2)/ε(1))A0{αexp(–k1t) − αexp(–k2t)},
| (10) |
| | |
A(4) = (ε(4)/ε(1))A0{βexp(–k1t) − βexp(–k3t)},
| (11) |
| | |
A(syn-3) = (ε(syn-3)/ε(1))A0{2–(k2/k1)αexp(–k1t) − (k3/k1)βexp(–k1t) + αexp(–k2t) + βexp(–k3t)},
| (12) |
| | |
A(anti-3) = (ε(anti-3)/ε(1))A0[1 − (k2/k1)αexp(–k1t) + αexp(–k2t)],
| (13) |
where
α and
β denote
k1/(
k2 −
k1) and
k1/(
k3 −
k1), respectively. The absorbance of thiazole (
1) at
t = 0,
A0, is fixed to 1 because the absorbance shown in
Fig. 7 is normalized using the
A0 value.
We have performed the least-squares fitting for the absorbance changes using the eqn (9)–(13) to determine three rate constants of k1, k2, and k3 and four ratios of absorption coefficients such as ε(4)/ε(1). The obtained rate constants are 0.00236 ± 0.00001, 0.112 ± 0.010, and 0.0228 ± 0.0040 min−1 for k1, k2, and k3, respectively, and the obtained ratios of absorption coefficients are 2.23 ± 0.18, 0.190 ± 0.028, 0.506 ± 0.013, and 0.536 ± 0.007 for ε(syn-(Z)-2)/ε(1), ε(4)/ε(1), ε(syn-3)/ε(1), and ε(anti-3)/ε(1), respectively, where the uncertainty represents one standard deviation. The rate constant k2 is five times larger than k3, meaning that the reactivity of syn-(Z)-2 is higher than that of (4). The ratio of absorption coefficients for syn-3 and anti-3 is calculated to be 0.506/0.536 = 0.944, which is almost consistent with the corresponding value obtained by the DFT calculation, 174.5/183.6 = 0.950 (see Table S3, ESI†). The calculated absorbance for each species is drawn by solid lines in Fig. 7, which reproduces the observed values satisfactorily, strongly supporting our proposed reaction pathways shown in Schemes 1–4.
3.3 Photodecompositions by two cleavages of the S1–C2 and N3–C4 bonds with hydrogen-atom migration
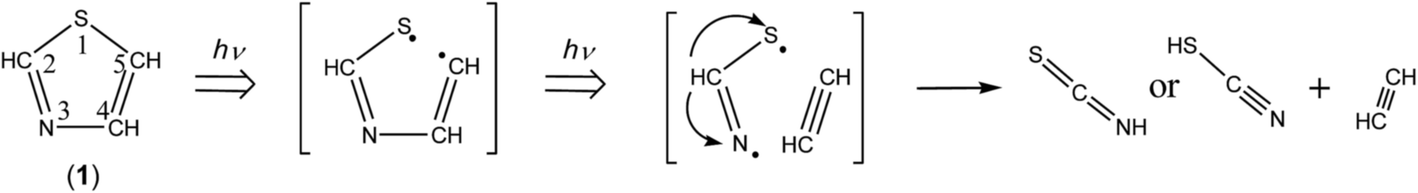
The ring-opening species initially produced by cleavage of the S1–C2 bond is ˙CHN–CHCH–S˙ biradical. In order to confirm that this biradical absorbs UV light, we have performed the time-dependent density-functional-theory (TD-DFT) calculations for vertical transition energy and oscillator strength. The obtained value is 293.15 nm with the oscillator strength of 0.167 as listed in Table 2, the wavelength being longer than that of thiazole (1), 221.54 nm, and the oscillator strength being two times larger than that of thiazole (1), 0.071. Therefore, we assume that ˙CHN–CHCH–S˙ immediately absorbs the second photon to cleave the N3–C4 bond, resulting in the co-production of HCN and ˙CHCH–S˙ biradical. The hydrogen atom on the center carbon atom of ˙CHCH–S˙ immediately migrates to the end carbon atom to form ethenethione (thioketene, CH2CS) or to the sulfur atom to form ethynethiol (HCC–SH), as shown in Scheme 5.
Table 2 Vertical transition energy and oscillator strength of biradicals produced by cleavage of the S1–C2, C5–S1, or N3–C4 bond in thiazole (1)a
| |
Vertical transition energy/nm |
Oscillator strength |
Obsd. wavelengthb/nm |
| Calculated at the TD-DFT/UB3LYP/aug-cc-pVTZ level. Confromation of biradicals is assumed to be unchanged from thiazole (1). Transition energy with the largest value of oscillator coefficient is listed. In solvents. Ref. 14. |
| Thiazole |
221.54 |
0.0715 |
∼235 |
| 199.38 |
0.0362 |
∼205 |
| (S1–C2 cleavage) ˙CHN–CHCH–S˙ |
293.15 |
0.1669 |
|
| (C5–S1 cleavage) ˙CHCH–NCH–S˙ |
251.48 |
0.1600 |
|
| (N3–C4 cleavage) ˙NCH–S–CHCH˙ |
195.14 |
0.1314 |
|
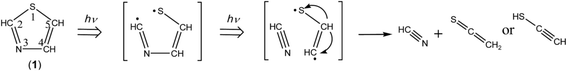 |
| | Scheme 5 Photodecomposition pathway to HCN and CH2CS or HCC–SH. | |
Several papers reported the bands of HCN in the gas phase30 and in the low-temperature matrices23,31–34 to be ∼3303 and ∼720 cm−1 for the C–H stretching and bending modes, respectively. The 3304 cm−1 and 715 cm−1 bands observed in Fig. 6 are consistent with the values of HCN (3306 and 721 cm−1) produced by the photolysis of s-tetrazine and s-triazine in argon matrices.35 Thus we conclude that HCN is produced by the cleavage of the N3–C4 following the cleavage of the S1–C2 bond according to Scheme 5.
There is an intense band at 1752 cm−1 marked with “TK” in Fig. 6. This band is characteristic of the CCS stretching mode of thioketene derivatives. For example, the bands of trimethylenethioketene and cyclopentylidenthioketene were observed at 1793 and 1790 cm−1 in argon matrices at 10 K,36,37 respectively, while the corresponding bands of methylthioketene (CH3–CHCS) and vinylthioketene (CH2CH–CHCS) were observed at 1777 and 1740 cm−1 in argon matrices at 12 K.38,39 Since the wavenumber of the 1752 cm−1 band observed in Fig. 6 is close to that of the CCS stretching mode of CH2CS in argon matrices,40 1755 cm−1, we conclude that CH2CS is produced in the photolysis of thiazole (1). The C–H stretching band of CH2CS, which is reported to be 3010 cm−1 in argon matrices, is also detected at 3011 cm−1. Six bands marked with “TK” in Fig. 6 are assignable to CH2CS. The observed and reported wavenumbers and the relative intensities are summarized in Table 3.
Table 3 Observed and reported wavenumbers and relative intensities of photofragments, CH2CS, HCC–SH, and HCN, in solid argon matrices
| Obsd. ν/cm−1 |
Intensitya |
Ar matrixb |
|
Assignment |
| Relative intensity is normalized to the most intense band. Ref. 40. Symbols of vs, s, m, w, and vw denote very strong, strong, medium, weak, and very weak, respectively. Overlapped with bands of syn-3 and anti-3. Undetectable. Ref. 32. Ref. 35. |
| Ethenethione (CH2CS) |
| 700c |
|
692 |
s |
CH2 wagging |
| 1330 |
11.5 |
1322 |
m |
|
| 1410 |
2.1 |
1410 |
w |
|
| 1752 |
100.0 |
1755 |
vs |
CCS stretching |
| 1843 |
3.4 |
1840 |
m |
|
| 3011 |
20.9 |
3010 |
m |
C–H stretching |
|
| Ethynethiol (HCC–SH) |
| d |
|
558 |
w |
C–H bending |
| 966 |
10.3 |
959 |
w |
S–H bending |
| d |
|
1112 |
m |
|
| 2062 |
19.5 |
2065 |
w |
CC stretching |
| 2532 |
10.9 |
2575 |
vw |
S–H stretching |
| 3313 |
100.0 |
3315 |
vs |
C–H stretching |
| Hydrogen cyanide (HCN) |
| Obsd. ν/cm−1 |
Intensitya |
Ar matrixe |
Armatrixf |
Assignment |
| 715 |
45.6 |
720.2 |
721 |
C–H bending |
| 2094, 2097 |
10.0 |
2093.4 |
2098 |
NC– stretching |
| 3304 |
100.0 |
3303.3 |
3306 |
C–H stretching |
HCC–SH was previously identified as a photoproduct of 1,2,3-thiadiazole,40 where the characteristic IR bands due to the C–H, S–H, and CC stretching modes were observed at 3315, 2575, and 2065 cm−1, respectively. The 3313 cm−1 band marked with “ET” in Fig. 6 is consistent with the reported 3315 cm−1 band. In addition, the bands due to the CC stretching and S–H bending modes are detected at 2062 and 966 cm−1, respectively (see Table 3), while the S–H stretching band is detected at 2532 cm−1. Thus we conclude that HCC–SH is co-produced from thiazole (1) with HCN by the cleavage of the N3–C4 bond following the cleavage of the S1–C2 bond like CH2CS. The detection of the IR bands for HCN, CH2CS and HCC–SH supports the photodecomposition pathway shown in Scheme 5.
It is known that HCC–SH is converted to CH2CS by hydrogen-atom migration upon UV irradiation.40 However, the absorbance of the bands for both CH2CS and HCC–SH increase linearly with no induction periods at the early irradiation state, as shown in Fig. 8. Thus we assume that CH2CS and HCC–SH are independently produced from thiazole (1) by cleavage of the N3–C4 bond following the cleavage of the S1–C2 bond with the hydrogen-atom migration according to Scheme 5. One may claim that HCN, CH2CS, and HCC–SH are produced by cleavage of the S1–C2 bond following the cleavage of the N3–C4 bond. However, the vertical transition energy of ˙NCH–S–CHCH˙, which is produced by cleavage of the N3–C4 bond, is calculated to be 195.14 nm with the oscillator strength of 0.1314, as listed in Table 2. Since the intensity of the light source of our SHPML with shorter wavelength than 200 nm is extremely weak, the secondary photolysis of ˙NCH–S–CHCH˙ is difficult in contrast to the hydrogen-atom migration in the primary photolysis described in Section 3.4.3.
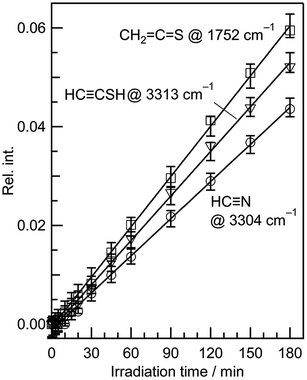 |
| | Fig. 8 Absorbance changes against irradiation time for HCN, CH2CS, and HCCSH. The absorbance is normalized using the absorbance of the band at 862 cm−1 for thiazole (1) before UV irradiation in Fig. 7. | |
3.4 Minor ring-opening reactions by cleavage of the S1–C2, C5–S1, or N3–C4 bond with hydrogen-atom migration
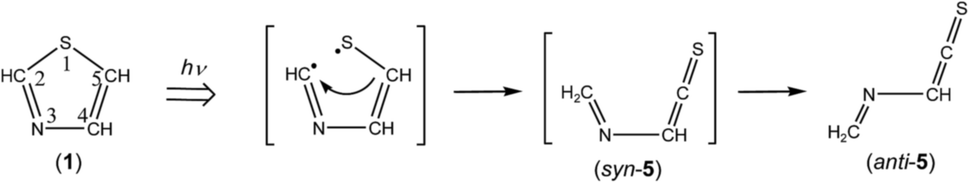
3.4.1. Identification of (methyleneamino)-ethenethione (CH2N–CHCS). The spectral region of the CCS stretching mode of Fig. 6 (1790–1720 cm−1) is enlarged in Fig. 9. One weak doublet band is detected at the higher-wavenumber side of the CH2CS band marked with “TK”. By comparison of the observed wavenumbers with the predicted values of many candidates obtained by the DFT calculation, we assume that the 1760 and 1763 cm−1 doublet band is due to the CCS stretching mode of (methyleneamino)-ethenethione (CH2N–CHCS) (5). This species is photoproduced by cleavage of the S1–C2 bond in thiazole (1) with the hydrogen-atom migration from C5 to C2 (see Scheme 6), instead of the hydrogen-atom migration from C2 to S1 to form syn-(Z)-2 (see Scheme 1) or from C2 to C5 to form (4) (see Scheme 2).
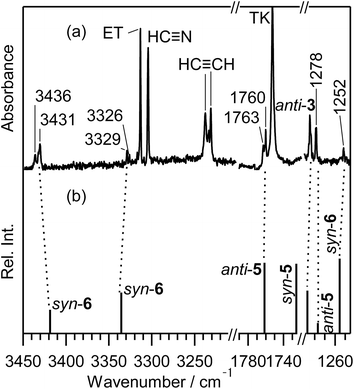 |
| | Fig. 9 Enlarged difference spectra (3450–3200, 1790–1720, and 1290–1250 cm−1) of Fig. 6. (a) Observed difference spectra. (b) Calculated spectral patterns of anti-2-isocyanoethanethial (anti-3), (methyleneamino)-ethenethione (anti-5 and syn-5), and syn-N-ethynylthioformamide (syn-6) at the DFT/UB3LYP/aug-cc-pVTZ level. Scaling factors of 0.96 and 0.98 are applied to regions over 2800 cm−1 and below 1900 cm−1, respectively. | |
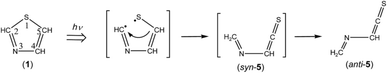 |
| | Scheme 6 Photoproduction pathway for anti-(methyleneamino)-ethenethione (anti-5). | |
This species (5) has two conformations around the N3–C4 single bond (syn-5 and anti-5). The optimized geometrical parameters and the relative energies are shown in Fig. 4. All the calculated wavenumbers and IR intensities of both anti-5 and syn-5 are listed in Table S5 of the ESI.† The conformation of (5) photoproduced initially could be syn-5 but not anti-5, because the molecular shape of syn-5 is more similar to that of the reactant, thiazole (1), than that of anti-5. However, the DFT calculation reveals that syn-5 is less stable than anti-5 by 10.04 kJ mol−1, probably because of the strong repulsion between π electrons on the CN and CCS double bonds like 1,3-butadiene.41,42 Note that the optimized structure of syn-5 is non-planar and the dihedral angle around CH2N–CHC is calculated to be 21.7° (see Fig. 4). The weak doublet band detected at 1760 and 1763 cm−1 in Fig. 9 is found to be consistent with the calculated value of the CCS stretching mode of anti-5, 1761.49 cm−1, but not with that of syn-5, 1725.47 cm−1. In addition, the C–H bending mode of anti-5 is detected at 1278 cm−1 in Fig. 9. Thus we conclude that anti-5 is produced from thiazole (1) via syn-5 initially photoproduced in an argon-matrix cage according to Scheme 6. The observed and calculated wavenumbers and the relative intensities of anti-5 are compared in Table 4.
Table 4 Observed and calculated wavenumbers, and relative intensities of (methyleneamino)-ethenethione (5) and N-ethynylthioformamide (6) in solid argon matrices
| Molecules |
Observed |
Calculateda |
Assignment |
| ν/cm−1 |
Intensityb |
ν/cm−1 |
Intensityb |
| Calculated at the DFT/UB3LYP/aug-cc-pVTZ level. Scaling factors of 0.96, 0.97, and 0.98 are applied to the regions over 2800 cm−1, between 2800 and 1900 cm−1, and below 1900 cm−1, respectively. Relative intensity is normalized to the most intense band. Overlapped with bands of the reactant. |
| anti-(Methyleneamino)-ethenethione (anti-5) |
1278 |
90.5 |
1276.71 |
14.77 |
C–H bending |
| 1760 |
100.0 |
1761.49 |
100.00 |
CC stretching |
| 1763 |
59.0 |
| syn-N-Ethynylthioformamide (syn-6) |
c |
|
1042.22 |
16.27 |
C–H bending |
| 1252 |
35.0 |
1255.41 |
77.36 |
C–H bending |
| c |
|
1389.64 |
100.00 |
C–H bending |
| 2167 |
19.8 |
2178.52 |
19.28 |
CC stretching |
| 2169 |
73.1 |
| 2989 |
0.9 |
2976.37 |
10.17 |
C–H stretching |
| 3326 |
36.7 |
3335.53 |
42.28 |
CC–H stretching |
| 3329 |
60.0 |
| 3431 |
100.0 |
3418.55 |
24.45 |
N–H stretching |
| 3436 |
51.1 |
3.4.2. Identification of N-ethynylthioformamide (HCC–NH–CHS). Four bands are detected at 3436, 3431, 3329, and 3326 cm−1 in the N–H or C–H stretching region of Fig. 9 besides the bands of HCC–SH (marked with “ET”), HCN, and HCCH; the assignment of HCCH is described in Section 3.6. As compared with the results obtained by the DFT calculations, we assign the 3436 and 3431 cm−1 doublet band and the 3329 and 3326 cm−1 doublet band to the N–H and the C–H stretching modes of N-ethynylthioformamide (HCC–NH–CHS) (6), respectively. The optimized structures and the geometrical parameters of two conformers, syn-6 and anti-6, are shown in Fig. 4. All the calculated wavenumbers and IR intensities are listed in Table S5 of the ESI.† It is possible to produce syn-6 from thiazole (1) by cleavage of the C5–S1 bond with hydrogen-atom migration from C4 to N3 to generate the CS double bond (see Scheme 7). The band due to the CC stretching mode of syn-6 is also detected as a weak doublet band at 2169 and 2167 cm−1 in Fig. 10, while the band due to the C–H bending mode is detected at 1252 cm−1 in Fig. 9. The observed and calculated wavenumbers and the relative intensities of syn-6 are compared in Table 4.
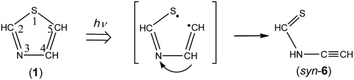 |
| | Scheme 7 Photoproduction pathway for syn-N-ethynylformamide (syn-6). | |
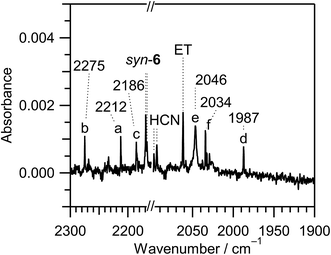 |
| | Fig. 10 Enlarged difference spectra (2300–2160 and 2100–1900 cm−1) of Fig. 6. The spectral region between 2160 and 2100 cm−1 is omitted because the intensity of the IR bands of syn-3 and anti-3 is too strong. Bands marked with “a”, “b”, “c”, “d”, “e”, and “f” are tentatively assigned to 2-cyanoethenethiol (NC–CHCH–SH), 2-cyanothiirane, NC–SH, HNCS, ˙CN radical, and HCNS, respectively. | |
We could not identify the other conformer, anti-6, although the relative energy of anti-6 estimated to be lower than that of syn-6 by 9.20 kJ mol−1. For example, neither the most intense band around 1250 cm−1 nor the second intense band around 1500 cm−1 is detectable in Fig. 6. We have performed the one-step optimization between anti-6 and syn-6, resulting in the barrier height of ∼80 kJ mol−1 at the dihedral angles of 105 and 285° (see Fig. S1, ESI†). Thus we assume that the barrier height is so high that the conversion from syn-6 initially produced from thiazole (1) to the more stable conformer, anti-6, is unable to occur in an argon-matrix cage.
It is noted that the absorbance of the bands for anti-5 and syn-6 is much weaker than that of anti-3, syn-3, HCN, CH2CS, and HCC–SH, as shown in Fig. 6. Thus we conclude that the photoreaction pathways to form anti-5 by the cleavage of the S1–C2 bond shown in Scheme 6 and to form syn-6 by the cleavage of the C5–S1 bond shown in Scheme 7 are minor ring-opening reactions.
3.4.3. Identification of thiocyanatoethene (NC–S–CHCH2). When the N3–C4 bond cleaves upon UV irradiation, ˙NCH–S–CHCH˙ biradical is produced, from which thiocyanatoethene (NC–S–CHCH2) could be produced by hydrogen-atom migration from C2 to C4 (see Scheme 8). The NC– stretching band of NC–S–CHCH2 is reported to be 2177 cm−1 in the vapor phase or 2172 cm−1 in argon matrices at 14 K,43 which is close to the two weak bands of syn-6 appearing at 2167 and 2169 cm−1 in Fig. 10. One of the two bands may be assigned to NC–S–CHCH2. However, no IR bands of NC–S–CHCH2 in other spectral regions are detected in Fig. 6 unlike anti-5 and syn-6. We do not have much confidence for the identification of NC–S–CHCH2.
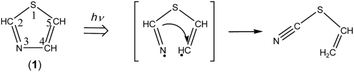 |
| | Scheme 8 Photoproduction pathway for thiocyanatoethene. | |
3.5 Minor photoconversions of isocyano group to cyano group in syn-(Z)-2 and (4)
2-Cyanoethenethiol (NC–CHCH–SH), which is known as a species of astrochemical interest,44 is one of the candidates of C3H3NS species including a cyano group. Only the direction of the NC group of 2-cyanoethenethiol (NC–CHCH–SH) is different from that of 2-isocyanoethenethiol (CN–CHCH–SH) identified in Section 3.2.1. The IR spectra of NC–CHCH–SH were measured previously, and the NC– stretching band is reported to be 2223.7 cm−1 in the gas phase44 or 2214 cm−1 in film45 at 77 K. We tentatively assign the weak band at 2212 cm−1 marked with “a” in Fig. 10 to NC–CHCH–SH. This species may be produced by detachment and recombination of the CN group in CN–CHCH–SH.
2-Cyanothiirane could be caused by detachment and recombination of the CN group in 2-isocianothiirane (4) like the photoconversion from CN–CHCH–SH to NC–CHCH–SH. The calculated wavenumbers and IR intensities of 2-cyanothiirane are shown in Table S4 of the ESI.† The NC– stretching band is predicted at 2276 cm−1, and the corresponding band is detected at 2275 cm−1, marked with “b” in Fig. 10. The most intense band due to the C–S symmetric stretching mode, 629 cm−1, and the thirdly intense band due to the CH2 waging mode, 1065 cm−1, are also detected at 658 and 1079 cm−1 in Fig. 6, respectively. Thus we conclude that a small amount of syn-(Z)-2 and (4), which are initially produced from thiazole (1), convert to the corresponding cyano compounds upon prolonged UV irradiation.
3.6 Minor photodecompositions by two cleavages of the C5–S1 and N3–C4 bonds with hydrogen-atom migration
As described in Section 3.4.2, the ring-opening species initially produced by cleavage of the C5–S1 bond is ˙CHCH–NCH–S˙ biradical. Since the vertical transition energy of ˙CHCH–NCH–S˙ is calculated to be 251.48 nm with the oscillator strength of 0.1600, as listed in Table 2, the N3–C4 bond can be cleaved in the secondary photolysis to co-produce CHCH and ˙NCH–S˙ biradical. The hydrogen atom of ˙NCH–S˙ immediately migrates to the nitrogen atom to form HNCS or to the sulfur atom to form NC–SH as shown in Scheme 9.
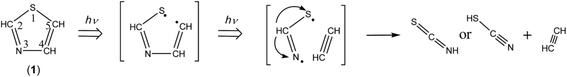 |
| | Scheme 9 Photodecomposition pathway to HCCH and NC–SH or HNCS. | |
A lot of papers have reported the wavenumbers of the vibrational modes for HCCH in low-temperature matrices46–50 and in the gas phase.51 We detected the 3238, 3234 and 3231 cm−1 triplet band in Fig. 6, which is in good agreement with the reported value for HCCH, 3240 cm−1. The NC– stretching mode of NC–SH52 and the NCS stretching mode of HNCS53 are reported to be 2182 cm−1 and 1981.8 cm−1 in low-temperature matrices, respectively. We detected the corresponding bands at 2186 cm−1 marked with “c” and at 1987 cm−1 marked with “d” in Fig. 10, respectively. The detection of HCCH, NC–SH, and HNCS supports the photodecomposition pathway shown in Scheme 9, although it is a minor reaction pathway.
If the hydrogen atom of ˙NCH–S˙ detaches but migrates neither to the sulfur atom nor to the nitrogen atom, ˙NCS radical could be co-produced with HCCH. The band of ˙NCS radical is reported to be 1942.2 cm−1 in free jet experiment.54,55 We tried to detect the corresponding band, but there is no band in the NCS stretching region between 1970 and 1900 cm−1 in Fig. 10. Instead of the band due to ˙NCS radical, we found a band at 2046 cm−1 marked with “e” in Fig. 10, which is consistent with the band of ˙CN radical produced from HCN by vacuum UV light in argon matrices at 14 K, 2046 cm−1.31
Venkatasubramanian and Krishnamachari claimed that ˙NCS radical changes to ˙CN radical in a flash photolysis experiment of thiazole (1).9 Note that our difference spectra were obtained by experiments without any short-cutoff glass filters, meaning that all the radiation coming from the SHPML is used to induce photoreactions. Thus one possibility for the detection of ˙CN radical is that the photodecomposition from ˙NCS radical to S and ˙CN radical immediately occurs even when ˙NCS radical is produced by detachment of the hydrogen atom from ˙NCH–S˙.
However, we propose another possibility that ˙CN radical is detached from the isocyano compounds, syn-(Z)-2 and (4). As explained in Section 3.5, a small amount of the cyano compounds are photoproduced from the corresponding isocyano compounds by detachment and recombination of ˙CN radical. If ˙CN radical detached from the isocyano compounds stays in a low-temperature argon-matrix cage without recombination, the IR band of ˙CN radical is detectable.
3.7 Other minor photofragments
There is an unassigned weak band at 2034 cm−1 marked with “f” in the CN− stretching region of Fig. 10. The wavenumber of this band is much lower than that of the normal isocyano compounds such as syn-(Z)-2, syn-3, anti-3, and (4), implying that it is characteristic of an accumulated multiple bond like HNCS. We tentatively assign this band to thiofulminic acid (HCNS), which is an isomer of HNCS and NC–SH. The wavenumber of the NC– stretching mode of HCNS is reported to be 2035 cm−1.56
4. Conclusions
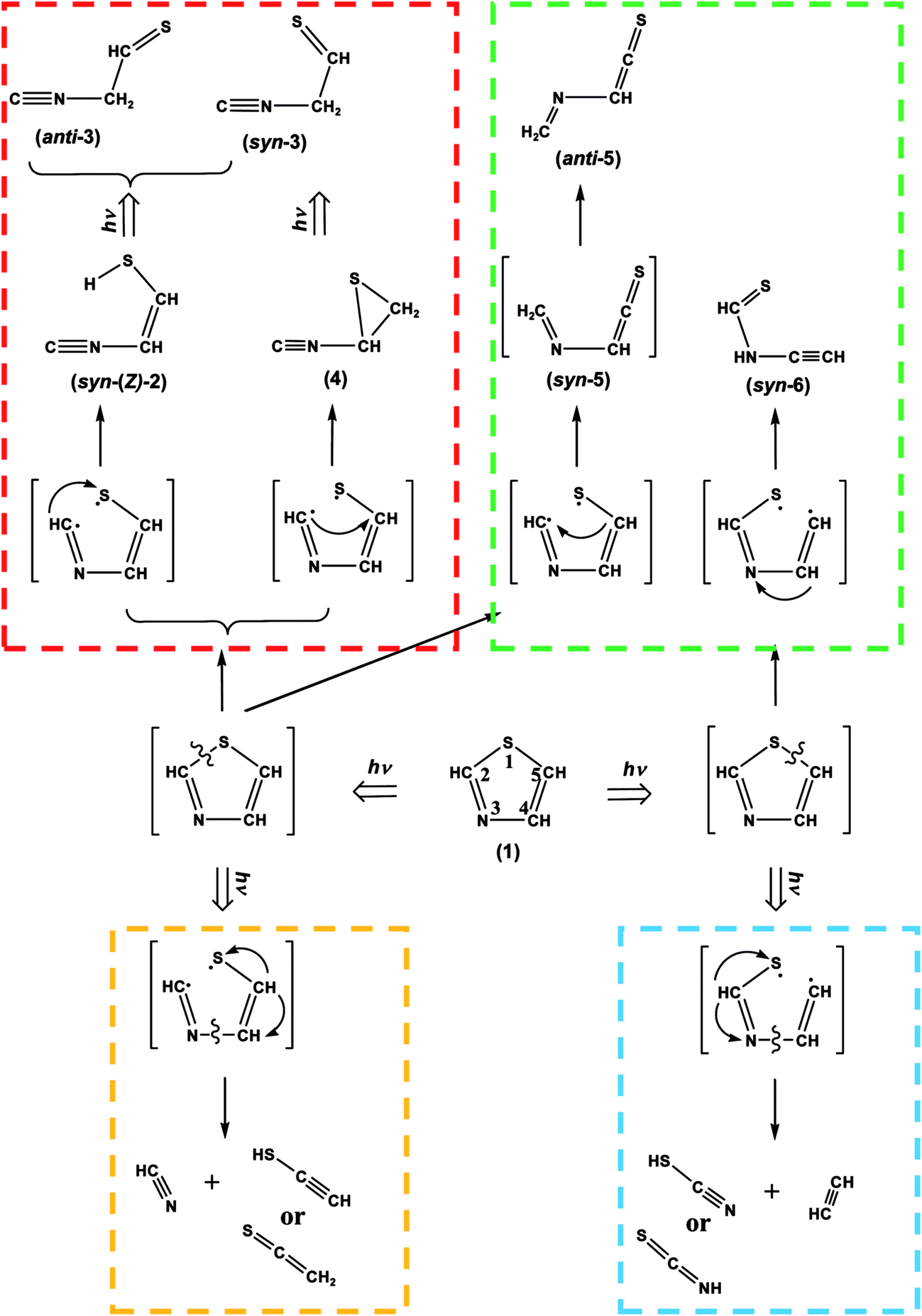
We have investigated the photolysis of thiazole (1) upon UV irradiation by a joint use of the low-temperature matrix-isolation IR spectroscopy and the DFT chemical calculations. The UV-induced photoreaction pathways of thiazole (1) isolated in solid argon matrices are summarized in Scheme 10. The major ring-opening photoreactions caused by the cleavage of the S1–C2 bond are initially induced at early irradiation stage to produce ˙CHN–CHCH–S˙ biradical. The hydrogen atom on the C2 atom of ˙CHN–CHCH–S˙ migrates to S1 to form syn-(Z)-2 or to C5 to form (4), as shown in the red area of Scheme 10. Syn-(Z)-2 changes to other undetected isocyano compounds, syn-3 and anti-3, in the secondary photolysis by the hydrogen-atom migration from S1 to C4, while (4) changes to syn-3. These photoreaction pathways are supported by the kinetic analysis of the absorbance changes of IR bands against the irradiation time. When the N3–C4 bond cleaves following the cleavage of the S1–C2 bond, HCN and ˙CHCH–S˙ biradical are photodecomposed from ˙CHN–CHCH–S˙. The hydrogen atom on the center carbon atom of ˙CHCH–S˙ immediately migrates to the end carbon atom to form CH2CS or to the sulfur atom to form HCC–SH, as shown in the orange area of Scheme 10.
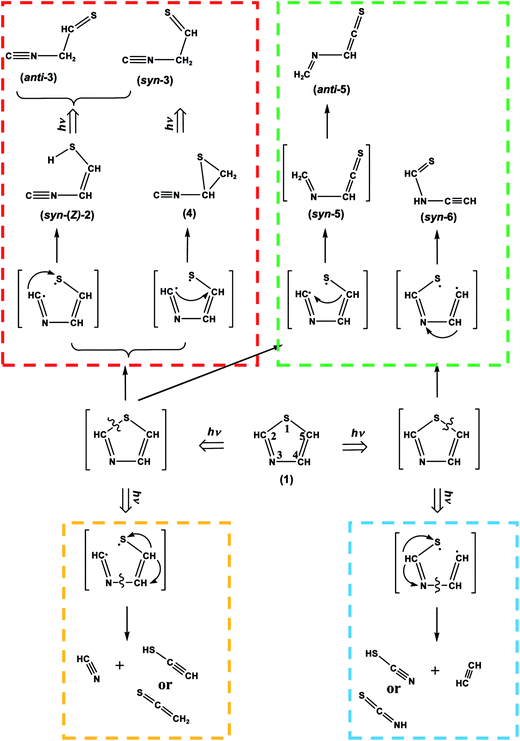 |
| | Scheme 10 Photoreaction pathways of thiazole (1). | |
On the other hand, another minor ring-opening photoreaction is caused by the cleavage of the S1–C2 bond upon prolonged UV irradiation. The hydrogen-atom migration from C5 to C2 in ˙CHN–CHCH–S˙ forms anti-5 via syn-5 initially produced in an argon-matrix cage, as shown in the green area of Scheme 10. Other minor ring-opening photoreaction is caused by the cleavage of the C5–S1 bond upon prolonged UV irradiation to produce ˙CHCH–NCH–S˙ biradical, from which syn-6 is produced by the hydrogen-atom migration from C4 to N3, where the syn conformation is kept in an argon-matrix cage without photoconversion to anti-6. The photoproducts of (2), (3), (4), (5) and (6) are identified for the first time in the present study by a joint use of IR spectroscopy and DFT calculations.
When the N3–C4 bond cleaves following the cleavage of the C5–S1 bond, HCCH and ˙NCH–S˙ biradical are photodecomposed from ˙CHCH–NCH–S˙. The hydrogen atom of ˙NCH–S˙ immediately migrates to the nitrogen atom to form HNCS or to the sulfur atom to form NC–SH, as shown in the blue area of Scheme 10. In addition, several weak bands in the difference spectra measured after prolonged UV irradiation are tentatively assigned to cyano compounds caused by photoconversion from isocyano compounds, ˙CN radical caused by detachment from isocyano compounds, and HCNS caused by photoisomerization from photofragments such as NC–SH.
In the present study, we investigated the photochemical reactivity and stability of thiazole isolated in solid argon matrices by a joint use of IR spectroscopy and DFT calculations. By examination of absorbance changes of IR bands against irradiation time, we identified intermediates and final products. Some of them are unknown isocyano compounds, and some of them are species interested in astrochemistry and astrophysics. We expect that the present results will contribute to develop these research fields and help to detect other simple but unknown species.
Conflict of interest
The authors declare no competing financial interest.
Acknowledgements
The authors are grateful to Prof. Igor Reva of University of Coimbra in Portugal for his valuable discussion and to emeritus Prof. Kozo Kuchitsu of the University of Tokyo in Japan for his helpful advice of English presentation. They are also grateful to Dr. Masaya Miyagawa of Chuo University for his helpful experimental supports.
References
- Z. Jin, Nat. Prod. Rep., 2011, 28, 1143–1191 RSC.
- Z. Jin, Nat. Prod. Rep., 2013, 30, 869–915 RSC.
- M. V. N. de Souza, J. Sulfur Chem., 2005, 26, 429–449 CrossRef CAS.
- C. A. Cole, N. J. Demarais, Z. Yang, T. P. Snow and V. M. Bierbaum, Astrophys. J., 2013, 779, 181 CrossRef.
- M. W. Powner, S.-L. Zheng and J. W. Szostak, J. Am. Chem. Soc., 2012, 134, 13889–13895 CrossRef CAS PubMed.
- M. W. Powner, B. Gerland and J. D. Sutherland, Nature, 2009, 459, 239–242 CrossRef CAS PubMed.
- A. Pawda, in Rearrangements in Ground and Excited States, Academic Press, 1980, pp. 501–549 Search PubMed.
- J. P. Catteau, A. Lablanche-Combier and A. Pollet, J. Chem. Soc. D, 1969, 1018 RSC.
- R. Venkatasubramanian and S. Krishnamachari, Pramana, 1988, 30, 529–533 CrossRef CAS.
- I. Couturier-Tamburelli, B. Sessouma and J. P. Aycard, J. Mol. Struct., 2001, 560, 197–203 CrossRef CAS.
- L. Williamson and B. Meyer, Spectrochim. Acta, Part A, 1970, 26, 331–336 CrossRef CAS.
- A. Halasa, I. Reva, L. Lapinski, M. J. Nowak and R. Fausto, J. Phys. Chem. A, 2016, 120, 2078–2088 CrossRef CAS PubMed.
- M. Sekine and M. Nakata, Chem. Phys. Lett., 2011, 508, 33–37 CrossRef CAS.
- M. Miyagawa, N. Akai and M. Nakata, J. Mol. Struct., 2015, 1086, 1–7 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision E.01, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- G. Sbrana, E. Castellucci and M. Ginanneschi, Spectrochim. Acta, Part A, 1967, 23, 751–758 CrossRef CAS.
- I. Reva, M. J. Nowak, L. Lapinski and R. Fausto, Phys. Chem. Chem. Phys., 2015, 17, 4888–4898 RSC.
- C. J. Evans, J. P. Carter, D. R. T. Appadoo, A. Wong and D. McNaughton, J. Mol. Spectrosc., 2015, 316, 32–37 CrossRef CAS.
- B. Ellis and P. J. F. Griffiths, Spectrochim. Acta, 1965, 21, 1881–1892 CrossRef CAS.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, in The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, ed. D. L.-V. B. C. G. F. G. Grasselli, Academic Press, San Diego, 1st edn, 1991, pp. 105–115 Search PubMed.
- G. A. McGibbon, J. Hrušák, D. J. Lavorato, H. Schwarz and J. K. Terlouw, Chem.–Eur. J., 1997, 3, 232–236 CrossRef CAS PubMed.
- W. A. Rendall, A. Clement, M. Torres and O. P. Strausz, J. Am. Chem. Soc., 1986, 108, 1691–1692 CrossRef CAS.
- C. M. Nunes, I. Reva, T. M. V. D. Pinho e Melo and R. Fausto, J. Org. Chem., 2012, 77, 8723–8732 CrossRef CAS PubMed.
- J. Miyazaki and Y. Yamada, J. Mol. Struct., 2004, 692, 145–153 CrossRef CAS.
- M. D'Auria, Tetrahedron, 2002, 58, 8037–8042 CrossRef.
- M.-D. Su, Phys. Chem. Chem. Phys., 2014, 16, 17030–17042 RSC.
- J. W. Pavlik, C. R. Pandit, C. J. Samuel and A. C. Day, J. Org. Chem., 1993, 58, 3407–3410 CrossRef CAS.
- A. Lablache-Combier and A. Pollet, Tetrahedron, 1972, 28, 3141–3151 CrossRef CAS.
- S. Califano, F. Piacenti and G. Sbrana, Spectrochim. Acta, 1964, 20, 339–344 CrossRef CAS.
- T. Nakagawa and Y. Morino, Bull. Chem. Soc. Jpn., 1969, 42, 2212–2219 CrossRef CAS.
- D. E. Milligan and M. E. Jacox, J. Chem. Phys., 1967, 47, 278–285 CrossRef CAS.
- C. M. King and E. R. Nixon, J. Chem. Phys., 1968, 48, 1685–1695 CrossRef CAS.
- J. Pacansky and G. V. Calder, J. Phys. Chem., 1972, 76, 454–456 CrossRef CAS.
- A. D. Abbate and C. B. Moore, J. Chem. Phys., 1985, 82, 1255–1262 CrossRef CAS.
- K. Satoshi, M. Takayanagi and M. Nakata, J. Mol. Struct., 1997, 413–414, 365–369 CrossRef CAS.
- E. Suzuki and F. Watari, Chem. Phys. Lett., 1990, 168, 1–4 CrossRef CAS.
- R. Schulz and A. Schweig, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1984, 39, 1536–1540 Search PubMed.
- V. A. Korolev and E. G. Baskir, Russ. Chem. Bull., 1995, 44, 448–454 CrossRef.
- E. Briard, J. Levillain, J.-L. Ripoll, Y. Dat, A. Marcual and C. Lange, Eur. J. Org. Chem., 1999, 1999, 869–874 CrossRef.
- A. Krantz and J. Laureni, J. Am. Chem. Soc., 1981, 103, 486–496 CrossRef CAS.
- K. B. Wiberg and R. E. Rosenberg, J. Am. Chem. Soc., 1990, 112, 1509–1519 CrossRef CAS.
- K. Kuchitsu, T. Fukuyama and Y. Morino, J. Mol. Struct., 1968, 1, 463–479 CrossRef CAS.
- J. A. Beukes, P. Klaeboe, H. Møllendal and C. J. Nielsen, J. Raman Spectrosc., 1995, 26, 799–812 CrossRef CAS.
- A. Bénidar, R. Georges, J.-C. Guillemin, O. Mó and M. Yáñez, J. Phys. Chem. A, 2010, 114, 9583–9588 CrossRef PubMed.
- A. Luna, O. Mó, M. Yáñez, J.-C. Guillemin, J.-F. Gal and P.-C. Maria, Int. J. Mass Spectrom., 2007, 267, 125–133 CrossRef CAS.
- G. A. Ozin, D. F. McIntosh, W. J. Power and R. P. Messmer, Inorg. Chem., 1981, 20, 1782–1792 CrossRef CAS.
- A. Engdahl and B. Nelander, Chem. Phys. Lett., 1983, 100, 129–132 CrossRef CAS.
- E. S. Kline, Z. H. Kafafi, R. H. Hauge and J. L. Margrave, J. Am. Chem. Soc., 1985, 107, 7559–7562 CrossRef CAS.
- E. S. Kline, Z. H. Kafafi, R. H. Hauge and J. L. Margrave, J. Am. Chem. Soc., 1987, 109, 2402–2409 CrossRef CAS.
- A. V. Golovkin, D. I. Davlyatshin, A. L. Serebrennikova and L. V. Serebrennikov, J. Mol. Struct., 2013, 1049, 392–399 CrossRef CAS.
- G. Herzberg, Molecular spectra and molecular structure II.: Infrared and Raman spectra of polyatomic molecules, Van Nostrand Reinhold, 1945 Search PubMed.
- M. Wierzejewska and Z. Mielke, Chem. Phys. Lett., 2001, 349, 227–234 CrossRef CAS.
- M. Wierzejewska and R. Wieczorek, Chem. Phys., 2003, 287, 169–181 CrossRef CAS.
- M. E. Jacox, J. Phys. Chem. Ref. Data, 2003, 32, 1–441 CrossRef CAS.
- F. J. Northrup and T. J. Sears, J. Chem. Phys., 1989, 91, 762–774 CrossRef CAS.
- T. Pasinszki, M. Krebsz, G. Bazsó and G. Tarczay, Chem.–Eur. J., 2009, 15, 6100–6102 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables listing observed wavenumbers and IR intensities of thiazole (1) isolated in solid argon matrices with references, and calculated wavenumbers and IR intensities of 2-isocyanoethenethiol (2), 2-isocyanoethenethial (3), 2-isocyanothiirane (4), (methyleneamino)-ethenethione (5), N-ethynylthiformamide (6), Dewar thiazole, and 2-cyanothiirane obtained at the DFT/UB3LYP/aug-cc-pVTZ level, and a figure showing calculated potential energy around the C–NH–CHS dihedral angle of N-ethynylthioformamide (6)obtained by the one-step optimization at an interval of 15°. See DOI: 10.1039/c6ra27496j |
|
| This journal is © The Royal Society of Chemistry 2017 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Hiroshi Takiyamab and
Munetaka Nakata
*ab,
Hiroshi Takiyamab and
Munetaka Nakata