
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly efficient visible-light-driven plasmonic photocatalysts based on graphene oxide mediated hybridization of graphite and Ag/AgBr†
Guangwei Gengab,
Bo Guanb,
Penglei Chen *abc,
Mingshan Zhub,
Changchun Yanga and
Minghua Liubc
*abc,
Mingshan Zhub,
Changchun Yanga and
Minghua Liubc
aCollege of Chemistry and Molecular Engineering, Zhengzhou University, 100 Science Road, Zhengzhou 450001, Henan, China
bBeijing National Laboratory for Molecular Science, Institute of Chemistry, Chinese Academy of Sciences, No. 2 Zhongguancun Beiyijie, Beijing 100190, China. E-mail: chenpl@iccas.ac.cn
cUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 3rd February 2017
Abstract
Graphite (Gr)-based composite materials have been attracting great interest from the scientific and engineering communities. While tremendous progress has been achieved in this field, it still remains a formidable challenge to hybridize pristine Gr with other components under ambient conditions without breaking the π-conjugated system of Gr. Herein, we report that Gr could be facilely hybridized with visible-light-driven plasmonic photocatalyst Ag/AgBr with the assistance of graphene oxide (GO) nanosheets under ambient conditions. We demonstrate that without the use of GO, Gr could not be well hybridized with Ag/AgBr, while a good hybrid Gr/GO/Ag/AgBr could be produced when GO nanosheets are introduced into the system. Our results show that compared to Ag/AgBr, GO/Ag/AgBr and Gr/Ag/AgBr species, thus-fabricated Gr/GO/Ag/AgBr hybrids could display substantially enhanced catalytic activity towards the photobleaching of methyl orange under visible-light irradiation. It is disclosed that the good hybridization between Gr and Ag/AgBr, which is mediated by GO, and the facilitated separation of photogenerated electrons and holes, which is promoted both by GO and Gr, contribute much to the boosted catalytic performances. Considering the excellent hybridization capability of GO, the exceptional electronic characteristics, versatile multifunctionality and low-cost of Gr, our protocol might be applicable not only to the Ag/AgBr-based plasmonic photocatalysts but also to a wide variety of other functional materials, wherein Gr/GO-based advanced materials of desired and reinforced functions or properties might be inaugurated.
1 Introduction
Graphene, which refers to a single-atom-thick two-dimensional sp2-bonded carbon atoms that are arranged in a hexagonal honeycomb lattice, has aroused numerous expectations.1–21 This stems from its unique electrical, mechanical and physicochemical attributes, which endow it with promising prospects in various areas of paramount importance.1–17 In the frontier field of graphene engineering, the hybridization of pristine graphene with other components is recognized to be one of the most important avenues to realize or to reinforce their function.1–7 The perfect π-conjugated system of pristine graphene helps it preserve its unique attributes, scientifically facilitating an investigation on the intrinsic nature of graphene, and technically favoring the construction of advanced hybrids of reinforced performances.1–7 Nevertheless, there generally exist two limitations with regard to this issue, which to some extent disfavor the development of pristine graphene-based hybrids. First, from the viewpoint of availability, pristine graphene is commonly produced via mechanical exfoliation, chemical vapor deposition, etc.18–21 Either the lack of an accurate reproducibility or the requirement of a careful control on the experimental parameters limits a scalable production.18–21 Second, from the viewpoint of hybridization, the lack of hybridizable sites on the skeleton of pristine graphene and its poor solution processability make it difficult to realize a facile and bulk hybridization under ambient conditions.1–7Practically, graphene oxide (GO) has gained much attention as a promising alternative of the pristine graphene.8–17 Structurally, GO is a polyaromatic fragment of pristine graphene with its skeleton being decorated with various oxygen-containing groups. Thanks to its scalable availability, good solution processability and the existence of fertile hybridizable sites on its skeleton, GO-based hybrids actually represent one of the main achievements of carbonaceous material engineering.8–17 However, the presence of oxygen-containing groups and defects on GO's skeleton destroy its π-conjugated system. This leads to a decrease in its electrical characteristics, and is generally undesired from the viewpoint of materials' performances.8–15,22,23 To overcome this, reduced GO (RGO), whose π-conjugated system is repaired to some extent, has been used to construct graphene-based hybrids, where enhanced performances could be realized.8–15,22,23 In most of the cases, various chemicals, electrolytes, dangerous gases, or high temperature are commonly required to obtain RGO,22–25 which in turn makes the manufacturing of RGO-based hybrids a relatively tedious work. Accordingly, a facile hybridization of these carbonaceous materials simultaneously characterized with easy availability, good π-conjugated system, and abundant hybridizable sites is a significant subject deserving to be explored intensively.
Among various carbonaceous materials, graphite (Gr), which consists of coherently stacked layers of pristine graphene, is among the most low-cost carbon sources.26,27 Its perfect π-conjugated system confers it with an electron mobility of as high as ca. 1.2 × 104 cm2 V−1 s−1.28 Similar as the case of pristine graphene, the lack of hybridizable sites on Gr's skeleton and its poor solution processability, however, limit its hybridization under ambient conditions,26,27 although it is strongly desired to make a full use of its π-conjugated system in the hybrids. Recently, it has been reported that driven by the π–π interactions between the π-conjugated aromatic domains of GO and the π system of Gr, powdery Gr could be easily dispersed in aqueous dispersion of GO under ambient conditions.29,30 This might provide researchers with great opportunities to fabricate Gr-based hybrids of good π-conjugated system via an introduction of GO nanosheets into the system.
Recently, silver/silver halide-based species (Ag/AgX, X = Cl, Br, I) have drawn great attention as promising plasmonic photocatalysts towards the photobleaching of organic pollutants.31–47 This is owing to their exceptional visible-light-responsive photocatalytic performances, although silver halides conventionally function as the primary source material in photographic plates and are unstable upon photoirradiation. Inspired by the above-addressed issues, we herein report that Gr, which originally has a poor hybridization capability, could be easily yet well hybridized with the Ag/AgBr plasmonic photocatalysts in the presence of GO nanosheets under ambient conditions. Compared to Ag/AgBr, the hybrids of GO and Ag/AgBr (GO/Ag/AgBr), Gr and Ag/AgBr (Gr/Ag/AgBr), the as-fabricated Gr/GO/Ag/AgBr displays distinctly boosted catalytic activity towards the photodegradation of methyl orange (MO) molecules under visible-light irradiation. We disclose that the GO-facilitated good hybridization between Gr and Ag/AgBr, and the efficient separation of photogenerated electrons and holes promoted by GO and Gr, play an important role for the enhanced catalytic activity. Taking into account the excellent hybridization capability of GO, its versatile multifunctionality and scalable accessibility, and together with the low-cost of Gr and its exceptional electronic characteristics, we anticipate that our efficient yet simple protocol might be applicable not only to Ag/AgBr-based plasmonic photocatalysts but also to a wide variety of others materials, wherein Gr/GO-based advanced materials of desired and improved functions might be facilely launched.
2 Experimental section
2.1 Chemicals and materials
Silver nitrate (AgNO3, Sigma-Aldrich, >99%), cetyltrimethylammonium bromide (CTAB, Alfa Aesar, 95%), graphite powder (Gr, Alfa Aesar, 325 mesh, >99.9%), methyl orange (MO, Alfa Aesar, >98%), ethanol (99.7%, Beijing Chemical Works), and NaCl (AR, Beijing Chemical Works) were used as received. P25-TiO2 (ca. 80% anatase and 20% rutile) species were purchased from Degussa and used as the reference photocatalysts for comparison. Indium tin oxide glasses (ITO) were purchased from Zhuhai Kaivo Optoelectronic Technology Co., Ltd. GO was synthesized via a modified Hummers' method.31 Ultrapure Milli-Q water (18.2 MΩ) was used for the preparation of various aqueous dispersions.2.2 Pretreatment of Gr powders and preparation of Gr/GO
The original Gr powders (1 g) were added into a mixture of Milli-Q water (40 mL) and ethanol (5 mL). The system was treated with an ultrasonic homogenizer (Ningbo Scientz Biotechnology Co., Ltd., Scientz-II D, frequency = 20 kHz, output power = 400 W) for 7 hours. During the ultrasonication, the system was maintained below 20 °C using an ice bath. Then, the suspension was repeatedly treated by centrifugation (11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 10 min) for 4 times using ethanol as solvent. The obtained precipitates were dried under vacuum at room temperature for 24 hours. For the preparation of the aqueous dispersion of Gr/GO, 20 mg of the as-pretreated Gr powders were added into an aqueous dispersion of GO (20 mL, 1 mg mL−1). Then, the system was treated with the ultrasonic homogenizer for 3 hours, after which the as-obtained dispersion was used for the preparation of Gr/GO/Ag/AgBr hybrids.
000 rpm, 10 min) for 4 times using ethanol as solvent. The obtained precipitates were dried under vacuum at room temperature for 24 hours. For the preparation of the aqueous dispersion of Gr/GO, 20 mg of the as-pretreated Gr powders were added into an aqueous dispersion of GO (20 mL, 1 mg mL−1). Then, the system was treated with the ultrasonic homogenizer for 3 hours, after which the as-obtained dispersion was used for the preparation of Gr/GO/Ag/AgBr hybrids.
2.3 Fabrication of Gr/GO/Ag/AgBr, Gr/Ag/AgBr, GO/Ag/AgBr and Ag/AgBr hybrids
To prepare Gr/GO/Ag/AgBr species, 0.5 mL of the above obtained Gr/GO dispersion was added dropwise into an aqueous solution of AgNO3 (10 mL, 5 × 10−3 mol L−1) under magnetic stirring. The mixture was stirred for 30 minutes, after which a chloroform solution of CATB (0.5 mL, 6 × 104 mg L−1) was injected into the suspension within ca. 5 minutes under ambient conditions. The system was stirred for 30 minutes for chloroform evaporation. Subsequently, the products was collected via centrifugation (11000 rpm, 10 minutes), and the obtained solids was washed by a mixture of Milli-Q water and ethanol (1:1) for 1 time, and by Milli-Q water for 3 times. Thus-fabricated solids were then subjected to various characterizations or uses. To prepare Gr/Ag/AgBr, GO/Ag/AgBr and Ag/AgBr species, nearly identical operations were conducted except that an aqueous dispersion of Gr (0.5 mL, 1 mg mL−1), GO (0.5 mL, 1 mg mL−1), or Milli-Q water (0.5 mL), respectively, but not the dispersion of Gr/GO, was added into the aqueous solution of AgNO3.
2.4 Photocatalytic performances
To estimate the photocatalytic activity of the above-prepared species, 9 mg of the respective structures was introduced into 9 mL aqueous solution of MO (60 mg L−1), where a quartz cuvette was used as photoreactor. For catalytic experiments, a 500 W xenon arc lamp (CHF-XM35-500 W, Beijing Trusttech Co. Ltd.), which was installed in a laboratory lamp housing system, was used as light source. Before entering the photoreactor, the light passed through a water filter of a thickness of 10 cm and a UV cutoff filter (>400 nm). The photoreaction system was kept in a dark room for 60 minutes to realize an equilibrium adsorption state before visible-light irradiation. The dark adsorption time was designed to be 60 minutes, since it was found that even when a longer adsorption time of 24 hours was applied, similar results were obtained. Aliquots of the dispersion (0.3 mL) were taken out from the photoreaction system for real-time sampling. The degradation of MOs was monitored by measuring their real-time UV-vis absorptions at 463 nm. For the evaluation of catalytic reactivity, C was the concentration of MO at a desired reaction time t, C0 was the concentration of the MO solution immediately before it was kept in dark.2.5 Photoelectrochemical measurements
For photoelectrochemical measurements, ITO glass electrodes were modified by our samples. To achieve this, the above-synthesized materials (5 mg) were dispersed in ethanol (2 mL), and the dispersion was drop-cast onto a pre-cleaned ITO electrode (2 cm × 4 cm). The as-modified ITO plates were dried under ambient conditions for 48 hours. The photocurrent experiments were conducted in a three-electrode system, where the as-treated ITO electrode, a platinum wire, and a saturated calomel electrode (SCE) were used as working electrode, counter electrode and reference electrode, respectively, while an aqueous solution of NaCl (0.1 M) was used as electrolyte. During the measurements, the ITO electrode was intermittently irradiated with visible-light, and the photocurrent-time characteristics were recorded with a CHI 660B electrochemical analyzer (Shanghai Chenhua Instrumental Co., Ltd.). The above set-up was also used for the measurement of electrochemical impedance spectra (EIS) of our samples, which were recorded with the help of ZPlot/ZView software under an ac perturbation signal of 5 mV over a frequency range of 0.05 Hz to 100 kHz at a potential of 0.1 V.2.6 Apparatus
The scanning electron microscopy (SEM) measurements were conducted using a Hitachi S-4800 system. The energy dispersive X-ray spectroscopy (EDX) of the samples was measured with a Horiba EMAX X-act energy dispersive spectroscopy that was attached to the Hitachi S-4800 system. The powder X-ray diffraction (PXRD) measurements were conducted on a PANalytical X'Pert PRO instrument with Cu Kα radiation. The UV-vis diffuse reflectance spectra of our samples were measured using an UV-vis spectrophotometer (Hitachi U-3010), wherein BaSO4 was used as reference. The degradation of MOs was monitored by measuring their real-time UV-vis spectra using a Hitachi U-3010 spectrometer. The Raman spectra were recorded on a Renishaw inVia plus Raman microscope using a 514.5 nm argon ion laser. X-ray photoelectron spectroscopy (XPS) was performed on an ESCALab220i-XL electron spectrometer from VG Scientific using 300 W Al Kα radiation. The binding energies were referenced to the C 1s line at 284.8 eV from adventitious carbon.3 Results and discussion
Experimentally, the Gr powders were pre-treated with an ultrasonic homogenizer before uses. As shown in Fig. 1a and b, the as treated Gr powders manifest themselves as irregular sheet-like bulk structures with a lateral dimension of 0.5–50 μm and a thickness of 3 μm to 50 nm. In practice, we tried to fabricate Gr/Ag/AgBr hybrids via a microemulsion protocol, in which CTAB and AgNO3 were employed as Br and Ag sources, respectively.31 To fulfill this, an aqueous dispersion of the as-treated Gr was injected dropwise into an aqueous solution of AgNO3 under vigorous stirring and ambient conditions. Then, a chloroform solution of CTAB was introduced into the dispersion. After the evaporation of chloroform, the products were collected and washed adequately with ultrapure Milli-Q water in terms of a couple of repeating centrifugation and redispersion. As reported in Fig. 1c and d, sheet-like bulk Gr structures and Ag/AgBr species are observed from the typical SEM images of as-fabricated samples. It should be noted that most of the Ag/AgBr species are evidently separated from the Gr sheets, while a large fraction of the surface of the Gr sheets display similar characteristics as that of the bare Gr powders shown in Fig. 1a and b, despite of the fact that merely a small percentage of the surface is occasionally decorated with Ag/AgBr nanospecies. These results suggest that the Gr could not form good composites with Ag/AgBr species. This could be a consequence of the lack of hybridizable sites on the surface of Gr, leading to a poor hybridization. | ||
| Fig. 1 The typical low-magnification (a and c) and high-magnification (b and d) SEM images of the Gr powders (a and b) and the Gr/Ag/AgBr structures. | ||
Then, we attempted to fabricate Gr/GO/Ag/AgBr hybrids with an expectation that Gr could be well hybridized with Ag/AgBr by taking the advantage of the excellent hybridization capability of GO nanosheets with Ag/AgBr and with Gr via the oxygen-containing functional groups31,32 and the π-conjugated polyaromatic domains,29,30 respectively. To achieve this, Gr/GO was first prepared by an ultrasonication treatment of a mixture of Gr and GO. As shown in Fig. 2a and b, the SEM image of thus-formulated products shows that sheet-like bulk Gr structures, whose surface are evidently covered with gauze-like GO nanosheets, could be observed, while nearly no free GO nanosheets, which stand away from the Gr structures could be perceived. On the one hand, it should be noted that the as-fabricated structures were collected and washed adequately via centrifugation and resuspension treatments several times. On the other hand, our results show that when the aqueous of GO nanosheets are treated under the similar conditions no precipitates could be observed from the bottom or the wall of the centrifuge tubes, suggesting that GO nanosheets could not be obtained simple by a centrifugation under our experimental conditions. These results basically imply the formation of Gr/GO composites.
 | ||
| Fig. 2 The typical low-magnification (a and c) and high-magnification (b and d) SEM images of the Gr/GO (a and b) and Gr/GO/Ag/AgBr hybrids. | ||
This could be verified by the Raman spectra of the samples as shown in Fig. 3, wherein those of the bare GO nanosheets and Gr powders are also reported for comparison. In the case of GO nanosheets, the characteristic G- and D-bands around 1604 and 1352 cm−1, respectively, could be observed. In addition, a combination of G- and D-bands at ca. 2947 cm−1, and a weakly smeared 2D-band around 2729 cm−1 could also be detected. These features are in accordance with the well-defined Raman spectra of GO nanosheets.48 The Raman spectrum of the bare Gr powders exhibits characteristic strong G-band around 1581 cm−1, weak D-band around 1352 cm−1, and 2D-band at ca. 2722 cm−1, respectively.49 In the case of the as-fabricated Gr/GO samples, D-, G-, overtone 2D-, and combination of D + G bands could be observed. Together with the observations of SEM image shown in Fig. 2a and b, these results validate the formation of Gr/GO composites, which is induced by the π–π interactions between these two components.29,30
 | ||
| Fig. 3 Raman spectra of Gr (black), GO (red) and Gr/GO (blue) species. | ||
Inspired by the successful fabrication of Gr/GO composites, we tried to synthesize Gr/GO/Ag/AgBr hybrids according to the similar procedures as those of Gr/Ag/AgBr. In this case, an aqueous dispersion of Gr/GO instead of that of the Gr powders was used. As presented in Fig. 2c and d, it can be seen from the SEM image of the as-fabricated products that Ag/AgBr species, which are distributed on the surface of Gr/GO species could be observed. Significantly, it is worth pointing out that only negligible amount of free Ag/AgBr species could be observed outside the Gr/GO structures. Accompanied by the results obtained from the Gr/Ag/AgBr systems (Fig. 1c and d), which are synthesized without the involvement of GO nanosheets, and together with the results of Raman spectra shown in Fig. 3, these present observations suggest that Gr could be facilely yet well hybridized with Ag/AgBr species under the assistance of GO nanosheets, wherein the anchoring of the Ag/AgBr species by the oxygen-containing functional groups of the GO component of the Gr/GO composites promotes the formation of the Gr/GO/Ag/AgBr species.31,32 It can be seen that Ag/AgBr could not be well hybridized with the pristine Gr without the assistance of GO nanosheets, while a good hybridization could be realized under the assistance of GO. Accordingly, we tentatively suggest that the Ag/AgBr species are most likely anchored on the surface of the GO species of the Gr/GO/Ag/AgBr hybrid, while the possibility of being anchored on the surface of the Gr species might be very low.
In addition to the Gr/Ag/AgBr and Gr/GO/Ag/AgBr species, GO/Ag/AgBr and Ag/AgBr structures (Fig. S1 and S2†) were also formulated by means of our microemulsion protocol, wherein an aqueous dispersion of GO or pure Milli-Q water, respectively, but not the dispersion of Gr or Gr/GO, was used during the synthesis. As described in the following sections, these materials were prepared to make a comparative investigation on their catalytic activity with regard to the effects of GO, Gr, and Gr/GO species.
The components of our Ag/AgBr, Gr/Ag/AgBr, GO/Ag/AgBr and Gr/GO/Ag/AgBr species were investigated by means of energy dispersive X-ray spectroscopy (EDX) analyses. As reported in Fig. S3,† in the case of the bare Ag/AgBr structures, only EDX signals attributed to Ag and Br elements could be evidently observed. The atomic ratio between Ag and Br is semiquantitatively estimated to be ca. 1.1:1. This value is larger than the theoretic stoichiometric atomic ratio between Ag and Br elements of AgBr, which should be 1:1. This could be owing to the generation of metallic Ag0 nanostructures, which is induced by the ambient light during the fabrication procedure, resulting in the formation of Ag/AgBr structures.31,32 For Gr/Ag/AgBr structures, nearly similar results with an Ag/Br ratio of ca. 1.1:1 are also obtained except that C element could also be detected distinctly. The appearance of C signals is in agreement with the existence of Gr in the samples. In the cases of the GO/Ag/AgBr and Gr/GO/Ag/AgBr structures, besides the Ag, Br and C signals, those attributed to O element could also be observed. This is owing to the presence of GO nanosheets in these systems. Similar as the results of the Ag/AgBr and Gr/Ag/AgBr structures, the atomic ratio between Ag and Br is also estimated to be ca. 1.1:1. Note that the atomic ratio between C and O is ca. 4.9 and 2.8, respectively, for Gr/GO/Ag/AgBr and GO/Ag/AgBr. This result is reasonable, since only C but negligible O exists in the skeleton of Gr, which increases and decreases the relative content of C and O, respectively.
To verify the production of Ag/AgBr-based species, our samples were investigated by means of powdery XRD (PXRD). As present in Fig. 4, all of our samples display distinct diffraction peaks (2θ) at ca. 26.9°, 31.1°, 44.4°, 52.7°, 55.2°, 64.7° and 73.4°, which could be well indexed to the typical diffractions of the (111), (200), (220), (311), (222), (400) and (331) facets of cubic phase AgBr (JCPDS file: 6-438).33,34 At the same time, a weak yet distinct diffraction peak around 38.3°, which could be ascribed to the (111) facet of cubic phase metallic Ag (JCPDS file: 65-2871),33 could also be observed. Together with the results of EDX analyses shown in Fig. S3,† these experimental facts validate the formation of Ag/AgBr species in our samples. It should be pointed out that negligible diffraction peaks ascribing to the GO or Gr could be observed from the PXRD patterns of our GO/Ag/AgBr, Gr/Ag/AgBr and Gr/GO/Ag/AgBr samples. This is a result of their low content.50,51
 | ||
| Fig. 4 The PXRD patterns of our Ag/AgBr (a), GO/Ag/AgBr (b), Gr/Ag/AgBr (c) and Gr/GO/Ag/AgBr (d) structures. The diffraction peaks indexed to AgBr and Ag0 species are marked with ▼ and ●, respectively. | ||
For visible-light-driven photocatalysts, it is essentially necessary that they should be visible-light active so as to fulfill an efficient energy supply and a high catalytic performance. The UV-visible diffuse reflectance spectra of our samples were examined, as shown in Fig. 5. It can be seen that besides the evident absorptions in the ultraviolet region, which could be ascribed to the AgBr species, broad and strong absorptions in the visible region in the range of 460–800 nm could also be distinctly observed. Generally, AgBr species could only exhibit evident absorptions in ultraviolet region but slight absorptions in visible region.33–35 Accompanied by the results of EDX, and PXRD presented in Fig. S3 and S4,† respectively, these results further indicate the existence of metallic Ag0 species in our products, which can arouse surface plasmon resonance (SPR) absorptions in visible region.36–38,52 As known, the plasmonic absorption of metallic Ag nanoarchitectures depends, to a great extent, on their size, shape etc., and those containing Ag nanostructures of diverse shapes or sizes could generally exhibit a broad SPR peak.37,38,52 Similar broad SPR absorptions in the visible region have been reported previously for the UV-vis spectra of Ag/AgBr-based materials.33,36 Experimentally, we attempted to disclose the shape and size of the metallic Ag0 structures of our samples via transmission electron microscopy (TEM). It is unfortunately found that the samples suffered a fast decomposition immediately after the focusing of the electron beam. This makes it difficult to identify the size and shape of Ag0 of the Ag/AgBr structures via TEM. Similar results has been reported previously for silver halide-based plasmonic photocatalysts.39–41 Nevertheless, it could be suggested that the broad SPR peaks observed from our samples could be due to the presence of Ag0 nanospecies of various sizes and shapes.36–38,52
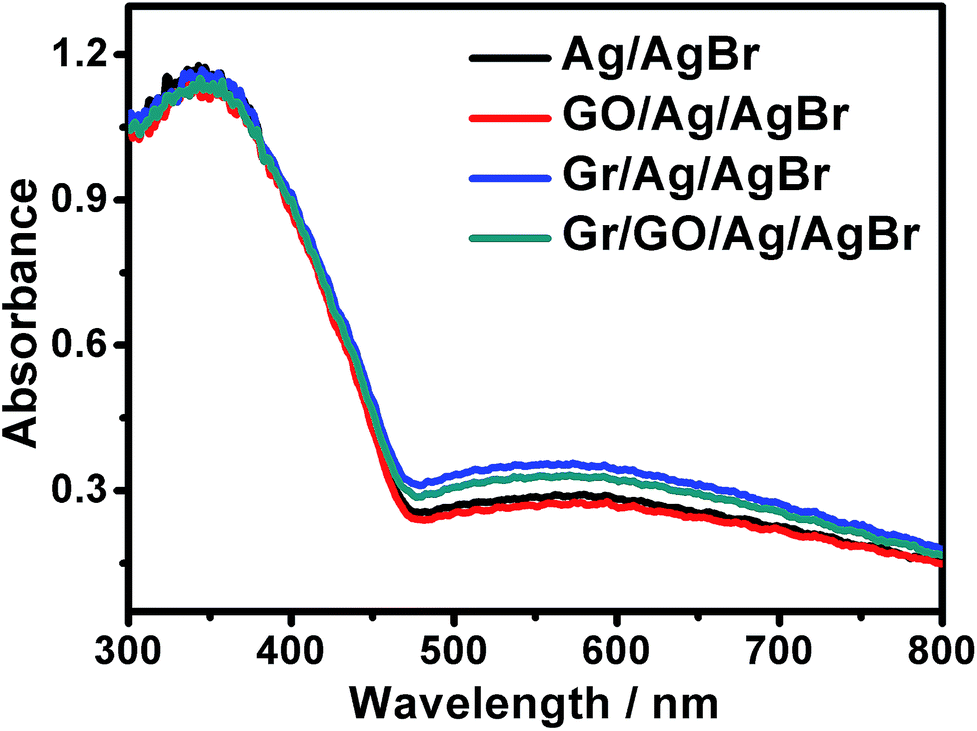 | ||
| Fig. 5 The UV-visible diffuse reflectance spectra of our Ag/AgBr-based materials. | ||
Motivated by the above-mentioned results, the photocatalytic performances of our Ag/AgBr-based structures toward the degradation of MO molecules under visible-light irradiations were studied. As plotted in Fig. 6a (magenta curve), when the photoreaction was conducted without the presence of our Ag/AgBr-based materials, negligible photodegradation of MOs could be observed, implying that the self-photosensitized bleaching of MOs under our experimental conditions could basically be ignored. When the bare Ag/AgBr structures are introduced into the photoreaction system, ca. 46.9% MOs are eliminated within 40 minutes under the similar experimental conditions (Fig. 6a, black curve), indicating that that our Ag/AgBr could work as visible-light-driven photocatalysts for the photodegradation of MOs.33,34,36–38 On the other hand, when the as-fabricated GO/Ag/AgBr and Gr/Ag/AgBr structures are employed as photocatalysts, ca. 67.0% (Fig. 6a, red curve) and 74.8% (Fig. 6a, blue curve) MOs are decomposed, respectively, while this value is further increased to as high as 94.8% (Fig. 6a, dark cyan curve) when our Gr/GO/Ag/AgBr species are introduced into the photoreaction system, as shown in Fig. 6a and summarized in Table 1. The rate constants of the photocatalytic performances of our Ag/AgBr-based materials could be derived by a kinetic linear simulation of the photocatalytic curves in terms of the correlation between ln(C/C0) and the reaction time (t). It can be seen from Fig. 6b that there is a good linear correlation between ln(C/C0) and t. This indicates that the photodegradation reaction of MOs over our Ag/AgBr-based photocatalysts follows the first-order kinetics: –dC/dt = kC, wherein C represents the concentration of MOs at a real-time, t and k stand for reaction time and rate constant, respectively.41
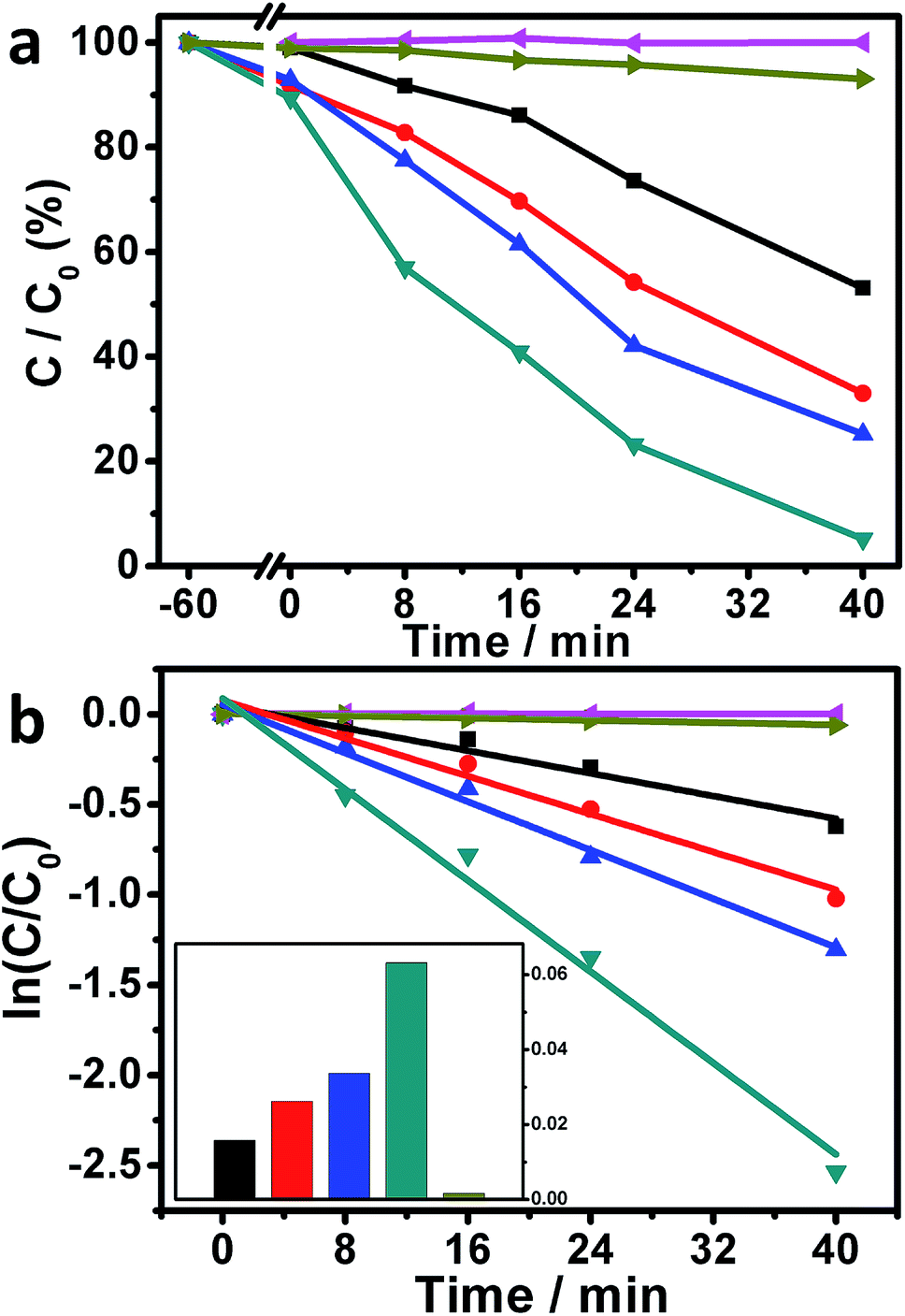 | ||
| Fig. 6 Photocatalytic performances (a), the corresponding kinetic linear simulation curves (b), and the histogram of the photoreaction rate constants (inset of b) of Ag/AgBr (black), GO/Ag/AgBr (red), Gr/Ag/AgBr (blue) and Gr/GO/Ag/AgBr (dark cyan) structures. The results of a blank experiment (magenta curves) and a controlled (dark yellow curves) experiment, wherein no catalysts and P25-TiO2 species are used, respectively, are also presented in the corresponding panels for comparison. The C/C0 located at −60 and 0 minutes are the results obtained before and after the dark adsorption experiments, respectively, which are conducted in a dark room without light irradiation for 60 minutes. | ||
| Catalyst | Degradation percentage (%) | Kinetic constant (min−1) | Determination coefficient (R2) |
|---|---|---|---|
| Ag/AgBr | 46.9 | 0.016 | 0.949 |
| Gr/Ag/AgBr | 67.0 | 0.026 | 0.971 |
| GO/Ag/AgBr | 74.8 | 0.034 | 0.987 |
| Gr/GO/Ag/AgBr | 94.8 | 0.063 | 0.985 |
As shown in Fig. 6b and summarized in Table 1, the rate constants of the photocatalytic degradation of MOs over our Ag/AgBr, GO/Ag/AgBr, Gr/Ag/AgBr, and Gr/GO/Ag/AgBr species are estimated to be 0.016, 0.026, 0.034, and 0.063 min−1, respectively, wherein an increasing tendency is obtained. It can be seen that compared to the Ag/AgBr, GO/Ag/AgBr, and Gr/Ag/AgBr counterparts, the catalytic activity of our Gr/GO/Ag/AgBr hybrids is evidently enhanced by a factor of 3.9, 2.4, and 1.9 times, respectively. These results suggest that among our Ag/AgBr-based structures, the Gr/GO/Ag/AgBr hybrids are relatively superior visible-light-driven photocatalysts towards the photobleaching of MOs. Moreover, the photoreaction was also conducted by using the commercially available P25-TiO2 as the photocatalyst. The results show that only ca. 6.9% MOs are bleached under the similar experimental conditions with a rate constant of as low as ca. 0.0016 min−1 (Fig. 6a and b, dark yellow curves). These values are evidently smaller than the corresponding data obtained from the photoreactions systems, wherein our Ag/AgBr-based species were employed as photocatalysts. This indicates the distinct advantages of our Ag/AgBr-based structures over the P25-TiO2.
On the one hand, it is widely known that the separation/transfer capability of photogenerated electrons and holes is one of the most important and fundamental issues that could affect the catalytic reactivity of semiconductor-based photocatalysts, wherein the more efficient of the electron–hole separation/transfer, the higher the catalytic activity of the catalysts, and vice versa.36–38,53 On the other hand, it has been demonstrated that the catalytic reactivity of semiconductor photocatalysts could be improved to some extent when they are hybridized with GO nanosheets.9,11–15,17 This could be owing to the GO-promoted separation/transfer of the photogenerated electrons and holes.9,11–15,17 In spite of the fact that GO nanosheets exhibit substantially depressed electron transport characteristic compared to the pristine graphene, their polyaromatic graphene segment could serve as an electron relay mediator to facilitate the charge separation/transfer, resulting in a boosted photocatalytic performance.9,11–15,17 To validate this issue in our present case, the transient photocurrent response behaviors of our Ag/AgBr and GO/Ag/AgBr species were examined with regard to their photocurrent–time response (I–t) curves, which were measured by means of a three-electrode photoelectrochemical cell. During the measurements, the indium tin oxide (ITO) glasses, which were covered by our Ag/AgBr-based materials, were used as the working electrodes and were intermittently irradiated by visible-light. As illustrated in Fig. 7 (black and red curves), our results show that the photocurrents of the ITO electrodes covered both by our Ag/AgBr and GO/Ag/AgBr species display an evident increase immediately when the light irradiation is switched on. In contrast, the intensity of the photocurrent recovers to its original value when the light irradiation is switched off. Such reversible photocurrent response behaviors could be reproducibly and repeatedly conducted over a couple of light switched-on/off cycles, implying the separation/transportation of the photogenerated electrons and holes in our Ag/AgBr and GO/Ag/AgBr structures upon light irradiations. At the same time, we note that for the ITO electrodes covered by our bare Ag/AgBr species (Fig. 7, black curve), relatively unconspicuous photocurrent responses with weak photocurrent intensity could be observed. In contrast, in the case of the ITO electrodes modified by our GO/Ag/AgBr structures (Fig. 7, red curve), much more perceptible photocurrent responses of a distinctly enhanced photocurrent intensity could be detected. These observations are in good accordance to our photocatalytic results (Fig. 6 and Table 1) that the compared to the bare Ag/AgBr species, the GO/Ag/AgBr counterparts are superior photocatalysts, wherein the GO-promoted separation of photogenerated electrons and holes play an important role.54,55
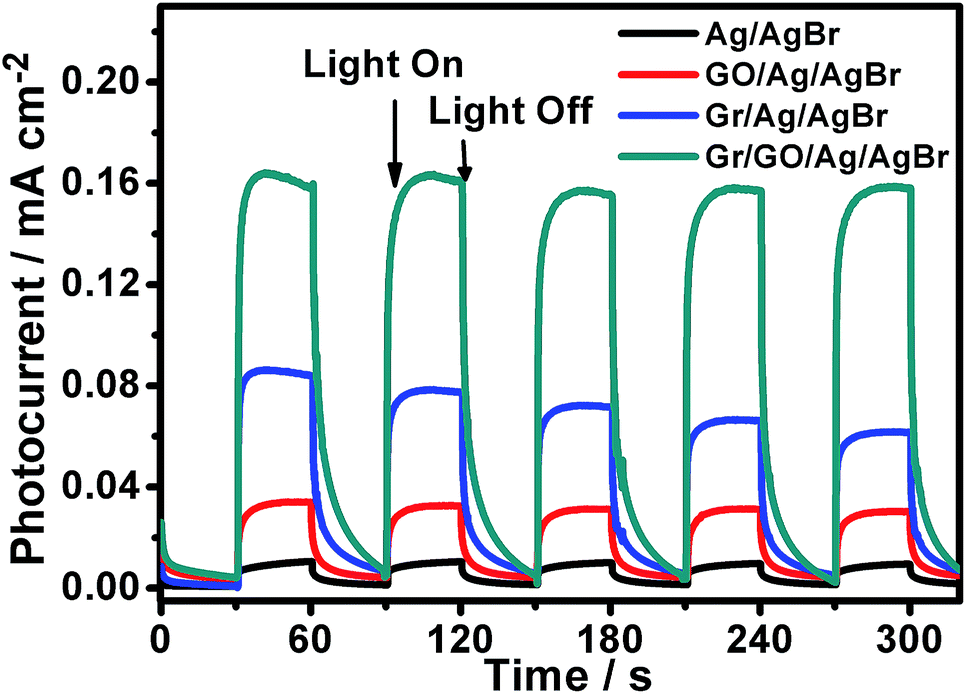 | ||
| Fig. 7 The transient photocurrent responses of our Ag/AgBr-based structures. The light irradiation was periodically interrupted every 30 seconds during the measurements. | ||
As addressed in the introduction section, Gr is structurally composed of coherently superimposed layers of pristine graphene sheets, in which the intact π-delocalized structure resulted from the sp2-hybridized carbon atoms, provides it with a good electron mobility of as large as 1.2 × 104 cm2 V−1 s−1.28 Such unique characteristics might make Gr excellent electron collector in photocatalysts to accept and transport the photogenerated electrons so as to promote a boosted photocatalytic performance, although the lack of enough hybridizable sites on its surface disfavors its hybridization with Ag/AgBr species (Fig. 1). As shown in Fig. 7 (blue curve), compared to those of the bare Ag/AgBr and GO/Ag/AgBr species, the ITO electrodes covered by our Gr/Ag/AgBr structures exhibit more obvious transient photocurrent responses with a further increased photocurrent intensity. These facts are in agreement with our catalytic results shown in Fig. 6 and summarized in Table 1, wherein compared to their Ag/AgBr and GO/Ag/AgBr counterparts, Gr/Ag/AgBr displays a further improved catalytic activity. This suggests that despite of its poor hybridization capability with Ag/AgBr (Fig. 1), Gr could promote the spatial separation/transfer of the photogenerated electrons and holes of the Ag/AgBr species, leading to the enhanced photocatalytic performances of the Gr/Ag/AgBr structures.
Based on the above mentioned experimental facts and analyses, it accordingly could be proposed that the separation/transfer efficiency of the photogenerated electrons and holes might be further promoted in the case of our Gr/GO/Ag/AgBr hybrids, since the Gr powders in this case are well hybridized with Ag/AgBr under the assistance of GO nanosheets (Fig. 2), and both of GO nanosheets and Gr could play a role of electron relay mediator to promote the charge separation/transfer (Fig. 7, red and blue curves). As shown in Fig. 7 (dark cyan curve), compared to their counterparts of the Ag/AgBr, GO/Ag/AgBr, and Gr/Ag/AgBr, the ITO electrodes covered by the Gr/GO/Ag/AgBr species display even more prominent transient photocurrent responses, from which an substantially enhanced photocurrent intensity could be observed. These results are essentially consistent with the fact that among our Ag/AgBr-based visible-light-energized photocatalysts, the Gr/GO/Ag/AgBr species display the highest catalytic reactivity (Fig. 6 and Table 1). Accompanied by the results obtained from the GO/Ag/AgBr and Gr/Ag/AgBr systems, this confirms that the synergistic effect of GO and Gr, wherein the former favors a good hybridization between Ag/AgBr and Gr,31,32 while both the former and the latter promote the separation/transfer of photogenerated electrons and holes,9,11–15,17 contribute much to the observed significantly boosted photocatalytic performances of the Gr/GO/Ag/AgBr species.
These proposals could be further experimentally corroborated by means of an investigation on the electrochemical impedance spectra (EIS) of our Ag/AgBr-based structures, which were measured with respect to Nyquist plots. As illustrated in Fig. 8, it can be seen that the EIS of the ITO electrodes covered by our Ag/AgBr, GO/Ag/AgBr and Gr/Ag/AgBr structures display a gradually decrease tendency in the radius of their semicircular arcs. Commonly, a smaller semicircular arc in the EIS implies a faster interfacial charge transfer and a more effective separation of the photogenerated electrons and holes.50,56,57 These results indicate a decrease in the solid state interface layer resistance and the charge transfer resistance on the surface of Ag/AgBr, GO/Ag/AgBr, and Gr/Ag/AgBr species gradually.50,55,56 Accompanied by the aforementioned results of transient photocurrent responses of the corresponding samples shown in Fig. 7 (black, red and blue curves), this further confirms that GO nanosheets and Gr could in faith play a role of electron reservoirs to facilitate charge separation and thus suppress the recombination of the photogenerated electrons and holes, resulting in enhanced catalytic performances. Significantly, it is found that the arc radius of the Nyquist plots of the electrodes cover by our Gr/GO/Ag/AgBr hybrids exhibit a further decrease compared to those modified by our GO/Ag/AgBr and Gr/Ag/AgBr structures. This verifies a more efficient charge separation in the Gr/GO/Ag/AgBr species, wherein the GO-medicated good hybridization between Gr and Ag/AgBr, and the synergistic effect of GO and Gr play an important role.
 | ||
| Fig. 8 The typical EIS spectra of the ITO electrodes modified by our Ag/AgBr-based species. | ||
The XPS spectra of the bare Ag/AgBr and Gr/GO/Ag/AgBr species were also investigated, as shown in Fig. S4.† In the case of the bare Ag/AgBr structures, two bands at ca. 373.4 and 367.4 eV, which could be ascribed to Ag 3d3/2 and Ag 3d5/2 binding energies, respectively, could be detected (Fig. S4a†). These two bands could be further deconvoluted into two groups of peaks at 373.4, 374.2 eV and 367.4, 368.3 eV, respectively. The bands located at 374.2 and 368.3 eV are attributed to the metallic Ag0 of Ag/AgBr, while those located at 373.4 and 367.4 eV are attributed to Ag+.31,32,41–47 It is interesting to note that compared to those of the bare Ag/AgBr species, these bands shift to higher binding energies in the XPS spectra of the Gr/GO/Ag/AgBr hybrids (Fig. S4a†). Together with the results of the transient photocurrent responses (Fig. 7) and the EIS spectra (Fig. 8) of the corresponding samples, this evident shift in the XPS spectra further indicate that the Ag/AgBr species of the Gr/GO/Ag/AgBr hybrids work as electron donor during the photocatalytic performance,31,32 leading to a facilitated charge separation and an enhanced photocatalytic performance (Fig. 6). On the other hand, as shown in Fig. S4b,† the XPS spectra of the Br species of these samples display binding energy of Br 3d3/2 and Br 3d5/2 at about 68.1 and 69.0 eV,43,45,47 respectively. On the basis of these XPS spectra, the mole ratio between Ag and Br elements, and between Ag0 and Ag+ could be semiquantitatively evaluated to be ca. 1.1:1, and 1:10.2, respectively, both for the bare Ag/AgBr and Gr/GO/Ag/AgBr structures. These results are in accordance with those of EDX (Fig. S3†), wherein the content of Ag0 in the Gr/GO/Ag/AgBr hybrid could be semiquantitatively assessed to be ca. 4.6%.
As known, the adsorptive capacity of a catalyst is another factor that could affect its catalytic activity. As shown in Fig. 6a, the adsorptive capacity of our Ag/AgBr-based structures towards the MO molecules follows Ag/AgBr < GO/Ag/AgBr ≈ Gr/Ag/AgBr < Gr/GO/Ag/AgBr. This could be attributed to the involvement of GO, Gr, and Gr/GO species in the latter three cases, wherein the noncovalent intermolecular π–π interactions between the MO molecules and the involved carbonaceous materials promote the adsorption of MOs.31,50 These facts indicate that the good adsorptive capacity of our Gr/GO/Ag/AgBr structures might also contribute partially to their evidently enhanced photocatalytic activity.
In addition to a high catalytic performance, the durability of a photocatalyst is another important criterion required by excellent catalysts. The recyclability of our Gr/GO/Ag/AgBr plasmonic photocatalyst was also evaluated in terms of conducting the catalytic performances repeatedly a couple of times. As shown in Fig. 9, the catalytic activity displays only a slight decrease after the catalytic performances are conducted five times continuously. These results suggest that our Gr/GO/Ag/AgBr hybrids might be used as highly efficient yet stable visible-light-driven plasmonic photocatalysts, indicating its bright future for potential uses.
 | ||
| Fig. 9 The consecutive cycling photocatalytic behaviors of Gr/GO/Ag/AgBr towards the degradation of MOs. | ||
4 Conclusions
In summary, we herein demonstrate that Gr, which originally has a poor hybridization capability owing to the lack hybridizable sites on its skeleton, could be easily and well hybridized with Ag/AgBr-based plasmonic photocatalysts with the assistance of GO nanosheets under ambient conditions. We show that, compared to the counterparts of the bare Ag/AgBr, GO/Ag/AgBr and Gr/Ag/AgBr species, the as-synthesized Gr/GO/Ag/AgBr composites display evidently enhanced photocatalytic activity towards the photodegradation of MOs under visible-light irradiations. Our results indicate that the good hybridization between Gr and Ag/AgBr, which is facilitated by GO nanosheets, and the promoted separation of photogenerated electrons and holes, which is favored both by GO and Gr, contribute much to the boosted photocatalytic performances of our Gr/GO/Ag/AgBr species. On the one hand, on account of the excellent hybridization capacity of GO nanosheets, and the great achievements accumulated in the field of GO-based hybrids, our protocol might not only be applicable to the hybridization of Gr with Ag/AgBr-based plasmonic photocatalysts but also to other functional species. On the other hand, considering the good π-conjugated system of Gr, which make it of great interest in material science, our protocol might provide Gr, which is among the most conventional, low cost, yet important carbonaceous materials, with new and varied opportunities for the construction of emerging advanced hybrid materials.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (Grants 21673253, 21372225, 20873159, 21321063, and 91027042), the National Key Basic Research Project of China (Grants 2013CB834504 and 2011CB932301), and the Chinese Academy of Sciences (Grants XDA09030200, XDB12020200 and 1731300500015).Notes and references
- J. Coraux, L. Marty, N. Bendiab and V. Bouchiat, Acc. Chem. Res., 2012, 46, 2193–2201 CrossRef PubMed.
- A. Mazid Munshi and H. Weman, Phys. Status Solidi RRL, 2013, 7, 713–726 CrossRef CAS.
- Z. Bo, S. Mao, Z. J. Han, K. Cen, J. Chen and K. Ostrikov, Chem. Soc. Rev., 2015, 44, 2108–2121 RSC.
- J. Park and M. Yan, Acc. Chem. Res., 2013, 46, 181–189 CrossRef PubMed.
- W. I. Park, C.-H. Lee, J. M. Lee, N.-J. Kim and G.-C. Yi, Nanoscale, 2011, 3, 3522–3533 RSC.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2013, 42, 3222–3233 RSC.
- W. Jie and J. Hao, Nanoscale, 2014, 6, 6346–6362 RSC.
- Y. Xu, G. Shi and X. Duan, Acc. Chem. Res., 2015, 48, 1666–1675 CrossRef CAS PubMed.
- N. Zhang, M.-Q. Yang, S. Liu, Y. Sun and Y.-J. Xu, Chem. Rev., 2015, 115, 10307–10377 CrossRef CAS PubMed.
- S. Mao, G. Lu and J. Chen, Nanoscale, 2015, 7, 6924–6943 RSC.
- N. Gao and X. Fang, Chem. Rev., 2015, 115, 8294–8343 CrossRef CAS PubMed.
- X. Fan, G. Zhang and F. Zhang, Chem. Soc. Rev., 2015, 44, 3023–3035 RSC.
- F. Perreault, A. F. de Faria and M. Elimelech, Chem. Soc. Rev., 2015, 44, 5861–5896 RSC.
- V. Georgakilas, J. N. Tiwari, K. C. Kemp, J. A. Perman, A. B. Bourlinos, K. S. Kim and R. Zboril, Chem. Rev., 2016, 116, 5464–5519 CrossRef CAS PubMed.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC.
- C. L. Su and K. P. Loh, Acc. Chem. Res., 2013, 46, 2275–2285 CrossRef CAS PubMed.
- Q. Xiang, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2012, 41, 782–796 RSC.
- I. Levchenko, K. Ostrikov, J. Zheng, X. Li, M. Keidar and K. Teo, Nanoscale, 2016, 8, 10511–10527 RSC.
- D. Geng, H. Wang and G. Yu, Adv. Mater., 2015, 27, 2821–2837 CrossRef CAS PubMed.
- A. V. Zaretski and D. J. Lipomi, Nanoscale, 2015, 7, 9963–9969 RSC.
- J. N. Coleman, Acc. Chem. Res., 2013, 46, 14–22 CrossRef CAS PubMed.
- T. Kuila, A. K. Mishra, P. Khanra, N. H. Kim and J. H. Lee, Nanoscale, 2013, 5, 52–71 RSC.
- O. C. Compton and S. T. Nguyen, Small, 2010, 6, 711–723 CrossRef CAS PubMed.
- S. Thakur and N. Karak, Carbon, 2015, 94, 224–242 CrossRef CAS.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291–312 RSC.
- R. Sengupta, M. Bhattacharya, S. Bandyopadhyay and A. K. Bhowmick, Prog. Polym. Sci., 2011, 36, 638–670 CrossRef CAS.
- D. Appy, H. Lei, C.-Z. Wang, M. C. Tringides, D.-J. Liu, J. W. Evans and P. A. Thiel, Prog. Surf. Sci., 2014, 89, 219–238 CrossRef CAS.
- F. L. Vogel, Synth. Met., 1979/1980, 1, 279–286 Search PubMed.
- J. Kim, L. J. Cote, F. Kim, W. Yuan, K. R. Shull and J. X. Huang, J. Am. Chem. Soc., 2010, 132, 8180–8186 CrossRef CAS PubMed.
- J. Kim, L. J. Cote and J. Huang, Acc. Chem. Res., 2012, 45, 1356–1364 CrossRef CAS PubMed.
- M. Zhu, P. Chen and M. Liu, ACS Nano, 2011, 5, 4529–4536 CrossRef CAS PubMed.
- M. Zhu, P. Chen and M. Liu, Langmuir, 2012, 28, 3385–3390 CrossRef CAS PubMed.
- P. Wang, B. Huang, X. Zhang, X. Qin, H. Jin, Y. Dai, Z. Wang, J. Wei, J. Zhan, S. Wang, J. Wang and M.-H. Whangbo, Chem.–Eur. J., 2009, 15, 1821–1824 CrossRef CAS PubMed.
- B. Li, H. Wang, B. Zhang, P. Hu, C. Chen and L. Guo, ACS Appl. Mater. Interfaces, 2013, 5, 12283–12287 CAS.
- H. Cheng, B. Huang, P. Wang, Z. Wang, Z. Lou, J. Wang, X. Qin, X. Zhang and Y. Dai, Chem. Commun., 2011, 47, 7054–7056 RSC.
- Z. Lou, Z. Wang, B. Huang and Y. Dai, ChemCatChem, 2014, 6, 2456–2476 CrossRef CAS.
- S. Sarina, E. R. Waclawik and H. Zhu, Green Chem., 2013, 15, 1814–1833 RSC.
- X. Zhou, G. Liu, J. Yu and W. Fan, J. Mater. Chem., 2012, 22, 21337–21354 RSC.
- M. Zhu, P. Chen and M. Liu, J. Mater. Chem., 2011, 21, 16413–16419 RSC.
- J. Jiang and L. Zhang, Chem.–Eur. J., 2011, 17, 3710–3717 CrossRef CAS PubMed.
- M. Zhu, P. Chen, W. Ma, B. Lei and M. Liu, ACS Appl. Mater. Interfaces, 2012, 4, 6386–6392 CAS.
- M. Cui, J. Yu, H. Lin, Y. Wu, L. Zhao and Y. He, Appl. Surf. Sci., 2016, 387, 912–920 CrossRef CAS.
- C.-C. Shen, Q. Zhu, Z.-W. Zhao, T. Wen, X. Wang and A.-W. Xu, J. Mater. Chem. A, 2015, 3, 14661–14668 CAS.
- L. Dong, Y. He, T. Li, J. Cai, W. Hu, S. Wang, H. Lin, M. Luo, X. Yi, L. Zhao, W. Weng and H. Wan, Appl. Catal., A, 2014, 472, 143–151 CrossRef CAS.
- W.-S. Wang, H. Du, R.-X. Wang, T. Wen and A.-W. Xu, Nanoscale, 2013, 5, 3315–3321 RSC.
- T. Li, Y. He, H. Lin, J. Cai, L. Dong, X. Wang, M. Luo, L. Zhao, X. Yi and W. Weng, Appl. Catal., B, 2013, 138–139, 95–103 CrossRef CAS.
- Q. Zhu, W.-S. Wang, L. Lin, G.-Q. Gao, H.-L. Guo, H. Du and A.-W. Xu, J. Phys. Chem. C, 2013, 117, 5894–5900 CAS.
- S. Wang, S. Dong, J. Wang, L. Zhang, P. Han, C. Zhang, X. Wang, K. Zhang, Z. Lan and G. Cui, J. Mater. Chem., 2012, 22, 21051–21056 RSC.
- X. D. Ren, R. Liu, L. M. Zheng, Y. P. Ren, Z. Z. Hu and H. He, Appl. Phys. Lett., 2016, 108, 071904 CrossRef.
- H. Zhang, X. Lv, Y. Li, Y. Wang and J. Li, ACS Nano, 2010, 4, 380–386 CrossRef CAS PubMed.
- V. Causin, C. Marega, A. Marigo, G. Ferrara and A. Ferraro, Eur. Polym. J., 2006, 42, 3153–3161 CrossRef CAS.
- A. R. Tao, S. Habas and P. Yang, Small, 2008, 4, 310–325 CrossRef CAS.
- C. Chen, W. Ma and J. Zhao, Chem. Soc. Rev., 2010, 39, 4206–4219 RSC.
- X. Zhang, M. Zhu, P. Chen, Y. Li, H. Liu, Y. Li and M. Liu, Phys. Chem. Chem. Phys., 2015, 17, 1217–1225 RSC.
- Y. Wang, P. Chen, Y. Shen, C. Chen, C. Yang and M. Liu, Phys. Chem. Chem. Phys., 2015, 17, 25182–25190 RSC.
- M. Adachi, M. Sakamoto, J. Jiu, Y. Ogata and S. Isoda, J. Phys. Chem. B, 2006, 110, 13872–13880 CrossRef CAS PubMed.
- C. Zhai, M. Zhu, F. Ren, Z. Yao, Y. Du and P. Yang, J. Hazard. Mater., 2013, 263, 291–298 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The SEM images of our Ag/AgBr and GO/Ag/AgBr structures. The EDX elemental analysis and XPS spectra of our Ag/AgBr and Gr/GO/Ag/AgBr structures. See DOI: 10.1039/c6ra27462e |
| This journal is © The Royal Society of Chemistry 2017 |