
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The influence of Na2O on the fast diffusion layer around diopside crystals
Wendi Fan,
Bo Liu*,
Jian Yang and
Shengen Zhang *
*
Institute for Advanced Materials and Technology, University of Science and Technology Beijing, Beijing 100083, P. R. China. E-mail: liubo@ustb.edu.cn; zhangshengen@mater.ustb.edu.cn; Fax: +86 82376835; Fax: +86 62333375; Tel: +86 82376835 Tel: +86 62333375
First published on 31st January 2017
Abstract
The establishment of a fast diffusion layer in glass–ceramics can reduce carbon emissions and manufacturing cost during their preparation. In this study a CaO–MgO–SiO2 (CMS) glass–ceramic was prepared by forming a fast diffusion layer doped with different Na+ contents, taking advantage of differential scanning calorimetry (DSC), Fourier transform infrared spectroscopy (FTIR), scanning electron microscopy (SEM) and energy dispersive spectroscopy line scanning (EDSLS). Upon increasing the Na+ content from 0.10 mol to 0.14 mol, the number of crystal particles increased by 350%. The Na+ content in the fast diffusion layer around the crystal particles decreased, which ultimately affected the crystal growth. This result may benefit the fast diffusion layer theory on the inhibitory effect of Na during crystal growth.
1. Introduction
Glass–ceramics combine crystalline and amorphous features,1,2 and are produced by melting the parent glass, followed by a heat treatment step. By adjusting the composition and regulating the heat treatment process, the properties of the glass–ceramic can be controlled,3,4 such as mechanical strength, chemical resistance and abrasive resistance.5,6 Glass–ceramics have been widely used in several fields, particularly with respect to applications in solid oxide fuel cells, optics, photonics, biomedicine materials and construction materials.7–11Usually, the production of glass–ceramics requires two heat treatment steps (nucleation and crystal growth) due to its poor crystallization ability. However, the two heat treatment steps consume a lot of energy and emit a large amount of carbon. Hence, improving the parent glass' nucleation and crystal growth capacities is a way of reducing the energy consumption and carbon emissions.
Recently, numerous studies on glass–ceramics have focused on the nucleation and crystal growth steps, which are related to the diffusion layer around the crystals.12–15 In multi-component systems, doping with small amounts of other components affects the crystal growth.2 Generally, during the crystallization of the glass–ceramic, both a nucleating agent and network modifiers are concentrated in the crystal nuclei. Moreover, the glass network formers (Al, Si) are concentrated on the glass layer between the residual glass and the crystal.16 Eventually, the glass layer limits the diffusion of ions in the crystal phase like Ca2+ and Mg2+. Wisniewski and co-authors17 described the crystallization process in which the formation of concentration gradients decelerates the crystal growth. This phenomenon leads to an increase in the viscosity of the diffusion layer and results in a smaller diffusion rate, which ultimately affects the crystal growth. In Yang's study, the melts contained an excess of alkali metal ions.18 During crystallization, the excess alkali metal ions lead to increasing amounts of Si–O–Na bonds in the residual glass phase, which result in an increase in the diffusion coefficients and the formation of a fast diffusion layer.
However, most studies have focused on the formation of the diffusion limiting layers during the crystallization process.19,20 Yang and co-authors described a phenomenon in which the diffusion limiting layers become a fast diffusion layer.18 By adding excess Na2O and CaF2, a fast diffusion layer was formed around the crystals to help increase the diffusion coefficient of the crystal phase ions. Thus, the crystal growth was accelerated. Nevertheless, studies on the effect of the Na+ content in the diffusion layer are rarely reported.18
In this study, an excess of Na2O was added in the CMS system. During crystallization, Na+ was considered as a glass modifier, which forms the diffusion layer. The crystallization kinetics index of the glass–ceramics was calculated using the Kissinger and Augis–Bennett equations. The results indicated that an excess of Na+ inhibits the crystal growth. The study presented herein directly tests the fast diffusion layer theory.
2. Experimental
The parent glass was prepared from the highly purified raw materials CaO, MgO, SiO2, Al2O3, Fe2O3, Cr2O3, CaF2 and Na2CO3 (purity 99%; China Medicine, Inc.). Prior to use, CaO and Na2CO3 were heated at 900 °C for 2 h. The thermal decomposition of CaO removed impurities such as CaCO3 and Ca(OH)2. Na2O was obtained via the decomposition of Na2CO3.A batch of raw materials was homogeneously mixed for 1 h in a ball mill. The mixed raw materials were packed in a corundum crucible and melted at 1450 °C for 2 h for complete melting and homogenization. The melt was quenched into a preheated iron mold (600 °C) for 30 min in order to remove the internal stress. Then, the parent glass was cut into small pieces. The glass–ceramic samples were obtained by annealing the parent glass pieces. The heat treatments were carried out at the temperatures around the Tp for 20 min, 30 min and 60 min, respectively. The glass compositions are given in Table 1.
| Sample | CaO | MgO | SiO2 | Al2O3 | Fe2O3 | Cr2O3 | CaF2 | Na2O |
|---|---|---|---|---|---|---|---|---|
| GC-1 | 20.1 | 13.3 | 46.6 | 4.4 | 5.3 | 1.2 | 7.9 | 0 |
| GC-2 | 20.1 | 13.3 | 46.6 | 4.4 | 5.3 | 1.2 | 7.9 | 7.4 (0.12 mol) |
| GC-3 | 20.1 | 13.3 | 46.6 | 4.4 | 5.3 | 1.2 | 7.9 | 8.7 (0.14 mol) |
Thermal analysis was performed using differential scanning calorimetry (DSC, NETZSCH STA 409C/CD) with the powdered parent glass sample (quenched into water) at heating rates of 10 °C min−1, 15 °C min−1, 20 °C min−1 and 25 °C min−1 from room temperature to 1000 °C. The glass crystallization temperatures (Tp) at the different heating rates were obtained from these measurements. After heating at the Tp, the glass ceramics were obtained.
The diffraction patterns of the glass–ceramics were obtained via X-ray diffraction (Philips APD-10, monochromatic Cu Kα radiation). The Fourier transform infrared (FTIR) spectra of the glass–ceramics were taken on Nicolet-is 10 spectrophotometer in the range of 400–4000 cm−1. KBr was mixed with the sample powders and palletized using a hydraulic press. The contribution of KBr was cancelled out by normalizing the spectrum of each sample to the spectrum of KBr. High resolution scanning electron microscopy (SEM) images of the glass–ceramic samples were recorded on Carl Zeiss EVO 18. The fractured surface was etched with diluted HF (5%) to prepare the samples for SEM.
3. Results and discussion
Fig. 1(a) and (b) show the DSC curves obtained for the parent glass powder at different heating rates. In Fig. 1(a), one crystallization exothermic peak was observed in the DSC curves at the heating rates of 10 K min−1, 15 K min−1, 20 K min−1 and 25 K min−1. The DSC curves showed the crystallization temperature (Tp) ranging from 1119.3 K to 1148.3 K. The crystallization exothermic peak temperature increased as the heating rate increased. Basically, as the heating rate slowed down, diffusion and atomic rearrangements transformed the structure from non-crystalline to crystalline. When the crystalline phase transformation rate was very slow, the crystallization exothermic peak of the glass was flat. However, as the heating rate increased, the crystallization of the glass transition was faster. The transformation rate increased upon increasing the crystallization temperature and the crystallization exothermic peak became sharp. | ||
| Fig. 1 (a) The DSC curves obtained at 10 K min−1, 15 K min−1, 20 K min−1 and 25 K min−1 for GC-2, (b) the DSC curves obtained at 10 K min−1, 15 K min−1, 20 K min−1 and 25 K min−1 for GC-3 and (c) the variation of ln(α/Tp2) 1000/Tp for the different CMS glasses. | ||
Nucleation shows endothermic effects, while crystallization shows exothermic effects. The endothermic and exothermic peaks in the DSC curves were due to nucleation and crystallization, respectively. The crystallization temperature depends on the exothermic temperature due to the increasing crystallization rate. When compared to GC-2 and GC-3, the increase in sodium content significantly reduces the Tp, indicating that Na+ accelerates the crystallization of the glass.
The kinetic parameters, such as the crystallization activation energy, depend on the obtained DSC curves. The Kissinger equation can be used to assess the crystallization dynamics:21
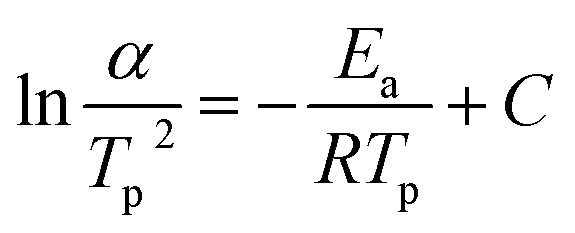 | (1) |
 | (2) |
| Ea, kJ mol−1 | n | R2 | |
|---|---|---|---|
| Tp1 | 498 | 6.0 | 0.97299 |
| Tp2 | 567 | 5.2 | 0.81175 |
Fig. 2 shows the XRD patterns of the samples heat-treated at the crystallization peak temperatures for 1 h. To confirm the nature of the obtained materials, X-ray diffraction studies were carried out. As seen in Fig. 2, the broad scattering peak in GC-1 suggests that no crystals precipitated in the glass matrix. The major phase of GC-2 and GC-3 was diopside, indicating that the addition of Na2O has no effect on the main crystalline phase. The secondary phase of GC-2 was nepheline. With the addition of Na2O, the secondary phase transferred to sodium alumosilicate (Na6Al4Si4O17, 76-2385#) in GC-3. This indicates that at the end of the crystallization process, Na+ was rich in the residual glass. The nepheline and sodium alumosilicate phases were formed by these Na+ ions, silicon and aluminum.
 | ||
| Fig. 2 The XRD patterns of the prepared glasses. | ||
In order to characterize the structural building units of the glass ceramics, infrared absorption measurement studies were performed on the glass ceramics. Fig. 3 presents the FT-IR spectra of the glass ceramics with different Na+ contents. As seen in the spectra, there were characteristic absorption bands ranging from 400 to 1200 cm−1, which were due to the main silicate network group vibrations with different bonding arrangements. The absorption band about 450 cm−1 was attributed to the bending vibrations of the Al–O–Al or Al–O–Si bonds.22 The absorption band located at about 490 cm−1 was associated with the Si–O–Si bending vibration mode. The broadened band at 750 cm−1 indicates the scission of the Si–O–Si chain in the glass network.23 The band located at about 940 cm−1 was assigned to Si–O with two non-bridging oxygens.24 The band located at about 1040 cm−1 is characteristic of the asymmetric stretching vibrations of Si–O–Si bonds with a three-dimensional network structure. Upon the addition of Na+, the Si–O–Si chain and non-bridging oxygens evidently increased. This led to a decrease in the integrity of the glass network.
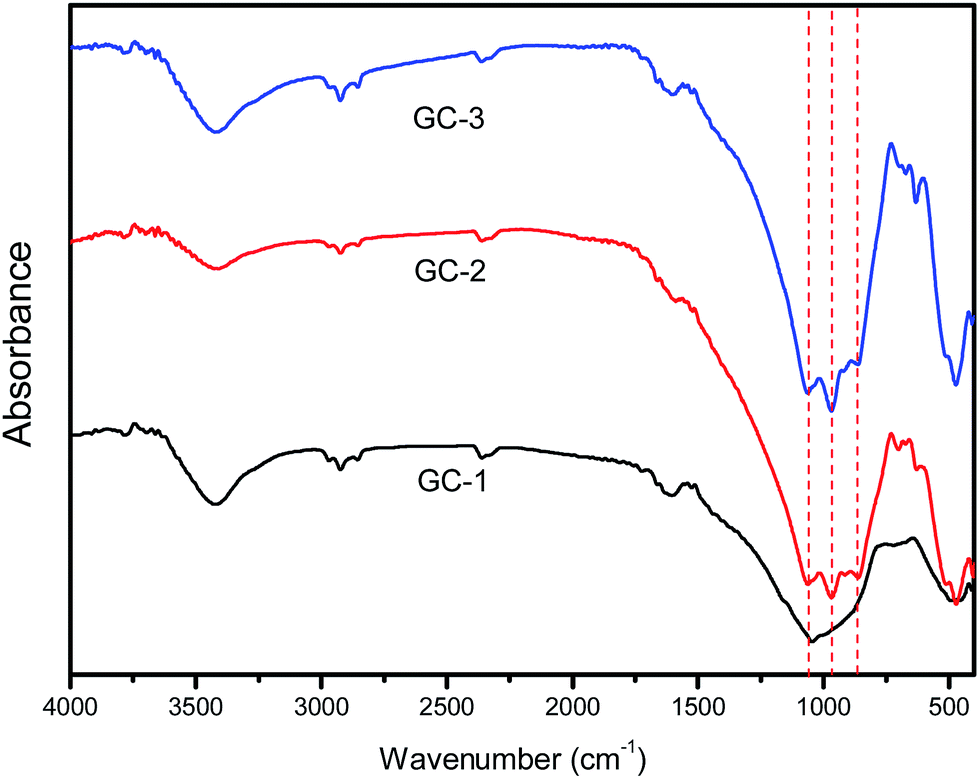 | ||
| Fig. 3 The FT-IR absorption spectra of the prepared glasses. | ||
The microstructures of the fractured surfaces of the glass ceramics prepared using different amounts of Na2O are shown in Fig. 4. GC-1 was heat-treated at 1120 K for 1 h and is shown in Fig. 4(a). No crystalline phase appears in the annealed glass, which may be due to the lack of Na2O. Fig. 4(b) and (c) display the GC-2 sample heat-treated at the targeted temperature for 20 min and 30 min. The addition of Na2O leads to crystal particles being formed in the glass ceramics. The black matrix is the glassy phase, which does not tend to crystallize further. Fig. 4(b) displays diopside crystals (very light grey) and the residual glassy phase (black). With the heat treatment time extended to 30 min, the crystal particles in the glass ceramics are apparently grown further. A similar microstructure was also evident for the GC-3 sample containing 0.14 mol Na+. Fig. 4(d)–(f) display the GC-3 sample heated for 5 min, 20 min and 30 min, respectively. According to Fig. 4(d), the addition of 0.14 mol Na+ results in small crystal particles formed in 5 min. Extending the heat treatment time lead to the continuously growth of the crystal particles, while the crystallite dimensions were evidently smaller than those of GC-2. In addition, the number of crystal particles in the field of view in GC-3 was significantly higher. Thus, it is reasonable to say that the addition of Na2O contributes to the crystal particles refinement and improves the crystal nucleus density.
 | ||
| Fig. 4 (a) GC-1; Na+ 0 mol, (b) GC-2; Na+ 0.10 mol (heated at 1119 K for 20 min), (c) GC-2; Na+ 0.14 mol (heated at 1119 K for 30 min), (d) GC-3; Na+ 0.14 mol (heated at 1130 K for 5 min), (e) GC-3; Na+ 0.14 mol (heated at 1130 K for 20 min) and (f) GC-3; Na+ 0.14 mol (heated at 1130 K for 30 min). | ||
Evaluation of the chemical composition of the glass–ceramics interface was performed via SEM and the energy dispersive X-ray spectroscopy (EDXS) line scan. Fig. 5(a) shows the micrograph of GC-3 and the EDXS line scan up the trace of the straight line shown in the micrograph. The distribution of sodium, magnesium and oxygen differs between the crystal particle and the residual glass phase. The Na+ content in the crystal particle is significantly lower than that in the residual glass. The Mg2+ and O2− ion contents in the crystal particle are higher than that in the residual glass. This phenomenon means that the Na+ ions diffused into the glass layer around the crystal during the crystallization process. While this process proceeds, the diffusion of Mg2+ and O2− to the crystal nucleus occurs at the same time.
 | ||
| Fig. 5 (a) A micrograph of the GC-3 sample heated at 1119 K for 60 min and EDXS line scanning along the trace of the straight line indicated in the micrograph. (b) The sodium ion content obtained via the EDXS line scan of GC-3; (c) the magnesium ion content obtained via the EDXS line scan of GC-3 and (d) the oxygen ion content obtained via the EDXS line scan of GC-3. | ||
During the crystallization process, the phase separation, nucleation and crystal growth take place at the same time at the Tp. In general, the amorphous glass will be divided into the residual glass phase and the phase-separated regions during the phase separation stage. The enriched elements in the phase separation region are similar to the resulting crystal nucleus. Thus, the formation of a crystal nucleus is easier. During the crystallization of the CMS glass–ceramics, the addition of Na leads to an increase in the number of Si–O–Na bonds and non-bridging oxygen atoms. This further promotes the diffusion of calcium ions and magnesium ions, thereby promoting the crystal growth. As a result, the nucleation activation energy is decreased.
In a previous study,18 the activation energy of CMSglass ceramics with 0 mol Na+ was found to be 301.2 kJ mol−1. The activation energy of the 0.1 mol Na+ added sample was 195.4 kJ mol−1. In this study, the activation energy after the addition of 0.12 mol Na+ was 498 kJ mol−1 and after the addition of 0.14 mol Na+ it was 567 kJ mol−1. A high activation energy value means that the nucleation and crystallization was difficult. The activation energy decreased with Na+ ranging from 0 mol to 0.1 mol. The activation energy increased with Na+ ranging from 0.12 mol to 0.14 mol. In brief, different sodium ion contents (0 mol, 0.1 mol, 0.12 mol and 0.14 mol) have a non-linear relationship with the activation energy of the CMS glass–ceramics.
FT-IR spectra showed that the addition of sodium ions affects the glass network (Fig. 3). The addition of sodium ions produces a large number of non-bridging oxygen atoms. This phenomenon was consistent with the sodium ions acting as a network former that influences the glass network. At the same time, it can be found that excessive sodium ions (0.14 mol) do not affect the new structures generated in the glass network. The XRD patterns showed that the main phase did not change upon increasing the sodium ion content.
The sodium layer was found between the nuclei and residual glass. The results of EDX-line scanning performed on the GC-2 and GC-3 samples show that the Na+ content around the crystals was gradually changed. This also means that the glass network integrity around the nuclei was different. This phenomenon affects the diffusion rate. During the growth of the nuclei, the diffusion of Ca2+ and Mg2+ occurs through the sodium ion layer around the crystal nuclei. When the sodium layer changes, it may have an effect on the Ca2+ and Mg2+ diffusion velocity.
By means of the SEM micrographs shown in Fig. 4, it can be seen that the increase in sodium ion content results in an increase in the crystal number. As shown in Fig. 4(b) and (e), the number of crystal particles in the same area by counting was 68 and 239 (increased by 350%), respectively. This might be assumed as a result of sodium ions increasing the nucleation density during the isothermal hold. In GC-3, the non-bridging oxygen makes the glass network open. Therefore, the crystal nucleus is easier to generate. This phenomenon leads to a decrease in the sodium ion content around the crystal nucleus, thereby reducing the diffusion rate of the calcium ions. When the nucleation ends, the relative lack of sodium ions formed around the crystal nucleus leads the Ca2+ and Mg2+ diffusion mobility to decrease. Ea reflects the crystallization process. A comparison of the crystal nuclei growth can also verify the abovementioned assumptions. During the first 5 minutes of the isothermal process at Tp, the crystals grow in GC-3 first. This phenomenon proves that an excess of Na improves the nucleation ability. However, the size of the crystal nuclei in GC-3 is smaller than that in GC-2, which is obtained from the isothermal heat treatment for 20 minutes. This phenomenon proves that the lack of sodium content around the crystal nuclei inhibits the crystal growth. The abovementioned evidence shows that the sodium ions make the crystallization process easier but limit the nuclei crystals' continued growth. In summary, the excessive sodium ion content makes the activation energy increase abnormally.
4. Conclusion
In this study, the CMS glass–ceramic was prepared via a one-step heat treatment process, during which a fast diffusion layer was formed. During the crystallization process, diopside crystals phase out of the homogeneous parent glass and concomitantly form a glass layer between the diopside and residual glass. As observed experimentally, the addition of sodium ions promoted the formation of Si–O–Na bonds. The Si–O–Na bonds in the glass layer will lead to an increase in the Ca2+ and Mg2+ diffusion mobility and further promotes the formation of diopside. Upon increasing the Na+ content from 0.10 mol to 0.14 mol, the number of crystal particles increased 3.5 fold. The Na+ content in the fast diffusion layer around the crystal particles decreased. This phenomenon lead to the number of Si–O–Na bonds around the crystal nucleus to decrease, thereby inhibit the crystal growth.Acknowledgements
This study was sponsored by the National Natural Science Foundation of China (Grants U1360202, 51472030, 51672024 and 51502014). The authors would like to thank the editor for editing the manuscript and the anonymous reviewers for their detailed and helpful comments.References
- R. Casasola, J. M. Rincón and M. Romero, J. Mater. Sci., 2011, 47, 553–582 CrossRef.
- N. Karpukhina, R. G. Hill and R. V. Law, Chem. Soc. Rev., 2014, 43, 2174–2186 RSC.
- M. Erol, S. Küçükbayrak and A. Ersoy-Meriçboyu, Chem. Eng. J., 2007, 132, 335–343 CrossRef CAS.
- A. Gawronski and C. Rüssel, J. Mater. Sci., 2013, 48, 3461–3468 CrossRef CAS.
- Z. Zhang, L. Zhang and A. Li, Waste Manag., 2015, 38, 185–193 CrossRef CAS PubMed.
- S. Agathopoulos, D. U. Tulyaganov, J. M. G. Ventura, S. Kannan, A. Saranti, M. A. Karakassides and J. M. F. Ferreira, J. Non-Cryst. Solids, 2006, 352, 322–328 CrossRef CAS.
- Q. Z. Chen, Y. Li, L. Y. Jin, J. M. Quinn and P. A. Komesaroff, Acta Biomater., 2010, 6, 4143–4153 CrossRef CAS PubMed.
- R. Harshe, C. Balan and R. Riedel, J. Eur. Ceram. Soc., 2004, 24, 3471–3482 CrossRef CAS.
- Y. Xu, X. Zhang, S. Dai, B. Fan, H. Ma, J.-l. Adam, J. Ren and G. Chen, J. Phys. Chem. C, 2011, 115, 13056–13062 CAS.
- A. J. Salinas and M. Vallet-Regí, RSC Adv., 2013, 3, 11116 RSC.
- N. Tanibata, K. Noi, A. Hayashi and M. Tatsumisago, RSC Adv., 2014, 4, 17120 RSC.
- C. Bocker, A. Herrmann, P. Loch and C. Rüssel, J. Mater. Chem. C, 2015, 3, 2274–2281 RSC.
- A. Wajda and M. Sitarz, J. Non-Cryst. Solids, 2016, 441, 66–73 CrossRef CAS.
- S. Ghosh and A. Ghosh, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 132204 CrossRef.
- C. Bocker, C. Rüssel and I. Avramov, Int. J. Appl. Glass Sci., 2013, 4, 174–181 CrossRef CAS.
- M. Storek, M. Adjei-Acheamfour, R. Christensen, S. W. Martin and R. Bohmer, J. Phys. Chem. B, 2016, 120, 4482–4495 CrossRef CAS PubMed.
- W. Wisniewski, R. Carl, G. n. Volksch and C. Rüssel, Cryst. Growth Des., 2011, 11, 784–790 CAS.
- J. Yang, B. Liu, S. Zhang and A. A. Volinsky, J. Alloys Compd., 2016, 688, 709–714 CrossRef CAS.
- K. Thieme and C. Rüssel, J. Eur. Ceram. Soc., 2014, 34, 3969–3979 CrossRef CAS.
- T. Höche, M. Mäder, S. Bhattacharyya, G. S. Henderson, T. Gemming, R. Wurth, C. Rüssel and I. Avramov, CrystEngComm, 2011, 13, 2550 RSC.
- A. A. Cabral, V. M. Fokin, E. D. Zanotto and C. R. Chinaglia, J. Non-Cryst. Solids, 2003, 330, 174–186 CrossRef CAS.
- Y. Li, K. Liang, J. Cao and B. Xu, J. Non-Cryst. Solids, 2010, 356, 502–508 CrossRef CAS.
- N. O. Dantas, W. E. Ayta, A. C. Silva, N. F. Cano, S. W. Silva and P. C. Morais, Spectrochim. Acta, Part A, 2011, 81, 140–143 CrossRef CAS PubMed.
- E. A. Mahdy and S. Ibrahim, J. Mol. Struct., 2012, 1027, 81–86 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2017 |