
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ansavaricins A–E: five new streptovaricin derivatives from Streptomyces sp. S012†
Zhiqiang Zhanga,
Juanli Zhangb,
Rentai Songc,
Zhixing Guoa,
Haoxin Wangc,
Jing Zhuc,
Chunhua Lua and
Yuemao Shen *ac
*ac
aKey Laboratory of Chemical Biology, Ministry of Education, School of Pharmaceutical Sciences, Shandong University, No. 44 West Wenhua Road, Jinan, Shandong 250012, P. R. China. E-mail: yshen@sdu.edu.cn; Tel: +86-531-88382108
bDepartment of Pharmacy, Xijing Hospital, The Fourth Military Medical University, Changle West Street 15, Xi'an, Shaanxi 710032, P. R. China
cState Key Laboratory of Microbial Technology, Shandong University, No. 27 South Shanda Road, Jinan, Shandong 250100, P. R. China
First published on 17th January 2017
Abstract
Five new polyketides of the ansamycin class, named ansavaricins A–E (1–5), together with three known streptovaricins 6–8, were isolated from the Streptomyces sp. S012 strain. The structures of 1–8 were determined by analysis of the 1D and 2D NMR spectroscopic and high-resolution ESIMS data, which showed plentiful structural variations in the naphthoquinoid moieties and ansa chains of ansavaricins A–E. Ansavaricins A–E showed almost no obvious toxicity to the test Gram positive and negative bacteria. However, compounds 5, 6 and 7 significantly inhibited the secretion of SPI-1 effectors of Salmonella enterica serovar Typhimurium, indicating a potential in the development of antivirulence antibacterial drugs.
Introduction
Ansamycins are a family of antibiotics with a high degree of druggability exemplified by the antituberculosis rifamycins,1 antitumor geldanamycin,2 and numerous other compounds as well as streptovaricins.3 Structurally, ansamycins are characterized by a macrocycle composed of a benzenic or naphthalenic chromophore bridged by an aliphatic ansa chain that terminates at the chromophore in an amide linkage.4 Despite the structural variations by carrying either benzenic (maytansine, geldanamycin) or naphthalenic (rifamycin) chromophores, genetic and feeding experiments have revealed that all the ansamycins share the same polyketide starter unit, 3-amino-5-hydroxybenzoic acid (AHBA).5–7 Therefore, the AHBA synthase gene can serve as a useful tool in the genetic screening for diverse AHBA-derived new natural products.Previously, we have approached to new ansamycins through PCR screening of the AHBA synthase genes of plant-associated, soil-dwelling and marine-derived actinomycetes,8 which afforded the characterization of the biosynthesis of hygrocins,8,9 divergolides,10,11 ansamycins juanlimycins12 and neoansamycins,13 and the isolation of ansatrienols B–H with diverse post-PKS modifications from the strain Streptomyces sp. XZQH13OEΔastC.14
Streptomyces sp. S012 isolated from rhizosphere soil of Nanjing Zhongshan Botanical Garden was identified as an AHBA synthase gene-positive strain, and revealed to have the potential of producing streptovaricins, the undecaketide-type ansamycins on the basis of genome scanning. Streptovaricins are a class of ansamycins that contain a naphthoquinone nucleus and a macrocyclic aliphatic ansa bridge. Streptovaricins, along with the related rifamycins, have been shown to have important biological activities including inhibiting nucleoside incorporation15 and anti-leukemia virus,16 especially, the antibacterial activities against Gram positive and negative microorganisms.17 This study reported the isolation, structure elucidation and biologic activity of eight streptovaricins from the S. sp. S012 strain (Fig. 1).
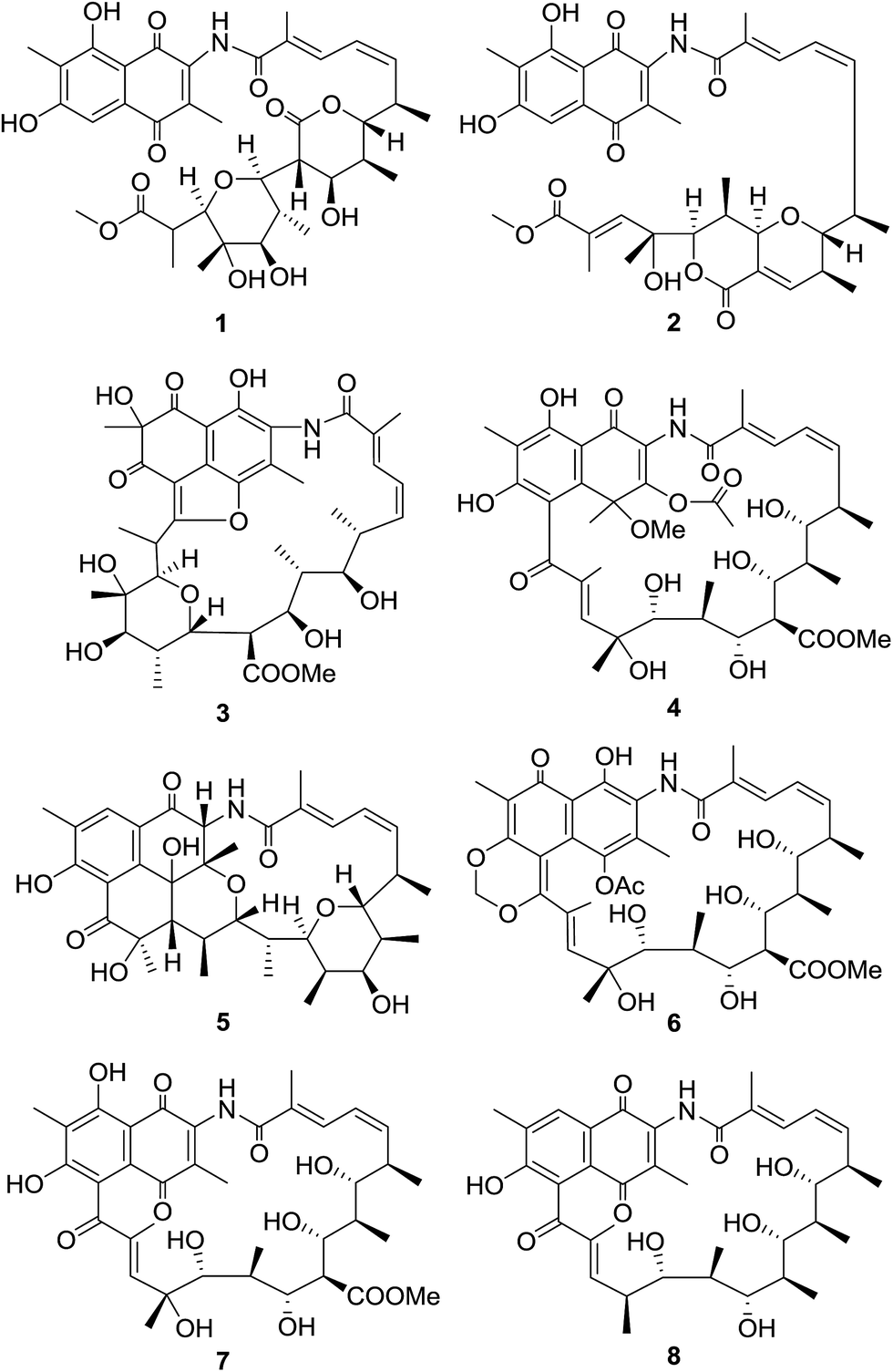 | ||
| Fig. 1 Structures of compounds 1–8. | ||
Results and discussion
The strain S. sp. S012 was cultured for 12 d on ISP3 agar plates at 28 °C. The 30 L fermentated agar cakes were diced and extracted three times overnight with EtOAc–MeOH (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20, v/v) at room temperature. After removing the organic solvents, the crude extract was partitioned between doubly-distilled water and EtOAc (1:1, v/v), and then the EtOAc extract was dried with sodium sulfate (anhydrous) and partitioned between 95% aqueous MeOH and petroleum ether (PE). The MeOH layer was concentrated under vacuum to afford the defatted MeOH extract (4.2 g), which was subjected to MPLC over reversed-phase C18 silica gel, column chromatography over Sephadex LH-20 and finally HPLC to yield compounds 1–8.
:20, v/v) at room temperature. After removing the organic solvents, the crude extract was partitioned between doubly-distilled water and EtOAc (1:1, v/v), and then the EtOAc extract was dried with sodium sulfate (anhydrous) and partitioned between 95% aqueous MeOH and petroleum ether (PE). The MeOH layer was concentrated under vacuum to afford the defatted MeOH extract (4.2 g), which was subjected to MPLC over reversed-phase C18 silica gel, column chromatography over Sephadex LH-20 and finally HPLC to yield compounds 1–8.
The molecular formula of compound 1 was assigned as C37H47NO13 on the basis of analysis of the high-resolution ESIMS data (a protonated molecular ion at m/z 714.3124 [M + H]+, calcd for C37H48NO13+, 714.3120) (ESI Fig. S57†). The planar structure of 1 was established from analysis of the NMR spectroscopic data (Table 1 and ESI Fig. S9–S14†). The 13C NMR spectroscopic data of 1 showed two carbonyl signals at δC 185.1 and 185.4, characteristic for the quinone carbonyls (Table 1). Additionally, the HMBC correlations from H-5 and Me-11 protons to the corresponding carbons revealed that 1 belonged to naphthoquinoid that was similar with the previously described chaxamycin A and C.18 The seventeen-carbon chain from C-12 to C-28 was established on the basis of 1H–1H COSY correlations, along with the HMBC correlations from the protons of Me-30, 31, 32, 34, 35 and 36 to the corresponding carbons. The C-22–C-26 pyran ring was identified by the key HMBC correlation from H-26 to C-22 (Fig. 2). The HMBC correlation from H-37 (δH 3.74, s, 3H) to C-28 revealed the presence of methoxyl group, confirming that the aliphatic chain (ansa chain) was not cyclized, which resembles the macrocycle openings in the seco-divergolides E and D.11 In consideration of the degrees of unsaturation, molecular formula and the similar downfield chemical shift of the H-18 (δH 4.80) with that of streptovaricin F owning the six-membered lactone (δH 4.62), an α-pyrone ring was proposed among C-18–C-21 and C-33 (δC 170.8) (Table 1).19 The relative configuration of 1 was deduced by analysis of ROESY correlations. The E-configuration of the C-13/14 double bond was proposed from the relative upfield shift of the allylic methyl group C-30 (δC 13.9) (Table 1). The geometry of the C-15/16 double bond was determined to be Z-form on the basis of the cis-1H–1H coupling constants between H-15 (J = 11.3 Hz) and H-16 (J = 10.6 Hz) (Table 1). The ROESY correlations (H-32/H-18/H-21, H-22/H-34/H-24/H-26 and H-23/H-35) (Fig. 2) suggested that 1 had the same relative stereochemistry as the known compound streptovaricin U in the ansa chain from C-18 to C-25.20,21 The stereochemistry of C-17 was assumed to be the same as that of other streptovaricins on the basis of biosynthetic logic (ESI Fig. S65†).22–24 The ROESY cross-peak observed from H-26 to H-24 proposed the relative configuration at the C-26 position, but the relative configuration of C-27 remained unknown for lack of related experimental data. Thus, 1 was deduced to be a new seco-streptovaricin, namely ansavaricin A (Fig. 2).
| Pos. | δH (mult., J Hz) | δC | 1H–1H COSY | HMBC |
|---|---|---|---|---|
| 1 | 185.4s | |||
| 2 | 140.8s | |||
| 3 | 138.2s | |||
| 4 | 185.1s | |||
| 5 | 7.53 (s) | 108.8d | C-1/C-4, C-6, C-7, C-9 | |
| 6 | 163.1s | |||
| 7 | 118.3s | |||
| 8 | 164.7s | |||
| 9 | 108.4s | |||
| 10 | 132.8s | |||
| 11 | 2.41 (s) | 9.0q | C-6, C-7, C-8 | |
| 12 | 168.4s | |||
| 13 | 132.2s | |||
| 14 | 7.93 (d, 11.7) | 130.8d | H-15 | C-12, C-16, C-30 |
| 15 | 6.70 (t, 11.3) | 125.7d | H-14, H-16 | C-14, C-17 |
| 16 | 6.20 (t, 10.6) | 139.0d | H-15, H-17 | C-14, C-18 |
| 17 | 3.25 (m) | 34.9d | H-16, H-18, H-31 | |
| 18 | 4.80 (dd, 1.7, 10.5) | 83.7d | H-17, H-19 | C-16, C-19, C-20, C-31 |
| 19 | 2.62 (m) | 36.0d | H-18, H-20, H-32 | C-18 |
| 20 | 4.34 (t, 2.3) | 74.0d | H-19, H-21 | C-18, C-32, C-33 |
| 21 | 3.38 (t, 2.2) | 51.6d | H-20, H-22 | C-19, C-20, C-22, C-33 |
| 22 | 3.73 (d, 1.7) | 85.5d | H-21, H-23 | C-20, C-33 |
| 23 | 3.03 (m) | 39.4d | H-22, H-24, H-34 | C-22, C-24, C-34 |
| 24 | 3.65 (d, 10.6) | 82.3d | H-23 | C-23, C-25 |
| 25 | 74.4s | |||
| 26 | 3.81 (d, 8.4) | 85.3d | H-27 | C-22, C-24, C-25, C-27, C-36 |
| 27 | 3.28 (m) | 40.8d | H-26, H-36 | C-26, C-28, C-36 |
| 28 | 176.1s | |||
| 29 | 2.30 (s) | 14.1q | C-2, C-3, C-1/C-4 | |
| 30 | 2.31 (s) | 13.9q | C-12, C-13, C-14 | |
| 31 | 1.20 (d, 6.9) | 19.1q | H-17 | C-16, C-17, C-18 |
| 32 | 1.29 (d, 6.9) | 13.8q | H-19 | C-18, C-19, C-20 |
| 33 | 170.8s | |||
| 34 | 1.31 (d, 6.4) | 14.5q | H-23 | C-22, C-23, C-24 |
| 35 | 1.75 (s) | 15.6q | C-24, C-25, C-26 | |
| 36 | 1.57 (d, 6.8) | 16.5q | H-27 | C-26, C-27, C-28 |
| 37 | 3.74 (s) | 52.1q | C-28 | |
| NH | 10.11 (s) |
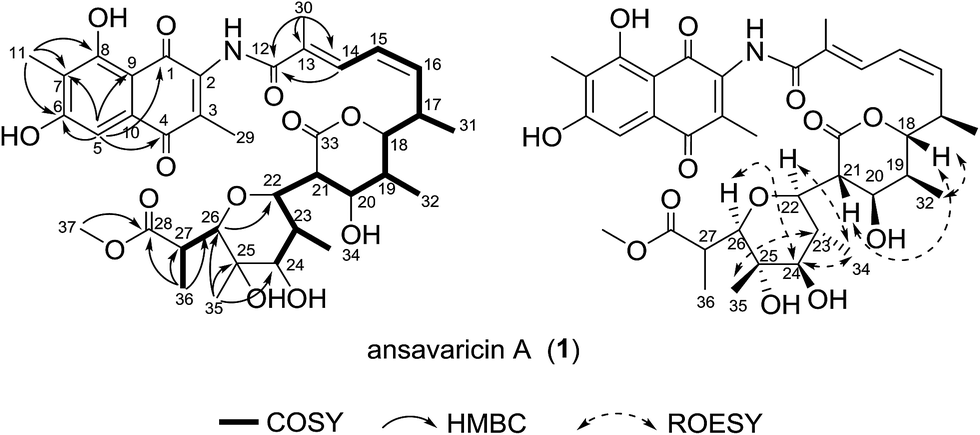 | ||
| Fig. 2 Selected 1H–1H COSY, HMBC and ROESY correlations for compound 1. | ||
Compound 2 was isolated as red powder. The high-resolution ESIMS gave a quasi-molecular ion at m/z 678.2908 [M + H]+ (696.3013 [M + NH4]+, ESI Fig. S58†) consistent with the molecular formula C37H43NO11 (calcd for C37H44NO11+, 678.2909). The comparison of its 1H and 13C NMR data (Table 2 and ESI Fig. S15–S20†) with those of 1 indicated that both structures showed a similar backbone fused with the ansa chain and naphthoquinone ring. The seventeen-carbon chain from C-12 to C-28 was established on the basis of 1H–1H COSY correlations, along with the HMBC correlations from the protons of the methyl groups and the tertiary carbons to the corresponding carbons (Fig. 3). Additionally, the presence of a pyran ring and a δ-lactone ring was prosed on the basis of the relative downfield chemical shifts of H-22 (δH 5.0) and H-24 (δH 4.65) in comparing with that of H-17 (δH 3.15), H-18 (δH 3.95), H-19 (δH 2.52) and H-23 (δH 3.15), and the degrees of unsaturation and molecular formula assumed (Table 2). The chemical shift of C-37 (δC 52.2) and HMBC correlation from H-37 (δH 3.74, s, 3H) to C-28 supported the presence of methoxyl group (Table 2). The relative configuration of 2 was proposed identical to that of 1. The geometries of the C-18, C-19, C-22 and C-23 were assumed on the basis of the ROESY correlations of H-32 to H-18 and H-34 to H-22. The relative configurations of C-17, C-24 and C-25 were deduced on the basis of the same biosynthetic origin (Fig. 3 and ESI Fig. S65†).23,24 Therefore, compound 2 was conjectured to be a new seco-streptovaricin, similar as compound 1, namely ansavaricin B.
| Pos. | δH (mult., J Hz) | δC | 1H–1H COSY | HMBC |
|---|---|---|---|---|
| 1 | 185.4s | |||
| 2 | 140.7s | |||
| 3 | 138.3s | |||
| 4 | 185.1s | |||
| 5 | 7.52 (s) | 108.8d | C-1/C-4, C-7 | |
| 6 | 163.1s | |||
| 7 | 118.3s | |||
| 8 | 164.7s | |||
| 9 | 108.4s | |||
| 10 | 132.2s | |||
| 11 | 2.41 (s) | 9.0q | C-6, C-7, C-8 | |
| 12 | 168.3s | |||
| 13 | 133.0s | |||
| 14 | 7.76 (d, 11.7) | 130.5d | H-15 | C-12, C-30 |
| 15 | 6.55 (t, 11.4) | 125.7d | H-14, H-16 | C-14, C-17 |
| 16 | 5.90 (t, 10.7) | 138.6d | H-15, H-17 | C-14 |
| 17 | 3.15 (m) | 34.5d | H-16, H-31 | |
| 18 | 3.95 (dd, 2.2, 10.7) | 87.0d | H-19 | C-16, C-31, |
| 19 | 2.52 (m) | 32.5d | H-18, H-32 | C-20 |
| 20 | 6.92 (br s) | 148.2d | C-18, C-19, C-22, C-32, C-33 | |
| 21 | 134.2s | |||
| 22 | 5.0 (br s) | 75.0d | H-23 | C-20, C-21, C-23, C-24, C-33 |
| 23 | 3.15 (m) | 38.4d | H-22, H-34 | C-22, C-34 |
| 24 | 4.65 (s) | 76.8d | C-22, C-23, C-26, C-34, C-35 | |
| 25 | 77.0s | |||
| 26 | 7.71 (s) | 147.9d | C-25, C-28, C-36 | |
| 27 | 128.1s | |||
| 28 | 170.1s | |||
| 29 | 2.31 (s) | 14.5q | C-2, C-3, C-1/C-4 | |
| 30 | 2.26 (s) | 13.8q | C-12, C-13, C-14 | |
| 31 | 1.09 (d, 6.7) | 18.4q | H-17 | C-16, C-17, C-18 |
| 32 | 0.96 (d, 7.0) | 16.6q | H-19 | C-18, C-19, C-20 |
| 33 | 165.4s | |||
| 34 | 1.54 (d, 6.9) | 13.6q | H-23 | C-22, C-23, C-24 |
| 35 | 1.76 (s) | 27.2q | C-24, C-25, C-26 | |
| 36 | 2.52 (m) | 14.2q | C-26, C-27, C-28 | |
| 37 | 3.67 (s) | 52.2q | C-28 | |
| NH | 10.03 (s) | C-2, C-12 |
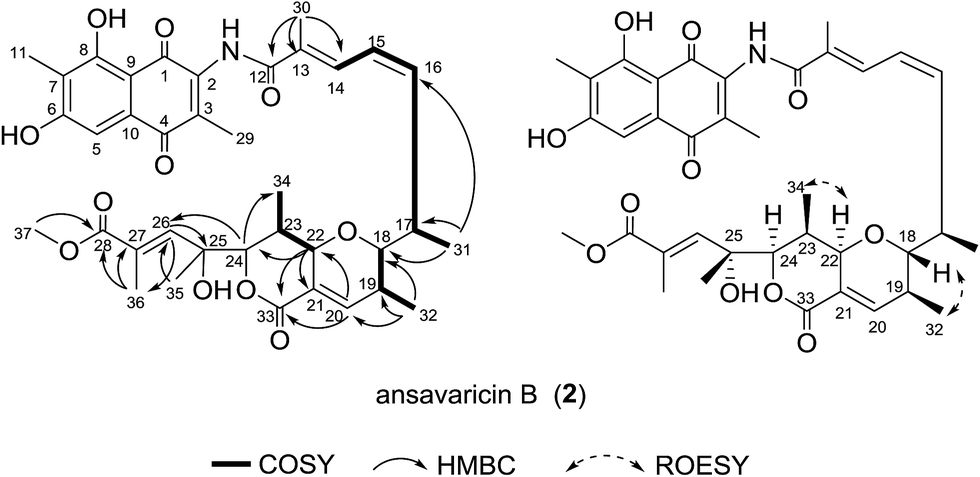 | ||
| Fig. 3 Selected 1H–1H COSY, HMBC and ROESY correlations for compound 2. | ||
Compound 3 was obtained as light yellowish powder. The molecular formula of compound 3 was determined to be C37H47NO13 by high-resolution ESIMS (m/z 714.3120 [M + H]+, calcd for C37H48NO13+, 714.3120) (ESI Fig. S59†). The thirteen-carbon fragment from C-12 to C-24 was assigned on the basis of interpretation of the 1H–1H COSY data, which was further confirmed by the HMBC correlations (Table 3, ESI Fig. S23 and S25†). The HMBC correlations from H-26, H-27, H-36 and H-37 to the corresponding carbons indicated the structure of the fragment from C-25 and C-28. Additionally, the HMBC correlation of H-22 to the C-26 confirmed the presence of the six-membered ether ring. The HMBC correlations from the methyl resonance at δH 1.59 (Me-11) to the carbon resonances at δC 86.0 (C-7), δC 193.9 (C-6), and δC 200.7 (C-8) showed that this methyl group was attached to a quaternary carbon (C-7) bearing a hydroxyl, which was in turn flanked by two ketones (Table 3). The presence of the 1,3-naphthoquinone moiety was corroborated by the HMBC correlations from H-29 to C-1, C-2, C-3 and C-4. Finally, to account for the remaining degrees of unsaturation and molecular formula, the ansa chain was deduced to be connected to C-5 of the naphthoquinone system via C-28, followed by the formation of a hemiacetal and subsequent dehydration to give an enol ether between C-28 and C-4 (Fig. 4). Considering the broad water peak at 5.2 ppm in the NMR spectra recorded in pyridine-d5, the ROESY spectra of 3 were measured in acetone-d6 (ESI Table S3†). The ROESY correlations (H-18/H-32/H-20/, H-32/H-21, H-35/H-24 and H-23/H-36) and the close similarity of biosynthetic logic of streptovaricins suggested that 3 had the same relative configurations as that of streptovaricin C (6), whose structure and absolute configuration were determined by X-ray analysis of a heavy atom derivative, in the ansa chain from C-17 to C-25 (ESI Fig. S26 and S44†).19,22,23 The stereochemistry of C-26 was assumed to be the same as that of 1 on the basis of the ROESY cross-peak observed from H-21 to H-26. The relative configurations of C-7 and C-27 remained unresolved due to lack of ROESY correlations. Hence, compound 3 was assumed to be a new streptovaricin, namely ansavaricin C.
| Pos. | δH (mult., J Hz) | δC | 1H–1H COSY | HMBC |
|---|---|---|---|---|
| 1 | 153.7s | |||
| 2 | 128.4s | |||
| 3 | 128.2s | |||
| 4 | 146.4s | |||
| 5 | 107.5s | |||
| 6 | 193.9s | |||
| 7 | 86.0s | |||
| 8 | 200.7s | |||
| 9 | 110.3s | |||
| 10 | 127.5s | |||
| 11 | 1.59 (s) | 27.9q | C-6, C-7, C-8 | |
| 12 | 175.0s | |||
| 13 | 131.4s | |||
| 14 | 6.91 (d, 11.0) | 128.8d | H-15, H-30 | C-12, C-16, C-30 |
| 15 | 6.40 (t, 10.9) | 124.1d | H-14, H-16 | C-13, C-17 |
| 16 | 5.85 (t, 10.3) | 147.1d | H-15, H-17 | C-14, C-18 |
| 17 | 3.37 (m) | 40.7d | H-16, H-18, H-31 | C-18, C-31 |
| 18 | 3.55 (d, 9.3) | 82.5d | H-17, H-19 | C-16, C-17, C-19, C-20, C-32 |
| 19 | 2.08 (m) | 36.7d | H-18, H-20, H-32 | C-20, C-32 |
| 20 | 4.83 (d, 10.4) | 72.9d | H-19, H-21 | C-19, C-21, C-22, C-32, C-33 |
| 21 | 3.17 (s) | 48.3d | H-20 | C-19, C-20, C-22, C-23, C-33 |
| 22 | 3.85 (d, 10.5) | 80.9d | H-23 | C-20, C-21, C-23, C-24, C-26, C-33, C-35 |
| 23 | 2.63 (m) | 39.7d | H-22, H-24, H-35 | C-22 |
| 24 | 3.74 (m) | 82.2d | C-23, C-25, C-35, C-36 | |
| 25 | 74.5s | |||
| 26 | 3.91 (s) | 88.3d | C-22, C-24, C-25, C-27, C-28, C-36, C-37 | |
| 27 | 4.99 (m) | 34.5d | H-37 | C-26, C-28, C-37 |
| 28 | 170.9s | |||
| 29 | 2.65 (s) | 12.7q | C-1 (weak), C-2, C-3, C-4 | |
| 30 | 2.34 (s) | 14.4q | C-12, C-13, C-14 | |
| 31 | 0.50 (d, 6.7) | 19.1q | H-17 | C-16, C-17, C-18 |
| 32 | 1.15 (d, 6.8) | 17.6q | H-19 | C-18, C-19, C-20 |
| 33 | 174.0s | |||
| 34 | 3.88 (s) | 52.1q | C-33 | |
| 35 | 1.34 (d, 6.5) | 14.7q | H-23 | C-22, C-23, C-24 |
| 36 | 1.82 (s) | 17.8q | C-24, C-25, C-26 | |
| 37 | 1.65 (d, 7.0) | 12.2q | H-27 | C-26, C-27, C-28 |
| NH | 9.89 (s) | |||
| HO-20 | 3.73 (br s) | H-20 | C-19, C-20, C-21 |
 | ||
| Fig. 4 Selected 1H–1H COSY, HMBC and ROESY correlations for compound 3. | ||
Compound 4 was obtained as a red powder. The molecular formula C40H53NO15 was deduced from the HRESIMS at m/z 786.3337 [M − H]− (calcd for C40H52NO15−, 786.3337) (ESI Fig. S60†). The 1H–1H COSY and HMBC correlations confirmed the connection of the ansa chain that was identical to that of the known compound damavaricin C (7) (Table 4, ESI Fig. S29 and S31†).25 The resonances at δC 144.6, 85.9 and 127.5 (Table 4 and ESI Fig. S28†), showed correlations to the protons of Me-31 (δH 2.01), were assigned to C-3, C-4 and C-10, respectively. Considering the HMBC correlations from H-30 (δH 2.36) to C-29 (δC 168.8) and the chemical shifts of C-3 (δC 144.6) and C-30 (δC 21.2), an acetoxyl group was assigned to be located at C-3, which was different from a methyl group at this position in compounds 1–3 (Table 4). On the other hand, the HMBC correlations from Me-11 protons to C-6, C-7 and C-8, and HO-8 to C-7, C-8 and C-9 established the connectivity from C-6 to C-9. The relative configuration of 4 was proposed to be identical to that of damavaricin C (7) by comparison of their ROESY spectrum from C-17 to C-25 (ESI Fig. S32 and S50†). The ROESY correlations (H-34/H-18/H-20, H-18/H-35 and H-22/H-38/H-26/H-39/H-24) suggested the relative configurations of C-17, C-18, C-19, C-20, C-22, C-23, C-24 and C-25. The relative configuration of C-21 was assumed to be the same as that of C-21 in 3 by comparison of their ROESY spectra (H-21/H-35 in 4, H-21/H-32 in 3) and the same biosynthetic origin (ESI Fig. S65†).23,24 The relative configuration of C-4 remained to be clarified. From the above findings and other correlations observed, the structure of 4, namely ansavaricin D, was deduced (Fig. 5).
| Pos. | δH (mult., J Hz) | δC | 1H–1H COSY | HMBC |
|---|---|---|---|---|
| 1 | 199.5s | |||
| 2 | 132.3s | |||
| 3 | 144.6s | |||
| 4 | 85.9s | |||
| 5 | 120.4s | |||
| 6 | 163.1s | |||
| 7 | 113.5s | |||
| 8 | 164.0s | |||
| 9 | 109.3s | |||
| 10 | 127.5s | |||
| 11 | 2.19 (s) | 9.5q | C-6, C-7, C-8 | |
| 12 | 171.8s | |||
| 13 | 131.1s | |||
| 14 | 7.47 (d, 10.9) | 130.3d | H-15 | C-12, C-16, C-33 |
| 15 | 6.50 (t, 11.0) | 125.6d | H-14, H-16 | C-13, C-17 |
| 16 | 5.91 (t, 10.4) | 142.1d | H-15, H-17 | C-14, C-34 |
| 17 | 3.10 (m) | 36.9d | H-16, H-34 | |
| 18 | 3.73 (dd, 1.8, 7.7) | 83.7d | H-19 | C-16, C-19, C-34, C-35 |
| 19 | 2.22 (m) | 37.5d | H-18, H-20, H-35 | C-17, C-18, C-20, C-35 |
| 20 | 4.736–4.76 (m) | 76.7d | H-19, H-21 | C-18, C-19, C-21, C-22, C-36 |
| 21 | 3.44 (m) | 49.4d | H-20 | C-19, C-20, C-22, C-36 |
| 22 | 4.45 (d, 10.6) | 71.7d | H-23 | C-20, C-23, C-24, C-36, C-38 |
| 23 | 3.57 (m) | 37.5d | H-22, H-24, H-38 | C-22, C-38 |
| 24 | 4.74–4.76 (m) | 79.1d | H-23 | C-25, C-26, C-38, C-39 |
| 25 | 77.6s | |||
| 26 | 7.51 (br s) | 151.0d | C-25, C-27, C-28, C-40 | |
| 27 | 137.9s | |||
| 28 | 200.0s | |||
| 29 | 168.8s | |||
| 30 | 2.36 (s) | 21.2q | C-29 | |
| 31 | 2.01 (s) | 11.6q | C-3, C-4, C-10 | |
| 32 | 3.51 (s) | 51.8q | C-4 | |
| 33 | 1.97 (s) | 14.0q | C-12, C-13, C-14 | |
| 34 | 1.26 (d, 6.5) | 19.4q | H-17 | C-16, C-17, C-18 |
| 35 | 1.06 (d, 6.6) | 16.7q | H-19 | C-18, C-20, C-23 |
| 36 | 174.0s | |||
| 37 | 3.50 (s) | 51.6q | C-36 | |
| 38 | 1.44 (d, 7.0) | 11.7q | H-23 | C-22, C-23, C-24 |
| 39 | 1.80 (s) | 30.5q | C-24, C-25, C-26 | |
| 40 | 2.80 (s) | 12.7q | C-26, C-27, C-28 | |
| NH | 10.61 (s) | C-12 | ||
| HO-8 | 13.91 (s) | C-7, C-8, C-9 |
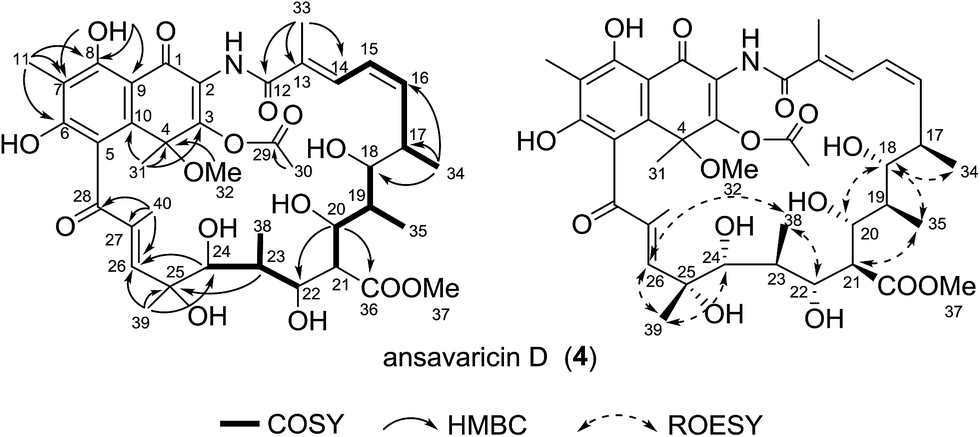 | ||
| Fig. 5 Selected 1H–1H COSY, HMBC and ROESY correlations for compound 4. | ||
Compound 5, giving the molecular formula C36H47NO9 by HRESIMS (m/z 638.3320 [M + H]+, calcd for C36H48NO9+, 638.3324) (ESI Fig. S61†), was obtained as a white amorphous powder. The seventeen-carbon fragment from C-12 to C-28 was constructed from the interpretation of 1H–1H COSY data and was further confirmed by HMBC correlations (Table 5, ESI Fig. S35 and S37†). The noticeable HMBC correlation from H-24 (δH 4.24) to C-3 permitted the ether linkage between C-24 and C-3. The cyclohexanone unit possessing C-28, C-5, C-10, C-4, C-26 and C-27 was established based on a ketone at δC 202.2 and the HMBC correlations from the protons of Me-36 to the carbons C-26, C-27 and C-28, and from H-26 to C-4, C-10 and C-28. In consideration of the degrees of unsaturation, molecular formula and structural stability (ESI Fig. S34†), a pyran ring was proposed between the carbon C-18 and C-22. The relative configuration of 5 was proposed to be the same as that of co-isolated prostreptovaricin I (8) from C-17 to C-25 by comparison of their ROESY correlations (ESI Fig. S38 and S56†) and biosynthesis of streptovaricins (ESI Fig. S65†).22–24,26 The ROESY correlations (H-26/H-24/H-2/H-29) indicated that H-26, H-24, H-2 and H-29 were on the same face. The ROESY cross-peak observed from H-36 to H-25 proposed the relative configuration at the C-27 position, but the relative configuration of C-4 remained unknown. Thus, 5 was a new natural product of the undecaketide-type polyketide, for which we proposed the name ansavaricin E (Fig. 6).
| Pos. | δH (mult., J Hz) | δC | 1H–1H COSY | HMBC |
|---|---|---|---|---|
| 1 | 193.5s | |||
| 2 | 6.00 (d, 6.9) | 59.7d | C-1, C-3, C-29 | |
| 3 | 76.8s | |||
| 4 | 81.4s | |||
| 5 | 112.7s | |||
| 6 | 164.7s | |||
| 7 | 129.6s | |||
| 8 | 7.86 (s) | 133.4d | C-1, C-6, C-10, C-11 | |
| 9 | 121.0s | |||
| 10 | 147.5s | |||
| 11 | 2.26 (s) | 15.6q | C-6, C-7, C-8 | |
| 12 | 172.4s | |||
| 13 | 132.6s | |||
| 14 | 7.70 (d, 10.6) | 129.9d | H-15 | C-12, C-16, C-30 |
| 15 | 6.58 (t, 11.2) | 125.3d | H-14, H-16 | C-13, C-17 |
| 16 | 5.76 (t, 10.0) | 140.8d | H-15, H-17 | C-14, C-31 |
| 17 | 2.75 (t, 7.9) | 40.0d | H-16, H-31 | C-15, C-16, C-19 |
| 18 | 3.73 (d, 5.3) | 83.9d | H-19 | C-16, C-19, C-31, C-32 |
| 19 | 2.50 (m) | 43.7d | H-18, H-20, H-32 | C-17, C-18, C-20, C-32 |
| 20 | 3.95 (d, 9.2) | 80.1d | H-19 | C-18, C-21, C-22, C-33 |
| 21 | 2.00 (m) | 35.1d | H-33 | C-20, C-33 |
| 22 | 4.70 (d, 9.3) | 70.1d | H-23 | C-23, C-24, C-20, C-33 |
| 23 | 2.17 (m) | 38.3d | H-22, H-34 | C-22, C-34 |
| 24 | 4.24 (d, 10.3) | 71.2d | H-25 | C-3, C-22, C-23, C-26, C-34 |
| 25 | 2.87 (m) | 29.8d | H-24, H-35 | C-24, C-26, C-27 |
| 26 | 2.93 (d, 9.2) | 64.0d | C-4, C-10, C-25, C-28, C-35 | |
| 27 | 88.9s | |||
| 28 | 202.2s | |||
| 29 | 1.59 (s) | 17.4q | C-2, C-3, C-4 | |
| 30 | 2.21 (s) | 14.4q | C-12, C-13, C-14 | |
| 31 | 1.19 (d, 6.8) | 22.4q | H-17 | C-16, C-17, C-18 |
| 32 | 1.07 (d, 6.7) | 16.9q | H-19 | C-19, C-20, C-18 |
| 33 | 1.30 (d, 7.0) | 11.3q | H-21 | C-20, C-21, C-22 |
| 34 | 1.17 (d, 7.0) | 9.5q | H-23 | C-22, C-23, C-24 |
| 35 | 0.85 (d, 6.6) | 16.1q | H-25 | C-24, C-25, C-26 |
| 36 | 1.65 (s) | 14.3q | C-26, C-27, C-28 | |
| NH | 8.69 (d, 6.5) | H-2 |
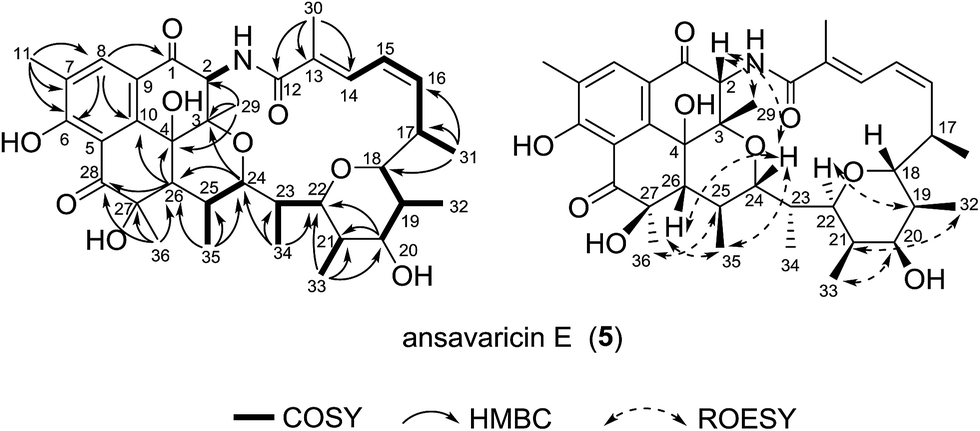 | ||
| Fig. 6 Selected 1H–1H COSY, HMBC and ROESY correlations for compound 5. | ||
The known compounds (6–8) were identified by 1D, 2D NMR, HRESIMS data (ESI Tables S6–S8, Fig. S39–S56 and S62–S64†) and comparison of their spectroscopic data with those reported in the literature, including streptovaricin C (6),27,28 damavaricin C (7)25,29 and prostreptovaricin I (8).26
In the antimicrobial experiment, only known compounds 6, 7 and 8 exhibited modest activity against Bacillus subtilis strain 86315 (diameters of inhibitory zones 15, 20 and 16 mm, respectively) and Staphylococcus aureus ATCC 25923 (diameters of inhibitory zones 25, 32 and 14 mm, respectively) at 20 μg per disc. In addition, 8 showed moderate activity against Mycobacterium smegmatis mc2 155 (diameters of inhibitory zones 16 mm, respectively). None of these compounds showed antibacterial activity against Pseudomonas aeruginosa PA01, Candida albicans 5314 and Salmonella enterica serovar Typhimurium UK-1 χ8956.
In view of lack of antimicrobial activity, especially for new ansamycins, the compounds 1–8 were further assayed for their activities of inhibiting the type three secretion system (T3SS) of Salmonella enterica. S. enterica is the causative agent of typhoid fever and is also a major cause of foodborne illness.30 It uses a type III secretion system (T3SS) to mediate translocation of virulence factors including specific effector proteins into host cells.31 This T3SS, which is an injector-like secretion, is becoming an attractive drug target for developing novel anti-virulence agents because of its highly conservation among different Gram-negative bacteria.32,33 Most of the secreted effector proteins are encoded in Salmonella pathogenicity island 1 (SPI-1), a horizontally acquired chromosomal region of ∼40 kbp.34 To exploit novel antibacterial agents targeting SPI-1, the effect of compounds 1–8 on secretion of SPI-1 effector proteins was assessed in vitro.35 The results indicated that the new compound 5, together with known compound 6 and 7, significantly inhibited the secretion of SPI-1 effectors including SipA/B/C/D and FliC. Meanwhile, 1–4 and 8 showed weaker or no inhibitory effect on SPI-1 at the concentration of 100 μM (Fig. 7A). Besides, compound 5 exhibited obviously concentration-dependent inhibition, especially at the concentration of 50 μM (Fig. 7B). Combining with antibacterial activity test, we supposed that 5, 6 and 7 could potentially reduce virulence of S. enterica serovar Typhimurium without causing bacteria death, thereby avoiding subsequent development of resistance.
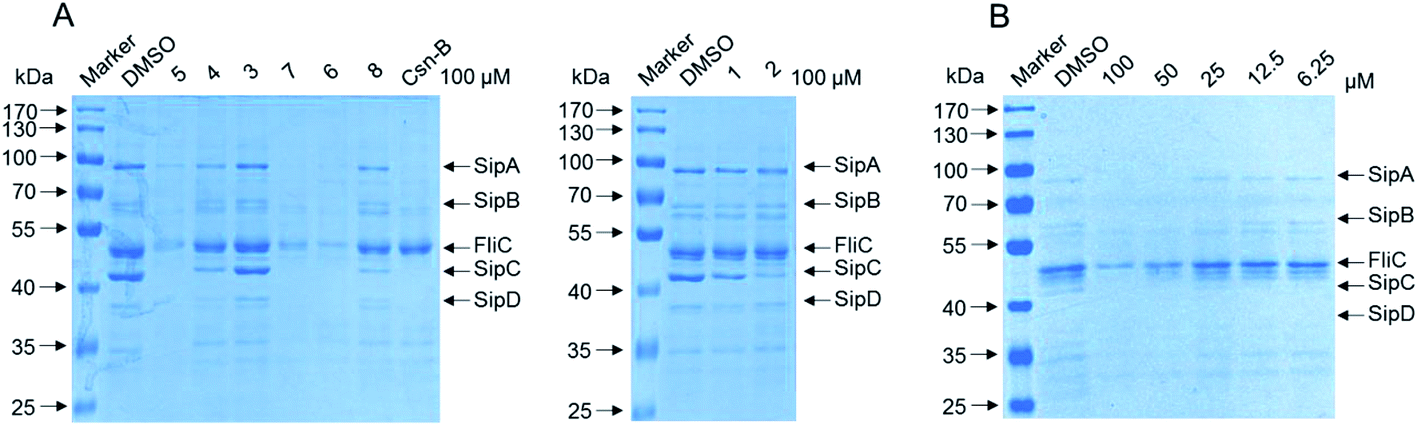 | ||
| Fig. 7 (A) Compounds 1–8 inhibited the secretion of invasion-associated SPI-1 effector proteins in vitro. S. enterica serovar Typhimurium UK-1 χ8956 (ΔP rpoS183: TT araC PBAD rpoS) was grown in LB medium supplemented with 0.2% L-arabinose in the presence of solvent control, compounds 1–8 and Csn-B as positive control at the final concentration of 100 μM, respectively. The effector proteins of SPI-1 in the supernatant of culture were precipitated by 10% TCA–acetone and analyzed by 10% SDS-PAGE followed by staining with coomassie blue. SipA/B/C/D, SPI-1 effector proteins; FliC, flagellin of Salmonella. (B) Compound 5 inhibited the secretion of FliC in a dose-dependent manner (at a range of concentrations from 50 to 100 μM) and did not affect growth of S. enterica serovar Typhimurium. | ||
Experimental section
General experimental procedures
NMR spectra were recorded on a Bruker DRX-600 MHz and DRX-400 MHz NMR spectrometer (Bruker Daltonics Inc., Billerica, Massachusetts) with tetramethylsilane (TMS) as an internal standard. HR-ESIMS were measured on an LTQ-Orbitrap XL. Sephadex LH-20 was obtained from the GE Amersham Biosciences (Pis-cataway, New Jersey). Reversed-phase C-18 silica gel for column chromatography was obtained from Merck (Darmstadt, Germany). Silica gel GF254 for thin-layer chromatography (TLC) was purchased from Qingdao Marine Chemical Ltd (Qingdao, China). HPLC separations were mainly performed on three HPLC systems: Waters 1525 Binary HPLC Pump equipped with Waters 996 Photodiode Array Detector using Agilent Eclipse XDB-C18 column (5 μm, 9.4 × 250 mm); Waters 2545 Pump equipped with Waters 2998 Detector using Sunfire Prep OBD-C18 column (5 μm, 19 × 150 mm); Agilent 1260 instrument equipped with Agilent Eclipse XDB-C18 column (5 μm, 9.4 × 250 mm).Amplification of 16S rRNA gene sequences and taxonomical identification
The DNA preparations were used as template for partial 16S rRNA gene PCR amplifications applying the universal primers 27f and 1492r. The reaction mixture (50 μL) includes 10–100 ng genomic DNA, 20 pmol of each universal primer (27f: 5′>AGAGTTTGATCMTGGCTCAG<3′, 1492r: 5′>TACGGYTACCTTGTTACGACTT<3′), 1.6 μL dNTP (2.5 mmol L−1 each), 1.25 U of rtaq (TaKaRa Biotechnology Co., Dalian, China), 5 μL 10× PCR buffer (Mg2+ Plus). The PCR cycling program was as following: initial denaturing for 10 min at 95 °C, 30 cycles of denaturation for 30 s at 94 °C, annealing for 30 s at 58 °C, extension for 60 s at 72 °C and final extension for 10 min at 72 °C. The amplicons were gel purified and bidirectional sequenced by Sanger method. The sequences of 16S rRNA were assigned into taxonomic ranks (genus) using Ribosomal Database Project (RDP) Classifier.36 Strain S012 was isolated from rhizosphere soil collected at Zhongshan Botanical Garden, Nanjing, China. It was identified as a Streptomyces species, according to the 16S rRNA sequence (GenBank accession no. KY411968).Amplification of AHBA gene sequences
For screening of AHBA synthase gene, PCR was performed in 1× GC buffer I (TaKaRa). Four degenerate primers (AHBA351, AHBA418, AHBA1080, AHBA1289) were designed to recover unexploited potential strains. The reaction mixture (20 μL) includes 10–100 ng genomic DNA, 5 pmol each primer (AHBA351: 5′>GCSGTCACSAACGGSACSCAYGCSCTSGA<3′, AHBA418: 5′>ATCGTSCCSGCSTTCACSTTCATCTC<3′, AHBA10805: ′>AYNCGGAACATSGCCATGTAGTG<3′, AHBA1289: 5′>SCGGTGGTGCAGCCASANGCMGT<3′), 1.6 μL dNTP (2.5 mmol L−1 each), 0.5 U of Ex-taq (TaKaRa). The PCR cycling program was as following: initial denaturation for 10 min at 95 °C, 35 cycles of denaturation for 30 s at 94 °C, annealing for 30 s at 58 °C, extension for 30 s at 72 °C and final extension for 10 min at 72 °C. To avoid redundant work, two rounds of PCR screening were conducted. The first round of PCR amplification was performed at ten-strain level to identify the positive groups. The individual strains of positive group were subjected to the second round to identify the positive one. Positive PCR products were cloned into T vector and then sequenced and compared.37Strain and fermentation
The mutant strain S012 was isolated from botanical garden soil collected at Nanjing, China. It was identified as an AHBA synthase gene-positive strain according to PCR screening and was cultured in triangular flasks with oatmeal medium (oatmeal 30 g, saline salt 1 mL, pH 7.2) to afford a seed culture. The fermentation (30 L) was performed on oatmeal medium for 12 d at 28 °C in Petri dishes. The saline salt contained FeSO4·7H2O 0.1 g, ZnSO4·7H2O 0.1 g, MnCl2·4H2O 0.1 g, dd H2O 100 mL.Extraction and isolation
To extract the metabolites, the culture was diced and extracted three times overnight with EtOAc/MeOH (80:20, v/v) at room temperature and partitioned between EtOAc and doubly-distilled water until the EtOAc layer was colorless. Then, the EtOAc soluble fraction was dried with sodium sulfate (anhydrous) and the solvent was removed under vacuum to afford the EtOAc extract. EtOAc extract was dissolved in 95% methanol and extracted five times with an equal volume of petroleum ether to afford the defatted MeOH extract (4.2 g).
The MeOH extract was fractionated by MPLC (145 g RP-18 silica gel; 30%, 50%, 70% MeOH and 100% MeOH, 1 L each, respectively). In accordance with the HPLC results, Fr. 1–4, Fr. 5–11, Fr. 12–15 and Fr. 16–30 were combined and marked as Fr. A, Fr. B, Fr. C and Fr. D, respectively. Fr. A was chromatographed over Sephadex LH-20 (60 g; MeOH) to obtain Fr. A1. Fr. A1 was purified by reversed-phase HPLC (Waters 2545 instrument; Sunfire Prep OBD-C18, 19 × 150 mm, 5 μm, 15 mL min−1, UV 254 nm) eluted with 40% CH3CN to obtain 7 (tR = 15.6 min, 3.2 mg), 6 (tR = 17.1 min, 4.2 mg) and ansavaricin D (4) (tR = 26 min, 4.0 mg).
Fr. B was chromatographed over Sephadex LH-20 (120 g; MeOH) to obtain Fr. B1. The resulting Fr. B1 was purified by MPLC (40 g RP-18 silica gel; 30%, 50%, 70% CH3CN and 100% MeOH, 600 mL each, respectively) to afford Fr. B1a–B1f. Fr. B1c was purified by reversed-phase HPLC (Agilent 1260 instrument; ZORBAX Eclipse XDB-C18 9.4 × 250 mm, 5 μm, UV 254 nm) eluted with 40% CH3CN at a flow rate of 4.0 mL min−1 to obtain ansavaricin A (1) (tR = 18.1 min, 3.0 mg).
Fr. C was chromatographed over Sephadex LH-20 (120 g; MeOH) to obtain Fr. C3. Fr. C3 was subjected to MPLC (145 g RP-18 silica gel; 30%, 50%, 70% CH3CN and 100% MeOH, 600 mL each, respectively) to afford Fr. C3a–C3e. Fr. C3d was further purified by MPLC (40 g RP-18 silica gel; 60%, 70%, 80% and 100% MeOH, 300 mL each, respectively) repeated two times and finally purified by HPLC (Waters 1525 instrument; ZORBAX Eclipse XDB-C18 9.4 × 250 mm, 5 μm, 50% CH3CN, 3.5 mL min−1, UV 254 nm) to obtain ansavaricin B (2) (tR = 18.8 min, 3.5 mg).
Fr. D was further purified by Sephadex LH-20 (120 g) eluted with MeOH to give Fr. D1–Fr. D4. The resulting Fr. D3 was further separated by Sephadex LH-20 (120 g) using MeOH to yield four fractions (Fr. D3a–Fr. D3d). The fourth fraction, Fr. D3d, was further purified by MPLC (145 g RP-18 silica gel; 50%, 70%, 80%, 90% and 100% MeOH, 600 mL each, respectively) to afford Fr. D3d1–Fr. D3d5. Fr. D3d2 was purified by HPLC to yield ansavaricin C (3) (tR = 19.6 min, 4 mg; Waters 1525 instrument; ZORBAX Eclipse XDB-C18 9.4 × 250 mm, 5 μm, 50% CH3OH, 3.5 mL min−1, UV 254 nm). Fr. D3d4 was further purified by Sephadex LH-20 (80 g; MeOH) and finally purified by HPLC (Waters 1525 instrument; ZORBAX Eclipse XDB-C18 9.4 × 250 mm, 5 μm, 80% CH3OH, 3.5 mL min−1, UV 254 nm) to yield 8 (tR = 19 min, 14.4 mg) and ansavaricin E (5) (tR = 25.6 min, 17.9 mg).
ε): 270.0 (2.31), 218.0 (2.33); 1H and 13C NMR data, see Table 1; HRESIMS: m/z 714.3124 [M + H]+ (calcd for C37H48NO13+, 714.3120).ε): 270.0 (2.33), 218.0 (2.48); 1H and 13C NMR data, see Table 2; HRESIMS: m/z 678.2908 [M + H]+ (calcd for C37H44NO11+, 678.2909).ε): 232.0 (2.40); 1H and 13C NMR data, see Table 3; HRESIMS: m/z 714.3120 [M + H]+ (calcd for C37H48NO13+, 714.3120).ε): 254.0 (2.96); 1H and 13C NMR data, see Table 4; HRESIMS: m/z 786.3337 [M − H]− (calcd for C40H52NO15−, 786.3337).ε): 260.0 (3.20), 214.0 (3.03); 1H and 13C NMR data, see Table 5; HRESIMS: m/z 638.3320 [M + H]+ (calcd for C36H48NO9+, 638.3324).Antimicrobial assay
The compounds 1–8 were tested for their antibacterial activity against Candida albicans 5314, Staphylococcus aureus ATCC 25923, Mycobacterium smegmatis mc2 155, Pseudomonas aeruginosa PA01, Bacillus subtilis 86315 and Salmonella enterica serovar Typhimurium UK-1 χ8956 purchased from China Center of Industrial Culture Collection (Beijing, China) and conserved in the laboratory of the authors with paper disc diffusion assay. Rifamycin SV and amphotericin were used as positive control. Test compounds were absorbed onto individual paper disks (6 mm diameter) at 20 μg per disc and placed on the surface of the agar. The assay plates were incubated at 37 °C for 24 h for antibacterial activity and at 28 °C for 48 h for antifungal activity and examined for the presence of a zone of inhibition.Anti-T3SS assay
Salmonella enterica serovar Typhimurium UK-1 χ8956 (ΔP rpoS183: TT araC PBAD rpoS) obtained from Roy Curtiss III (School of Life Sciences, Arizona State University) was cultivated on LB agar plates with 0.2% L-arabinose at 37 °C or 25 °C. The assay of isolation and detection of effector proteins of SPI-1 (Salmonella Pathogenicity Island-1) were performed as described previously.23 The temperature of bacterial culture was changed to induce SPI-1 to promote the secretion of effector proteins. In brief, bacteria culture was incubated overnight at 25 °C with agitation. Compounds 1–8 at 100 μM and solvent control were added into the diluted cultures next day and the cultures were continued to grow at for 4 h 37 °C with agitation. Cytosporone B (Csn-B) was selected as the positive control.35 The secreted proteins were precipitated by 10% trichloroacetic acid (TCA)–acetone. Sediments were dissolved in 1× sample buffer and heated for 5 min at 95 °C to make protein denaturation. The protein samples were analysed by 10% SDS-PAGE followed staining with coomassie blue.Conclusions
In summary, we isolated and characterized 5 new undecaketides-type ansamycins, and 3 known ones from the Streptomyces sp. S012 strain. All new compounds showed no obvious cytotoxicities against the growth of the test bacteria. However, the preliminary screening of the SPI-1 effector proteins of Salmonella showed that compounds 5, 6 and 7 had evident inhibitory activity against the secretion of SPI-1 effector proteins SipA/B/C/D and FliC. Specially, new compound 5 was discovered apparent inhibitory activity on the SPI-1 effector proteins in a dose-dependent manner without having effect on the growth of bacteria, which was a promising candidate for discovering new natural compounds against antimicrobial-resistant. Moreover, this study demonstrates the structural diversity and biological activity of streptovaricins and proves that PCR screening of AHBA synthase is an efficient approach to discover new ansamycins from microbial resources.Acknowledgements
This work was supported by the National Natural Science Foundation of China (81530091, 81673317, 81602979), the Independent Innovation Foundation of Shandong University (IIFSDU, 2014JC027) and the Program for Changjiang Scholars and Innovative Research Team in University (IRT13028).Notes and references
- H. G. Floss and T. W. Yu, Chem. Rev., 2005, 105, 621–632 CrossRef CAS PubMed.
- Y. Fukuyo, C. R. Hunt and N. Horikoshi, Cancer Lett., 2010, 290, 24–35 CrossRef CAS PubMed.
- R. McCune, K. Dettschle, C. Jordahl, R. Des Pbez, C. Muschenheim and W. McDermott, Am. Rev. Tuberc. Pulm. Dis., 1957, 75, 659–666 CAS.
- V. Prelog and W. Oppolzer, Helv. Chim. Acta, 1973, 56, 2279–2287 CrossRef CAS.
- J. J. Kibby, I. A. McDonald and R. W. Rickards, J. Chem. Soc., Chem. Commun., 1980, 768–769, 10.1039/C39800000768.
- O. Ghisalba and J. Nüesch, J. Antibiot., 1981, 34, 64–71 CrossRef CAS PubMed.
- K. Hatano, S.-I. Akiyama, M. Asai and R. W. Rickards, J. Antibiot., 1982, 35, 1415–1417 CrossRef CAS PubMed.
- C. Lu, Y. Li, J. Deng, S. Li, Y. Shen, H. Wang and Y. Shen, J. Nat. Prod., 2013, 76, 2175–2179 CrossRef CAS PubMed.
- S. Li, C. Lu, J. Ou, J. Deng and Y. Shen, RSC Adv., 2015, 5, 83843–83846 RSC.
- S.-R. Li, G.-S. Zhao, M.-W. Sun, H.-G. He, H.-X. Wang, Y.-Y. Li, C.-H. Lu and Y.-M. Shen, Gene, 2014, 544, 93–99 CrossRef CAS PubMed.
- G. Zhao, S. Li, Z. Guo, M. Sun and C. Lu, RSC Adv., 2015, 5, 98209–98214 RSC.
- J. Zhang, Z. Qian, X. Wu, Y. Ding, J. Li, C. Lu and Y. Shen, Org. Lett., 2014, 16, 2752–2755 CrossRef CAS PubMed.
- S. Li, Y. Li, C. Lu, J. Zhang, J. Zhu, H. Wang and Y. Shen, Org. Lett., 2015, 17, 3706–3709 CrossRef CAS PubMed.
- X. Li, J. Zhu, G. Shi, M. Sun, Z. Guo, H. Wang, C. Lu and Y. Shen, RSC Adv., 2016, 6, 88571–88579 RSC.
- K. Tan and B. McAuslan, Biochem. Biophys. Res. Commun., 1971, 42, 230–236 CrossRef CAS PubMed.
- W. W. Brockman, W. A. Carter, L.-H. Li, F. Reusser and F. R. Nichol, Nature, 1971, 230, 249–250 CrossRef CAS PubMed.
- P. Siminoff, R. M. Smith, W. T. Sokolski and G. Savage, Am. Rev. Tuberc. Pulm. Dis., 1957, 75, 576–583 CAS.
- M. E. Rateb, W. E. Houssen, M. Arnold, M. H. Abdelrahman, H. Deng, W. T. A. Harrison, C. K. Okoro, J. A. Asenjo, B. A. Andrews, G. Ferguson, A. T. Bull, M. Goodfellow, R. Ebel and M. Jaspars, J. Nat. Prod., 2011, 74, 1491–1499 CrossRef CAS PubMed.
- A. H. J. Wang, I. C. Paul, K. L. Rinehart and F. J. Antosz, J. Am. Chem. Soc., 1971, 93, 6275–6276 CrossRef CAS PubMed.
- W. M. Knöll, J. Rinehart, L. Kenneth, P. F. Wiley and L. H. Li, J. Antibiot., 1980, 33, 249–251 CrossRef.
- W. Torres, R. R. Rodríguez and J. A. Prieto, J. Org. Chem., 2009, 74, 2447–2451 CrossRef CAS PubMed.
- B. Milavetz, K. Kakinuma, K. L. Rinehart, J. P. Rolls and W. J. Haak, J. Am. Chem. Soc., 1973, 95, 5793–5795 CrossRef CAS PubMed.
- A. L. Staley and K. L. Rinehart, J. Antibiot., 1991, 44, 218–224 CrossRef CAS PubMed.
- Q. Kang, Y. Shen and L. Bai, Nat. Prod. Rep., 2012, 29, 243–263 RSC.
- K. L. J. Rinehart, F. J. Antosz, P. V. Deshmukh, K. Kakinuma, P. K. Martin, B. I. Milavetz, K. Sasaki, T. R. Witty, L. H. Li and F. Reusser, J. Antibiot., 1976, 29, 201–203 CrossRef CAS PubMed.
- P. V. Deshmukh, K. Kakinuma, J. J. Ameel, K. L. Rinehart Jr, P. F. Wiley and L. H. Li, J. Am. Chem. Soc., 1976, 98, 870–872 CrossRef CAS PubMed.
- K. L. Rinehart, P. K. Martin and C. E. Coverdale, J. Am. Chem. Soc., 1966, 88, 3149–3150 CrossRef CAS.
- A. H. Wang and I. C. Paul, J. Am. Chem. Soc., 1976, 98, 4612–4619 CrossRef CAS PubMed.
- W. R. Roush, D. S. Coffey and D. J. Madar, J. Am. Chem. Soc., 1997, 119, 11331–11332 CrossRef CAS.
- J. A. Crump, S. P. Luby and E. D. Mintz, Bull. W. H. O., 2004, 82, 346–353 Search PubMed.
- Y.-H. Sun, H. G. Rolán and R. M. Tsolis, J. Biol. Chem., 2007, 282, 33897–33901 CrossRef CAS PubMed.
- L. M. Stamm and M. B. Goldberg, Science, 2011, 331, 1147–1148 CrossRef CAS PubMed.
- L. J. Worrall, E. Lameignere and N. C. J. Strynadka, Curr. Opin. Microbiol., 2011, 14, 3–8 CrossRef CAS PubMed.
- C. P. Lostroh and C. A. Lee, Microbes Infect., 2001, 3, 1281–1291 CrossRef CAS PubMed.
- J. Li, C. Lv, W. Sun, Z. Li, X. Han, Y. Li and Y. Shen, Antimicrob. Agents Chemother., 2013, 57, 2191–2198 CrossRef CAS PubMed.
- Q. Wang, G. M. Garrity, J. M. Tiedje and J. R. Cole, Appl. Environ. Microbiol., 2007, 73, 5261–5267 CrossRef CAS PubMed.
- H. X. Wang, Y. Y. Chen, L. Ge, T. T. Fang, J. Meng, Z. Liu, X. Y. Fang, S. Ni, C. Lin, Y. Y. Wu, M. L. Wang, N. N. Shi, H. G. He, K. Hong and Y. M. Shen, J. Appl. Microbiol., 2013, 115, 77–85 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data and other relevant information for compounds 1–8. See DOI: 10.1039/c6ra27405f |
| This journal is © The Royal Society of Chemistry 2017 |