
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stimuli-responsive hyperbranched poly(amidoamine)s integrated with thermal and pH sensitivity, reducible degradability and intrinsic photoluminescence†
Chen Zhana,
Xiao-Bin Fub,
Yefeng Yaob,
Hua-Ji Liu*a and
Yu Chen *a
*a
aTianjin Key Laboratory of Molecular Optoelectronic Science, Department of Chemistry, School of Sciences, Tianjin University, Collaborative Innovation Center of Chemical Science and Engineering (Tianjin), Tianjin 300354, People's Republic of China. E-mail: liuhuaji@tju.edu.cn; chenyu@tju.edu.cn
bDepartment of Physics & Shanghai Key Laboratory of Magnetic Resonance, East China Normal University, North Zhongshan Road 3663, Shanghai 200062, People's Republic of China
First published on 17th January 2017
Abstract
A series of hyperbranched poly(amidoamine)s (HPAs) were synthesized from the Michael addition copolymerization of tris(2-aminoethyl) amine (TAEA) and two bisacrylamide monomers N,N′-cystamine bisacrylamide (CBA) and N,N′-hexamethylene bisacrylamide (HMBA) at room temperature. The further modification with isobutyric anhydride led to isobutyramide terminated HPA (HPA-C4). 1H NMR and 15N NMR characterizations proved the successful preparation of these polymers. Moreover, beside the contents of TAEA, CBA and HMBA units in the composition, 15N NMR spectrometry could supply more structural information than 1H NMR spectrometry, such as the ratio of different amine groups in polymers, the transformation efficiency of reactive primary and secondary amines into C4 groups. GPC measurements not only gave the information of molecular weight and polydispersity, but also proved that all the HPA-C4s containing disulfide bonds could be degraded after being treated with dithiothreitol (DTT). Turbidimetry measurements showed that HPA-C4s had thermoresponsive property in water. The cloud point temperature (Tcp) of HPA-C4s was pH-dependent. Moreover, DTT could only affect the thermoresponsive property of HPA-C4s containing disulfide bonds due to the induced polymer degradation. Although no traditional fluorophores existed in HPA-C4s, HPA-C4s could emit blue fluorescence centered at ca. 455 nm. The fluorescence intensity was influenced pronouncedly by polymer concentration, pH, oxidizing time.
Introduction
During the past decade polymers with stimuli-responsive properties, such as reversible and fast conformational or phase changes in response to variations in temperature/pH have attracted plenty of interest in many aspects.1,2 One of the most appealing stimuli-responsive species is the thermoresponsive polymer with a lower critical solution temperature (LCST) in aqueous solution, which show a large decrease in solubility in water above a specific temperature.3 To date, the most studied thermoresponsive materials have been those with linear structure displaying LCST properties.3–5 In other words, even though the thermoresponsive materials have some specific topology, such as hydrogels,6 star or grafted polymeric structures7–9 and polymeric micelles,10 their LCST properties are originated from their thermoresponsive linear polymer components.Since 2004, thermoresponsive dendritic polymer as a new member of the family of thermoresponsive polymers has occurred.11–23 Compared to the traditional linear thermoresponsive polymers that usually adopt a loose coil conformation in solution, the dendritic polymer has a compacted sphere-like structure. As a consequence of such a structure, the thermoresponsive dendritic polymer usually only has a minor conformation adjustment during the transition (i.e., the globule-to-globule transition), different from the coil-to-globule transition occurring during the transition of the traditional thermoresponsive linear polymer.24,25 Meanwhile, the thermoresponsive dendritic polymers exhibit obvious difference in properties compared to the traditional thermoresponsive linear polymers.26–29 For instance, the phase transition temperature of thermoresponsive dendritic polymers is addressed to be more sensitive to the addition of salts (including anions and cations) than those of thermoresponsive linear ones.28,29 To date, research concerning a single thermo-stimulus has been extended to dual11,15 and even triple stimuli.30–32 Besides temperature, other typical stimuli include pH,11,15,30 light31,32 and ionic strength.28–30
Hyperbranched poly(amidoamine)s (HPAs) have attracted considerable attention in a variety of chemical and biological fields, such as nanotechnology and nanomedicine.33–36 HPAs are normally prepared from the polymerizations of Ax + By monomers in one pot, where Ax monomer is multifunctional amines and By monomer is multifunctional acrylamide or multifunctional ester. The types of these monomers are so versatile that the obtained HPAs might have different properties. For example, HPAs chemically analogous to PAMAM dendrimers have been prepared from the amidation polymerization of tris(2-aminoethyl) amine (TAEA, A3) and tris(2-di(methyl acrylate)-aminoethyl)amine (hexaester, B6).37 Such HPAs and their gluconamide derivatives have been successfully used to prepare and stabilize gold nanoparticles.33,34 The introduction of isobutyramide groups to the terminals could endow thermal and pH dual-stimuli to HPAs in water.30 Disulfide-functionalized hyperbranched HPAs were synthesized by Michael addition polymerization of 1-(2-aminoethyl)piperazine (AEPZ, A3) and N,N′-cystaminebisacrylamide (B2). Such HPAs displayed bright fluorescence, and they are also degradable through the reductive cleavage of disulfide bonds.38 Moreover, such HPAs have also been used as bio-reducible high efficient nonviral gene delivery vectors.35 Michael addition and amidation polymerizations between AEPZ (A3) and methyl acrylate (B2) led to HPAs with intrinsic fluorescence.39 The further alkylation of the residual amino protons of such HPAs with propylene oxide enhanced their fluorescence.40 HPAs with vinyl terminals have been prepared via the Michael addition polymerization of AEPZ (A3) and N,N′-methylene bisacrylamide (MBA, B2), subsequently, AEPZ was added to change the vinyl terminals to primary amines. Such HPAs showed weak fluorescence, however, fluorescence was strengthened after short polyethylene glycol chains were linked onto HPA via Michael addition reaction.41 When an equal feeding mole ratio of AEPZ/MBA was polymerized through the Michael addition polymerization, HPAs with amine terminals could be obtained, which exhibited the aggregation-induced emission.42 A series of reducible HPAs as high efficient nonviral gene delivery vectors have been synthesized by Michael addition copolymerization of N,N-dimethylaminodipropylenetriamine (A3) and two bisacrylamide monomers N,N′-hexamethylene bisacrylamide (HMBA, B2) and N,N′-cystamine bisacrylamide (CBA, B2).36
In this work, we prepared a series of new HPA derivatives that were not only thermal and pH sensitive in water, but also have the properties of reducible degradability and intrinsic photoluminescence. To our best knowledge, stimuli-responsive polymers possessing so many functionalities simultaneously are very scarce.
Experimental
Materials
Cystamine dihydrochloride (96%) was obtained from Tianjin Heowns Biochem Company and used directly. Tris(2-aminoethyl) amine (TAEA, 97%) was obtained from Alfa Aesar and was distilled under reduced pressure before use. Triethyl amine (A.R., TEA) was purchased from Tianjin University Kewei Chemical Company and distilled before use. 1,6-Hexanediamine (A. R.) was purchased from Tianjin Kemiou Chemical Reagent Company. Sodium hydroxide (NaOH, A. R.), methanol (A. R.), and dimethyl sulfoxide (DMSO, A. R.) were obtained from Tianjin University Kewei Chemical Company. N,N′-cystaminebisacrylamide (CBA) and N,N′-hexmethylenebisacrylamide (HMBA) were synthesized according to the literature.43 Dithiothreitol (DTT, 99%) was purchased from Shanghai Aladdin biological technology Company and used directly.Syntheses of hyperbranched poly(amidoamine) (HPA)
A series of HPAs containing different amount of disulfide groups were synthesized by Michael addition reaction between TAEA and different molar ratio of HMBA/CBA. Under N2 atmosphere, a solution of TAEA (2.2 mmol) in 2 mL of methanol was added dropwise into the mixture of CBA/HMBA (2.0 mmol in total) in 2 mL of methanol at 0–5 °C. The polymerization lasted for 4 days at room temperature under vigorous stirring. Subsequently, the pH of the polymerization solution was adjusted to be about 2 using 2 M of HCl solution. This acidified solution was dropped into a large amount of acetone under vigorous stirring. The collected solid product was dried overnight at 40 °C in vacuo. Pure hydrochloride salt of HPA was obtained.Syntheses of isobutyric amide (IBAm) terminated HPA (HPA-C4)
Under nitrogen atmosphere, isobutyric anhydride (0.38 g, 2.4 mmol) was added dropwise to the mixture of HPA hydrochloride (0.60 g) and triethylamine (0.51 g, 5.0 mmol) in 5 mL of DMSO at 0 °C with vigorous stirring. Subsequently, the reaction mixture was kept and carried out at room temperature for 24 h. After the reaction complete, the precipitated triethylamine hydrochloride was filtered off and the polymer was purified by dialysis against water using a benzoylated cellulose membrane (MWCO 3500 Da) for 2 days. The product was obtained after lyophilizing.Characterization
1H NMR spectra were recorded on a Bruker 400 MHz spectrometer. The chemical shifts are given in parts per million (ppm). 15N NMR spectra were acquired at 25 °C on a Bruker 50.68 MHz spectrometer by using 5–7 kHz spectral width, 4 μs (45°) pulse width, a recycle delay of 4 s, and 938 scans. The data were processed with an exponentially decaying window function with a line broadening factor of 2 Hz. UV-vis spectra were obtained from a Purkinje General (China) T6 UV-Vis Spectrophotometer. The molecular weight and molecular weight distributions were determined by gel permeation chromatography (GPC) equipped with a Viscotek GPC270 system. Freshly distilled N,N-dimethyl formamide was used as an eluent at 35 °C. The flow rate was set to be 0.8 mL min−1. Fluorescence spectra were recorded using a Varian Cary Eclipse photoluminescence spectrometer with a scan rate of 600 nm min−1. Dynamic light scattering (DLS) measurement was performed using the Malvern Nano ZS instrument at different temperatures with a 633 nm He–Ne laser light, and light collection at 90°. The particle size was calculated from the CONTIN method.Turbidity measurement
HCl (4 M) or NaOH (2 M) was used to adjust the pH of the aqueous solution of polymer. Light transmittance of the solution was measured on a temperature-controlled Purkinje General (China) T6 UV-Vis Spectrophotometer at 660 nm, and the heating rate was 0.2 °C/2 min. The cloud-point temperature (Tcp) was taken from the intersection of the maximal slope tangent and the initial horizontal tangent in the resulting transmittance versus temperature curve. The temperature error is ±0.1 °C.Degradation experiment
Light transmittance of the solution of HPA-C4s (5 mg mL−1) was measured on UV-Vis spectrophotometer every 12 seconds at 660 nm, and the temperature was kept at 37 °C during the whole measurement. DTT was added after 3 min. Light transmittance was recorded until the it did not increase any more. After the measurement, molecular weight of the DTT treated HPA-C4s were measured by GPC after the solution was lyophilized and re-dissolved in freshly distilled N,N-dimethyl formamide.Results and discussion
Syntheses of HPAs and their derivatives
HPAs were synthesized from the Michael addition copolymerization of TAEA and two bisacrylamide monomers CBA and HMBA at room temperature (Scheme 1). These two bisacrylamide monomers belong to B2 monomers. TAEA has three primary amines that are initially changed into secondary amines after being reacted with acrylamide groups through Michael addition. Theoretically, the formed secondary amines can further react with acrylamide groups to form the unreactive tertiary amines. It is known that the formed secondary amines are far less reactive than primary amines,44 especially at room temperature, leading that the formed secondary amines have almost no chance to react with the acrylamide groups due to the use of excess primary amines in this polymerization (the ratio of primary amines to acrylamides is 1.65![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Hence TAEA can be regarded as A3 monomer. In each batch of the copolymerizations, the polymerization time, temperature and ratio of TAEA/bisacrylamide are fixed to be 4 days, room temperature and 1.1:1, respectively. The only variable in the copolymerization is the ratio of CBA/HMBA, through which the content of the reducible disulfide units is modulated. Since excess TAEA is used in the copolymerization, the resulting HPAs should be terminated with plenty of primary amine groups (Scheme 1). When the obtained HPAs are directly purified through precipitation without HCl treatment, some insoluble materials are formed during the drying or storing stage. Finally, it is found that HPAs in the HCl salt form can be stably stored. The further modification of HPA with isobutyric anhydride results in IBAm terminated HPA (HPA-C4).
:1). Hence TAEA can be regarded as A3 monomer. In each batch of the copolymerizations, the polymerization time, temperature and ratio of TAEA/bisacrylamide are fixed to be 4 days, room temperature and 1.1:1, respectively. The only variable in the copolymerization is the ratio of CBA/HMBA, through which the content of the reducible disulfide units is modulated. Since excess TAEA is used in the copolymerization, the resulting HPAs should be terminated with plenty of primary amine groups (Scheme 1). When the obtained HPAs are directly purified through precipitation without HCl treatment, some insoluble materials are formed during the drying or storing stage. Finally, it is found that HPAs in the HCl salt form can be stably stored. The further modification of HPA with isobutyric anhydride results in IBAm terminated HPA (HPA-C4).
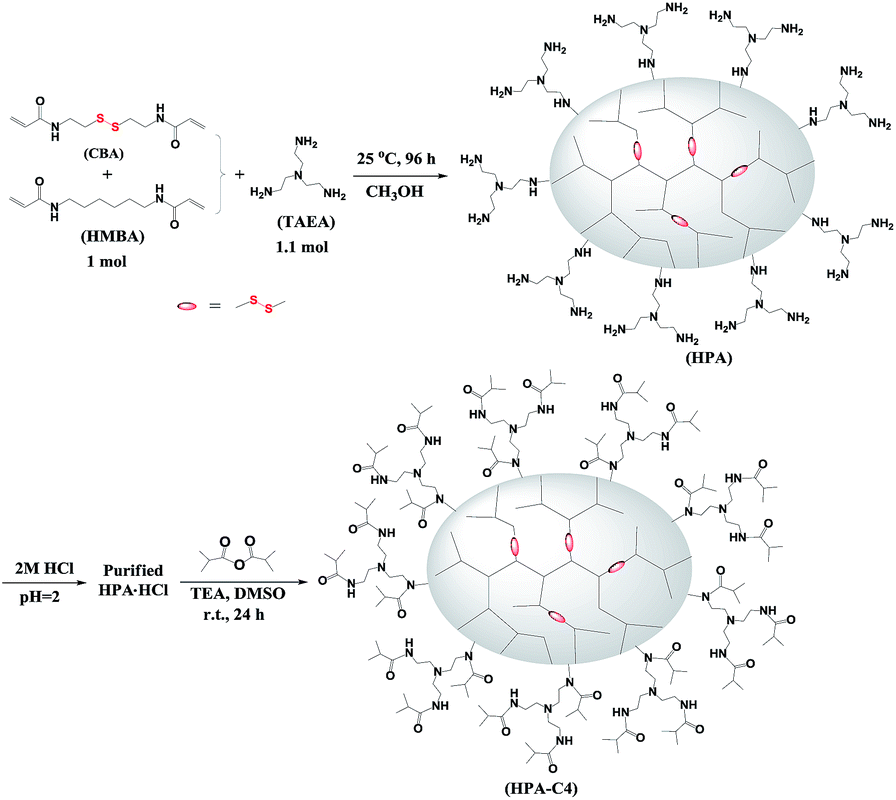 | ||
| Scheme 1 Synthetic procedure for HPA-C4. | ||
NMR characterizations
The obtained HPAs with different ratio of CBA/HMBA were characterized by 1H NMR spectrometry (Fig. 1). In the range of 6–7 ppm no signals coming from the unreacted double bonds can be observed (ESI, Fig. S1†), indicating that the obtained HPAs are terminated with amine groups just as designed. In the 1H NMR spectra, the signals coming from CBA, HMBA and TAEA units can be observed below 4 ppm, however, the partial overlapping of some signals makes it difficult to assign all the signals in detail (ESI, Fig. S2†). As for CBA and HMBA, typical signals that are separated well from others exist, such as the methylene units connected with amide groups of CBA units (CONH–C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) 2–CH2–S–S–CH2–C2–NHCO, around 3.51 ppm), the signals of HMBA located at 1.47 and 1.29 ppm (CONH–CH2–
2–CH2–S–S–CH2–C2–NHCO, around 3.51 ppm), the signals of HMBA located at 1.47 and 1.29 ppm (CONH–CH2–![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) 2–2–2–2–CH2–NHCO). Comparing the integrals of these typical signals, the ratio of CBA/HMBA units in the obtained HPAs can be calculated. In this work we prepared five HPA samples. HPA-S100 does not contain HMBA units, while HPA-S0 does not contain CBA units. The other three samples, HPA-S75, HPA-S50 and HPA-S25 contain both CBA and HMBA units, but the ratios of CBA/HMBA are different and the results are listed in Table 1. It is clear that the ratios of CBA/HMBA in polymers are similar or a little higher than the corresponding feed ratios. Moreover, minus the integrals coming from the CBA and HMBA units in the range of 2.6–3.4 ppm, the integrals of TAEA units can be obtained; hence the final molar contents of these three units are deduced and the results are also listed in Table 1. However, the direct interpretation of the type of amine groups from the 1H NMR spectra of HPAs is impossible.
2–2–2–2–CH2–NHCO). Comparing the integrals of these typical signals, the ratio of CBA/HMBA units in the obtained HPAs can be calculated. In this work we prepared five HPA samples. HPA-S100 does not contain HMBA units, while HPA-S0 does not contain CBA units. The other three samples, HPA-S75, HPA-S50 and HPA-S25 contain both CBA and HMBA units, but the ratios of CBA/HMBA are different and the results are listed in Table 1. It is clear that the ratios of CBA/HMBA in polymers are similar or a little higher than the corresponding feed ratios. Moreover, minus the integrals coming from the CBA and HMBA units in the range of 2.6–3.4 ppm, the integrals of TAEA units can be obtained; hence the final molar contents of these three units are deduced and the results are also listed in Table 1. However, the direct interpretation of the type of amine groups from the 1H NMR spectra of HPAs is impossible.
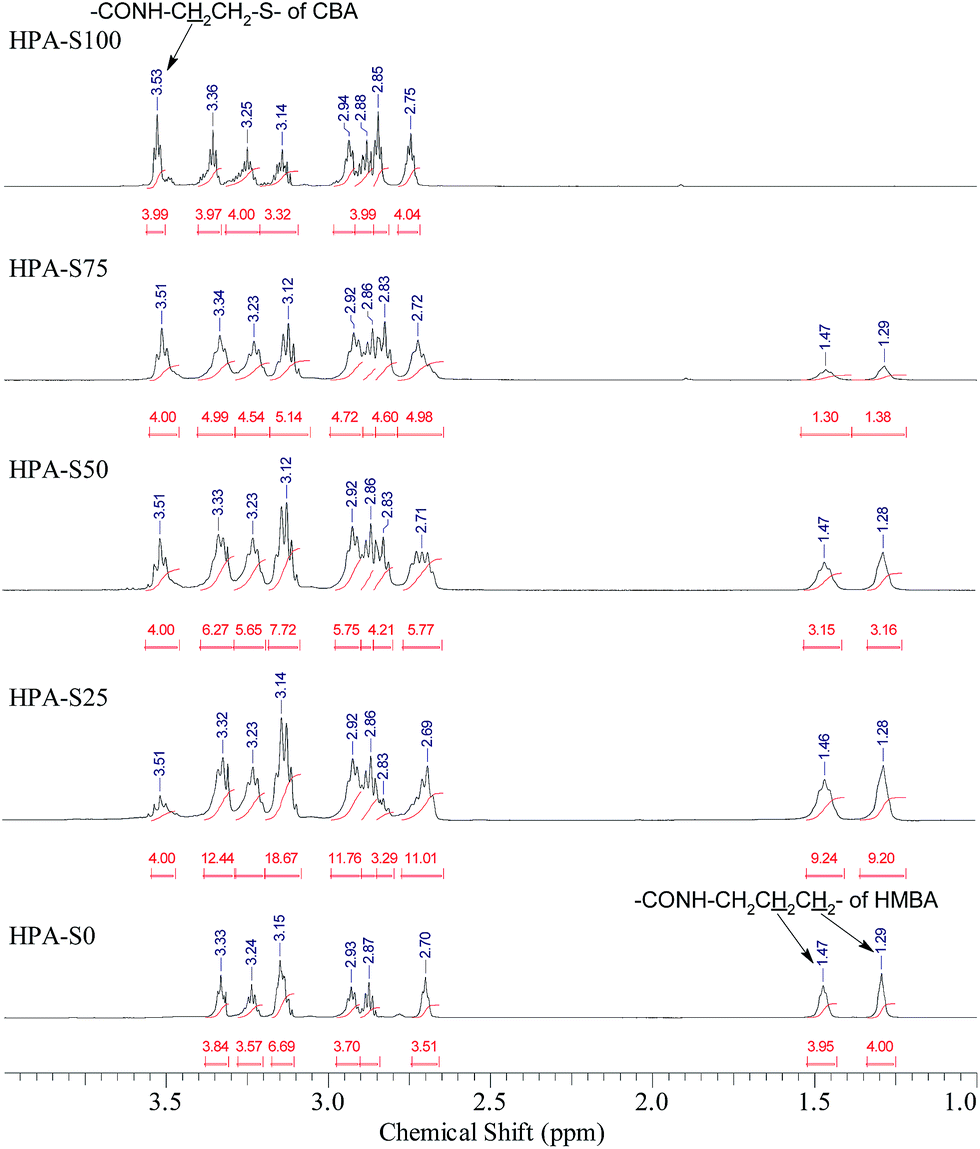 | ||
| Fig. 1 1H NMR spectra of HPA HCl. | ||
| Polymer | Feed ratio of CBA/HMBA | [CBA]/[HMBA] in polymer | Content of CBA (mol%) | Content of HMBA (mol%) | Content of TAEA (mol%) |
|---|---|---|---|---|---|
| HPA-S100 | 100:0 |
100:0 |
49.1 | 0 | 50.9 |
| HPA-S75 | 75:25 |
74.9:25.1 |
37.0 | 12.4 | 50.6 |
| HPA-S50 | 50:50 |
55.9:44.1 |
30.4 | 24.0 | 45.6 |
| HPA-S25 | 25:75 |
30.3:69.7 |
15.6 | 36.0 | 48.4 |
| HPA-S0 | 0:100 |
0:100 |
0 | 52.5 | 47.5 |
All the three units of the prepared HPAs contain nitrogen elements, thus 15N NMR spectrometry was employed not only to verify the structural information interpreted from 1H NMR spectra, but also to acquire additional structural information (Fig. 2). The signals coming from the amide nitrogens of CBA and HMBA units can be observed clearly at ca. −271 and −268 ppm, respectively, while those coming from the secondary, tertiary and primary amines of TAEA units can be found at ca. −354, −362 and −369 ppm, respectively. Since these signals are separated well from each other, all the structural information interpreted from the 1H NMR spectra can be also obtained from 15N NMR spectra (Table 2). Comparing the data in Table 1 with those in Table 2, it is clear that the difference of the data interpreted from 1H NMR and 15N NMR spectra is minor. From 15N NMR spectra the ratio of primary, secondary and tertiary amines can be deduced, which cannot be interpreted from 1H NMR spectra.
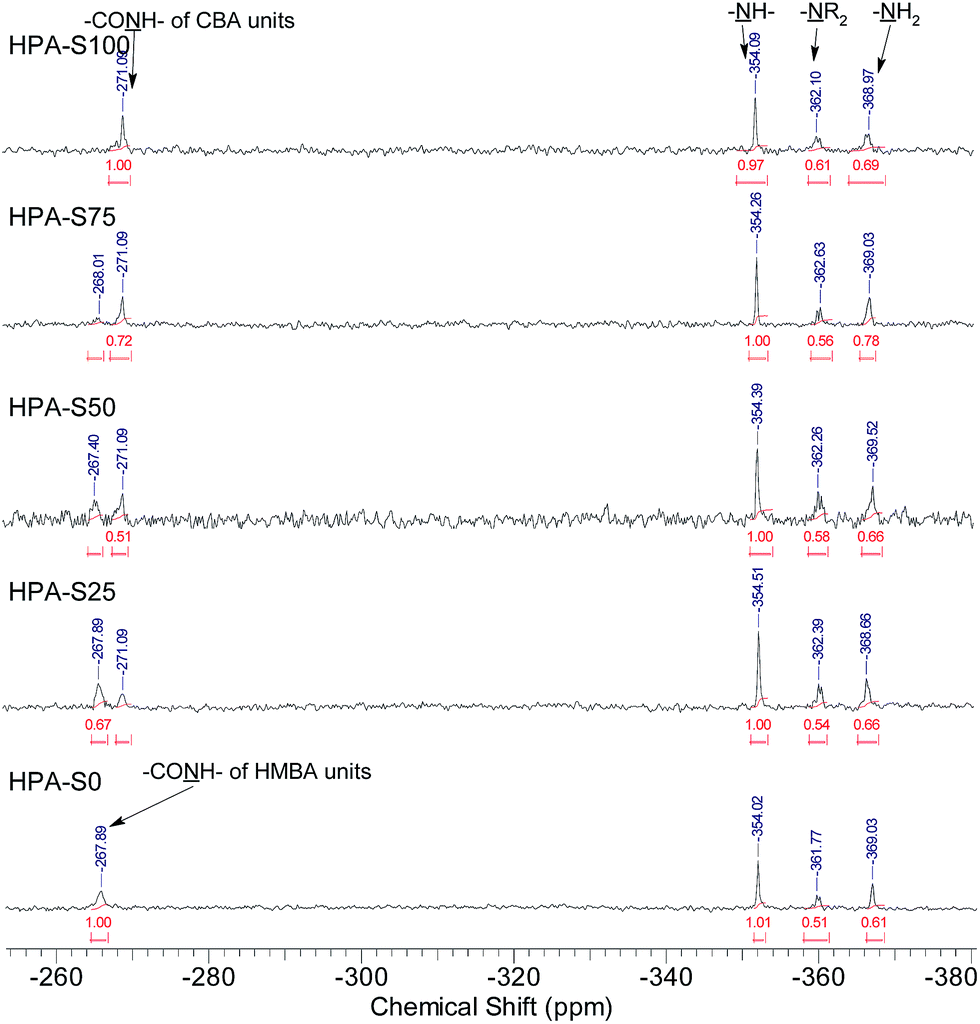 | ||
| Fig. 2 15N NMR spectra of HPAs. | ||
| Polymer | Feed ratio of CBA/HMBA | [CBA]/[HMBA] in polymer | Content of CBA (mol%) | Content of HMBA (mol%) | Content of TAEA (mol%) | [–NR2]:[–NH–]:[–NH2] |
|---|---|---|---|---|---|---|
| HPA-S100 | 100:0 |
100:0 |
46.8 | 0 | 53.2 | 1:1.59:1.13 |
| HPA-S75 | 75:25 |
71.9:28.1 |
33.5 | 13.1 | 53.4 | 1:1.79:1.39 |
| HPA-S50 | 50:50 |
52.6:47.4 |
24.4 | 22.0 | 53.6 | 1:1.72:1.14 |
| HPA-S25 | 25:75 |
33.0:67.0 |
15.7 | 31.9 | 52.4 | 1:1.85:1.22 |
| HPA-S0 | 0:100 |
0:100 |
0 | 48.4 | 51.6 | 1:1.98:1.20 |
The HPA derivatives, HPA-C4s were characterized by 1H NMR spectrometry (Fig. 3 and ESI, Fig. S3†). Compared with the 1H NMR spectra of HPA precursors, the new broad signal at around 1.03 ppm shown in Fig. 3 is assigned as the methyl protons of C4 moieties, which is a strong evidence of the successful attachment of C4 groups to the HPAs. The signal of methine proton of C4 moieties is located at around 2.5 ppm, which are overlapped partially with the HPA scaffold. The methylene protons of HPA skeleton connected directly to C4 groups are located in the range of 3.0–3.9 ppm, which are overlapped partially with the other signals of HPA scaffold. It is impossible to learn the information on the conversion of the amino terminals of HPAs into the C4 groups from 1H NMR spectra.
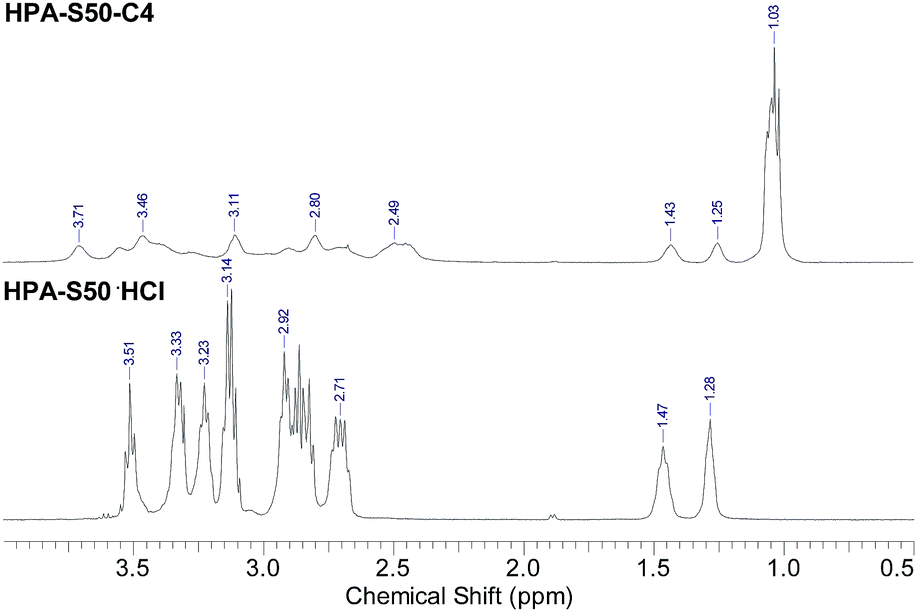 | ||
| Fig. 3 Comparison of the 1H NMR spectra of HPAs and HPA-C4s (HPA-S50 and HPA-S50–C4 as the representatives). | ||
HPA-C4s were further characterized by 15N NMR spectrometry (Fig. 4 and ESI, Fig. S4†). Compared with the 15N NMR spectra of HPAs, it is clear that only the signals coming from the amide and tertiary amine nitrogens can be observed in the 15N NMR spectra of HPA-C4s. Beyond the amide-nitrogen signals of HPAs, isobutyramide-nitrogen signals appear. When the integrals of tertiary amine nitrogens are kept to be the same, the incremental integral value of amide nitrogens in the 15N NMR spectra of HPA-C4s is similar to the integral values of primary and secondary amine nitrogens in the 15N NMR spectra of HPAs. Therefore, it can be deduced that under the modification condition adopted here, nearly all the reactive primary and secondary amines of HPAs are transformed into C4 groups.
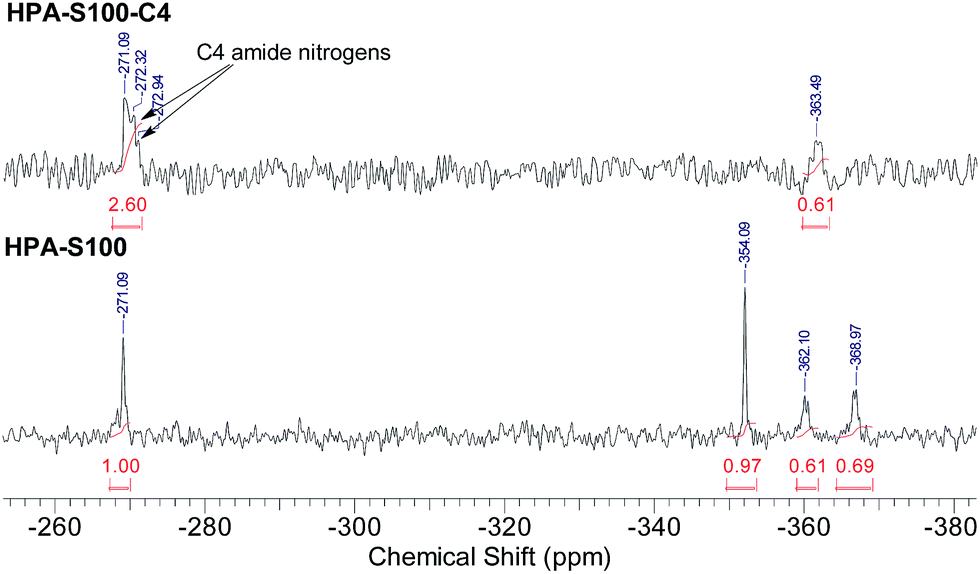 | ||
| Fig. 4 Comparison of the 15N NMR spectra of HPAs and HPA-C4s (HPA-S100 and HPA-S100–C4 as the representatives). | ||
GPC characterizations
All the HPA and HPA-C4 samples are measured by GPC. No signal can be observed in the GPC diagrams of HPA samples, which is ascribed to the strong hydrogen-bonding interaction between the amine end groups of HPAs and the GPC columns. As for HPA-C4 samples, strong signals can be observed in GPC diagrams (ESI, Fig. S5†), which is because the amine end groups that have strong interaction with GPC columns have been transformed into the C4 groups. From GPC the molecular weight of PDI values of all the HPA-C4s are deduced and the results are listed in Table 3.| Polymer | Mn/103 | Mw/103 | PDI |
|---|---|---|---|
| HPA-S100–C4 | 4.70 | 11.5 | 2.45 |
| HPA-S75–C4 | 4.12 | 10.5 | 2.56 |
| HPA-S50–C4 | 5.09 | 15.5 | 3.04 |
| HPA-S25–C4 | 5.25 | 15.3 | 2.92 |
| HPA-S0–C4 | 5.27 | 26.6 | 5.04 |
Disulfide bonds can be reduced into thiol groups in the presence of reducing agent, such as DTT. When disulfide bonds are located in the backbone of polymer, the reduction will lead to the disruption of polymer into small segments. Except of HPA-S0–C4, the other HPA-C4s contain different amount of disulfide bonds in the backbone. Whether DTT can effectively degrade these HPA-C4s through reducing and cleaving the disulfide bonds are characterized by GPC (Fig. 5). It is clear that all the HPA-C4s containing disulfide bonds degrade into lower molecular weight segments after being treated with enough DTT and the molecular weights of these segments are normally less than 1000 Da for the samples with higher amount of disulfide bonds. The HPA-S0–C4 without disulfide bonds cannot degrade.
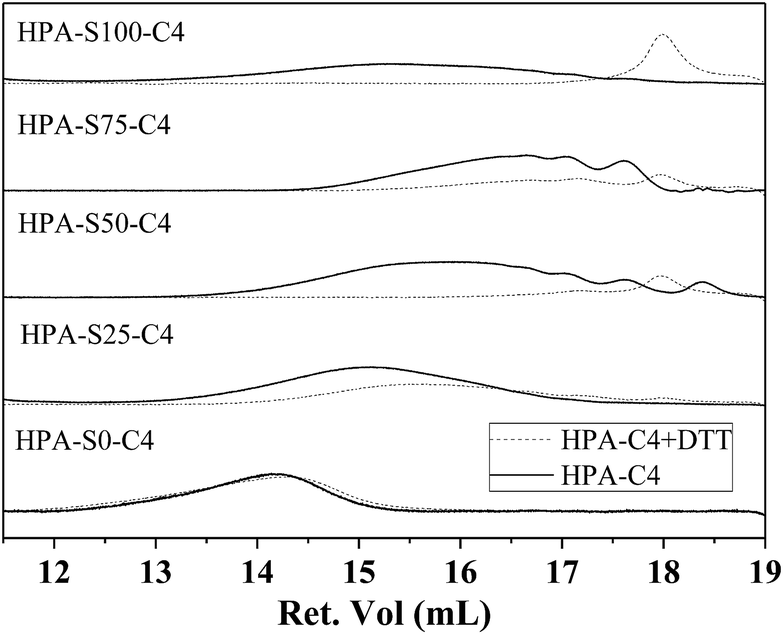 | ||
| Fig. 5 GPC diagrams of HPA-C4s before and after being treated with DTT. | ||
Thermoresponsive property in water
The obtained HPAs and HPA-C4s are soluble in water. The aqueous solutions of HPAs were always transparent at any temperature. However, the aqueous solutions of HPA-C4s became turbid after being heated above certain temperatures, and went transparent again when they were cooled down. This indicated that the obtained HPA-C4s in water were thermoresponsive, whereas the unmodified HPAs were not. It is well-known that polymers exhibit thermoresponsive properties in water only when the ratio of hydrophilic to hydrophobic units lies in a specific range, and the more hydrophilic one shows a higher LCST than the more hydrophobic counterpart.3 HPAs are too hydrophilic to exhibit thermoresponsive property in water. The introduction of hydrophobic C4 groups reduces the hydrophilicity of polymers, thus favoring the occurrence of thermoresponsive property in water.Turbidimetry was adopted to measure the thermoresponsive behavior of these HPA-C4s in water. Obvious phase transition can be observed in both the heating and cooling processes (Fig. 6A). Furthermore, the heating and cooling cycles do not overlap and the obvious hysteresis can be attributed to the strong hydrogen-bonds among macromolecules.45 The phase transition temperature, here called as the cloud point temperature (Tcp), is taken from the intersection of the maximal slope tangent and the initial horizontal tangent in the heating phase transition curve. Fig. 6B shows the concentration dependence of the Tcp of these five HPA-C4 samples. Initially, the Tcp value decreases with the increase of polymer concentration, but varies insignificantly when the concentration is above 5 mg mL−1. The Tcp values of these HPA-C4 samples at the concentration of 5 mg mL−1 are compared, and from Fig. 6C it can be seen that HPA-C4 with a higher CBA content shows a lower Tcp value. It is known that increasing the hydrophobicity of thermoresponsive polymers decreases the phase transition temperature, whereas increasing the hydrophilicity increases it.46 The above phenomenon indicates that disulfide units might be more hydrophobic than the ethylene units.
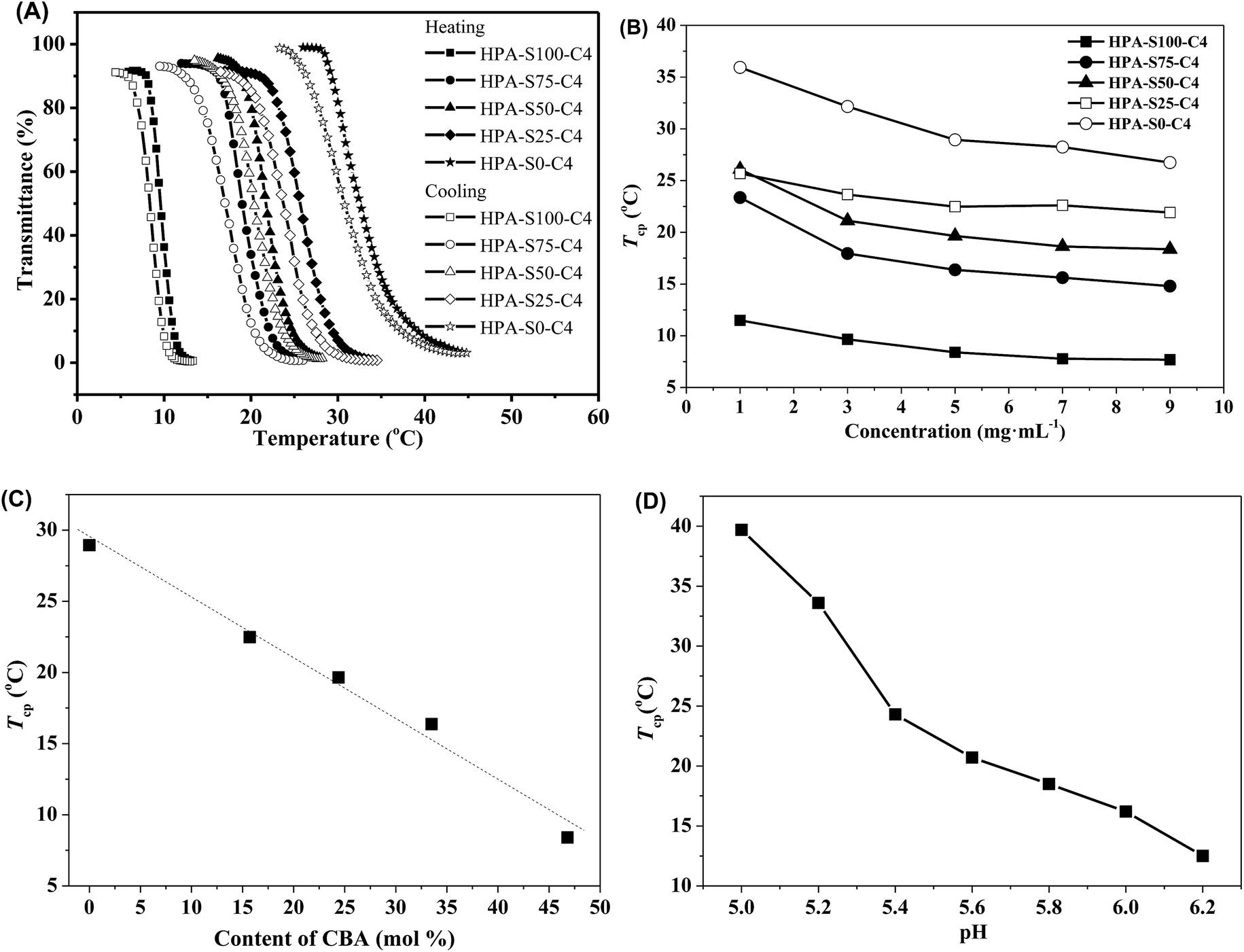 | ||
| Fig. 6 (A) Typical temperature-dependent light transmittance of the aqueous solutions of HPA-C4s (concentration of polymer is 5 mg mL−1); (B) effect of the concentration of HPA-C4s in de-ionized water on the Tcp; (C) effect of the CBA content in HPA-C4s on the Tcp (concentration of polymer is 5 mg mL−1) and (D) effect of pH on the Tcp (HPA-S100–C4 as the representative). | ||
Fig. 6D shows that the thermoresponsive property of HPA-C4 is pH-sensitive. In the pH range of 5.0 to 6.2, the Tcp of HPA-C4 increases markedly with lowering the pH. Above pH 6.2, the Tcp is so low that it is difficult to measure the phase transition. Below pH 5.0, the Tcp is too high to be measured. This phenomenon can be explained as follows: the less polar amine groups can be transformed into the more polar ammonium groups in acidic condition. The lower the pH is, the more the ammonium groups are generated, thus the higher the polarity of HPA-C4 is. This leads to the significant increase of Tcp of HPA-C4 at more acidic condition.
DLS was used to monitor the size variation of HPA-C4s during the phase transition (ESI, Fig. S6†). HPA-S25–C4 was used as the polymer representative. Below the transition temperature, the average diameter of HPA-S25–C4 in water is around 80 nm, indicating the existence of stable small HPA-S25–C4 aggregates. Above the phase transition temperature, large aggregates quickly form.
The influence of DTT on the thermoresponsive property of HPA-C4s in water is further studied. Temperature is kept above the Tcp of the aqueous solutions of HPA-C4s, thus all the solutions are turbid. The aqueous solution of HPA-S0–C4 that has no disulfide bond is always turbid after a maximal 5 mg of DTT is added after 140 min (Fig. 7). As for the HPA-C4s that contain disulfide bonds, the addition of DTT leads to the gradual increase of the solution's transmittance with the time (Fig. 7). When enough DTT is added, the turbid solutions become transparent after certain time. These phenomena indicate that DTT can only influence the thermoresponsive property of HPA-C4s containing disulfide bonds. When the same amount of DTT is added, the solution of HPA-C4 with more reducible disulfide bonds becomes transparent faster. The influence of DTT on the thermoresponsive property of HPA-C4s with disulfide bonds can be explained as follows: disulfide bonds can be reduced into thiol groups by DTT. Since disulfide bonds are located in the backbone of HPA-C4s, the reduction by DTT will lead to the disruption of HPA-C4s into small segments, which has been verified by the GPC measurements (Fig. 5). It has been reported that the phase transition temperature of thermoresponsive dendrimers and hyperbranched polymers is sensitive to molecular weight alteration of polymer, and the dendritic polymer with a lower molecular weight normally has a higher phase transition temperature.11,15 The degradation of HPA-C4s by DTT results in a severe decrease of molecular weight, thus a pronounced increase of Tcp occurs, embodied with the increase of the solution's transmittance with the time when the temperature is fixed.
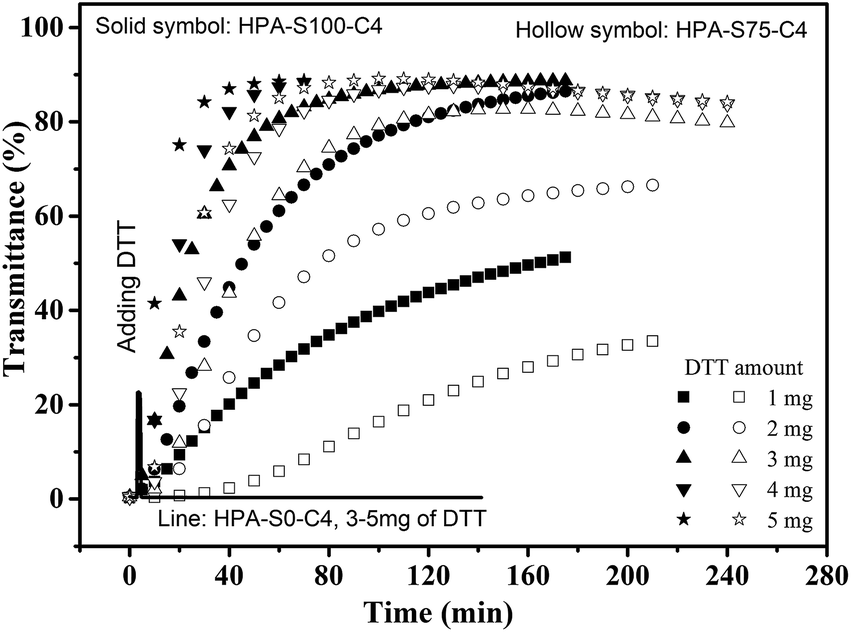 | ||
| Fig. 7 Time-dependent transmittance of the aqueous solutions of HPA-S100–C4, HPA-S75–C4 and HPA-S0–C4 upon the addition of different amount of DTT (polymer concentration is 5 mg mL−1, 37 °C). | ||
Photoluminescence property of HPA-C4s
It is found that the aqueous solutions of these HPA-C4s can emit blue light under the UV irradiation; therefore, the photoluminescence properties of HPA-C4s are studied. HPA-S100–C4, HPA-S50–C4 and HPA-S0–C4 are used as the representatives of these polymers. All these HPA-C4s show an excitation peak centered at ca. 370 nm and an obvious fluorescence emission centered at ca. 455 nm (Fig. 8A). Their fluorescence intensities increase with raising the polymer concentration (Fig. 8B and ESI, Fig. S7†). Because of restrict of polymer solubility in water, the fluorescence at higher polymer concentration is not acquired.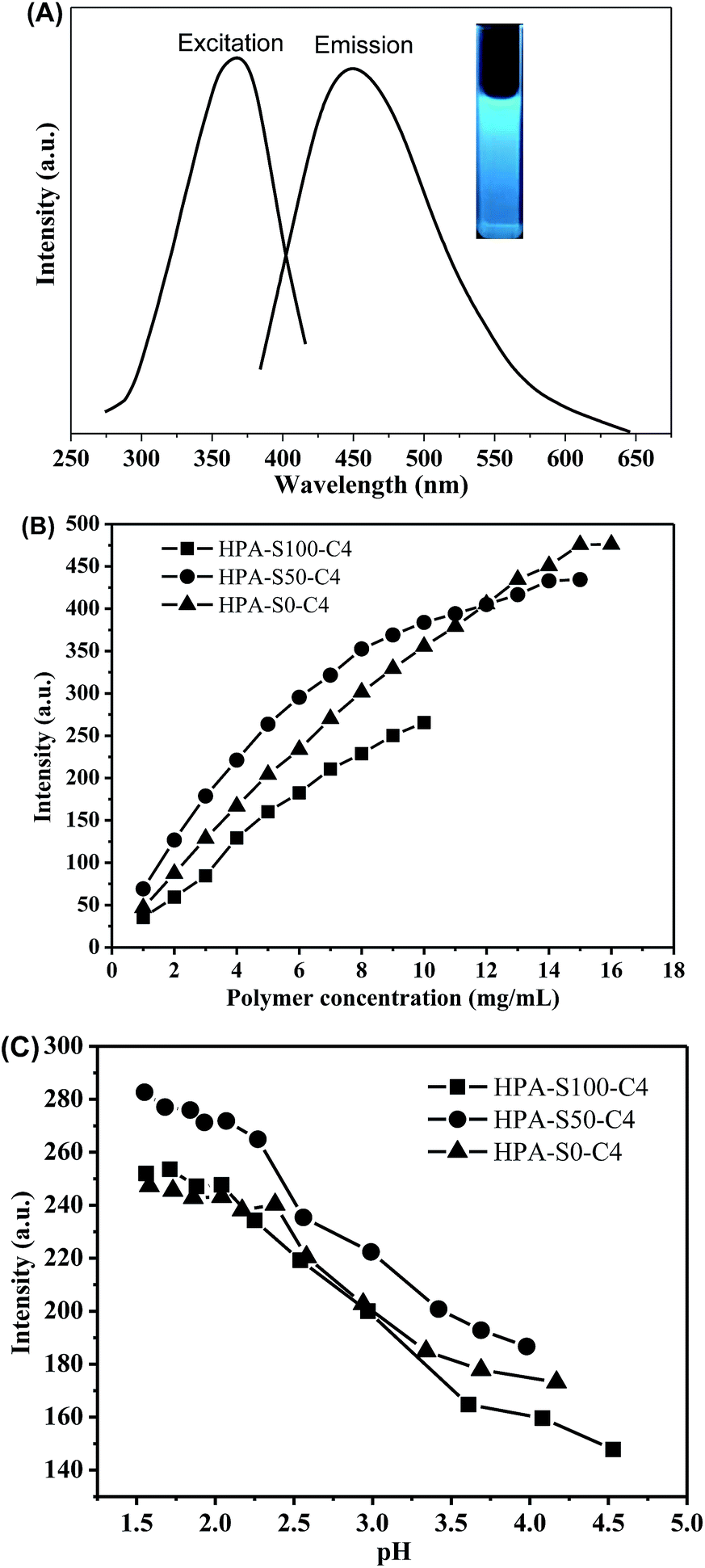 | ||
| Fig. 8 (A) Typical fluorescence excitation and emission spectra of HPA-C4s in water; (B) influence of polymer concentration on fluorescence emission intensity and (C) influence of pH on fluorescence emission intensity of HPA-C4s (polymer concentration is 5 mg mL−1; excitation wavelength is 370 nm; slit widths of excitation and emission are set to be 10 nm and 10 nm, respectively). | ||
The pH influence on fluorescence of these HPA-C4s is further studied. It is found that the variation of pH has almost no influence on the maximal excitation and emission wavelength of HPA-C4s (ESI, Fig. S8†), indicating that the type of fluorophore in HPA-C4s is not affected by pH. However, the fluorescence intensity is sensitive to the pH variation (Fig. 8C). Initially the fluorescence intensity of HPA-C4s can be effectively enhanced through decreasing the pH, but it is almost leveled off when pH is reduced below 2. This pH dependent fluorescent property can be tentatively explained as follows: in HPA-C4s, tertiary amines and luminogens coexist. Tertiary amine is well known as a fluorescence quencher. Adjusting pH to the acidic condition transforms tertiary amine into ammonium salt, which reduces the number of fluorescence quenchers, resulting in the strengthened emission in acidic condition. Moreover, in acidic condition, HPA-C4s becomes more rigid due to the strong charge–charge repulsion among the quaternized tertiary amines in the core, which decreases the non-emission energy transfer. From Fig. 6D it is known that HPA-C4s still keep their thermoresponsive property in water at pH 5.0, but lose it below pH 5.0. Thus, the quantum yields of all these five HPA-C4s are measured at pH 5. Using quinine sulfate as a reference, the PL quantum yields of all these five HPA-C4s are found to be similar, which is in the range of 10–14% (ESI, Fig. S9†).
HPA-C4s do not contain any traditional fluorophore, but bear many tertiary amine and amide groups. The photoluminescence of tertiary aliphatic amines have been extensively studied and strong fluorescence has been observed only in vapor phase.47 Recently, it was addressed that the tertiary amine moiety located in the branching points of hyperbranched poly(amine-ester) could display relatively strong fluorescence.48 However, this conclusion cannot be commonly applied in all the dendritic polymer systems with the N-branching moieties since certain polymers with the N-branching moieties, such as hyperbranched polyethylenimine and PAMAM dendrimer, emit weakly without special treatment.49–51 Moreover, it is known that HPA-C4s has the same type fluorophore at different pH, whereas pH 2 can change nearly all the amine groups of HPEI into ammonium salt.52 Thus, it can be deduced that tertiary amines in the branching point are not the real deciding functional groups contributing to the fluorescence of HPA-C4s. It is known that tertiary amine can be oxidized into tertiary amine oxide (![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N → O) that can contribute to the fluorescence.53 To verify whether tertiary amine oxide is the luminogen, all these HPA-C4s are oxidized by air at 95 °C for different time and their fluorescence properties are measured (Fig. 9 and ESI, Fig. S10†). With the increase of oxidation time, the maximal excitation and emission wavelength do not vary, indicating that HPA-C4s before and after oxidation have the same fluorophore. Oxidation by air leads to the strengthened emission intensity that reaches the maximum around 6–8 h at 95 °C. As for HPG, oxidation has no influence on its UV-vis and fluorescence spectra. These oxidation experiments verify that the luminogen of HPA-C4s is the tertiary amine oxide, not tertiary amine and oxidation increases the number of tertiary amine oxides in HPA-C4s.
N → O) that can contribute to the fluorescence.53 To verify whether tertiary amine oxide is the luminogen, all these HPA-C4s are oxidized by air at 95 °C for different time and their fluorescence properties are measured (Fig. 9 and ESI, Fig. S10†). With the increase of oxidation time, the maximal excitation and emission wavelength do not vary, indicating that HPA-C4s before and after oxidation have the same fluorophore. Oxidation by air leads to the strengthened emission intensity that reaches the maximum around 6–8 h at 95 °C. As for HPG, oxidation has no influence on its UV-vis and fluorescence spectra. These oxidation experiments verify that the luminogen of HPA-C4s is the tertiary amine oxide, not tertiary amine and oxidation increases the number of tertiary amine oxides in HPA-C4s.
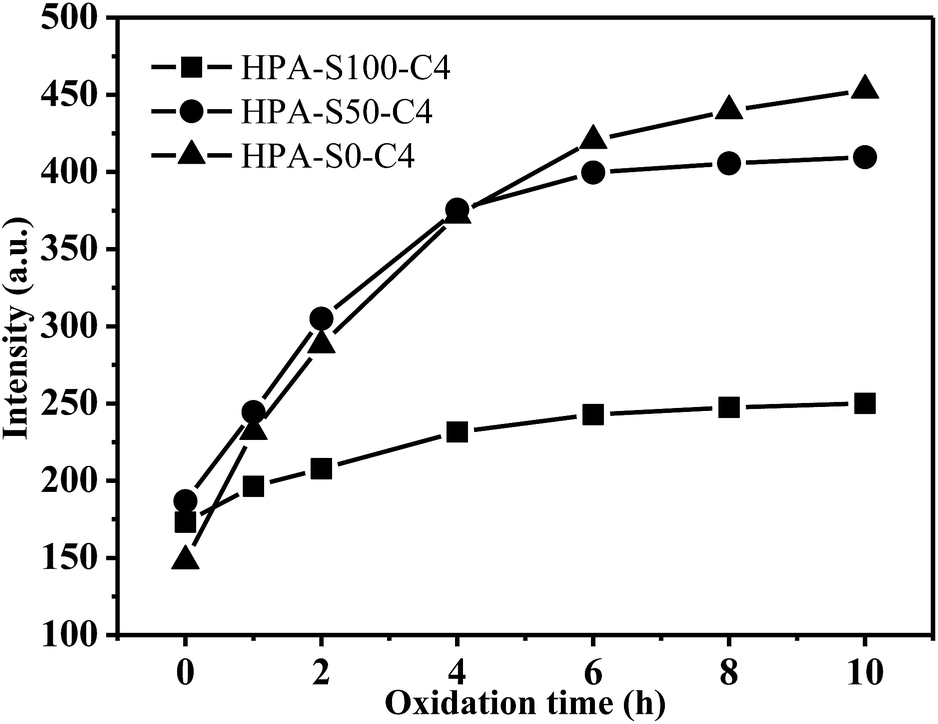 | ||
| Fig. 9 Influence of oxidation time on fluorescence emission intensity of HPA-C4s (polymer concentration is 5 mg mL−1; excitation wavelength is 370 nm; slit widths of excitation and emission are set to be 10 nm and 10 nm, respectively). | ||
Conclusions
Stimuli-responsive HPA-C4s integrated with thermal and pH sensitivity, reducible degradability and intrinsic photoluminescence were successfully prepared through the Michael addition copolymerization of TAEA and two bisacrylamide monomers CBA and HMBA and the subsequent modification with isobutyric anhydride. 15N NMR spectrometry was a more useful tool than 1H NMR spectrometry to determine the structural information of the obtained polymers. Those HPA-C4s with disulfide bonds were degradable upon being treated with reducing agent, such as DTT. HPA-C4s exhibited thermoresponsive property in water and their Tcp values were pH-dependent. Moreover, the thermoresponsive property of HPA-C4s with disulfide bonds could be also influenced by DTT due to polymer degradation. HPA-C4s without the traditional fluorophores could emit blue fluorescence centered at ca. 455 nm. The fluorescence intensity could be enhanced through lowering the pH and oxidizing the polymer by air. The luminogen of HPA-C4s was assumed to be tertiary amine oxide.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (20804027 and 21274106).Notes and references
- I. Y. Galaev and B. Mattiasson, Trends Biotechnol., 1999, 17, 335–340 CrossRef CAS PubMed.
- H. J. Moon, D. Y. Ko, M. H. Park, M. K. Joo and B. Jeong, Chem. Soc. Rev., 2012, 41, 4860–4883 RSC.
- C. Weber, R. Hoogenboom and U. S. Schubert, Prog. Polym. Sci., 2012, 37, 686–714 CrossRef CAS.
- C. D. l. H. Alarcón, S. Pennadam and C. Alexander, Chem. Soc. Rev., 2005, 34, 276–285 RSC.
- B. Wang, H.-J. Liu, T.-T. Jiang, Q.-H. Li and Y. Chen, Polymer, 2014, 55, 6036–6043 CrossRef CAS.
- T. Yoshida, T. Aoyagi, E. Kokufuta and T. Okano, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 779–787 CrossRef CAS.
- M. Kimura, M. Kato, T. Muto, K. Hanabusa and H. Shirai, Macromolecules, 2000, 33, 1117–1119 CrossRef CAS.
- J. Xu, S. Luo, W. Shi and S. Liu, Langmuir, 2006, 22, 989–997 CrossRef CAS PubMed.
- D. A. Z. Wever, E. Riemsma, F. Picchioni and A. A. Broekhuis, Polymer, 2013, 54, 5456–5466 CrossRef CAS.
- M. Nakayama and T. Okano, Biomacromolecules, 2005, 6, 2320–2327 CrossRef CAS PubMed.
- Y. Haba, A. Harada, T. Takagishi and K. Kono, J. Am. Chem. Soc., 2004, 126, 12760–12761 CrossRef CAS PubMed.
- S. V. Aathimanikandan, E. N. Savariar and S. Thayumanavan, J. Am. Chem. Soc., 2005, 127, 14922–14929 CrossRef CAS PubMed.
- Z. Jia, H. Chen, X. Zhu and D. Yan, J. Am. Chem. Soc., 2006, 128, 8144–8145 CrossRef CAS PubMed.
- Y. Haba, C. Kojima, A. Harada and K. Kono, Macromolecules, 2006, 39, 7451–7453 CrossRef CAS.
- H. Liu, Y. Chen and Z. Shen, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1177–1184 CrossRef CAS.
- Y. Shen, M. Kuang, Z. Shen, J. Nieberle, H. Duan and H. Frey, Angew. Chem., Int. Ed., 2008, 47, 2227–2230 CrossRef CAS PubMed.
- M. Schömer, J. Seiwert and H. Frey, ACS Macro Lett., 2012, 1, 888–891 CrossRef.
- R.-C. Wang, X.-B. Fu, X. Liu, H.-J. Liu, Y. Chen and J. Cui, RSC Adv., 2013, 3, 17016–17020 RSC.
- X. Tao, K. Liu, W. Li and A. Zhang, Polymer, 2014, 55, 3672–3679 CrossRef CAS.
- H.-J. Liu, R.-H. Dong and Y. Chen, Chin. J. Polym. Sci., 2014, 32, 961–968 CrossRef CAS.
- H.-W. Qin, H.-J. Liu and Y. Chen, Chin. J. Polym. Sci., 2014, 32, 1338–1347 CrossRef CAS.
- Y. Liu, W. Li, L. Hou and P. Wu, RSC Adv., 2014, 4, 24263–24271 RSC.
- J.-G. Tong, Z.-Y. Wei, H.-L. Yang, Z.-Y. Yang and Y. Chen, Polymer, 2016, 84, 107–116 CrossRef CAS.
- C. Wu and X. H. Wang, Phys. Rev. Lett., 1998, 80, 4092–4094 CrossRef CAS.
- X. H. Wang, X. P. Qiu and C. Wu, Macromolecules, 1998, 31, 2972–2976 CrossRef CAS.
- Y. Haba, C. Kojima, A. Harada and K. Kono, Angew. Chem., Int. Ed., 2007, 46, 234–237 CrossRef CAS PubMed.
- X.-R. Mu, J.-G. Tong, Y. Liu, X.-Y. Liu, H.-J. Liu and Y. Chen, Polymer, 2013, 54, 2341–2346 CrossRef CAS.
- X.-Y. Liu, X.-R. Mu, Y. Liu, H.-J. Liu, Y. Chen, F. Cheng and S.-C. Jiang, Langmuir, 2012, 28, 4867–4876 CrossRef CAS PubMed.
- X. Liu, F. Cheng, H. Liu and Y. Chen, Soft Matter, 2008, 4, 1991–1994 RSC.
- Y. Liu, X.-Y. Liu, H.-J. Liu, F. Cheng and Y. Chen, Macromol. Res., 2012, 20, 578–584 CrossRef CAS.
- B. Wang, H. J. Liu, X. B. Fu, Y. Yao and Y. Chen, Chem.–Asian J., 2015, 10, 1690–1697 CrossRef CAS PubMed.
- J. Zhang, H.-J. Liu, Y. Yuan, S. Jiang, Y. Yao and Y. Chen, ACS Macro Lett., 2013, 2, 67–71 CrossRef CAS.
- N. Pérignon, J.-D. Marty, A.-F. Mingotaud, M. Dumont, I. Rico-Lattes and C. Mingotaud, Macromolecules, 2007, 40, 3034–3041 CrossRef.
- N. Pérignon, A.-F. Mingotaud, J.-D. Marty, I. Rico-Lattes and C. Mingotaud, Chem. Mater., 2004, 16, 4856–4858 CrossRef.
- Y. Ping, D. Wu, J. N. Kumar, W. Cheng, C. L. Lay and Y. Liu, Biomacromolecules, 2013, 14, 2083–2094 CrossRef CAS PubMed.
- J. Chen, C. Wu and D. Oupický, Biomacromolecules, 2009, 10, 2921–2927 CrossRef CAS PubMed.
- P. R. Dvornic, J. Hu, D. J. Meier, R. M. Nowak and P. L. Parham, Hyperbranched polyurea, polyurethane, polyamidoamine, polyamide or polyester, US6534600–B2, 2002.
- W. Yang and C. Y. Pan, Macromol. Rapid Commun., 2009, 30, 2096–2101 CrossRef CAS PubMed.
- G. Yu, S. W. Yin, Y. Q. Liu, J. S. Chen, X. J. Xu, X. B. Sun, D. G. Ma, X. W. Zhan, Q. Peng, Z. G. Shuai, B. Z. Tang, D. B. Zhu, W. H. Fang and Y. Luo, J. Am. Chem. Soc., 2005, 127, 6335–6346 CrossRef CAS PubMed.
- G. Jiang, X. Sun, Y. Wang and M. Ding, Polym. Chem., 2010, 1, 1644–1649 RSC.
- D. Wang, Z.-Q. Yu, C.-Y. Hong and Y.-Z. You, Eur. Polym. J., 2013, 49, 4189–4194 CrossRef CAS.
- R.-B. Wang, W.-Z. Yuan and X.-Y. Zhu, Chin. J. Polym. Sci., 2015, 33, 680–687 CrossRef CAS.
- E. Emilitri, E. Ranucci and P. Ferruti, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 1404–1416 CrossRef CAS.
- Y.-Z. You, C.-Y. Hong and C.-Y. Pan, Macromolecules, 2009, 42, 573–575 CrossRef CAS.
- H. Wang, S. Sun and P. Wu, J. Phys. Chem. B, 2011, 115, 8832–8844 CrossRef CAS PubMed.
- M. M. Bloksma, D. J. Bakker, C. Weber, R. Hoogenboom and U. S. Schubert, Macromol. Rapid Commun., 2010, 31, 724–728 CrossRef CAS PubMed.
- A. M. Halpern and T. Gartman, J. Am. Chem. Soc., 1974, 96, 1393–1398 CrossRef CAS.
- M. Sun, C. Y. Hong and C. Y. Pan, J. Am. Chem. Soc., 2012, 134, 20581–20584 CrossRef CAS PubMed.
- W. I. Lee, Y. Bae and A. J. Bard, J. Am. Chem. Soc., 2004, 126, 8358–8359 CrossRef CAS PubMed.
- L. Pastor-Pérez, Y. Chen, Z. Shen, A. Lahoz and S.-E. Stiriba, Macromol. Rapid Commun., 2007, 28, 1404–1409 CrossRef.
- S. G. Liu, N. Li, Y. Ling, B. H. Kang, S. Geng, N. B. Li and H. Q. Luo, Langmuir, 2016, 32, 1881–1889 CrossRef CAS PubMed.
- M. Borkovec and G. J. M. Koper, Macromolecules, 1997, 30, 2151–2158 CrossRef CAS.
- Y. Fan, Y.-Q. Cai, X.-B. Fu, Y. Yao and Y. Chen, Polymer, 2016, 107, 154–162 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: It includes: NMR spectra of HPA and HPA-C4; GPC diagrams of HPA-C4s; fluorescence excitation and emission spectra of polymers. See DOI: 10.1039/c6ra27390d |
| This journal is © The Royal Society of Chemistry 2017 |