
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Engineering a favourable osteogenic microenvironment by heparin mediated hybrid coating assembly and rhBMP-2 loading†
Haoqi Tan ab,
Honglei Wangab,
Yanjun Chaiab,
Yuanman Yuab,
Hua Hongab,
Fei Yangc,
Xue Qu*ab and
Changsheng Liu*ab
ab,
Honglei Wangab,
Yanjun Chaiab,
Yuanman Yuab,
Hua Hongab,
Fei Yangc,
Xue Qu*ab and
Changsheng Liu*ab
aKey Laboratory for Ultrafine Materials of Ministry of Education, East China University of Science and Technology, Shanghai 200237, China. E-mail: quxue@ecust.edu.cn; liucs@ecust.edu.cn; Fax: +86-21-64251358; Tel: +86-21-64251358
bThe State Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai, 200237, China
cState Key Laboratory of Polymer Physics and Chemistry, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
First published on 15th February 2017
Abstract
With the development of bone tissue engineering, it is a great challenge to fabricate a bioactive surface which can provide a favourable microenvironment for cell adhesion, migration and osteo-differentiation. Coating the as-prepared substrates with bioactive components is an effective method to induce osteogenesis. In this study, we designed a multi-layered hybrid coating containing hydroxyapatite nanoparticles, heparin and chitosan. Heparin is used to mediate the electrostatic assembly of hydroxylapatite nanoparticles with chitosan, as well as the subsequent recombinant human bone morphogenetic protein-2 (rhBMP-2) loading. Several independent pieces of evidence were provided to indicate the successful fabrication of the hybrid coating. Sustained release of rhBMP-2 by this coating was also achieved. The in vitro biological studies reveal that the hybrid coating system has excellent biocompatibility and can improve the osteo-differentiation especially for rhBMP-2 present on the coating surface. The results demonstrate that the proposed rhBMP-2 loaded hybrid coating can provide a favourable osteogenic microenvironment and has the potential to improve in situ bone repair due to its superior osteoconductivity as well as osteoinductivity.
1. Introduction
Bone defects and injuries caused by trauma or diseases represent major clinical problems.1–3 Currently, implanting bone grafts for tissue engineering scaffolds has been widely used to address this issue.4,5 The graft surface is the first contact with the organism and initiates subsequent biological responses. Therefore, engineering a favourable surface microenvironment for cell adhesion, migration and differentiation is critical for improving tissue repair and regeneration processes. In the case of bone regeneration, surface molecular engineering utilizes the specific molecules for instance collagen,6,7 hydroxylapatite (HAp),8,9 polysaccharide,10,11 growthfactors12,13 and bone therapeutic drugs14 to conceive a favourable microenvironment to modulate implant–tissue interactions. HAp is a naturally occurring crystalline calcium phosphate Ca10(PO4)6(OH)2. As one of the principal inorganic components of human hard tissues such as vertebrae and teeth, HAp has been proven to offer better osteoconductivity,15,16 leading to ultimate osteointegration when applied onto biomedical implant surfaces. Recombinant human bone morphogenetic protein-2 (rhBMP-2) is an important osteoinductive growth factor. rhBMP-2 is clinically used to treat persistent non-union, open tibial fractures and spine fusion because it can trigger bone generation by initiating the signal transduction pathway for osteo-differentiation inside cells.17,18 Introducing both HAp and rhBMP-2 onto biomaterials surface is highly recommended to improve the microenvironment for osteogenesis related cells growth, and thus can achieve better osteogenesis and osteointegration.19In this context, various strategies have been developed to modify the biomaterials' surface with HAp and rhBMP-2. Han et al. co-precipitated apatite crystals with BMP-2 nano-complex onto titanium surface via a simultaneous mineralization.20 However, this process was time consuming, and also would lead to unanticipated protein denaturation. Tsai et al. developed a facile “one-pot” deposition method based on dopamine polymerization in the presence of HAp, rhBMP-2 and RGD conjugate, and thus can introduce these active components onto titanium quickly.21 Although this method generated a cell-adhesive, osteoconductive, and osteoinductive implants surface, the dissoluble polydopamine coating would entrap these active components forever. The layer-by-layer (LbL) assembly of polyelectrolyte multilayered film represents an attractive approach to coat the substrate because of its simpleness of preparation, versatility in composition and substrates, and fine control over architecture.22–26 Importantly, LbL film deposition does not require any organic media and therefore can incorporate sensitive biomacromolecules.27 Recently, we have shown that well designed LbL coating assembled by polymer nanoparticles and polysaccharide can load and sustained release BMP-2 and dexamethasone, through which synergistic osteoinductivity was successfully achieved.28
In this study, we propose to assemble a hybrid LbL coating electrostatically to incorporate HAp and rhBMP-2 for bone regeneration. Here heparin is used to mediate the coating formation and rhBMP-2 loading. The strategy is illustrated in Scheme 1. (1) First HAp nanoparticles are conferred with negative charges by surface modification with heparin via dopamine chemistry. (2) And then heparinized HAps and polycation chitosan are assembled alternatively to form a hybrid coating in a LbL manner. (3) rhBMP-2 is finally introduced into this coating via the intermolecular binding with heparin, since heparin is known to interact with rhBMP-2 through sulfated group, thus can be used to load and stabilize rhBMP-2.29–32 Planar PLGA polymers were used as a model substrate to build up this composite coating. The focus of our study is to demonstrate that the surface incorporation of HAp and rhBMP-2 can be easily achieved by utilizing the inherent characteristics of heparin (i.e. charge and sulfate unit). We envision this engineered surface structure can offer a good osteogenic microenvironment for bone development and regeneration. Moreover, since layer-by-layer strategy is easy to be applied onto varying substrates, our proposed surface engineering strategy can be easily introduced onto varying biomedical implant surfaces to improve their osteogenetic performance.
 | ||
| Scheme 1 Heparin mediates the assembling of HAp nanoparticles and chitosan and subsequent rhBMP-2 loading for bone regeneration. (1) Heparin is modified onto HAp nanoparticles by dopamine chemistry to endow HAp with negative charges. (2) The hybrid coating is assembled in a layer-by-layer manner by using anionic HAp nanoparticles and cationic chitosan. (3) rhBMP-2 is post loaded into the coating via the interaction with heparin. This coating can provide good osteoconductivity and osteoinductivity for bone repair. | ||
2. Experimental
2.1 Materials
Poly(lactide-co-glycolide) (PLGA, LA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :GA = 75:25, Mw = 100 kDa) was purchased from Jinan Daigang Biomaterial Co., Ltd. (Shandong, China). Hydroxyapatite nanoparticles (HApNPs) were synthesized in our researchgroup. rhBMP-2 was kindly supplied by Rebone Biomaterials Co., Ltd. (Shanghai, China). Chitosan (CS, medium molecular weight) and heparin (HEP) were purchased from Sigma-Aldrich (CA, USA). CCK-8 assay kit, BCA protein assay kit and rhBMP-2 ELISA assay kit were purchased from Beyotime Biotech (Jiangsu, China).
:GA = 75:25, Mw = 100 kDa) was purchased from Jinan Daigang Biomaterial Co., Ltd. (Shandong, China). Hydroxyapatite nanoparticles (HApNPs) were synthesized in our researchgroup. rhBMP-2 was kindly supplied by Rebone Biomaterials Co., Ltd. (Shanghai, China). Chitosan (CS, medium molecular weight) and heparin (HEP) were purchased from Sigma-Aldrich (CA, USA). CCK-8 assay kit, BCA protein assay kit and rhBMP-2 ELISA assay kit were purchased from Beyotime Biotech (Jiangsu, China).
2.2 Synthesis of HEP–Dopa conjugate
HEP was grafted with dopamine (Dopa) using EDC and sulfo-NHS as coupling agents as presented in ESI.†2.3 Heparin-modified hydroxyapatite nanoparticles
Ellipse-like shaped HApNPs (synthetic method was presented in ESI†) were dispersed in the Tris–HCl buffer (0.01 M pH = 8.0) at a concentration of 5 mg mL−1, followed by addition of HEP–Dopa (2 mg mL−1). Ultrasonication was then applied to the prepared solution for 10 min using a Ultrasonic cleaner. After magnetic stirring at 60 °C for 12 h, the nanoparticles were collected by centrifugation, followed by rinsing with ultrapure water and centrifugation for 3 times to completely remove unbound HEP–Dopa molecules.2.4 Layer-by-layer assembly of multilayered coatings on PLGA substrate
PLGA film (Mw = 100 kDa, size 10 × 10 cm2 with a thickness of 100 μm) was firstly cleaned by alcohol/water (1/1 v/v) solution for 30 min under ultrasonication, and dried under nitrogen. The PLGA film was subsequently immersed in PEI aqueous solution at a concentration of 10 mg mL−1 for 1 h to obtain stable positively charged surface, then rinsed with ultrapure water.The alternate assembly of HApNPs modified with HEP–Dopa (HEP–HApNPs) and chitosan (CS) was performed on the above film. Two solutions of HEP–HApNPs and CS were prepared by dispersion in HAc–NaAc buffer (0.2 M pH = 5) at a concentration of 5 mg mL−1 and 2 mg mL−1 respectively. For multilayer build-up, in brief, aminolysed PLGA film (denoted as bare substrate) was immersed in HEP–HApNPs suspension for 30 min under gentle agitating, then washed with ultrapure water and then dried under N2 gas. Next, the substrate was immersed in CS solution for 30 min, followed by a washing and drying step. This alternate adsorption process was repeated to obtain the desired number of layers. The uppermost layer was always deposited with CS to confer biocompatibility.
2.5 Characterization of HApNPs modified with HEP–Dopa and LBL multilayer coatings
Surface charge and hydrodynamic size of HEP–HApNPs were measured using a Zetasizer (Nano ZS, Malvern Instruments, UK). The dispersion stability of pure HApNPs, HApNPs/HEP mixture and HEP–HApNPs in PBS solution were recorded by camera. The morphology of the HApNPs and HEP–HApNPs were examined by transmission electron microscope (TEM, JEOL JEM-2100, Nikon, Japan) and field-emission scanning electron microscopy (FE-SEM, Hitachi S4800, Nikon, Japan). The amount of sulfo group in the surface of HApNPs was measured by barium chloride-gelatin turbidimetry.33Quartz Crystal Microbalance with Dissipation Monitoring (QCM, Q-sense E1, Biolin, Sweden) was used to monitor the LBL self-assembly process. In brief, an aminolysed coating was firstly fabricated on the gold surface of the quartz crystal by PEI (4 mg mL−1) solution. Assembly of the multi-layer on the crystal was carried on by contacting HEP–HApNPs suspension (5 mg mL−1) and CS (2 mg mL−1) solution alternately at a flow rate of 50 μL min−1. To remove the loosely-bound molecules, buffer rinsing was employed for 5–10 min after the equilibrium state was reached for each absorption. Morphological characterization of top view and lateral view of the multilayered coatings were performed with SEM. In addition, to confirm the surface chemical composition, X-ray photoelectron spectroscopy (XPS, PH1500C, Japan) equipped with an argon ion gun was used.
2.6 rhBMP-2 loading and release
rhBMP-2 was last loaded by dropping the solution containing 2 μg of rhBMP-2 onto the as-prepared coatings (1 cm2). Stand the films for 2 h to let rhBMP-2 absorbed fully before subsequent freeze-drying step. The control bare substrate (PLGA) containing the same amount of rhBMP-2 was prepared by the same operation method.For evaluation of rhBMP-2 releasing profile, the bare substrate and substrate with LBL coatings loaded rhBMP-2 were placed into a tube containing 2 mL PBS respectively. At predetermined time of rhBMP-2 releasing profile, 0.3 mL incubation solution was collected from the tube for measurement and another 0.3 mL fresh PBS was supplemented. The amount of released rhBMP-2 was determined by using a Human rhBMP-2 ELISA Kit (n = 3).
2.7 Cell evaluation
Murinemyoblast cells (C2C12) purchased from the American Type Culture Collection (ATCC, VA, USA) and rat bone marrow stromal cells (rBMSCs) extracted from rat bone marrow were used for evaluating osteogenesis vitality of the multilayer-coating. This study was performed in strict accordance with National Institutes of Health (NIH) guidelines for the care and use of laboratory animals (NIH Publication no. 85-23 Rev. 1985) and all procedures were approved by the Research Center for Laboratory Animals of Shanghai University of Traditional Chinese Medicine (Shanghai, China). The cell culture and cell implantation are presented in ESI.†| ALP activity = absorbance of 405 nm/(protein content × color reaction time) |
3. Results and discussion
3.1 Fabrication of heparin-modified HAp nanoparticles
In order to electrostatically assemble HApNPs into the LbL coating, the pristine HApNPs need to be modified to enhance their surface charges and colloidal stability. Since pure HApNPs only have a poor surface charge and are easily aggregated to precipitate from aqueous solution,34 several surface techniques have been proposed to address this issue.35,36 Dopamine chemistry that inspired by the adhesive proteins of mussels for attachment to wet surfaces has been developed widely to modify the solid surface properties.37,38 Heparin (HEP) is well known to play an important role in regulating adhesion, growth of cells39,40 and has highly negative charge and steric repulsion effects. In addition, HEP can be used to stabilize rhBMP-2 and improves its activity. In this regard, we propose to develop HEP modified HApNPs using dopamine chemistry to increase their surface charges and colloidal stability.We first synthesized dopamine grafted heparin (HEP–Dopa) by using EDC/NHS chemistry. The reaction mechanism is roughly as below: firstly, EDC activates carboxyl of HEP to form an unstable reactive O-acylisourea ester, then NHS stabilizes this intermediate by converting it to an amine-reactive NHS ester. This NHS-ester subsequently reacts with nucleophilic free amine group of dopamine to form amide bond. The chemical structure of obtained HEP–Dopa was confirmed by 1H NMR spectroscopy. The results are shown in Fig. S1.† Compared to the spectrum of HEP, the spectrum of HEP–Dopa indicates a group of new peaks in 6.5–7.0 ppm, which can be ascribed to the characteristic peaks of benzene ring on dopamine. The approximately 34.2% degree of substitution (DS) can be determined by calculating the relative peak intensity ratio between anomeric protons in heparin (5.0–5.6 ppm) and aromatic protons in dopamine (6.5–7.0 ppm).41
After mixing HApNPs and HEP–Dopa for 12 h, the heparin moieties can be easily anchored onto HApNPs through the formation of polydopamine coating. The obtained nanoparticles were denoted as HEP–HApNPs. Four pieces of evidence are provided to demonstrate the successful heparin modification.
First, zeta potential results in Fig. 1A show that the surface charge of HApNPs increased significantly from −3 mV to −26 mV after contacting HEP–Dopa solution. Quantitative description of the modified HApNPs size was provided by DLS measurement in Fig. 2B. The heparin modified HApNPs have an average size of about 180 ± 20 nm, while the pristine controls are significantly aggregated due to their weak surface charges, and thus cannot give a nano-scaled particle size (data are not shown here). For dispersion stability test, the samples of pristine HApNPs, heparin physically adsorbed HApNPs and HEP–Dopa modified HApNPs were individually dispersed in PBS solution (Fig. 1C). The colloidal stability was monitored by photography during 24 h incubation. The pristine HApNPs and heparin physically adsorbed HApNPs were observed to be mostly precipitated after 6 h incubation, while HEP–Dopa modified HApNPs display good suspending stability during long time placement. The morphologies of nanoparticles before and after heparin modification were characterized by SEM and TEM in Fig. 1D. The pristine HApNPs were observed to have a fibrous structure and aggregate strongly. After heparin modification, the particles seem to be fat and dispersed very well on the sample stages. The HEP–HApNPs although covered with a heparin coating still have a nano-scaled size. The involved heparin content on HApNPs was calculated as 22 μg HEP per 1 mg HEP–HApNPs.
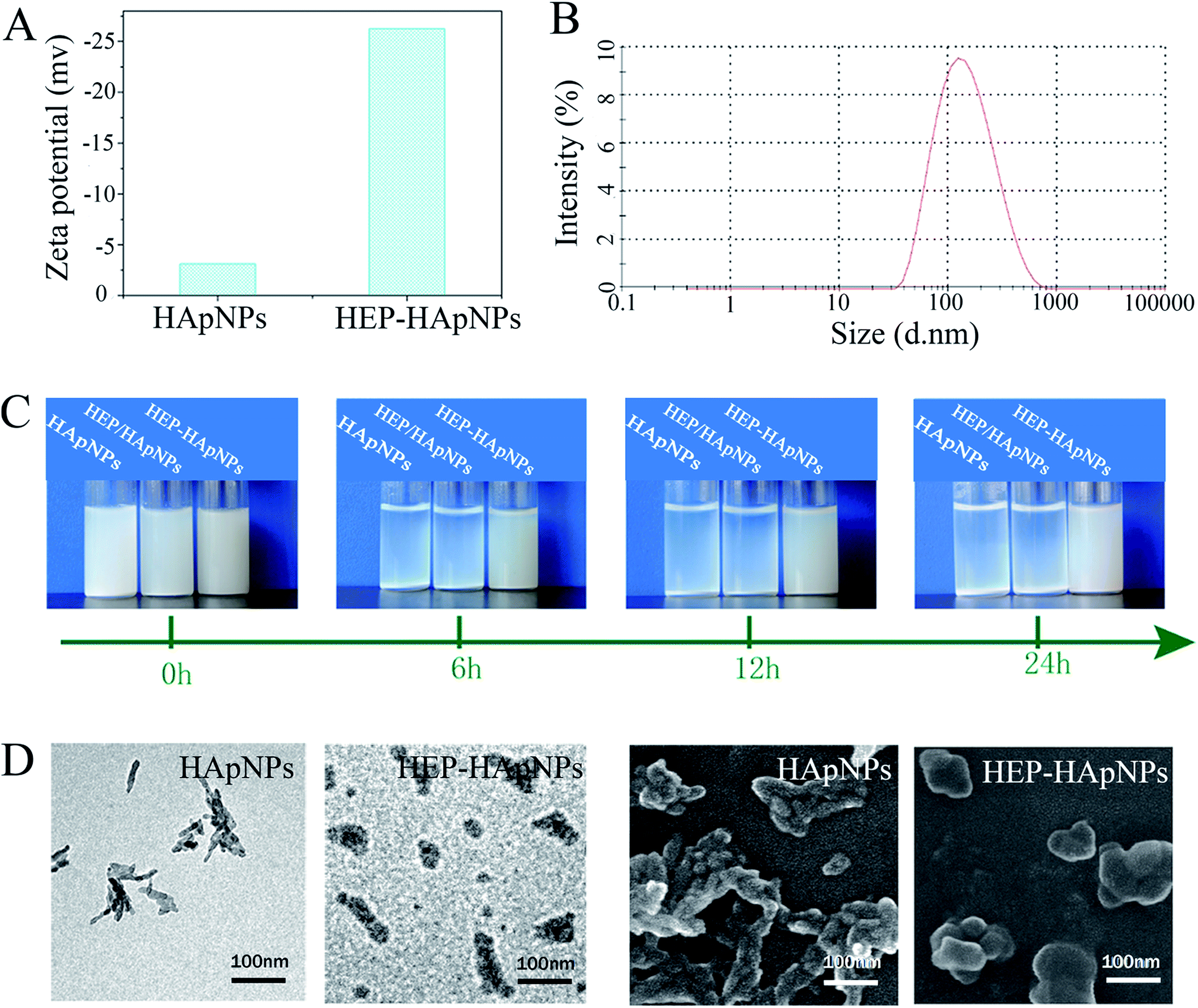 | ||
| Fig. 1 Characterization of the pristine HApNPs and heparin modified HApNPs (HEP–HApNPs). (A) Zeta potential of HApNPs and HEP–HApNPs. (B) The size distribution of HEP–HApNPs. (C) Time-dependent colloidal stability of pristine HApNPs, heparin physically adsorbed HApNPs (HEP/HApNPs) and HEP–Dopa modified HApNPs (HEP–HApNPs). (D) TEM (left two) and SEM (right two) images of HApNPs and HEP–HApNPs. | ||
 | ||
| Fig. 2 Characterization of hybrid multi-layer coatings. (A) QCM monitor the layer-by-layer assembly of HEP modified HApNPs (HEP–HApNPs) and chitosan (CS). (B) SEM images of the front view of the bare substrate (PEI aminolysed PLGA film) and the substrates covered by HEP–HApNPs/CS with 4 bilayers, 12 bilayers, and 20 bilayers, respectively. The inserts show the cross-sections of the corresponding samples. (C) XPS spectra of the bare substrate (PEI aminolysed PLGA film) and the [HEP–HApNPs/CS]10 coating that is formed on the substrate. | ||
The above results demonstrate that HEP–Dopa molecules can be easily anchored onto the HApNPs surface via a dopamine mediated surface coating. The obtained multilayered structure based on the electrostatics interactions.
3.2 Fabrication of hybrid multi-layer coating
In this study, alternate assembly of two oppositely charged species, i.e. HEP–Dopa modified HApNPs (HEP–HApNPs) and chitosan (CS), is anticipated to fabricate an inorganic/organic hybrid coating [HEP–HApNPs/CS]n (n represent the bilayer number of the HEP–HApNPs/CS) on the PLGA model surface.We first performed quartz crystal microbalance measurement (QCM) to demonstrate self-assembly of this multi-layer coating.42,43
The gold surface of the QCM crystal was initially treated with positively charged polyethyleneimine (PEI). This surface allows the initial layer of HEP–HApNPs to be bound to the sensor through electrostatic interactions. We then sequentially contacted the PEI-coated sensor with solutions of HEP–HApNPs and CS. The QCM measurements in Fig. 2A show decreases in resonant frequency after each contacting step. This observation indicates that multi-layers of HEP–HApNPs and chitosan can be assembled on the basis of electrostatic interactions.
Secondly, the HEP–HApNPs/CS multi-layer coatings with 4, 12 and 20 bilayer number were individually built up on the PLGA film for topography observation. The Fig. 2B reveals that after 4 bilayers deposition, the formed HEP–HApNPs/CS assembly masks most of the original appearance of the substrate. A much more condense and rugged surface was yielded for the coating with 12 bilayers. An unexpected scratch on this coating reveals an obvious morphologic difference between the coated and uncoated substrate surface. HEP–HApNPs/CS assembled with 20 bilayers has the most rugged topography that is due to plenty of HAp nanoparticles are introduced into this coating. The cross-section images for each coating are shown in the inserts. The evolution of coating thickness alone with the increase of the deposition cycle was quantified. At least five different area for one sample were measured to obtain an averaged value. The coating thicknesses of [HEP–HApNPs/CS]4, [HEP–HApNPs/CS]12 and [HEP–HApNPs/CS]20 were measured to be about 310 ± 16 nm, 596 ± 50 and 1068 ± 93 nm, respectively, which shows a positive correlation to the deposited layer number.
Finally, we analyzed the elemental composition of the coating by XPS measurements. Fig. 2C shows that compared to the bare PLGA substrate, the generated [HEP–HApNPs/CS]10 coating shows new peaks of N 1s, Ca 2p, S 2p, P 2p at 400 eV, 348 eV and 168 eV, 135 eV, respectively. Ca 2p and P 2p peak are assigned to HApNPs. And the signal of S 2p peak can be attributed to the sulfate group on the heparin. The XPS results provide chemical evidence for the successful assembly of HEP–HAps incorporated coating.
These results demonstrate that a hybrid coating composed of chitosan and heparinized HAp nanoparticles can be easily fabricated using electrostatic interactions as driving force. And also the hybrid coating thickness is finely controlled by the assembly parameters we used for instance the bilayer numbers.
3.3 Loading and release of rhBMP-2
We used a coating assembled by 10 bilayers of HEP–HApNPs and CS to evaluate the loading and release behaviors of rhBMP-2. rhBMP-2 was post-loaded into the coating by dropping a certain amount of rhBMP-2 solution onto the as-prepared coating followed by a freeze-drying step. The initial loading amount for rhBMP-2 was about 2.0 μg cm−2. The coating loaded with rhBMP-2 is defined as rhBMP-2@[HEP–HApNPs/CS]10 and the initial loading percentage is defined as 100% (2 μg cm−2).We further examined the release dynamics of rhBMP-2 from the assembled coating. Bare PLGA substrate loaded with rhBMP-2 at the same level as that on coated substrate was used as control. Fig. 3 shows the release profiles of rhBMP-2 from the two substrates. The control surface exhibits a rapid burst release of over 70% for rhBMP-2 within one day. However, in the presence of hybrid coatings, the release of rhBMP-2 was significantly delayed. Despite of a minor “burst release” occurred at the beginning, a sustained donation of rhBMP-2 was observed over the period of 2 weeks incubation. The initial rapid release is presumably because of some physically adsorbed rhBMP-2 on the outmost surface of coatings. The sustained release of rhBMP-2 in the next stage could be attributed to the incorporated heparin moieties inside the coating that can bind rhBMP-2 via their heparin binding domains,29–32 which is supported by many literatures in that the non-covalent interactions between BMP and heparin have been demonstrated via the clusters of positively charged basic amino acids on BMP-2 and specific, spatially grouped, negatively charged sulfate groups on heparin,44 and this binding is reversible.45
 | ||
| Fig. 3 Cumulative release of rhBMP-2 in PBS from bare substrate and substrate with [HEP–HApsNPs/CS]10 coatings, respectively. | ||
After 2 weeks incubation, about 55% of initially loaded rhBMP-2 was observed to be released into the incubation buffer. It is known that a localized and sustained delivery is most favourable for rhBMP-2 when applied in vivo.46 The proposed hybrid coating could be served as an rhBMP-2 reservoir for continuous donation of rhBMP-2 with bioactivity into the defect sites.
3.4 Cell viability and morphology on hybrid coatings
A cell-favourable microenvironment should first support cell adhesion and spreading. Cell growth is a prerequisite for further cell growth and development.47,48The viability of the cells cultured on [HEP–HApNPs/CS]10 was evaluated by CCK-8 assay. As shown in Fig. 4A, the cell viability exposed to the coating surface is comparable to that on the bare surface, which indicates that the coating has no obvious cytotoxicity.
 | ||
| Fig. 4 Cell viability and morphology of the C2C12 cells on bare substrate and substrate with [HEP–HApsNPs/CS]10. (A) Cell viability on substrates with or without coating after 3 days incubation, determined by CCK-8 assay. (B) CLSM and SEM observations of the cell morphology adhered on substrates with or without coating for 24 h. | ||
The morphology of cells that attached onto the [HEP–HApNPs/CS]10 coating and bare substrate was evaluated by CLSM and SEM, respectively. The upper panel in Fig. 4B shows the SEM observations of cells after 24 h incubation. Cells spread well on the coating surface and exhibited normal filopodia extensions as indicated by the green arrows. A parallel morphology analysis by a nuclei (blue)–cytoskeleton (green) fluorescent staining assay were performed and the CLSM observations are shown in the lower panel in Fig. 4B. The cells all showed elongated and well spread morphology on the coating, and the density of cells were found to be higher on coating surface in comparison to the PLGA bare substrate without any treatment, which could be ascribed to the incorporated biocompatible macromolecules and osteophilic hydroxylapatites in the coating.
These results indicate that the coating shows excellent cytocompatibility and provide a desirable environment for cell attachment and growth.
3.5 In vitro cell differentiation on the [HEP–HApNPs/CS]10 coating
In this study, rhBMP-2 is loaded into the hybrid coating for induction of osteo-differentiation. In order to evaluate the bio-performance of the incorporated rhBMP-2, we first measured the alkaline phosphatase (ALP) expression of cells that are stimulated by rhBMP-2 loaded hybrid coating i.e. rhBMP-2@[HEP–HApNPs/CS]10. ALP is a standard early marker of osteogenesis in several osteo-progenitor cells. C2C12 myoblast has osteoblastic potential when stimulated by rhBMP-2, therefore we use C2C12 to do this study. The cells were seeded on bare substrate, substrate with hybrid coating and substrate with rhBMP-2 loaded coating, respectively. After 3 days incubation, the cells were harvested and lysed for ALP quantification. The result is shown in Fig. 5A. Compared to the cells that cultured in bare substrate and hybrid coating, the cells exposed to rhBMP-2 loaded coating surface show a significant increase in ALP activity, which indicates the rhBMP-2@[HEP–HApNPs/CS]10 coating contains bioactive rhBMP-2 and thus has excellent osteoinductive ability.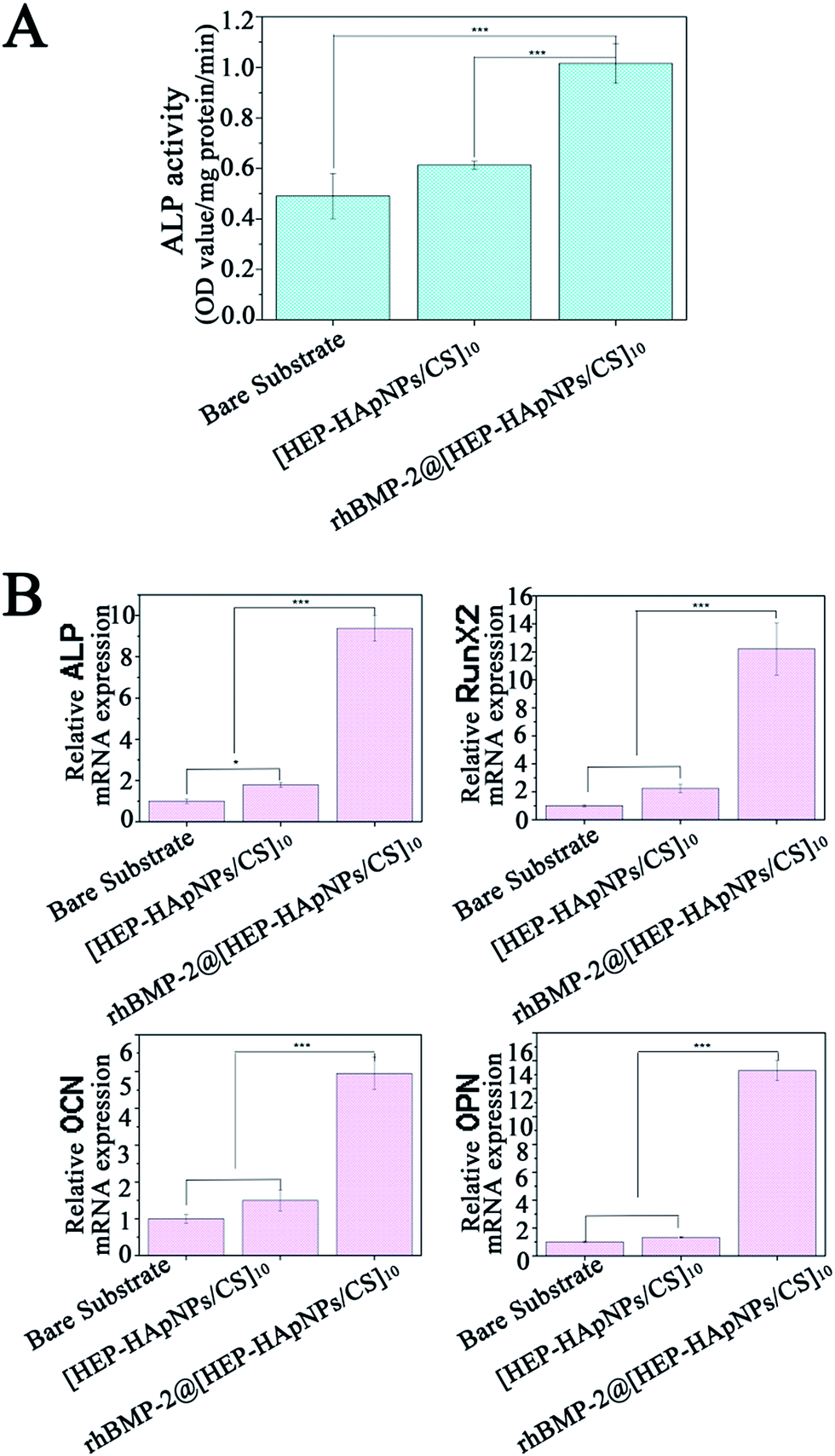 | ||
| Fig. 5 In vitro osteogenic differentiation of cells cultured on bare substrate, substrate with hybrid coating and substrate with rhBMP-2 loaded hybrid coating for 3 days. (A) ALP activity of C2C12 cells. (B) The mRNA expression of osteogenic gene markers ALP, OCN, OPN and RunX2 from BMSC cells by real-time PCR analysis (s.d., n = 3, ***P < 0.001; **P < 0.01; *P < 0.05). | ||
We further investigate the gene expression cascade involved in osteogenesis. Rat BMSC cells were cultured onto the three different substrates individually for three days and the osteogenesis related gene expression that is influenced by the microenvironment of substrate surface are evaluated by real-time PCR analysis. ALP gene is expressed early during the process of osteogenic differentiation, OCN and OPN is expressed later than ALP. RunX2 is a transcription factor that has an effect on the expression of bone-related genes by binding to the promoter of genes. The result in Fig. 5B reveals that compared to the cells cultured on bare substrate, cells that contact the hybrid coating surface slightly up-regulated the four genes that related osteogenesis. This phenomenon could be attributed to the hydroxylapatite component in the hybrid coating. Studies have demonstrated that moderate amount of extracellular calcium ions play an important role in regulating proliferation and osteogenic differentiation of bone mesenchymal stem cells.49 We thus hypothesize that the electrostatically assembled hybrid coating could also release calcium ions from the incorporated hydroxylapatite nanoparticles through which osteogenesis of the BMSC cells is facilitated. A much more significant gene expression of cells cultured on the rhBMP-2@[HEP–HApNPs/CS]10 coating is also observed, which is mainly triggered by the sustained released rhBMP-2 from the coating.
All the results demonstrate that the hybrid coating system can incorporate active rhBMP-2 and hydroxylapatite, thus can have the ability to induce cell differentiation towards osteoblasts which facilitates the final osteogenesis.
4. Conclusions
The repair of damaged bone tissue requires a favourable biological environment for osteogenesis related cell growth. In this work, we demonstrate that a well designed hybrid coating containing hydroxylapatite nanoparticles, chitosan and rhBMP-2 has been fabricated successfully. Heparin is used to mediate the assembly of hydroxylapatite nanoparticles with chitosan, as well as the subsequent rhBMP-2 loading into the coating. Biological studies indicate our coating system has biocompatibility and can improve the osteo-differentiation especially for rhBMP-2 present coating surface. The results demonstrate that the proposed rhBMP-2 loaded hybrid coating can provide a favourable osteogenic microenvironment and have the potential to improve the in situ bone repair due to the superior osteoconductivity as well as osteoinductivity.Acknowledgements
This work was supported by National Natural Science Foundation of China (51573047, E0213), Innovation Program of Shanghai Municipal Education Commission (14ZZ060), Shanghai Science and Technology Development Funds (14QA1401000), and the 111 project (B14018).Notes and references
- P. S. Lienemann, M. P. Lutolf and M. Ehrbar, Adv. Drug Delivery Rev., 2012, 64, 1078–1089 CrossRef CAS PubMed.
- E. S. Hara, M. Ono, H. T. Pham, W. Sonoyama, S. Kubota, M. Takigawa, T. Matsumoto, M. F. Young, B. R. Olsen and T. Kuboki, J. Bone Miner. Res., 2015, 30, 1585–1596 CrossRef CAS PubMed.
- J. M. Holzwarth and P. X. Ma, Biomaterials, 2011, 32, 9622–9629 CrossRef CAS PubMed.
- S. Song, E. J. Kim, C. S. Bahney, T. Miclau, R. Marcucio and S. Roy, Acta Biomater., 2015, 18, 100–111 CrossRef CAS PubMed.
- K. Chen, X. F. Lin, Q. Zhang, J. H. Ni, J. M. Li, J. Xiao, Y. Wang, Y. H. Ye, L. Chen, K. K. Jin and L. Chen, Acta Biomater., 2015, 19, 46–55 CrossRef CAS PubMed.
- F. Goncalves, R. Bentini, M. C. Burrows, A. C. O. Carreira, P. M. Kossugue, M. C. Sogayar and L. H. Catalani, Materials, 2015, 8, 408–423 CrossRef.
- A. Lopez-Noriega, E. Ruiz-Hernandez, E. Quinlan, G. Storm, W. E. Hennink and F. J. O'Brien, J. Controlled Release, 2014, 187, 158–166 CrossRef CAS PubMed.
- P. B. Zhang, H. T. Wu, H. Wu, Z. W. Lu, C. Deng, Z. K. Hong, X. B. Jing and X. S. Chen, Biomacromolecules, 2011, 12, 2667–2680 CrossRef CAS PubMed.
- J. Xie, C. Peng, Q. H. Zhao, X. L. Wang, H. H. Yuan, L. L. Yang, K. Li, X. X. Lou and Y. Z. Zhang, Acta Biomater., 2016, 29, 365–379 CrossRef CAS PubMed.
- T. Jiang, S. P. Nukavarapu, M. Deng, E. Jabbarzadeh, M. D. Kofron, S. B. Doty, W. I. Abdel-Fattah and C. T. Laurencin, Acta Biomater., 2010, 6, 3457–3470 CrossRef CAS PubMed.
- Y. C. Wu, S. Y. Shaw, H. R. Lin, T. M. Lee and C. Y. Yang, Biomaterials, 2006, 27, 896–904 CrossRef CAS PubMed.
- K. X. Zhang, S. M. He, S. F. Yan, G. F. Li, D. Q. Zhang, L. Cui and J. B. Yin, J. Mater. Chem. B, 2016, 4, 2628–2645 RSC.
- Z. S. Patel, S. Young, Y. Tabata, J. A. Jansen, M. E. K. Wong and A. G. Mikos, Bone, 2008, 43, 931–940 CrossRef CAS PubMed.
- C. T. Wu, W. Fan, M. Gelinsky, Y. Xiao, P. Simon, R. Schulze, T. Doert, Y. X. Luo and G. Cuniberti, Acta Biomater., 2011, 7, 1797–1806 CrossRef CAS PubMed.
- D. Gopi, E. Shinyjoy, A. Karthika, S. Nithiya, L. Kavitha, D. Rajeswari and T. T. Tang, RSC Adv., 2015, 5, 36766–36778 RSC.
- R. Rajesh and Y. D. Ravichandran, RSC Adv., 2015, 5, 41135–41143 RSC.
- S. F. Zhang, C. Kucharski, M. R. Doschak, W. Sebald and H. Uludag, Biomaterials, 2010, 31, 952–963 CrossRef CAS PubMed.
- S. F. Zhang, M. R. Doschak and H. Uludag, Biomaterials, 2009, 30, 5143–5155 CrossRef CAS PubMed.
- C. M. Xie, X. Lu, K. F. Wang, F. Z. Meng, O. Jiang, H. P. Zhang, W. Zhi and L. M. Fang, ACS Appl. Mater. Interfaces, 2014, 6, 8580–8589 CAS.
- S. E. Bae, J. Choi, Y. K. Joung, K. Park and D. K. Han, J. Controlled Release, 2012, 160, 676–684 CrossRef CAS PubMed.
- C. Y. Chien and W. B. Tsai, ACS Appl. Mater. Interfaces, 2013, 5, 6975–6983 CAS.
- Y. H. Jiao, Y. Li, S. Wang, K. Zhang, Y. G. Jia and Y. Fu, Langmuir, 2010, 26, 8270–8273 CrossRef CAS PubMed.
- J. Hong, N. J. Shah, A. C. Drake, P. C. DeMuth, J. B. Lee, J. Z. Chen and P. T. Hammond, ACS Nano, 2012, 6, 81–88 CrossRef CAS PubMed.
- M. L. Macdonald, R. E. Samuel, N. J. Shah, R. F. Padera, Y. M. Beben and P. T. Hammond, Biomaterials, 2011, 32, 1446–1453 CrossRef CAS PubMed.
- N. J. Shah, M. L. Macdonald, Y. M. Beben, R. F. Padera, R. E. Samuel and P. T. Hammond, Biomaterials, 2011, 32, 6183–6193 CrossRef CAS PubMed.
- S. G. Caridade, C. Monge, J. Almodovar, R. Guillot, J. Lavaud, V. Josserand, J. L. Coll, J. F. Mano and C. Picart, Acta Biomater., 2015, 15, 139–149 CrossRef CAS PubMed.
- N. J. Shah, J. Hong, M. N. Hyder and P. T. Hammond, Adv. Mater., 2012, 24, 1445–1450 CrossRef CAS PubMed.
- X. Qu, F. He, H. Q. Tan, Y. M. Yu, A. Axrap, M. Wang, K. Dai, Z. Zhang, F. Yang, S. G. Wang, J. Kohn and C. S. Liu, J. Mater. Chem. B, 2016, 4, 4901–4912 RSC.
- F. Blanquaert, D. Barritault and J. P. Caruelle, J. Biomed. Mater. Res., 1999, 44, 63–72 CrossRef CAS PubMed.
- H. Lin, Y. Zhao, W. Sun, B. Chen, J. Zhang, W. Zhao, Z. Xiao and J. Dai, Biomaterials, 2008, 29, 1189–1197 CrossRef CAS PubMed.
- T. Hu, S. A. Abbah, M. Wang, S. Y. Toh, R. W. M. Lam, M. Naidu, G. Bhakta, S. M. Cool, K. Bhakoo, J. Li, J. C. H. Goh and H. K. Wong, Spine, 2015, 40, 613–621 CrossRef PubMed.
- C. Ma, Y. Jing, H. Sun and X. Liu, Adv. Healthcare Mater., 2015, 4, 2699–2708 CrossRef CAS PubMed.
- T. Takada, T. Katagiri, M. Ifuku, N. Morimura, M. Kobayashi, K. Hasegawa, A. Ogamo and R. Kamijo, J. Biol. Chem., 2003, 278, 43229–43235 CrossRef CAS PubMed.
- M. N. Salimi, R. H. Bridson, L. M. Grover and G. A. Leeke, Powder Technol., 2012, 218, 109–118 CrossRef CAS.
- W. M. Chirdon, W. J. O'Brien and R. E. Robertson, J. Biomed. Mater. Res., Part B, 2003, 66, 532–538 CrossRef PubMed.
- T. G. Kim, S.-H. Park, H. J. Chung, D.-Y. Yang and T. G. Park, J. Mater. Chem., 2010, 20, 8927 RSC.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- J. H. Cho, K. Shanmuganathan and C. J. Ellison, ACS Appl. Mater. Interfaces, 2013, 5, 3794–3802 CAS.
- Y. Yang, P. K. Qi, F. Wen, X. Y. Li, Q. Xia, M. F. Maitz, Z. L. Yang, R. Shen, Q. F. Tu and N. Huang, ACS Appl. Mater. Interfaces, 2014, 6, 14608–14620 CAS.
- L. Ma, H. Qin, C. Cheng, Y. Xia, C. He, C. X. Nie, L. R. Wang and C. S. Zhao, J. Mater. Chem. B, 2014, 2, 363–375 RSC.
- T. G. Kim, H. Lee, Y. Jang and T. G. Park, Biomacromolecules, 2009, 10, 1532–1539 CrossRef CAS PubMed.
- N. Aggarwal, N. Altgarde, S. Svedhem, K. Zhang, S. Fischer and T. Groth, Langmuir, 2013, 29, 13853–13864 CrossRef CAS PubMed.
- N. Aggarwal and T. Groth, J. Biomed. Mater. Res., Part A, 2014, 102, 4224–4233 Search PubMed.
- I. Capila and R. J. Linhardt, Angew. Chem., Int. Ed., 2002, 41, 391–412 CrossRef PubMed.
- M. H. Hettiaratchi, T. Miller, J. S. Temenoff, R. E. Guldberg and T. C. McDevitt, Biomaterials, 2014, 35, 7228–7238 CrossRef CAS PubMed.
- R. H. Li and J. M. Wozney, Trends Biotechnol., 2001, 19, 255–265 CrossRef CAS PubMed.
- T. Hoshiba, N. Kawazoe, T. Tateishi and G. P. Chen, J. Biol. Chem., 2009, 284, 31164–31173 CrossRef CAS PubMed.
- H. Ceylan, A. B. Tekinay and M. O. Guler, Biomaterials, 2011, 32, 8797–8805 CrossRef CAS PubMed.
- R. Mentaverri, S. Yano, N. Chattopadhyay, L. Petit, O. Kifor, S. Kamel, E. F. Terwilliger, M. Brazier and E. M. Brown, FASEB J., 2006, 20, 2562–2564 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27308d |
| This journal is © The Royal Society of Chemistry 2017 |