
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfluence of epoxidized natural rubber on the phase structure and toughening behavior of biocarbon reinforced nylon 6 biocomposites
Emmanuel O. Ogunsonaab,
Manjusri Misra *ab and
Amar K. Mohanty*ab
*ab and
Amar K. Mohanty*ab
aSchool of Engineering, University of Guelph, Thornbrough Building, Guelph, N1G 2W1, Ontario, Canada. E-mail: mmisra@uoguelph.ca; mohanty@uoguelph.ca
bBioproducts Discovery and Development Center, Department of Plant Agriculture, University of Guelph, Ontario Agriculture College, Guelph, ON N1G 2W1, Canada
First published on 27th January 2017
Abstract
Key drivers for automotive material applications require light weight and sustainable materials. This paper reports the engineering properties of hybrid nylon biocomposites as a possible replacement for some currently used filled nylon systems in the automotive industry. Nylon reinforced with 20 wt% of biocarbon showed tensile and flexural strengths superior to that of neat nylon by 19.5 and 31% respectively. However, the addition of biocarbon had a detrimental effect on its impact strength and elongation at break. Therefore, epoxidized natural rubber (ENR) was incorporated at low concentrations as an impact modifier due to its compatibility with nylon. An increase in impact strength of 55% was observed even in the presence of 20 wt% biocarbon when 7 phr of ENR was incorporated. The encapsulation of some of the biocarbon particles by the ENR increased the absorbance of the impact energy as a result as observed from the morphological analysis of the microstructure of the biocomposites. However, the tensile and flexural strengths showed a relatively marginal decrease due to the presence of ENR at the interface of nylon and biocarbon. Fourier transform infrared spectroscopic (FTIR) analysis revealed possible interactions/compatibility of ENR with nylon and biocarbon.
Introduction
Nylon and its composites are some of the most studied amongst engineering polymers in polymer composite science and engineering. Their applications span a very wide degree depending particularly on their mechanical properties. However, they are very sensitive to notching as cracks start and easily propagate through those notches or stress concentration points during impact. Due to their sensitivity to notching and brittleness, they typically possess poor impact properties. The restriction of the polymer chain by the filler prevents it from plastically deforming and absorbing the impact energy subsequently causing brittle fracture. Several methods to improve the impact strength have been studied. The incorporation of spherical fillers to hinder crack propagation have been explored and observed to improve the impact strength. Another well-established method is the addition of a very low modulus material to the composite; typically the addition of elastomers. This method has been explored by many researchers using different types of elastomers. Modification of these elastomers to improve adhesion with the matrix has also been studied and shown to aid impact strength.In nylon composites, elastomers such as natural rubber (NR), ethylene propylene diene monomer (EPDM), ethylene propylene rubber (EPR),1,2 styrene ethylene butylene styrene (SEBS)1,3 just to mention a few have shown improvements in the impact strength but at the cost of a relatively high concentration of the elastomers. It has also been shown that modifying these elastomers with functionalities which can react with nylon end groups can further improve the impact strength by increasing the adhesion between the nylon and elastomer leading to better energy transfer and absorption. Maleic anhydride grafted elastomers (MAH-elastomers) have been used in nylons and shown to be a better impact modifier in comparison to their unmodified counterpart.4 Epoxide groups are capable of reacting with the carboxyl, amine and amide groups on the backbone of the nylon chains. Epoxidized natural rubber (ENR) is the derivative of the modification of natural rubber (NR) by the grafting of epoxide groups onto its backbone. Blends of ENR and nylon have been well studied but not as much when used in nylon composites. It was chosen based on the fact that it is a biobased elastomer, highly reactive with nylon and also capable of interacting with the biocarbon used in this work. ENR has shown to form nylon grafted ENR copolymer due to their compatibility.5 Several mechanisms have also been proposed for the interaction between nylon and ENR through the interaction of the epoxide group on the ENR with the COOH and amine groups in the nylon.6,7 It has also been suggested that the bond created between these materials are stronger than the individual materials and can lead to increases in the melting temperature of the blends.8
The addition of toughening agents such as elastomers to polymer composites materials have typically resulted in the separate dispersion of the filler and elastomer in the matrix. It has also resulted in the dispersion of both filler and elastomer within the matrix with interaction between the elastomer and filler as well leading to partial or complete encapsulation of the filler by the elastomer.9–11 Nylon–ENR based composites using other filler types have also been studied. Nouparvar et al.12 characterized nanocomposites made from nylon 6, ENR and organo-modified clay. It was found that by increasing the clay content, the ENR phase within the composite became smaller and more dispersed suggesting that the clay was increasing the viscosity of the nylon phase which in turn caused further dispersion the ENR from increased shearing. However, the maximum impact strength obtained was approximately 5.5 kJ m−2 at 10 wt% of ENR within the composite. Beyond that, a decrease was observed. In another study, halloysite nanotubes were used to reinforce nylon 6 with ENR used as toughening agent.13 It was observed that the addition of ENR to an already optimized nylon–halloysite composite resulted to a decrease in the tensile and flexural properties but a successive improvement in impact strength with increasing ENR content. At 5 and 20 wt% ENR loading, the impact strengths were at approximately 5 and 10 kJ m−2 respectively in comparison to that of the composite of approximately 25 kJ m−2. It was suggested that the interactions between the ENR and nylon and ENR and nanofibers resulted in enhanced impact strengths.
Considering the importance of nylon in various industrial applications, it is therefore necessary to explore new, alternative and bio-renewable resources to curb the use of already diminishing petroleum resources. To the best of our knowledge, the use of biocarbon in conjunction with elastomers and their interactions in nylon composites has never been reported. Biocarbon was chosen because of its advantages similar to those of natural fibers such as its low cost and density, sustainability, renewability and carbon sequestration.14 However, it was chosen over natural fibers due to its greater thermal stability and capability of withstanding the high processing temperature of nylon without degradation.15 Likewise, it is less hydrophilic than natural fibers; therefore, the concern of hydrolysis of the nylon chains during processing is reduced.
Currently, there are no studies on biocarbon reinforced nylons except for our previously published works.16–18 The first study investigated the effect of biocarbon loading on nylon 6.16 It showed that the mechanical properties of the resulting composites were diminished stemming from the inherently defective nature of the biocarbons. In the second study, the effect particle size on the mechanical properties of nylon 6 was investigated.17 It was found that by milling the biocarbon over increasing time periods, the particle size reduced. It was also shown that by reducing the particle size, the mechanical properties especially the impact strength was improved. The third study investigated the effect of biocarbon particle size and ranges on the mechanical and thermomechanical properties of nylon 6, 10.18 It showed that the strengths and moduli of nylon were enhanced when pre-treatment of the biocarbon such as milling was performed. It also showed that by reducing the biocarbon particle size and using a lower particle size range, the impact strength of the biocomposite can be improved and is insignificantly different from the neat nylon. However, in this study, our objectives were to fabricate sustainable hybrid toughened biocomposites from biocarbon reinforced nylon 6. Knowledge obtained from our previous studies was applied in this study by utilizing the biocarbon particle size range which resulted in maximum strength and stiffness. The novelty in this work was aimed at achieving significantly enhanced impact strength of the biocomposites by utilizing a relatively low concentration of ENR unlike high amounts used in previous studies by encapsulating the biocarbons to reduce rigidity and improve toughness. Typically, to achieve toughening of polymer composites with elastomers, the concentrations have to be as much as 10–20 wt% and with well dispersed small domains within the matrix. In order to achieve these, mechanical tests were performed such as tensile, flexural and impact analysis, morphological analysis using scanning electron microscopy to observe the composite structure, Fourier transform infrared spectroscopy to determine interactions between the components and differential scanning calorimetry to analyze their thermal properties.
Materials and methods
Materials
Nylon 6 (Ultramid B3K) manufactured by BASF was purchased from Entec Polymers, USA. Biocarbon used in this study was produced and supplied by Genesis Industries, CA, USA from Miscanthus fibers obtained from New Energy Farms, Leamington, ON, Canada. The pyrolysis conditions are known to be around 500 °C. The physical, chemical and mechanical properties of this biocarbon have been well characterized in our previously published work and is denoted as “MB”.19 The work characterized the elemental composition as well as the surface functional groups present on the biocarbon. Results from the analysis showed that it has a CHNO composition of 66.5 2.9, 1.59 and 22.9 wt% content respectively. FTIR analysis revealed that carboxyl groups are present and significant from the vibration of the carbonyl group peak within the carboxyl group present at 1700 cm−1. The biocarbon particles are heterogeneous in shape and size and therefore the aspect ratio could not be determined nor specified. The epoxidized natural rubber with 25 mol% epoxidation, under the trade name of Epoxyprene is a product of Muang Mai Guthrie Public Company Limited of Thailand and supplied by Sanyo Corporation of America. Laboratory grade toluene was purchased and supplied by Sigma Aldrich and was used as a solvent for etching ENR from the fractured surface of the composites.Composite fabrication process
The biocarbon was subjected to preprocessing conditions by placing 25 g of each in a 500 mL stainless steel ball mill container and milled for 1 h at 300 rpm using a milling machine (Retsch PM100, Germany). A combination of 65 zirconium dioxide balls and 2 stainless steel balls were used as milling media. The zirconium dioxide balls were 10 mm in diameter and weighed 3.34 g each, while the stainless steel balls were 40 mm in diameter and weighed 256 g. The milled biocarbon was collected and dried at 105 °C until constant weight prior to further use.The ENR was cut into long strips and masticated at 120 °C and 4 rpm on a two roll mill (Prep-Mill, manufactured by C.W. Brabender Instruments Inc., NJ, USA) to help break down and flatten it into thin sheets of around 1 to 2 mm in thickness. The sheets were then cut into small pieces approximately 5 × 5 mm to be used in the composite fabrication. All three materials were simultaneously compounded in a one-step process. All composites were processed at 250 °C and at a screw speed of 100 rpm for 120 s using a HAAKE MiniLab II Micro Compounder (Thermo Scientific, MA, USA) and then injection molded with an injection pressure, injection time, injection back pressure, injection back pressure time and mold temperature of 400 bars, 10 s, 150 bars, 5 s and 70 °C respectively using a HAAKE MiniJet Injection molding machine (Thermo Scientific, MA, USA). An illustration of the process is shown in Fig. 1. For simplicity and ease of identification, acronyms have been given to the samples and are listed in Table 1. Preliminary results show that incorporation of ENR below 3 phr has no significant effect on the impact strength of the composite while above 7 phr, the strengths and modulus are significantly reduced in comparison to the biocarbon reinforced nylon biocomposite. Therefore, ENR loading between 3 to 7 phr was used. A 20 wt% loading was chosen for two main reasons based on preliminary studies; above 20 wt% loading, the wetting of the biocarbon by the nylon is significantly reduced and the viscosity of the composites make it difficult to be injection molded.
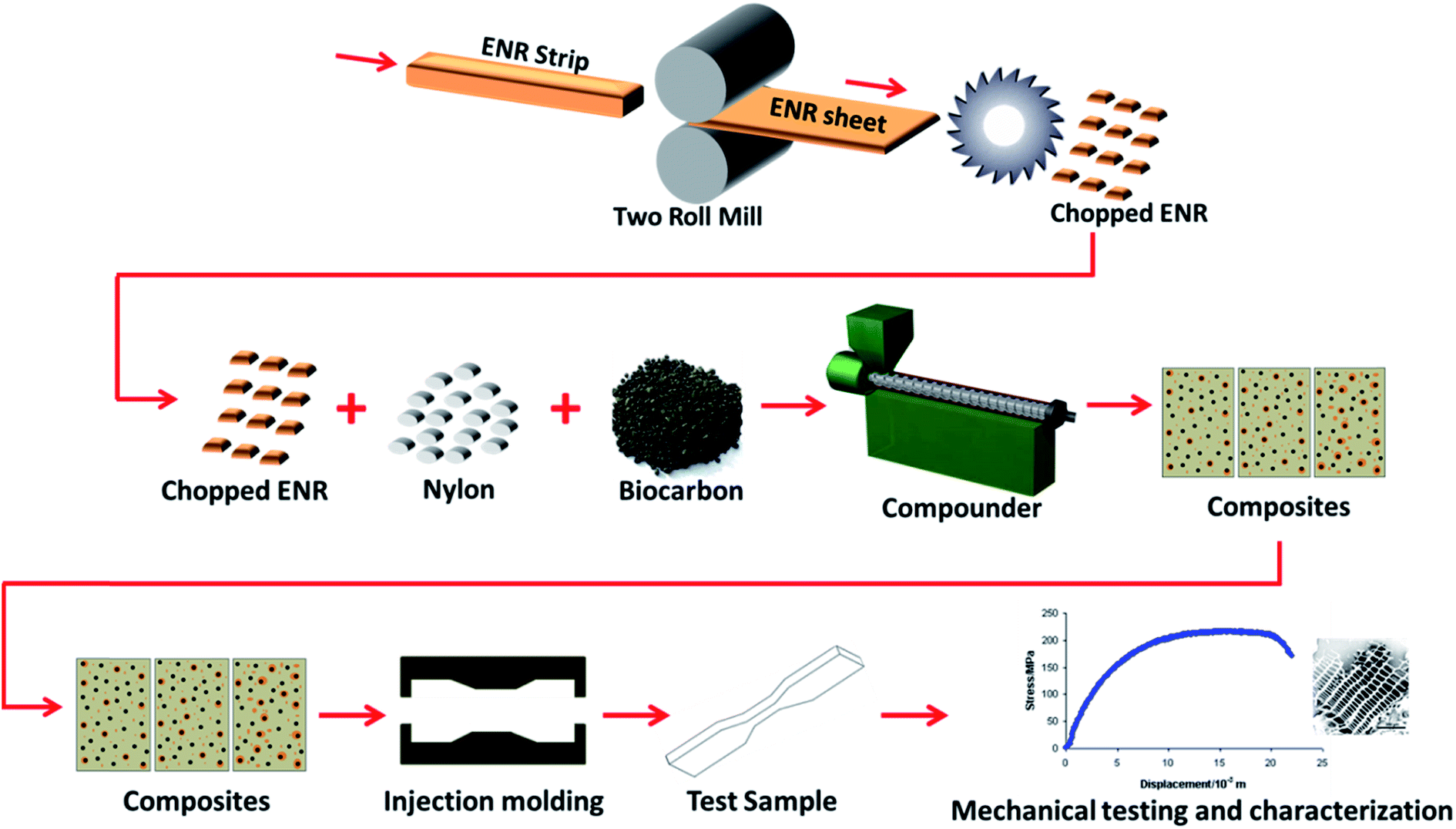 | ||
| Fig. 1 Preparation, processing and fabrication of hybrid nylon/biocarbon/ENR biocomposite. | ||
| Sample designation | Weight (%) | ENR (phr) | |
|---|---|---|---|
| Nylon 6 | Biocarbon | ||
| a N – nylon 6, B – biocarbon, E – ENR. | |||
| Nylon | 100 | 0 | 0 |
| NB | 80 | 20 | 0 |
| NBE3 | 80 | 20 | 3 |
| NBE5 | 80 | 20 | 5 |
| NBE7 | 80 | 20 | 7 |
Etching of the rubber phase was done by placing the impact fractured surfaces of the composite samples in a test tube with excess toluene. The rubber phase was left to dissolve for 48 h at room temperature while periodically agitating it to dislodge any rubber from the composites. After dissolving the rubber, the samples were removed, washed with toluene and acetone and then dried in a vacuum oven at 80 °C for 24 h.
Morphological analysis
The micrographs of the impact fractured surfaces of the composites were captured and analyzed using a scanning electron microscopy (SEM, FEI Inspect S50, OR, USA). All samples were first sputter gold coated under argon gas for 30 seconds using a Cressington Sputter Coater 108 auto (Cressington Scientific Instruments, UK).Material interactions
Possible interactions between the nylon, biocarbon and ENR were analyzed through a Fourier transform infrared spectroscopy (FTIR) Thermo Scientific, Nicolet 6700. A total of 32 scans, between a transmittance range of 4000 and 400 cm−1 and at a resolution of 4 cm−1 were collected for each of the aforementioned samples. Samples were prepared by grinding the composites to particles less than 1 mm in size to expose and disperse the interfaces in the composites.Thermal analysis
Thermal behaviors and transitions of the neat and composite materials were examined by differential scanning calorimeter (DSC Q200), manufactured by TA Instruments, USA under nitrogen flow at a rate of 50 mL min−1. Flat pieces removed from the same parts of the molded samples ranging between 5 to 10 mg in weight were placed in an aluminum pan and heated from 0 to 250 °C at a rate of 10 °C min−1, held at 250 °C for 5 min, cooled to 0 °C at a rate of 5 °C min−1, held at 0 °C for 5 min and then scanned again to 250 °C at the rate of 10 °C min−1. The melting temperature (Tm), crystallization temperature (Tc), crystallization enthalpy (ΔHc) and melt enthalpy (ΔHm) were determined from these scans. The first heating cycle was discarded as it was used to remove the thermal history of the samples.The possible miscibility and interactions occurring within the composites were investigated by studying the viscoelastic behavior of the samples using a Dynamic Mechanical Analyzer (TA Instruments) in dual cantilever mode with a frequency of 1 Hz and a strain amplitude of 25 μm. Testing was done within a temperature range of −80 to 150 °C with a ramp rate of 3 °C min−1 and a pre-load of 1 N. Samples with an approximate width and thickness of 12.6 mm and 3.35 mm were used.
Water absorption
Water uptake of the samples was performed in accordance with ASTM D570 for water absorption of plastics. Injection molded disc shaped coupons with a diameter and thickness of 25 mm and 15 mm respectively were used. Three replicates per sample were used and dried to constant weight prior to testing. Samples were placed in room temperature deionized water bath at the same depth with the water level, and the level was kept constant to prevent pressure changes. Each sample was weighed every 24 h using a precision weigh balance with a precision of 0.0001 mg. The water uptake was calculated using eqn (1):
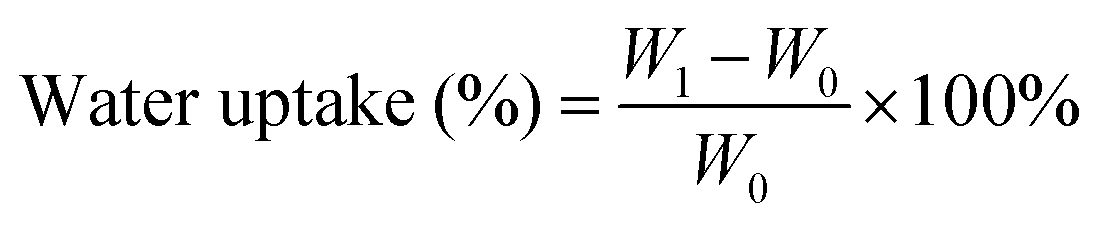 | (1) |
Mechanical properties
All samples were conditioned at 23 ± 2 °C and 50 ± 5% relative humidity for a period of 40 h before any test was done. Tensile properties were measured using samples prepared according to Type V specimen of ASTM standard D638. Flexural properties were measured using samples prepared according to procedure B of ASTM standard D790. The tensile and flexural tests were conducted on a Universal Testing Machine (Instron, Norwood, MA, USA).Impact strengths of the samples were measured using samples prepared in accordance to ASTM standard D256 with sample notch depths of 2 mm made using a TMI Notching Cutter. The tests were conducted on a TMI Monitor Impact Tester (Testing Machines Inc., DE, USA).
Results and discussion
Interactions between components of hybrid composites
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration from the formation of ester groups. Ester groups are formed in the blend from the esterification reaction between epoxide groups (1248 and 880 cm−1) in the ENR and COOH end groups (1650 cm−1) in nylon (carboxylic acid linkage) as shown in the schematic representation in Fig. 5. The peak at 3300 cm−1 of the nylon–ENR blend is representative of the N–H vibration of the amide group in nylon. This peak widens and forms a huge shoulder when compared to that of neat nylon. Also, an increase in the intensity is observed but not dominant in NBE7. This is due to the formation of hydroxyl groups from the formation of esters which vibrate around 3200 to 3400 cm−1, therefore overlapping with the N–H bonds. It could also be due to the increase in the number of N–H bonds formed from the interaction between the epoxide group in ENR and the amine end groups in nylon (amine linkage) as shown in Fig. 5. This would therefore imply an increase in the amount of nitros groups (N–O) in the nylon and should be observed through changes in the N–O peaks at 1370 and 1540 cm−1. It can be observed that the intensities are significantly increased as compared to that of neat nylon and NBE7. The peak at 1420 cm−1 was elaborated in the nylon–ENR blend whereas not as much in neither nylon nor the composites. This peak corresponds to the vibration of C–H bonds and is exaggerated due to the vibration of the same bonds also present in the ENR structure. The presence of the CO peak, the increase in intensity of the N–O peaks and broadening of the amide group at 3300 cm−1 suggests the interaction of ENR with nylon through amine and carboxylic acid linkages. Xie et al.5 suggested that blends of ENR and nylon form a graft copolymer (ENR-g-nylon) due to the interactions between the epoxide groups in the ENR and functional groups in nylon. There is good agreement between results found in this study and those by Xie et al. The changes in the spectra of the nylon–ENR blend which are not observed in that of NBE7 when compared to nylon can be attributed to the low concentrations of ENR and therefore fewer interactions between the epoxide groups in ENR with nylon. For these reasons, the ENR–biocarbon and nylon–ENR blends were made.
O vibration from the formation of ester groups. Ester groups are formed in the blend from the esterification reaction between epoxide groups (1248 and 880 cm−1) in the ENR and COOH end groups (1650 cm−1) in nylon (carboxylic acid linkage) as shown in the schematic representation in Fig. 5. The peak at 3300 cm−1 of the nylon–ENR blend is representative of the N–H vibration of the amide group in nylon. This peak widens and forms a huge shoulder when compared to that of neat nylon. Also, an increase in the intensity is observed but not dominant in NBE7. This is due to the formation of hydroxyl groups from the formation of esters which vibrate around 3200 to 3400 cm−1, therefore overlapping with the N–H bonds. It could also be due to the increase in the number of N–H bonds formed from the interaction between the epoxide group in ENR and the amine end groups in nylon (amine linkage) as shown in Fig. 5. This would therefore imply an increase in the amount of nitros groups (N–O) in the nylon and should be observed through changes in the N–O peaks at 1370 and 1540 cm−1. It can be observed that the intensities are significantly increased as compared to that of neat nylon and NBE7. The peak at 1420 cm−1 was elaborated in the nylon–ENR blend whereas not as much in neither nylon nor the composites. This peak corresponds to the vibration of C–H bonds and is exaggerated due to the vibration of the same bonds also present in the ENR structure. The presence of the CO peak, the increase in intensity of the N–O peaks and broadening of the amide group at 3300 cm−1 suggests the interaction of ENR with nylon through amine and carboxylic acid linkages. Xie et al.5 suggested that blends of ENR and nylon form a graft copolymer (ENR-g-nylon) due to the interactions between the epoxide groups in the ENR and functional groups in nylon. There is good agreement between results found in this study and those by Xie et al. The changes in the spectra of the nylon–ENR blend which are not observed in that of NBE7 when compared to nylon can be attributed to the low concentrations of ENR and therefore fewer interactions between the epoxide groups in ENR with nylon. For these reasons, the ENR–biocarbon and nylon–ENR blends were made.
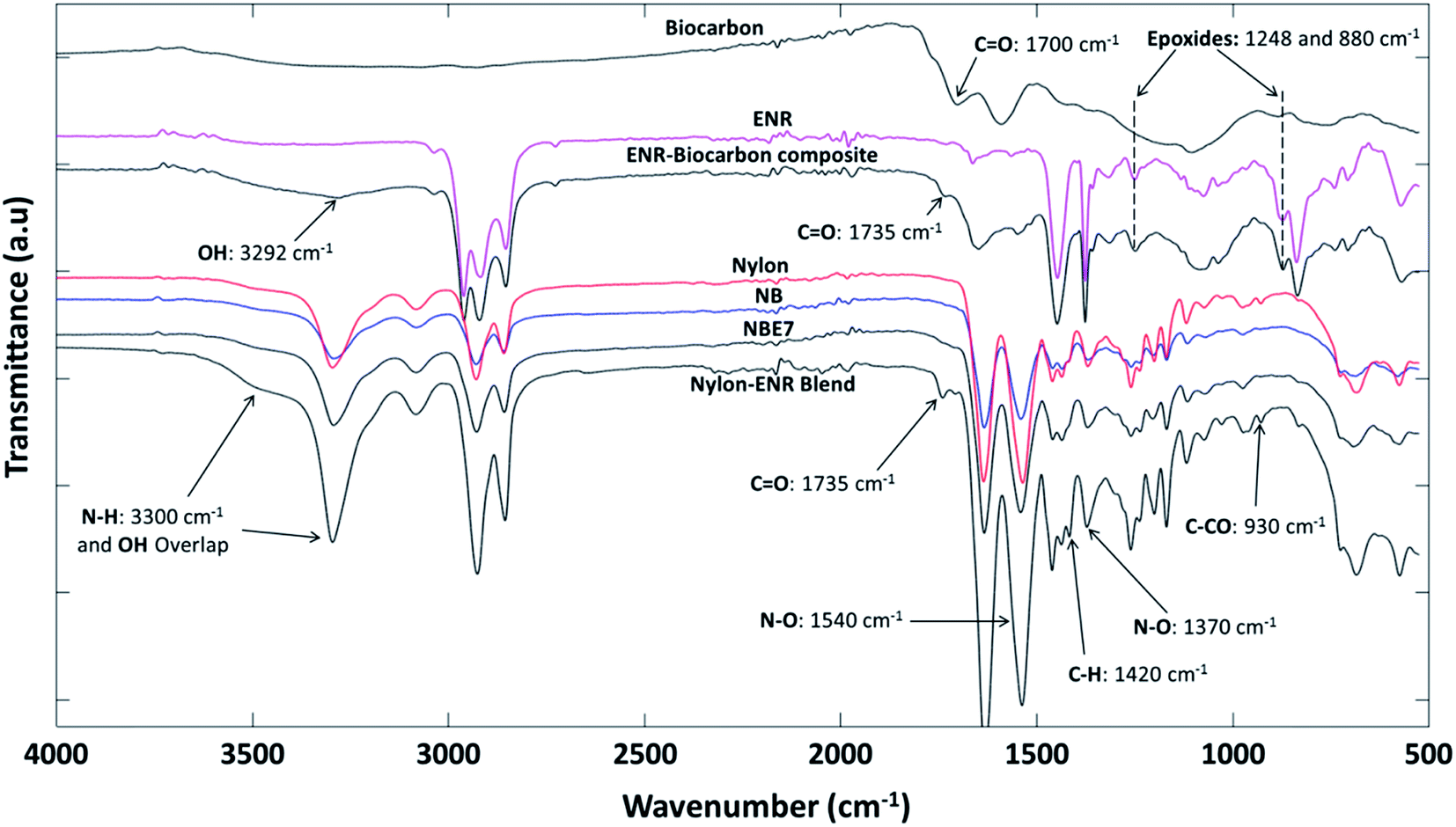 | ||
| Fig. 2 FTIR spectra (from top to bottom) of: biocarbon, ENR, ENR/biocarbon composite, nylon, NB, NBE7 and nylon/ENR blend. | ||
Morphological analysis
The micrograph of the impact fractured surfaces of all the composites are shown in Fig. 3. It can be observed that the surface of NB is relatively flat which is indicative and typical of a brittle fracture. It can also be observed that the biocarbons are well embedded in the matrix and to some degree visible but have a layer of matrix covering them. No voids or gaps can be observed at the interface between the biocarbon and nylon. The absence of voids and presence of a layer of matrix on the biocarbon suggests that the fracture propagated through the matrix and therefore having good adhesion to the biocarbon.21 With the addition of ENR (NBE3), the fracture surface is seen to be irregular with a rough topology. It is even more difficult to distinguish between the nylon, ENR and biocarbon. This is due the ENR acting as an interphase between the nylon and biocarbon partially encapsulating the biocarbon while in some cases, completely encapsulating it. This results to a thicker layer on the biocarbon surface making it difficult to visually identify it. It also corroborates with the results from the tensile and impact strengths, discussed in mechanical properties section. | ||
| Fig. 3 Micrographs of impact fractured surfaces of neat nylon (A), NBE3 (B), NBE5 (C) and (D) NBE7. | ||
Upon further addition of ENR (NBE5 and NBE7) the fractured surfaces become more uneven and the distinction between biocarbon particles and matrix is equally difficult. This is due to the same effect as observed in NBE3 but with more biocarbons being encapsulated with thicker layers of ENR. Evidence of the encapsulation of biocarbon by ENR and fracture of the ENR containing composite through the ENR phase is given in Fig. 4.
 | ||
Fig. 4 Micrographs of etched impact fractured surfaces of NBE3 (A), NBE5 (B) and (C) NBE7 (lower roll are magnification of upper images, 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 511 to 30000 times). 511 to 30000 times). | ||
In Fig. 4, micrographs of the impact fractured surfaces of composites containing ENR after etching with toluene to remove the rubber phase are presented. In NBE3, it can be observed that well distributed and relatively uniform voids are present within the matrix. This suggests that the ENR is well dispersed in the matrix and not forming large island phases. It can also be observed that the biocarbons are now visible and are lodged in some of the voids. The visibility of the biocarbons and presence of gaps around the biocarbons after etching out ENR show that the ENR encapsulated the biocarbon and acted as an interphase between the biocarbon and nylon. Similar trend is observed with further increase in ENR concentration; encapsulation of the biocarbons and a well dispersed rubber phase within the matrix is observed. However, it is noticed that the voids in the nylon are larger in size while the gaps between the nylon and biocarbon is thicker. Wider gaps indicate that a thicker layer of ENR was encapsulating the biocarbon particles while larger and irregularly shaped voids could indicate the removal of biocarbon particles which have no adhesion to the nylon after etching out of the ENR.
In summary, the presence of epoxide groups on the rubber makes it reactive with both nylon and the biocarbons and therefore drives it to their interface. The epoxide group can react with the nylon through carboxylic acid and amine end group linkages12,22 while also reacting with the COOH groups on the biocarbons. Similar results were observed by Hornsby et al.11 where rubber encapsulated glass beads were seen in polypropylene/rubber/glass beads composites when maleic anhydride grafted ethylene propylene rubber (MA-EPR) was used. Complete separation of the phases, with no interaction was observed when un-functionalized ethylene propylene rubber was used. Encapsulation of the glass beads by the MA-EPR was due to the reaction between the OH groups and maleic anhydride therefore driving it to the interface between the polypropylene and glass beads.
Thermal analysis
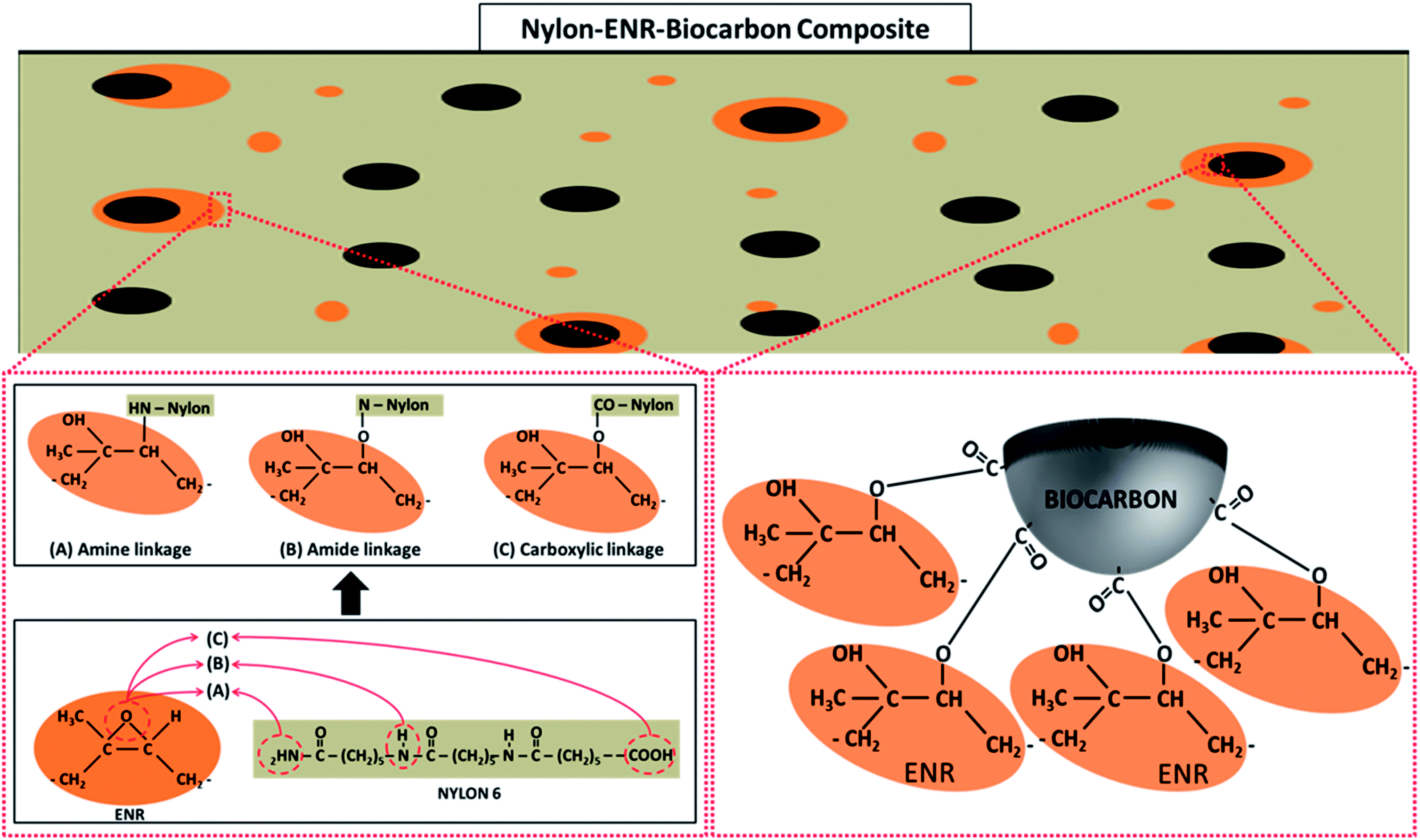 | ||
| Fig. 5 Representation of plausible interactions between nylon, biocarbon and ENR. | ||
 | ||
| Fig. 6 DSC heating cycle of nylon and the composites. | ||
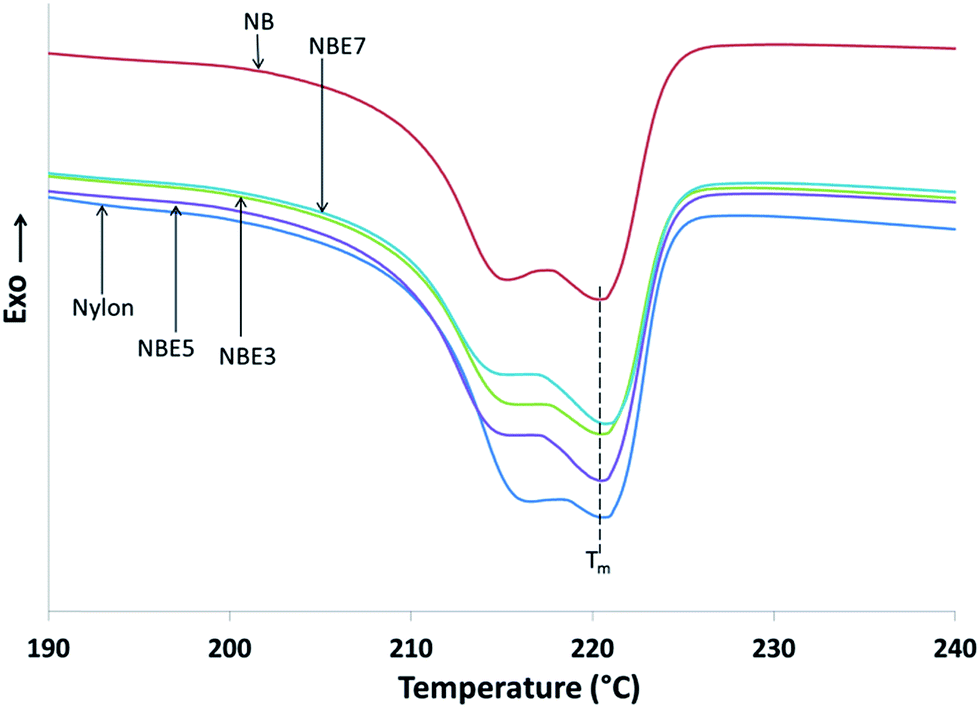
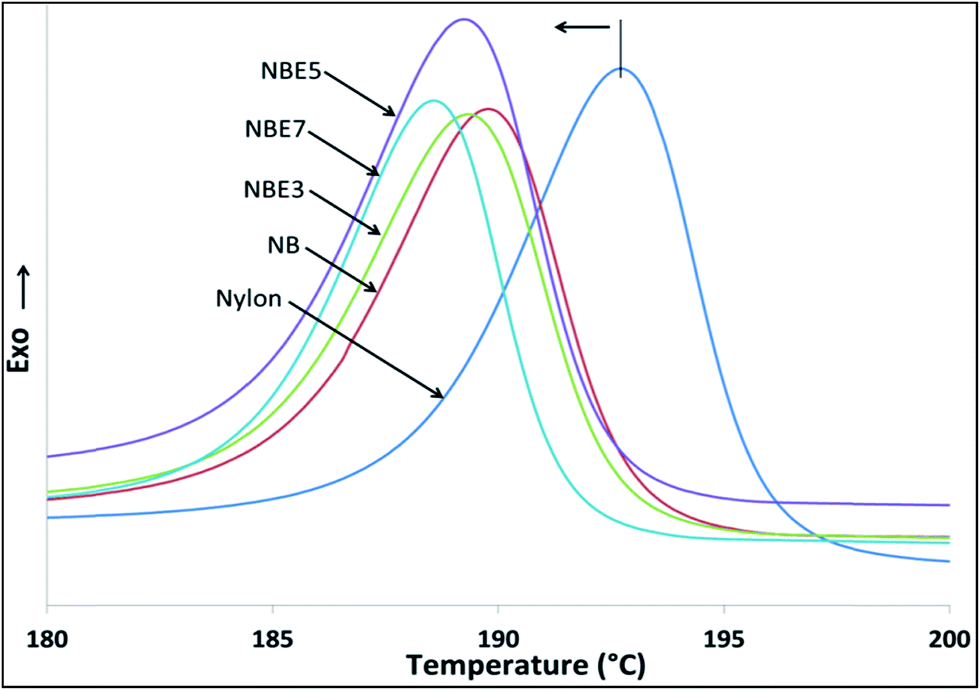
The cooling cycle of nylon and its composites are shown in Fig. 7. Table 3 summarizes the thermal attributes of all the samples. The melt crystallization curves of the composites compared to neat nylon is observed to shift to a lower temperature indicating that there is restriction of the nylon chains from crystallizing. The addition of biocarbon to nylon (NB) reduces the temperature from 192.7 °C to 189.8 °C implying that biocarbon restricts the mobility and therefore hindering crystal formation. When NBE3, NBE5 and NBE7 are observed, it can be noticed that the melt crystallization temperatures remains mostly the same as NB with no trend, indicating no effect on enhancing or retarding nucleation of crystals.
 | ||
| Fig. 7 DSC cooling cycle of nylon and the composites. | ||
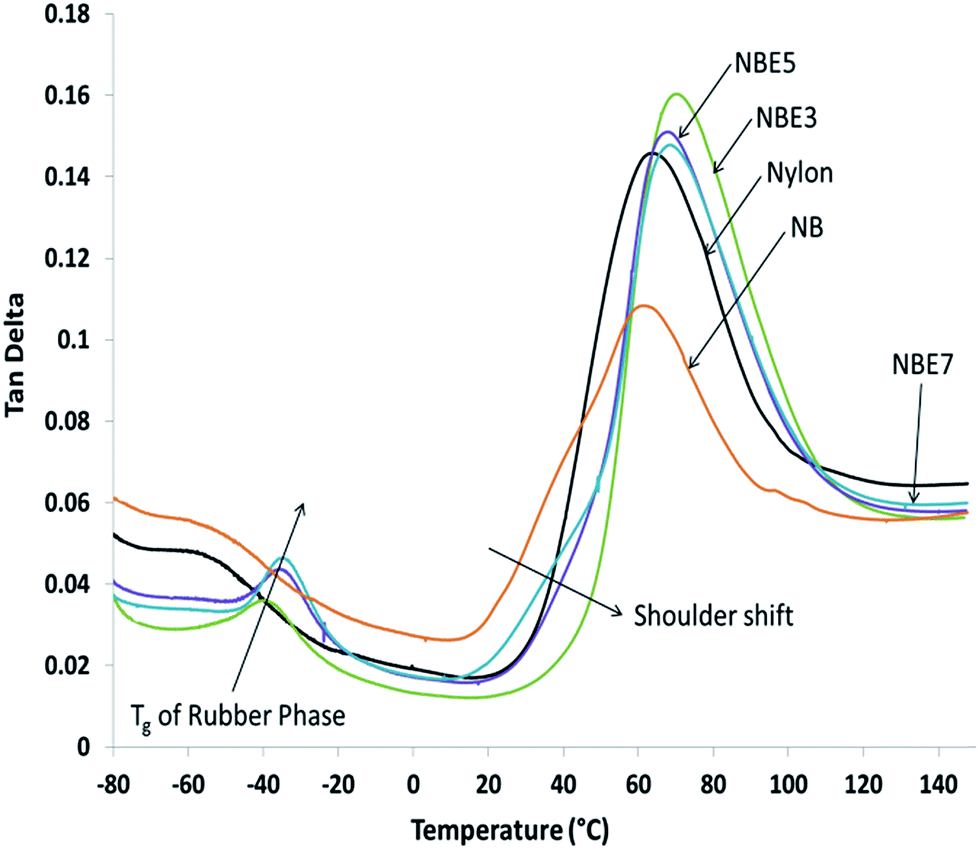
The viscoelastic behavior of the composites compared to neat nylon is represented in Fig. 8 and Table 2. The slight vibrations of the long molecular chains of the amorphous phase in nylon as detected by DMA was used to determine the Tg of both the nylon and ENR phases and also to observe any possible effect they have on each other. Two distinct sets of peaks can be observed; a set between −40 °C and −20 °C and another between 55 °C and 75 °C representing the Tg of ENR and nylon respectively. The nylon peak is at approximately 64 °C as determined from the tan delta curve apex. When biocarbon is added to nylon (NB), the intensity of the peak is reduced and a shift to a lower temperature of 60 °C is observed. Reduction in the intensity is due to the restricting effect of biocarbon on the nylon chains thereby reducing its ability to absorb energy. Addition of ENR to the composite (NBE3) shows an increase in the peak intensity and a shift to a higher temperature of 70.58 °C when compared to NB. Further increase of ENR shows no significant changes to the peak with the Tg remaining mostly the same. This behavior could be due to two mechanisms; encapsulation of the biocarbon and the formation ENR-g-nylon at the ENR–nylon interface. Encapsulation of some the biocarbon particles increases the ability of the nylon to absorb energy as it acts like a particle with a stiff core but rubbery outer layer. The formation of ENR-g-nylon has been reported in literature to be more thermally stable than both nylon or ENR8 and also stronger with higher flow resistance in the molten state.27 Therefore, the presence of a more thermally stable, stronger and flow resistant material grafted to nylon chains can cause a shift of the Tg to a higher temperature. Nouparvar et al.8 studied the thermal properties of nylon 6 and ENR blend composites with clay and found that the formation of ENR-g-nylon at the interface led to improved melting temperature of the nylon due to its higher thermal stability. A progressive shoulder shift or onset away from NB to higher temperatures can also be observed in NBE3, NBE5 and NBE7. This shows that the chains require more energy or heat to cause vibration and motion possibly due to the ENR-g-nylon.
 | ||
| Fig. 8 DMA graph showing the tan delta peaks of PA6 and its composites. | ||
| Samples | Tc (°C) | ΔHc (J g−1) | Tm (°C) | ΔHm (J g−1) | Tg, DMA–nylon (°C) | Tg, DMA–rubber (°C) |
|---|---|---|---|---|---|---|
| Nylon | 192.7 | 69.29 | 220.63 | 55.66 | 65.49 (2.41) | — |
| NB | 189.77 | 48.99 | 220.44 | 40.33 | 60.24 (1.42) | — |
| NBE3 | 189.93 | 47.57 | 221.08 | 44.43 | 70.58 (0.61) | −39.43 (0.24) |
| NBE5 | 188.19 | 44.72 | 220.86 | 44.61 | 69.85 (2.93) | −35.95 (0.59) |
| NBE7 | 188.56 | 47.42 | 220.74 | 43.39 | 69.36 (1.23) | −35.39 (0.45) |
Mechanical properties
 | ||
| Fig. 9 Tensile strength and modulus of nylon 6 and composites. | ||
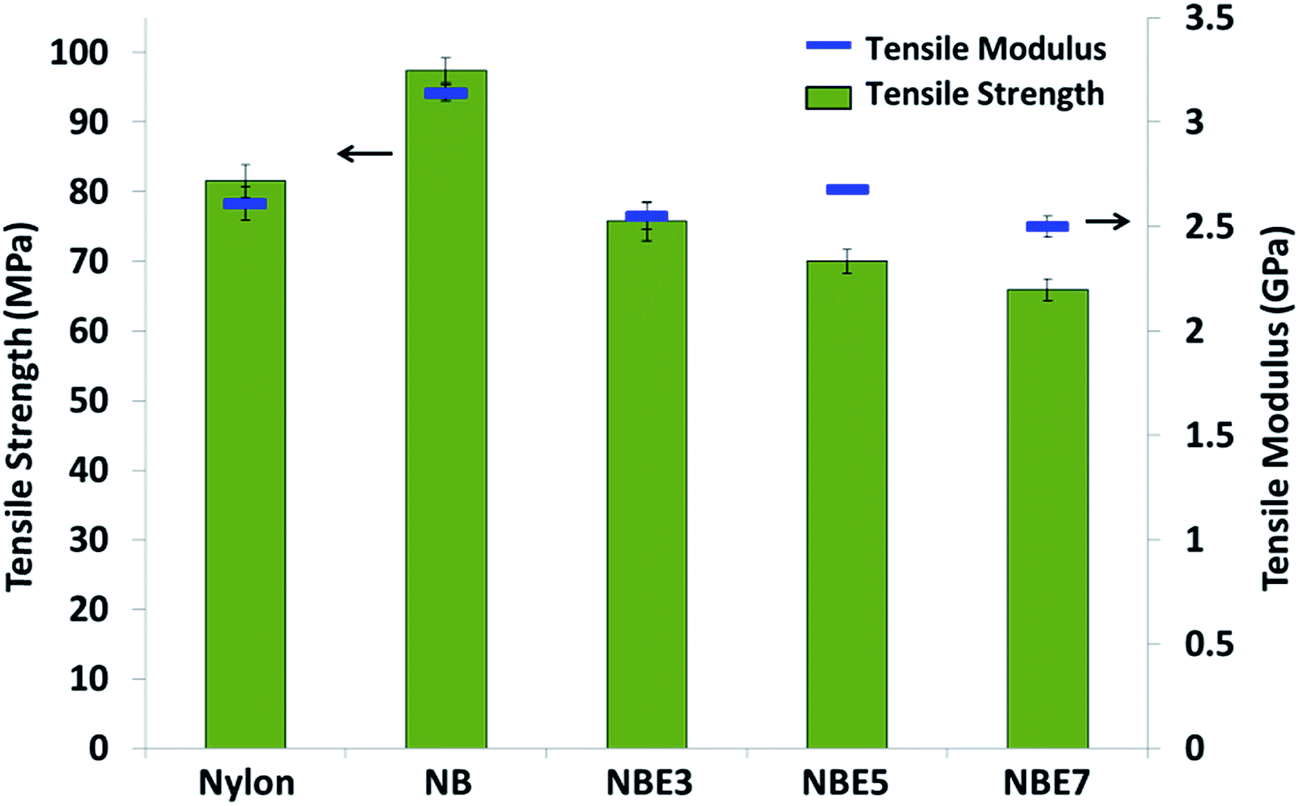
The tensile moduli of the composites also follow the same trend as the strength. There is an initial increase with the addition of biocarbon (NB) and then a decrease with the addition of ENR and subsequent addition thereafter (NBE3, NBE5 and NBE7). Addition of stiff filler to a polymer typically causes the modulus to increase as the filler acts to restrict the polymer chains. In our previous study, addition of biocarbon to nylon 6, 10 resulted in improvements in the moduli of the composites.18 Polycaprolactone filled with carbonized lignin particles also exhibited an increase in the composite modulus.31 ENR has a very low modulus; inferior to that of nylon and biocarbon and therefore decrease the modulus of the composites with increasing concentration as it is dispersed in the nylon. The sharp decrease noticed from NB to NBE3 can be attributed to the encapsulation of the biocarbon with ENR. A change in the phase structure from biocarbon–nylon to ENR–nylon interfaces prevents the biocarbon from restricting the mobility of nylon chains but acting as an elastic particle with a hard core. In the same work by Hornsby et al., comparable results were observed in the modulus. There was a decrease resulting from the rubber encapsulation of the glass beads.
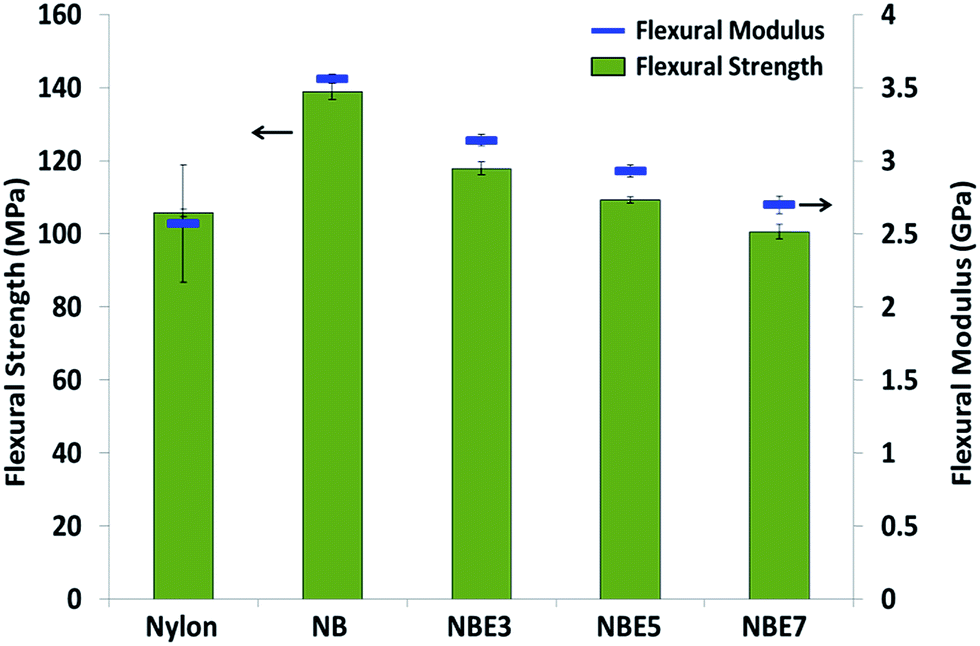 | ||
| Fig. 10 Flexural strength and modulus of nylon and its hybrid composites. | ||
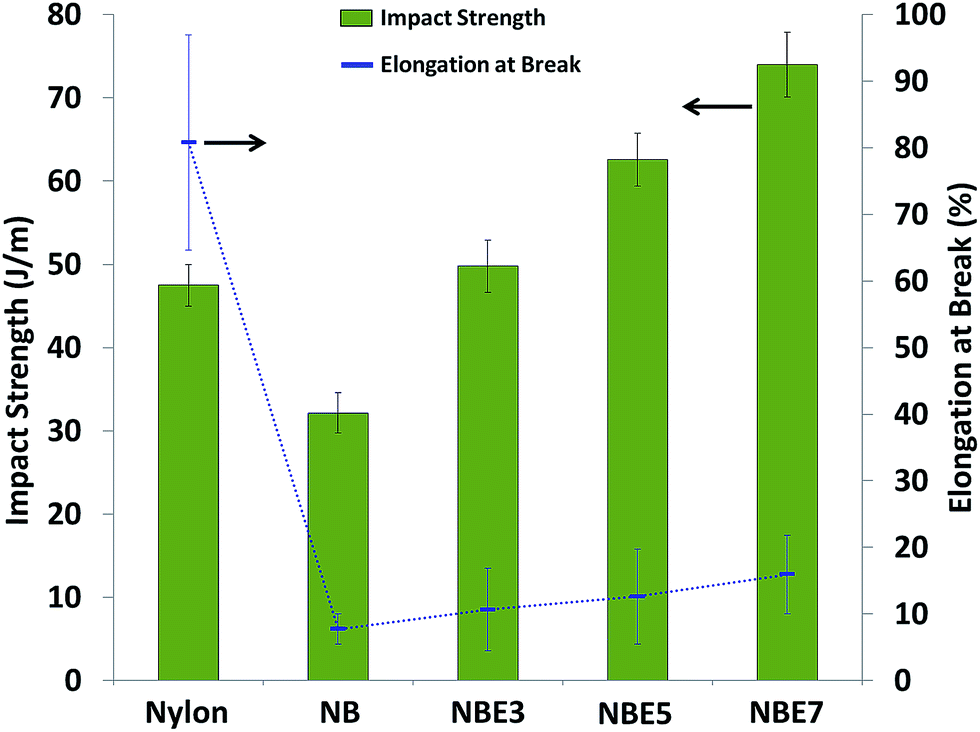 | ||
| Fig. 11 Impact strength and elongation of nylon 6 and its composites. | ||
| Properties | Unit | 20 wt% biocarbon filled nylon 6a | 20 wt% biocarbon + 3 phr ENR filled nylon 6b | 20 wt% mineral filled nylon 6c | 20 wt% natural fiber filled nylon 6d |
|---|---|---|---|---|---|
| a Data from this study.b Data from this study.c Datasheet from commercially available mineral filled nylon 6 (RTP 225 A).d Experimental data of APV3 from Santos et al.34 | |||||
| Tensile strength | MPa | 97 | 76 | 69 | 67 |
| Tensile modulus | GPa | 3.14 | 2.55 | 4.482 | 3.8 |
| Elongation at break | % | 7.75 | 11 | 3–4 | 2.9 |
| Flexural strength | MPa | 139 | 118 | 110 | 109 |
| Flexural modulus | GPa | 3.6 | 3.14 | 3.792 | 3.4 |
| Specific tensile strength | MPa (gcm−3)−1 | 84 | 65 | 53.91 | — |
| Specific tensile modulus | GPa (gcm−3) −1 | 2.7 | 2.19 | 3.5 | — |
| Specific flexural strength | MPa (gcm−3)−1 | 119 | 101 | 85.94 | — |
| Specific flexural modulus | GPa (gcm−3) −1 | 3.08 | 2.7 | 2.96 | — |
| Impact strength | J m−1 | 32 | 50 | 43 | 100 |
| Density | g cm−3 | 1.165 | 1.165 | 1.28 | — |
Water absorption
The water absorption of the composites was tested and the results showed very interesting behaviors. There was little to no difference between the neat nylon and the composites at equilibrium. It was expected that the presence of a hydrophobic phase (ENR) dispersed within the hydrophilic phase (nylon) and the encapsulation of the biocarbon by the ENR will drastically reduce the water uptake. However, the contrary is observed when the curves of the equilibrium water uptake region are magnified (Fig. 12). As observed, there is an erratic decrease and increase of the curves of all the samples at the equilibrium region. This is due to sorption and desorption which occurs at very minute levels when equilibrium is reached. We observe a trend; NBE7 showed the highest water uptake and decreases with reduction in the ENR content followed by NB then neat nylon. If a trend line was to be fitted to the curve, it will show clearer difference between the samples. This behavior can be attributed to the presence of hydroxyl groups which were formed from the formation of esters through the reaction of epoxide and carboxylic groups. With increase in the ENR content, there is an increase in the formation of esters which also produce hydroxyl groups as by products. It is well known that hydroxyl groups are highly hydrophilic and bond to water molecules very easily hence resulting in slightly higher water uptake than neat nylon and NB.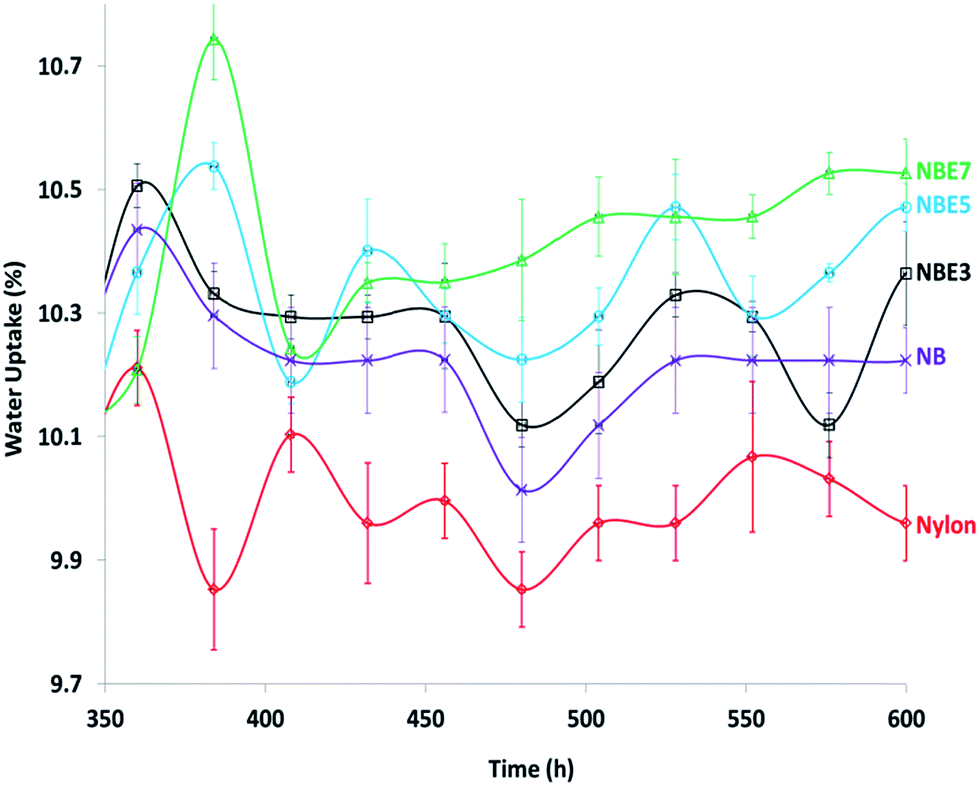 | ||
| Fig. 12 Expanded equilibrium region of the water uptake of nylon 6 and its composites. | ||
Conclusion
The study of the reinforcing and impact modifying effects of biocarbon and ENR, respectively, on nylon showed behaviors which were directly related to the microstructure formations of the composites as observed from the morphological properties and ENR and nylon. It was shown that the chemical interactions between ENR and biocarbon resulted to the encapsulation of the biocarbon by the ENR while also being dispersed in the nylon. FTIR confirmed the interactions between ENR–biocarbon and ENR–nylon by the presence of new peaks which were attributed to the CO vibration from the formation of ester groups from the interactions between epoxide groups on the ENR and carboxylic groups on the nylon and biocarbon. These resulted to drastic improvements in the impact strength of the composites when compared to that of NB at relatively low concentrations of ENR. The interactions between the biocarbon and nylon also resulted to good interfaces and increased tensile and flexural properties. The formation of an interface phase (ENR-g-nylon) was suggested to play an intrinsic role in the thermal properties such as the improvements in the glass transition temperatures of the ENR and nylon. The use of biocarbon and ENR to reinforce and modify the impact property of nylon respectively, showed a synergistic effect resulting to well-balanced properties. Based on the stiffness-strength-toughness balance in comparison to the other impact modified biocomposites and bench marking against commercially available mineral filled nylon 6 (RTP 225 A) used in the automotive industry, biocarbon reinforced nylon 6 using 3 phr of ENR provides the best or optimum balance of mechanical properties within the concentrations studied in this work. The properties are either superior or equal to those of the neat nylon 6 and RTP 225 A. It is believed that knowledge gained from this work can be used to further explore and engineer biocomposites derived from biocarbons geared towards automotive applications especially.
Acknowledgements
The authors acknowledge the financial support by the Ontario Ministry of Agriculture, Food and Rural Affairs (OMAFRA) – University of Guelph Product Development and Enhancement through Value Chains Research Theme (Project # 200399), the Natural Sciences and Engineering Research Council of Canada (NSERC) (Project # 401111) and Ontario Research Fund, Research Excellence Program; Round-7 (ORF-RE07) from the Ontario Ministry of Research and Innovation (MRI), currently known as the Ontario Ministry of Research, Innovation and Science (MRIS) (Project # 052644 and # 052665).References
- D. Laura, H. Keskkula, J. Barlow and D. Paul, Effect of rubber particle size and rubber type on the mechanical properties of glass fiber reinforced, rubber-toughened nylon 6, Polymer, 2003, 44, 3347–3361 CrossRef CAS.
- D. Laura, H. Keskkula, J. Barlow and D. Paul, Effect of glass fiber surface chemistry on the mechanical properties of glass fiber reinforced, rubber-toughened nylon 6, Polymer, 2002, 43, 4673–4687 CrossRef CAS.
- S. Tjong, Short glass fiber-reinforced polyamide 6,6 composites toughened with maleated SEBS, Compos. Sci. Technol., 2002, 62, 2017–2027 CrossRef CAS.
- M. G. Segatelli, I. V. P. Yoshida and M. D. C. Gonçalves, Natural silica fiber as reinforcing filler of nylon 6, Composites, Part B, 2010, 41, 98–105 CrossRef.
- B. Xie, M. Yang, S. Li, Z. Li and J. Feng, Studies on polyamide-6/polyolefin blend system compatibilized with epoxidized natural rubber, J. Appl. Polym. Sci., 2003, 88, 398–403 CrossRef CAS.
- M. Narathichat, C. Kummerlöwe, N. Vennemann and C. Nakason, Thermoplastic natural rubber based on polyamide-12: influence of blending technique and type of rubber on temperature scanning stress relaxation and other related properties, J. Appl. Polym. Sci., 2011, 121, 805–814 CrossRef CAS.
- M. Narathichat, C. Kummerlöwe, N. Vennemann, K. Sahakaro and C. Nakason, Influence of epoxide level and reactive blending on properties of epoxidized natural rubber and nylon-12 blends, Adv. Polym. Technol., 2012, 31, 118–129 CrossRef CAS.
- H. Nouparvar, A. Hassan, Z. Mohamad, M. U. Wahit and F. Roozbahani, The effect of organoclay contents on morphological characterization, mechanical and thermal properties of epoxidized natural rubber-50 toughened polyamide 6 nanocomposites, J. Polym. Eng., 2014, 34, 59–68 CAS.
- J. Jancar and A. Dibenedetto, Effect of morphology on the behaviour of ternary composites of polypropylene with inorganic fillers and elastomer inclusions, J. Mater. Sci., 1995, 311, 1601–1608 CrossRef.
- C. G. Ma, M. Q. Zhang and M. Z. Rong, Morphology prediction of ternary polypropylene composites containing elastomer and calcium carbonate nanoparticles filler, J. Appl. Polym. Sci., 2007, 103, 1578–1584 CrossRef CAS.
- P. R. Hornsby and K. Premphet, Influence of phase microstructure on the mechanical properties of ternary phase polypropylene composites, J. Appl. Polym. Sci., 1998, 70, 587–597 CrossRef CAS.
- H. Nouparvar, A. Hassan, Z. Mohamad and M. U. Wahit, Epoxidized natural rubber-50 toughened polyamide 6 nanocomposites: the effect of epoxidized natural rubber-50 contents on morphological characterization, mechanical and thermal properties, J. Elastomers Plast., 2012, 46, 269–283 CrossRef.
- N. F. A. Sharif, Z. Mohamad, A. Hassan and M. U. Wahit, Novel epoxidized natural rubber toughened polyamide 6/halloysite nanotubes nanocomposites, J. Polym. Res., 2011, 19, 9749 CrossRef.
- A. Mohanty, M. Misra and L. Drzal, Natural fibers, biopolymers, and biocomposites, 2005 Search PubMed.
- X. Cao and W. Harris, Properties of dairy-manure-derived biochar pertinent to its potential use in remediation, Bioresour. Technol., 2010, 101, 5222–5228 CrossRef CAS PubMed.
- A. K. Mohanty, S. Vivekanandhan, A. Anstey and M. Misra, Sustainable Composites From Renewable Biochar And Engineering Plastic, in: 20th International Conference on Composite Materials, Copenhagen, 2015 Search PubMed.
- T. Huber, M. Misra and A. K. Mohanty, Biochar and its Size Effects on Polyamide 6/Biochar Composites, in: Proceedings of The American Society for Composites, 2015 Search PubMed.
- E. O. Ogunsona, M. Misra and A. K. Mohanty, Sustainable biocomposites from biobased polyamide 6,10 and biocarbon from pyrolyzed Miscanthus fibers, J. Appl. Polym. Sci., 2016, 134, 44221 Search PubMed.
- E. Behazin, E. Ogunsona, A. Rodriguez-Uribe, A. K. Mohanty, M. Misra and A. O. Anyia, Mechanical, Chemical, and Physical Properties of Wood and Perennial Grass Biochars for Possible Composite Application, BioResources, 2016, 11, 1334–1348 CAS.
- A. K. Manna, P. P. De, D. K. Tripathy, S. K. De and M. K. Chatterjee, Chemical Interaction between Surface Oxidized Carbon Black and Epoxidized Natural Rubber, Rubber Chem. Technol., 1997, 70, 624–633 CrossRef CAS.
- S. Ahmed and F. Jones, A review of particulate reinforcement theories for polymer composites, J. Mater. Sci., 1990, 25, 4933–4942 CrossRef CAS.
- C. S. L. Baker, I. R. Gelling and R. Newell, Epoxidized Natural Rubber, Rubber Chem. Technol., 1985, 58, 67–85 CrossRef CAS.
- T.-C. Li, J. Ma, M. Wang, W. C. Tjiu, T. Liu and W. Huang, Effect of clay addition on the morphology and thermal behavior of polyamide 6, J. Appl. Polym. Sci., 2007, 103, 1191–1199 CrossRef CAS.
- N. S. Murthy, Hydrogen bonding, mobility, and structural transitions in aliphatic polyamides, J. Polym. Sci., Part B: Polym. Phys., 2006, 44, 1763–1782 CrossRef CAS.
- J. Jiang, D. Zhang, Y. Zhang, K. Zhang and G. Wu, Influences of Carbon Nanotube Networking on the Conductive, Crystallization, and Thermal Expansion Behaviors of PA610-Based Nanocomposites, J. Macromol. Sci., Part B: Phys., 2013, 52, 910–923 CrossRef CAS.
- J. D. Menczel, L. Judovits, R. B. Prime, H. E. Bair, M. Reading and S. Swier, Differential Scanning Calorimetry (DSC), in: Thermal Analysis of Polymers, John Wiley & Sons, Inc., 2008, pp. 7–239 Search PubMed.
- C. Nakason, M. Narathichat, C. Kummerlowe and N. Vennemann, Thermoplastic natural rubber based on polyamide-12 blended with various types of natural rubber: effect of processing oils and plasticizer, J. Elastomers Plast., 2012, 45, 47–75 CrossRef.
- C. Komalan, Dynamic mechanical analysis of binary and ternary polymer blends based on nylon copolymer/EPDM rubber and EPM grafted maleic anhydride compatibilizer, eXPRESS Polym. Lett., 2007, 1, 641–653 CrossRef CAS.
- Z. You and D. Li, Highly filled bamboo charcoal powder reinforced ultra-high molecular weight polyethylene, Mater. Lett., 2014, 122, 121–124 CrossRef CAS.
- X. Li, B. Lei, Z. Lin, L. Huang, S. Tan and X. Cai, The utilization of bamboo charcoal enhances wood plastic composites with excellent mechanical and thermal properties, Mater. Des., 2014, 53, 419–424 CrossRef CAS.
- W. Gindl-Altmutter, C. Fürst, A. Raj Mahendran, M. Obersriebnig, G. Emsenhuber and M. Kluge, et al., Electrically conductive kraft lignin-based carbon filler for polymers, Carbon, 2015, 89, 161–168 CrossRef CAS.
- R. J. M. Borggreve, R. J. Gaymans and J. Schuijer, Impact behaviour of nylon–rubber blends: 5. Influence of the mechanical properties of the elastomer, Polymer, 1989, 30, 71–77 CrossRef CAS.
- S. Y. Hobbs, R. C. Bopp and V. H. Watkins, Toughened nylon resins, Polym. Eng. Sci., 1983, 23, 380–389 CAS.
- P. A. Santos, M. A. S. Spinacé, K. K. G. Fermoselli and M.-A. De Paoli, Polyamide-6/vegetal fiber composite prepared by extrusion and injection molding, Composites, Part A, 2007, 38, 2404–2411 CrossRef.
- RTP Co., Product Datasheet and General Processing Conditions RTP 225 A Nylon 6 (PA) Mineral, http://web.rtpcompany.com/info/data/0200A/RTP225A.htm, Extracted on 15th Nov, 2016.
| This journal is © The Royal Society of Chemistry 2017 |