
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A nitrogen-doped three-dimensional carbon framework for high performance sodium ion batteries†
Ying Wanga,
Caiyun Wangc,
Huinan Guob,
Yijing Wang *b and
Zhenguo Huang*d
*b and
Zhenguo Huang*d
aSchool of Chemistry and Chemical Engineering, Jiangsu Key Laboratory of Green Synthetic Chemistry for Functional Materials, Jiangsu Normal University, Xuzhou, Jiangsu 221116, China
bCollaborative Innovation Centre of Chemical Science and Engineering (Tianjin), Key Laboratory of Advanced Energy Materials Chemistry (MOE), Nankai University, Tianjin 300071, China. E-mail: wangyj@nankai.edu.cn
cIntelligent Polymer Research Institute, ARC Centre of Excellence for Electromaterials Science, University of Wollongong, Wollongong, NSW 2522, Australia
dInstitute for Superconducting and Electronic Materials, University of Wollongong, Wollongong, NSW 2522, Australia. E-mail: zhenguo@uow.edu.au
First published on 5th January 2017
Abstract
A three-dimensional nitrogen-doped carbon framework (NCF) has been synthesized by annealing a zeolitic imidazolate framework (ZIF-67), followed by removing the residual metal ions. The NCF shows a polyhedral outline with strong interconnected hollow nanospheres as its subunits. It is highly porous, with a large surface area of 623 m2 g−1, and a high nitrogen content of 12.3 wt%. When applied as an anode material for a sodium ion battery, the NCF exhibits an excellent electrochemical performance with a high reversible capacity (327 mA h g−1, and stable at 300 mA h g−1), good cycling stability, and excellent rate capability (175 mA h g−1 at 2000 mA g−1). The presence of N in the NCF is believed to provide more active sites for enhancing the electrochemical performance, and provide more defects and nanovoids to trap more Na+ ions.
1. Introduction
High energy density storage devices, particularly lithium ion batteries (LIBs), hold the key to the development of portable electronic devices.1–3 Their application in electric vehicles and large stationary energy storage, however, has aroused concerns about the supply of lithium. Sodium ion batteries (SIBs) have therefore become the most promising candidate for large-scale energy storage due to the abundance of sodium.4 The fundamental working principles of SIBs are similar to those of LIBs. Thus some highly active anode materials in LIBs also display good electrochemical performance in SIBs, such as the conversion-based materials.5–8 The search for effective insertion/de-insertion-based anode materials, on the other hand, is still challenging due to the larger radius of Na+ (0.102 nm) compared to that of Li+ (0.076 nm) and its thermodynamic properties.9,10Carbon materials show huge potential as anode materials for SIBs owing to their low cost, high chemical stability, and high electrical conductivity. To date, hard carbon has been the most investigated anode material, due to its generally larger d-spacing.11–15 It has shown considerable Na+ storage capacity (100–200 mA h g−1), but with unsatisfactory cycling stability and rate capability. Improved electrochemical performance could be delivered by using nanostructured carbon materials such as hollow spheres,16,17 nanosheets,18,19 and nanofibers.20 The porous nature of these materials enables better contact between the electrode and electrolyte, and shortened diffusion paths for Na+ ions, and thus improved the electrochemical performance.
The performance of carbon materials can be further optimized by the introduction of heteroatoms (e.g. N, B, P). Heteroatom doped carbon shows higher specific capacities and better cycling stability.21–26 Nitrogen has a smaller atomic diameter and higher electronegativity than carbon, and has been considered as a good choice for heteroatom doping. For example, N-doped hollow carbon showed a high capacity of 89.9 mA h g−1 at 5 A g−1, outperforming the non-doped counterparts.27 Tradition fabrication of N-doped carbon materials normally requires the addition of a nitrogen source (ammonia, melamine etc.) or carbonizing polypyrrole, dopamine, or polyacrylonitrile.27–29 And the doping level is normally below ∼10 wt%.30,31 Therefore, it would be highly desirable to fabricate nanostructured carbon materials with a higher N-doping level.32,33
In this work, we report a highly efficient way to synthesize a three-dimensional (3D) nitrogen-doped carbon framework (NCF) with 12.3 wt% nitrogen (doping level) derived from a zeolitic imidazolate framework (ZIF-67). The obtained 3D NCF possesses the polyhedral outline of its precursor and consists of hollow nanospheres. Its unique structure facilitates good Na+ storage performance with a high reversible capacity (327 mA h g−1), good cycling stability (86% capacity retention after 200 cycles at 100 mA g−1), and excellent rate capability (175 mA h g−1 at a high current density of 2000 mA g−1).
2. Experimental
2.1 Synthesis of 3D NCF
2-Dimethylimidazole and Co(NO3)2·6H2O were purchased from Sigma-Aldrich and used as received. ZIF-67 was synthesized according to a previous report with minor modification.34 To obtain the NCF, the ZIF-67 powders were first heated to 950 °C (heating rate: 3 °C min−1, dwell time: 120 min) under Ar. After naturally cooling to room temperature, the obtained black powders were treated in 6 M HCl at 70 °C for 12 h. This process was repeated three times to completely remove the residual cobalt. The NCF solid powders were collected after centrifugation, rinsed with deionized water, and then dried in an oven at 80 °C.2.2 Characterization
X-ray diffraction (XRD, Rigaku MiniFlexII, Cu Kα radiation), X-ray photoelectron spectroscopy (XPS, VG Scientific ESCALAB 2201XL, Al Kα X-ray radiation), nitrogen sorption isothermal analysis (NOVA 2200e, Quantachrome), and Raman spectroscopy (Jobin Yvon HR800 Raman spectrometer with a 10 mW helium/neon laser at 632.8 nm excitation) were used to determine the structure and composition. Element analysis was carried out on an Elementar vario EL CUBE. Scanning electron microscopy (SEM, JEOL JSM7500FA), transmission electron microscopy (TEM), and high-resolution TEM (HR-TEM, JEOL JEM-2010FEF) were used for morphological and structural investigations.2.3 Electrochemical measurements
Standard 2032-type coin cells were assembled to determine the electrochemical performance. The working electrodes were fabricated with 80 wt% active materials, 10 wt% acetylene black (Super P), and 10 wt% sodium carboxymethyl cellulose (CMC). A sodium disc was used as the counter electrode. The electrodes were separated by a glass fiber separator. 1 M NaClO4 in ethylene carbonate and diethyl carbonate with the addition of 5% fluorinated ethylene carbonate (EC/DEC/FEC) was used as electrolyte. The cells were assembled in an Ar filled glove box with the oxygen and moisture content below 1 ppm. Galvanostatic charge–discharge tests were carried out on a battery testing system (Shenzhen NEWARE Battery) over a voltage range of 0.001 to 3.00 V. Electrochemical impedance spectroscopy (EIS) and cyclic voltammetry (CV) measurements were performed on a CHI660E electrochemical workstation.3. Results and discussion
The fabrication procedure for 3D NCF is illustrated in Scheme 1. During the thermal annealing, the organic linkers in ZIF-67 are carbonized into interconnected nitrogen-doped carbon shells, while the Co2+ is reduced to cobalt metal, as previously reported.5 Hydrochloric acid was then used to etch the residual cobalt metal.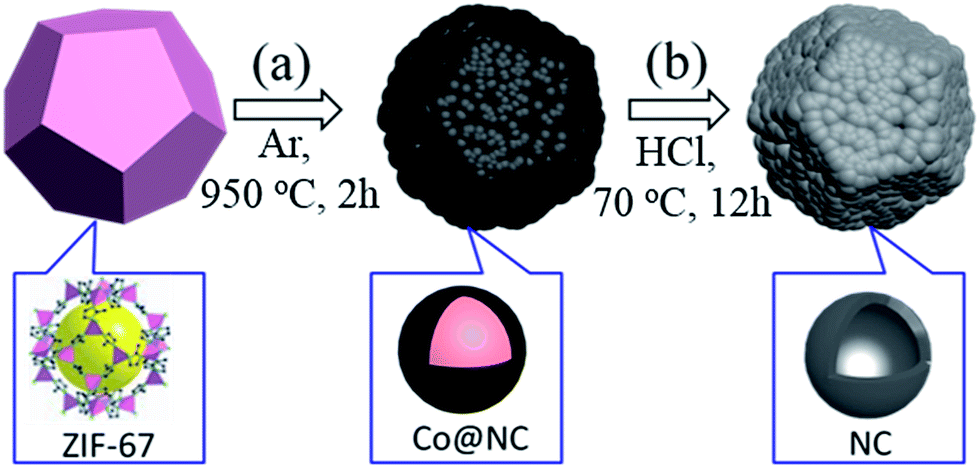 | ||
| Scheme 1 Schematic illustration of the formation procedure of 3D NCF via calcining ZIF-67 (a), and then etching to remove the Co (b). The figures in the bottom row illustrate the subunit structure of each material. | ||
The NCF basically inherits the polyhedral structure of ZIF-67, as shown in Fig. 1a. The partially broken polyhedra (inset to Fig. 1b) show their hollow interior. After the calcination the smooth shell of ZIF-67 pyrolyzed into numerous nanoparticles. The TEM images (Fig. 1c) and the HR-TEM image (Fig. 1d) illustrate the hollow nature of the NCF subunit (red circle in Fig. 1c), and the sizes of these hollow nanospheres vary from 3 to 5 nm. The interlayer spacing of NCF is 0.39 nm, which is large enough for sodiation/de-sodiation during the electrochemical process.
 | ||
| Fig. 1 SEM images of the ZIF-67 precursor (a), and the as-obtained 3D NCF (b), with the inset showing the hollow structure of the polyhedra. TEM (c) and HR-TEM (d) images of the NCF. The inset to (c) is an enlargement of the indicated area, and the red circles in (d) mark the hollow centers of the nanospheres. | ||
The XRD pattern of the NCF (Fig. 2a) shows two strong diffraction peaks of graphite. The C (002) diffraction at 23.0° corresponds to an interlayer spacing of 0.39 nm. Such an enlarged interlayer space has been deemed to be effective for Na+ insertion/de-insertion.11,26 In its Raman spectrum (Fig. 2b), the D band (located at 1332 cm−1) is associated with structural defects, and the G (1590 cm−1) band is due to sp2 carbon. The intensity ratio of the D band to the G band (ID/IG) is inversely proportional to the crystallization. The high ID/IG value (1.04, compared with ∼0.1 for fully graphitized carbon)35,36 implies that the nitrogen doping induces more defects in this NCF, which can facilitate electron transport and trap more Na+ ions during the electrochemical reactions.27,37
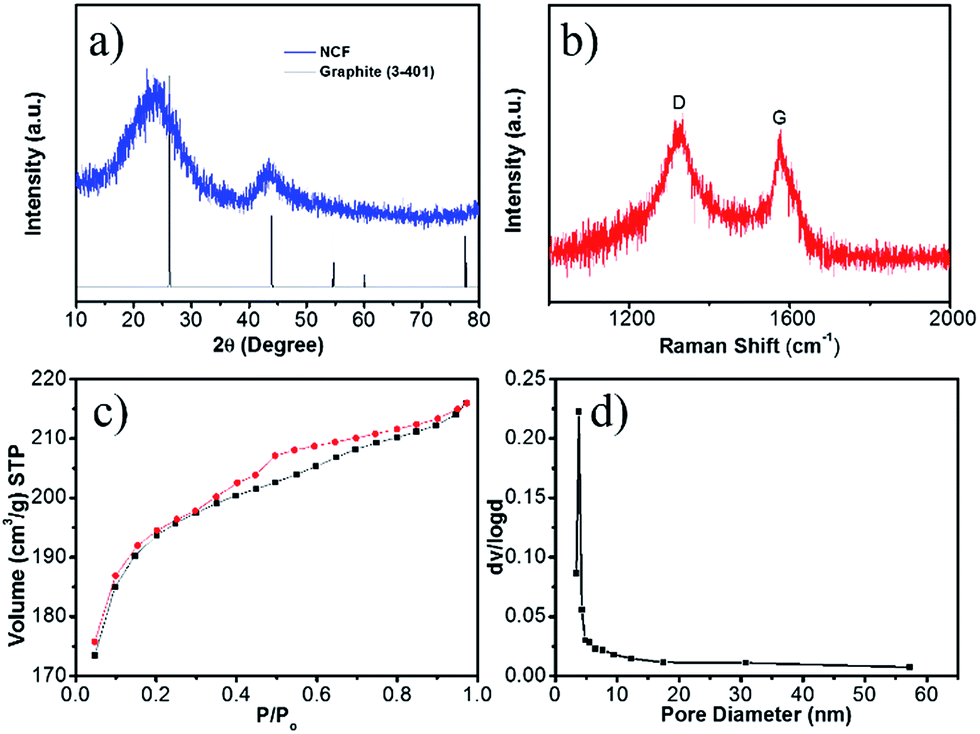 | ||
| Fig. 2 XRD pattern (a), Raman spectrum (b), nitrogen adsorption isothermal curves (c), and the corresponding pore-size distribution (d) of the 3D NCF. | ||
Surface area and pore size are important factors for efficient Na+ ion storage. The nitrogen adsorption isotherms of NCF (Fig. 2c) are a typical IV-type, indicative of a mesoporous structure. The NCF shows a high Brunauer–Emmett–Teller (BET) surface area of 623 m2 g−1, due to its porous hierarchical hollow structure. The pore size calculated using the Barrett–Joyner–Halenda method (BJH) (Fig. 2d) varies from 4 to 58 nm, demonstrating the presence of both micropores and mesopores. This high surface area and porous structure can effectively tolerate the volume changes during the continuous sodiation/de-sodiation.38 The mesopores also enable better contact between the electrode and the electrolyte, which is essential for good rate capability.17
The elemental analysis confirmed that content of element N in the NCF is 12.3 wt%, higher than the previously reported N-doped carbon materials for SIBs applications.27–29 The XPS spectra indicate the chemical states of the elements in NCF (Fig. 3). The C 1s band can be deconvoluted into three peaks. The peak at 284.4–284.6 eV is ascribed to sp2 carbon, including C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and CN bonds. The higher energy peak at 286.0 eV is associated with sp3 carbon, including C–C and C–N bonds. The N 1s band can be deconvoluted into three peaks, pyridinic N, pyrrolic N, and quaternary N. Pyridinic N substitutes for a carbon atom in the six-membered ring; pyrrolic N contributes two electrons to the five-membered ring π system; quaternary N substitutes a carbon atom in the graphite lattice. The content of each type of N has been calculated based on the XPS fitting results (Fig. 3c), which is 51.5% pyridinic N, 45.3% pyrrolic N, and 3.2% quaternary N. Fig. 3d illustrates the N distribution and bonding states in the NCF. The high contents of pyridinic N and pyrrolic N create more defects and active sites for Na+ insertion within the NCF, and therefore, facilitate ion transportation.37,39
C and CN bonds. The higher energy peak at 286.0 eV is associated with sp3 carbon, including C–C and C–N bonds. The N 1s band can be deconvoluted into three peaks, pyridinic N, pyrrolic N, and quaternary N. Pyridinic N substitutes for a carbon atom in the six-membered ring; pyrrolic N contributes two electrons to the five-membered ring π system; quaternary N substitutes a carbon atom in the graphite lattice. The content of each type of N has been calculated based on the XPS fitting results (Fig. 3c), which is 51.5% pyridinic N, 45.3% pyrrolic N, and 3.2% quaternary N. Fig. 3d illustrates the N distribution and bonding states in the NCF. The high contents of pyridinic N and pyrrolic N create more defects and active sites for Na+ insertion within the NCF, and therefore, facilitate ion transportation.37,39
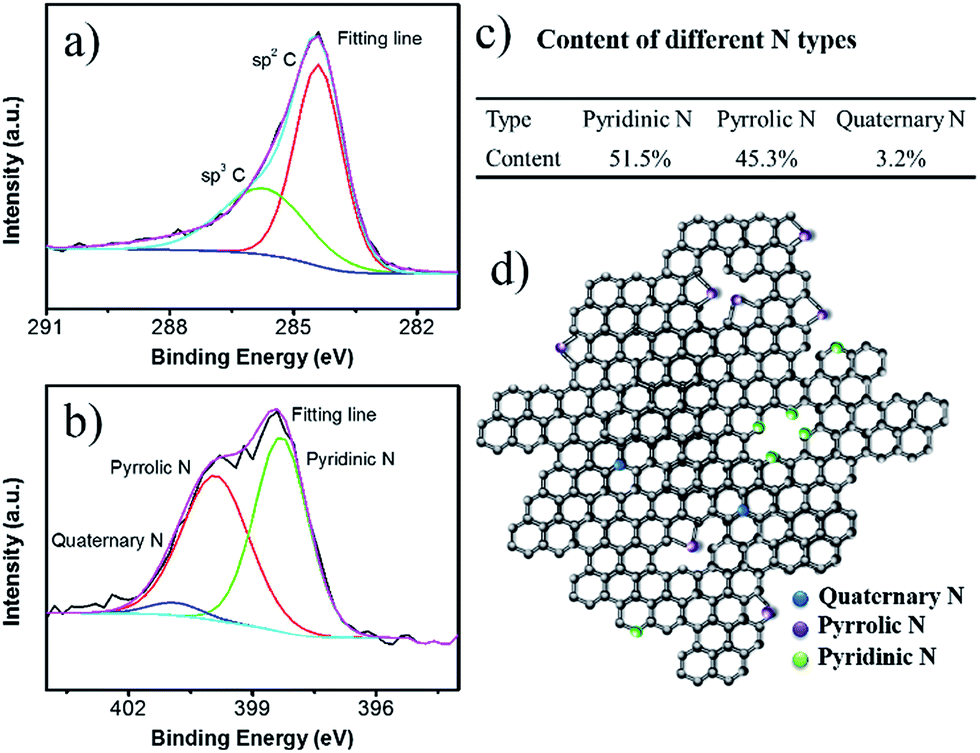 | ||
| Fig. 3 High resolution XPS spectra for C 1s (a) and N 1s (b). The proportions of each N type (c), and a structural illustration of the carbon shell in NCF (d). | ||
The sodium storage properties of NCF were characterized after been assembled into 2032 Na/NCF half cells. The initial four cyclic voltammograms (CVs) are presented in Fig. 4. The irreversible peaks around 0.01 V and 0.86 V could be ascribed to irreversible Na+ traps and the formation of a solid-electrolyte interphase (SEI) film40,41 In the second cycle, the broad peak around 0.5 V could be attributed to sodium ion insertion. During the anodic process, the broad peak at 0.7 V is associated with the extraction of Na+ from NCF.40 From the second cycle onward, the CV curves almost overlap with each other, indicating good cycle stability of the 3D NCF during sodiation/desodiation process.
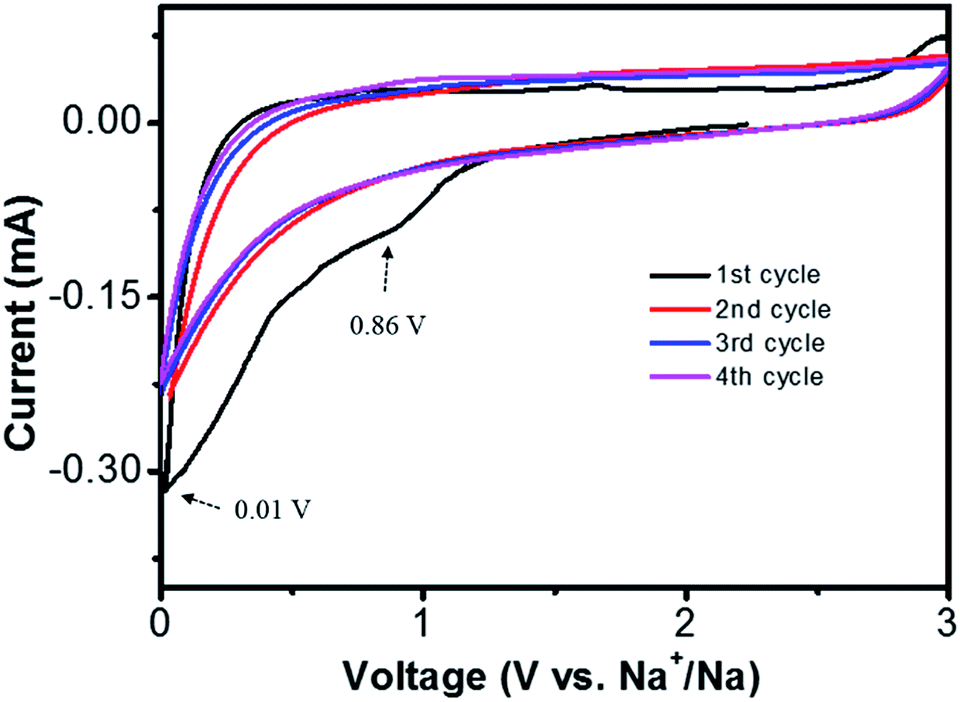 | ||
| Fig. 4 Cyclic voltammograms of the Na/NCF half cells at 0.5 mV s−1 scan rate, voltage 0.001–3 V. | ||
The discharge–charge experiments were performed at a current density of 100 mA g−1 (Fig. 5a). During the electrochemical process, the voltage profiles appear to be sloping lines without distinct plateaus, indicating the continuous insertion/de-insertion of Na+, which is consistent with the CV results. The first charge and discharge capacity of NCF is 327 and 864 mA h g−1, corresponding to 37% coulombic efficiency (CE). This capacity loss is likely caused by the irreversible side reactions, such as formation of the SEI film, decomposition of the electrolyte, and the irreversible reaction between sodium and surface functional groups in NCF.40 After several cycles' stabilization, the NCF showed a stable charge and discharge capacity of 300 and 323 mA h g−1 at the 4th cycle. This high first cycle reversible capacity (327 mA h g−1) and stable capacity (300 mA h g−1) was higher than the theoretical capacity (279 mA h g−1 for carbon) and most reported results for N-doped carbon materials,16,27–29 demonstrating the good electrochemical performance of the NCF.
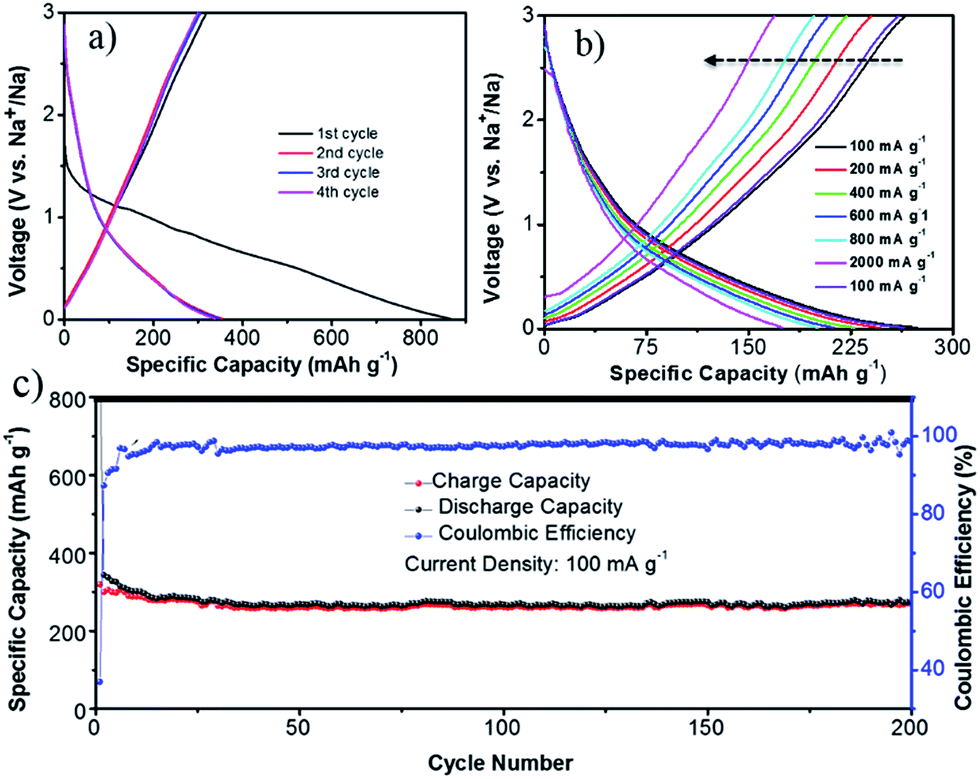 | ||
| Fig. 5 Sodium storage performance of the NCF: discharge–charge profiles for the first 4 cycles at a current density of 100 mA g−1 (a); rate capability at various current densities (100–2000 mA g−1) (b); and cycling performance (c). | ||
Rate capability is another crucial factor for SIBs. The discharge–charge curves of the NCF at various current densities were carried out after stabilization (seen cycling performances of the NCF at different current densities in Fig. S1 in the ESI†). The NCF only showed a slightly decreased capacity within the current density range of 100 to 2000 mA g−1 (Fig. 5b), e.g. a discharge capacity of 247, 227, 213, and 201 mA h g−1 obtained at 200, 400, 600, and 800 mA g−1, respectively. The NCF can still deliver a high capacity of 175 mA h g−1, even at a high current density of 2000 mA g−1, corresponding to 64% capacity retention. Such excellent rate capability is rarely reported for carbon-based electrode in SIBs.11,29,42–44 Importantly, when the current density was returned to 100 mA g−1, its capacity was nearly fully restored, back to 264 mA h g−1 with a high 96% capacity retention. This clearly indicates the good integrity of our NCF electrode.
The long-life cycling stability of the NCF is shown in Fig. 5c. From the 12th cycle onwards, the CE of NCF stays above 97%. Even after 200 cycles of sodiation/de-sodiation, the electrode still exhibited a high capacity of 274 mA h g−1 (86% capacity retention), outperforming other reports.12,45,46 SEM images of the cycled NCF was given in Fig. S2,† which showed a certain degree of aggregation, but the polyhedral outline of NCF was well maintained, which still composed of numerous nano-subunits, this together with the electrochemical results demonstrated the good integrity of the NCF.
The good rate capability can be supported by the results from electrochemical impedance spectroscopy (EIS, Fig. 6). The semicircle and sloping line in the EIS curves are related to the resistance and Warburg impedance, respectively. To better explain the charge-transfer resistance during the cycling, equivalent circuits were used to fit these EIS plots. The components Rs, Rct, CPE, and Wo stand for the electrolyte resistance, charge-transfer resistance, constant phase element, and Warburg impedance, respectively. After five cycles, the formation of SEI film resulting a new resistance RSEI, therefore, the equivalent circuit of the cycled cells is different (Fig. 6c). The fitting results show that the Rs for the as-assembled cell and the cell after 5 cycles is similar, 12 and 10 Ω, respectively. RSEI in the cycled cells is 26 Ω. Rct for the as-assembled cell and the cell after 5 cycles is 730 and 670 Ω, respectively, which is lower than for graphene (839.8 Ω), and N-doped graphene (744.8 Ω).47 The decreased Rct indicated better electron transfer and therefore, better rate capability.
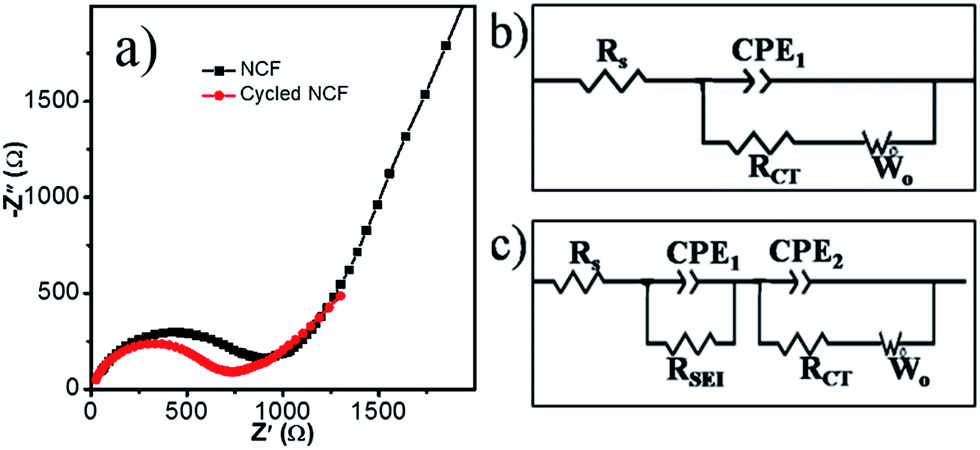 | ||
| Fig. 6 Nyquist plots of the Na/NCF half cells in different states (a), the corresponding equivalent circuits for an as-fabricated half-cell (b) and for the half-cell after five charge/discharge cycles (c). | ||
4. Conclusions
In summary, a 3D nitrogen-doped carbon framework with a high nitrogen content of 12.3 wt% was obtained via a facile two-step method (calcination and etching) using a ZIF-67 as precursor. The 3D NCF consists of numerous hollow spheres (3–5 nm in size), has a high surface area (632 m2 g−1), and is highly porous. The hollow sphere subunits in NCF can tolerate the volume changes during electrochemical cycling, resulting in improved cycling stability. The decreased Rct and defect-rich structure caused by the N-doping all contribute to the good electrochemical performance of NCF, such as the high reversible capacity, good cycling stability (86% capacity retention after 200 cycles at 100 mA g−1), and exceptional excellent rate capability (175 mA h g−1 at 2000 mA g−1 current density).Acknowledgements
This work was supported by the National Science Foundation of China (NSFC) (51471089), the Ministry of Education (MOE) (IRT13R30), the 111 Project (B12015), and the Priority Academic Program Development Fund of Jiangsu Higher Education Institutions. The authors would like to thank the Australian National Fabrication Facility-Materials node (ANFF) for equipment use.Notes and references
- Y. Wang, Z. G. Huang and Y. J. Wang, J. Mater. Chem. A, 2015, 3, 21314–21320 CAS
.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4301 CrossRef CAS PubMed
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed
- M. Sawicki and L. L. Shaw, RSC Adv., 2015, 5, 53129–53154 RSC
- Y. Wang, C. Y. Wang, Y. J. Wang, H. K. Liu and Z. G. Huang, J. Mater. Chem. A, 2016, 4, 5428–5435 CAS
- Y. Fu, Q. Wei, X. Wang, G. Zhang, H. Shu, X. Yang, A. C. Tavares and S. Sun, RSC Adv., 2016, 6, 16624–16633 RSC
- B. Nageswara Rao, P. Ramesh Kumar, O. Padmaraj, M. Venkateswarlu and N. Satyanarayana, RSC Adv., 2015, 5, 34761–34768 RSC
- J. P. Huang, D. D. Yuan, H. Z. Zhang, Y. L. Cao, G. R. Li, H. X. Yang and X. P. Gao, RSC Adv., 2013, 3, 12593–12597 RSC
- K. Nobuhara, H. Nakayama, M. Nose, S. Nakanishi and H. Iba, J. Power Sources, 2013, 243, 585–587 CrossRef CAS
- K. Hyungsub, K. Haegyeom, D. Zhang, L. Myeong Hwan, L. Kyungmi, Y. Gabin and K. Kisuk, Adv. Energy Mater., 2016, 6, 1600943 CrossRef
- Y. Cao, L. Xiao, M. L. Sushko, W. Wang, B. Schwenzer, J. Xiao, Z. Nie, L. V. Saraf, Z. Yang and J. Liu, Nano Lett., 2012, 12, 3783–3787 CrossRef CAS PubMed
- Y. Li, S. Xu, X. Wu, J. Yu, Y. Wang, Y.-S. Hu, H. Li, L. Chen and X. Huang, J. Mater. Chem. A, 2015, 3, 71–77 CAS
- Q. Wei, Y. Fu, G. Zhang, Y. Wang, X. Wang, M. Mohamedi and S. Sun, RSC Adv., 2016, 6, 84149–84154 RSC
- H. Liu, M. Jia, M. Wang, R. Chen, N. Sun, Q. Zhu, F. Wu and B. Xu, RSC Adv., 2016, 6, 78235–78240 RSC
- D. Qin, F. Zhang, S. Dong, Y. Zhao, G. Xu and X. Zhang, RSC Adv., 2016, 6, 106218–106224 RSC
- K. L. Zhang, X. N. Li, J. W. Liang, Y. C. Zhu, L. Hu, Q. S. Cheng, C. Guo, N. Lin and Y. T. Qian, Electrochim. Acta, 2015, 155, 174–182 CrossRef CAS
- K. Tang, L. J. Fu, R. J. White, L. H. Yu, M. M. Titirici, M. Antonietti and J. Maier, Adv. Energy Mater., 2012, 2, 873–877 CrossRef CAS
- N. R. Kim, Y. S. Yun, M. Y. Song, S. J. Hong, M. Kang, C. Leal, Y. W. Park and H. J. Jin, ACS Appl. Mater. Interfaces, 2016, 8, 3175–3181 CAS
- J. Ding, H. L. Wang, Z. Li, A. Kohandehghan, K. Cui, Z. W. Xu, B. Zahiri, X. H. Tan, E. M. Lotfabad, B. C. Olsen and D. Mitlin, ACS Nano, 2013, 7, 11004–11015 CrossRef CAS PubMed
- J. Jin, S. J. Yu, Z. Q. Shi, C. Y. Wang and C. B. Chong, J. Power Sources, 2014, 272, 800–807 CrossRef CAS
- R. Boyang, W. Jun, S. Dongqi, X. Yanfei, C. shulei, L. Huakun and W. Jiazhao, J. Mater. Chem. A, 2015, 3, 19011–19017 Search PubMed
- Y. Jianchuan, Z. Jun, T. Zhaowu, Z. Ming-sen and D. Quan-Feng, J. Mater. Chem. A, 2016, 4, 13223–13227 Search PubMed
- J. Zhang, Z. Zhang and X. Zhao, RSC Adv., 2015, 5, 104822–104828 RSC
- Z. Wang, Y. Li and X.-J. Lv, RSC Adv., 2014, 4, 62673–62677 RSC
- S. Vadahanambi, H.-H. Chun, K. H. Jung and H. Park, RSC Adv., 2016, 6, 38112–38116 RSC
- Y. Wang, C. Wang, Y. Wang, H. Liu and Z. Huang, ACS Appl. Mater. Interfaces, 2016, 8, 18860–18866 CAS
- Y. Wen, B. Wang, B. Luo and L. Wang, Eur. J. Inorg. Chem., 2016, 2051–2055 CrossRef CAS
- Z. Wang, L. Qie, L. Yuan, W. Zhang, X. Hu and Y. Huang, Carbon, 2013, 55, 328–334 CrossRef CAS
- L. Fu, K. Tang, K. Song, P. A. van Aken, Y. Yu and J. Maier, Nanoscale, 2014, 6, 1384–1389 RSC
- D. Wei, Y. Liu, Y. Wang, H. Zhang, L. Huang and G. Yu, Nano Lett., 2009, 9, 1752–1758 CrossRef CAS PubMed
- Z.-H. Sheng, L. Shao, J.-J. Chen, W.-J. Bao, F.-B. Wang and X.-H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed
- J. Zhou, N. Lin, W. l. Cai, C. Guo, K. Zhang, J. Zhou, Y. Zhu and Y. Qian, Electrochim. Acta, 2016, 218, 243–251 CrossRef CAS
- Y.-J. Li, J.-M. Fan, M.-S. Zheng and Q.-F. Dong, Energy Environ. Sci., 2016, 9, 1998–2004 CAS
- Y. Wang, B. Wang, F. Xiao, Z. Huang, Y. Wang, C. Richardson, Z. Chen, L. Jiao and H. Yuan, J. Power Sources, 2015, 298, 203–208 CrossRef CAS
- Y. Wang, C. An, Y. Wang, Y. Huang, C. Chen, L. Jiao and H. Yuan, J. Mater. Chem. A, 2014, 2, 16285–16291 CAS
- C. An, Y. Wang, Y. Xu, Y. Wang, Y. Huang, L. Jiao and H. Yuan, ACS Appl. Mater. Interfaces, 2014, 6, 3863–3869 CAS
- Y. C. Liu, N. Zhang, C. M. Yu, L. F. Jiao and J. Chen, Nano Lett., 2016, 16, 3321–3328 CrossRef CAS PubMed
- I. Elizabeth, B. P. Singh, S. Trikha and S. Gopukumar, J. Power Sources, 2016, 329, 412–421 CrossRef CAS
- Z. Zhu, S. Wang, J. Du, Q. Jin, T. Zhang, F. Cheng and J. Chen, Nano Lett., 2014, 14, 153–157 CrossRef CAS PubMed
- H. Hou, C. E. Banks, M. Jing, Y. Zhang and X. Ji, Adv. Mater., 2015, 27, 7861–7866 CrossRef CAS PubMed
- Y. Li, Y.-S. Hu, M.-M. Titirici, L. Chen and X. Huang, Adv. Energy Mater., 2016, 6, 1600659 CrossRef
- Z. Zhang, J. Zhang, X. Zhao and F. Yang, Carbon, 2015, 95, 552–559 CrossRef CAS
- S. Komaba, W. Murata, T. Ishikawa, N. Yabuuchi, T. Ozeki, T. Nakayama, A. Ogata, K. Gotoh and K. Fujiwara, Adv. Funct. Mater., 2011, 21, 3859–3867 CrossRef CAS
- J. Ding, H. Wang, Z. Li, A. Kohandehghan, K. Cui, Z. Xu, B. Zahiri, X. Tan, E. M. Lotfabad, B. C. Olsen and D. Mitlin, ACS Nano, 2013, 7, 11004–11015 CrossRef CAS PubMed
- W. Li, L. Zeng, Z. Yang, L. Gu, J. Wang, X. Liu, J. Cheng and Y. Yu, Nanoscale, 2014, 6, 693–698 RSC
- H.-g. Wang, Z. Wu, F.-l. Meng, D.-l. Ma, X.-l. Huang, L.-m. Wang and X.-b. Zhang, ChemSusChem, 2013, 6, 56–60 CrossRef PubMed
- J. T. Xu, M. Wang, N. P. Wickramaratne, M. Jaroniec, S. X. Dou and L. M. Dai, Adv. Mater., 2015, 27, 2042–2048 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27088c |
| This journal is © The Royal Society of Chemistry 2017 |