
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlled synthesis of dendritic gold nanostructures by graphene oxide and their morphology-dependent performance for iron detection in coastal waters
Haitao Han a,
Dawei Pan*ab,
Chenchen Wanga and
Rilong Zhu*cd
a,
Dawei Pan*ab,
Chenchen Wanga and
Rilong Zhu*cd
aKey Laboratory of Coastal Environmental Processes and Ecological Remediation, Yantai Institute of Coastal Zone Research (YIC), Chinese Academy of Sciences (CAS), Shandong Provincial Key Laboratory of Coastal Environmental Processes, YICCAS, Yantai, Shandong 264003, P. R. China. E-mail: dwpan@yic.ac.cn; Fax: +86-535-2109155; Tel: +86-535-2109155
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cCollege of Water Sciences, Beijing Normal University, Beijing 100875, P. R. China. E-mail: zrlden@hotmail.com
dHunan Environmental Monitoring Center Station, State Environmental Protection Key Laboratory of Monitoring for Heavy Metal Pollutants, Changsha, 410019, P. R. China
First published on 10th March 2017
Abstract
A facile and effective method was introduced for the controlled synthesis of dendritic gold nanostructures on the electrode via a simple electrochemical route in which graphene oxide (GO) was used as a morphology-controlling agent. The oxygen-containing groups on GO controlled not only the diffusion of AuCl4− ions but the subsequent generation of gold nuclei at the initial stage and the growth direction of the gold nanocrystals in the electrochemical synthesis process. The morphology-dependent electrochemical sensing ability of gold nanostructures was discussed in detail on the basis of control experiments related to the concentration of GO, HAuCl4, and the electrodeposition time. It could be a straightforward and controllable method for improving the nanoparticles' electrochemical performance. The unique nanostructure and excellent electro-catalytic properties of the dendritic gold nanostructures made the modified electrode an excellent choice for differential pulse voltammetric determination of Fe(III). The modified electrode exhibited a linear response to Fe(III) in the 7 nM to 1 μM concentration range, with a lower detection limit of 1.5 nM. The modified electrode was successfully applied to the sensitive determination of iron in local coastal waters.
Introduction
In recent years, inorganic materials with nanoscale dimensions have attracted broad interest because of their unique physical and chemical properties and extensive applications in various fields of science and technology, including catalysis and sensors.1–3 With recent advances in nanoscience, the properties and performance of nanomaterials have been found to strongly depend on their morphology.4,5 In particular, a great deal of attention has been shown toward the morphological control of gold nanoparticles (AuNPs), which possess fascinating morphology-dependent electrochemical and catalytic properties.6 Au nanostructures possess plenty of excellent electrochemical properties that make them star materials for electrochemical sensors. Because of their large surface-to-volume ratio, Au nanostructures can provide more effective surface area and reactive sites. The excellent conductivity and strong adsorption ability of Au nanostructures can facilitate the electrons transfer between species and electrode to increase the detection sensitivity of the electrochemical sensors. Moreover, Au nanostructures possess excellent electro-catalytic performance towards lots of analytes, which is essential for the electrochemical sensors.Several methods have been presented to control the morphology of Au nanostructures during the synthesis process. A traditional wet-chemical method has been widely used to achieve this purpose, and Au nanostructures with different morphologies have been obtained.7–10 In this wet-chemical method, Au nanostructures were synthesized separately after a series of processes such as reduction, centrifugation, washing, drying and so on. To fabricate the electrochemical sensor, Au nanostructures were re-dispersed and casted on the surface of the electrode, and then dried. However Au nanostructures prepared in this method were usually easy to exfoliate during the electrochemical experiments. Thus, immobilizing Au nanostructures on electrodes became the difficulty during the fabrication of electrochemical sensors. Therefore, to solve this problem, researchers have adopted a direct electrodeposition method to fabricate electrodes modified with Au nanomaterials with different morphologies.6 Numerous Au nanostructures, such as nanocubes, nanoplates, nanorods, branches, porous sheets, and dendrites have been synthesized to modify electrodes under this approach.11–18 Among these Au nanostructures used to modify electrodes, considerable attention has been devoted to dendritic Au nanostructures (DAuNs) because of their wide potential applications in electrochemical sensing and electro-catalysis.18,19
Most of the electrodeposition methods used to obtain the DAuNs involved the use of different additives such as iodide, N-methylimidazole, cysteine, and Fe(III) ions; such additives play important roles during the electrochemical growth of DAuNs.3,16–18,20,21 However, a question about this method had arisen because almost all of the DAuNs were obtained with the help of other additives in the electrolyte. Different strategies were necessary to remove these additives before used. In this work, GO was proposed as a morphology-controlling agent directly on the electrode for the electrochemical obtainment of DAuNs. On the other hand, GO had also been used widely as the electrode modifier due to its fascinating electrochemical properties. Thus the main advantage of this method lied on the elimination of the process for the removing of additives.
As an environmentally and biologically relevant metal ion, iron plays a critical role for all living organisms by participating in a wide diversity of metabolic processes. Iron is responsible for chlorophyll synthesis, nitrate reduction, detoxification of reactive oxygen species, and the growth of microorganisms and phytoplankton species.22 Thus, the quantitative determination of iron is important for biology and environment. Among the detection techniques, voltammetry bas been recognized as an extremely sensitive electrochemical method for iron determination.3
In present work, GO was deposited onto the surface of the glassy carbon electrode (GCE) as a morphology-controlling agent for the subsequent electrodeposition of DAuNs. Our results demonstrated the function of GO as the morphology-controlling agent for the direct electrodeposition of DAuNs. The morphology-dependent performance of DAuNs for iron detection was investigated in detail. The DAuNs modified GCE (DAuNs/GO/GCE) exhibited a linear response to Fe(III) ions in the 7 nM to 1 μM concentration range and a limit of detection (LOD) of 1.5 nM. Furthermore, the DAuNs/GO/GCE was successfully applied to the sensitive determination of Fe(III) in real coastal waters with satisfactory results.
Experimental
Reagents and materials
Iron standard solution was acquired from Acros Organics. GO was provided by Nanjing XFNANO Materials Tech Co., Ltd (Nanjing, China). Graphene (GR) was obtained via the electrochemical reduction of GO. HAuCl4 was supplied by Sinopharm Chemical Reagent (Shanghai, China). All other chemicals were analytical reagents and used without further purification. Deionized water (specific resistance 18.2 MΩ cm) obtained from a Pall Cascada laboratory water system was used throughout the experiments.Apparatus
The morphology of the Au nanostructures was characterized by scanning electron microscopy (SEM, Hitachi S-4800 microscope, Japan). The UV-digestion of coastal waters was conducted with a UV digester (Metrohm MVA-UV 705, Switzerland). Inductively coupled plasma-mass spectrometry (ICP-MS, ELAN DRC, Perkin Elmer Instruments, USA) was used for comparative testing. All the electrochemical experiments were carried out in a conventional three-electrode cell controlled by a CHI 660E electrochemical workstation (Chenhua Instruments, Shanghai, China). The modified GCE (3 mm in diameter) was used as the working electrode, with an Ag/AgCl electrode and platinum foil serving as the reference and counter electrodes, respectively.Fabrication of the DAuNs/GO/GCE
Prior to use, a bare GCE (BGCE) was polished mechanically with aqueous alumina slurry (0.3 and 0.05 μm), washed thoroughly with deionized water, and ultrasonicated for 1 min. The GCE was then subjected to potential cycling from −0.6 V to 0.6 V (50 mV s−1) in 0.5 M H2SO4 until reproducible cyclic voltammograms were obtained. The suspension of GO with different concentrations were obtained by dispersing the corresponding powders with deionized water and ultrasonicating for 10 min. 7 μL of the obtained GO suspension was dropped onto the surface of the GCE and dried under an infrared lamp to obtain the GO modified GCE (GO/GCE).Then, the GO/GCE was immersed in a 0.5 M H2SO4 solution containing 1 mM HAuCl4 and electrodeposited at a potential of −0.2 V for 60 s under stirring (when the morphology-dependent electro-catalytic performance was investigated, the parameters were modified accordingly). After careful cleaning, the DAuNs/GO/GCE was obtained. For comparison, the AuNPs/GCE was fabricated by direct electrodeposition of AuNPs on a GCE. The schematic for the fabrication of the modified electrodes is shown in Scheme 1.
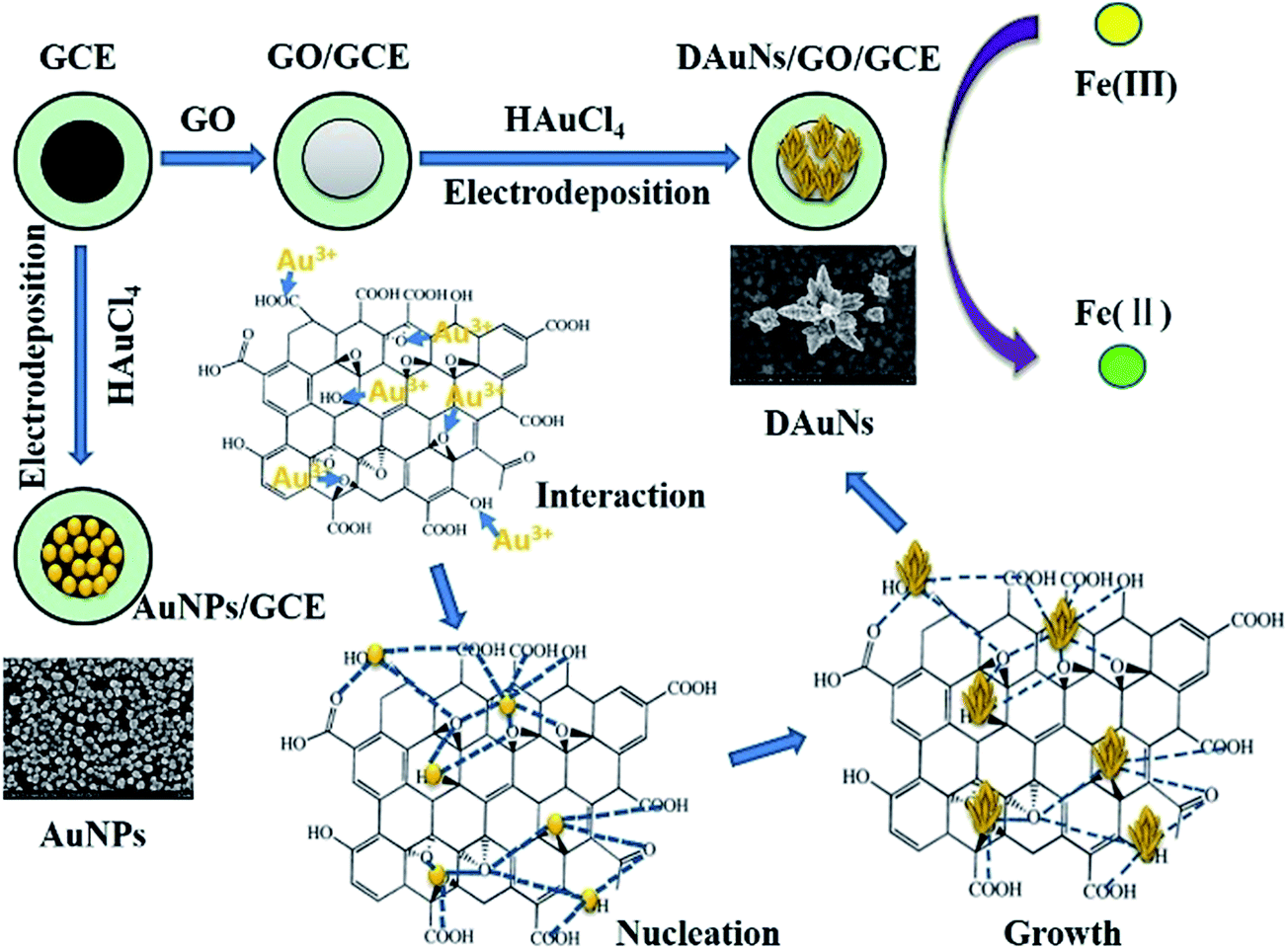 | ||
| Scheme 1 Schematic for the growth of DAuNs and fabrication of the DAuNs/GO/GCE. The DAuNs/GO/GCE was fabricated by electrodepositing at −0.2 V for 60 s under stirring with GO/GCE as the working electrode which obtained through dropping GO on the surface of GCE. The oxygen-containing groups on GO interacted with Au3+, subsequently controlled the generation of Au nuclei and the growth direction of the Au nanocrystals in the electrochemical synthesis process. | ||
Analysis procedure
Unless otherwise stated, the experiments were performed in 0.1 M HCl electrolyte. Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) methods were used. In the CV experiments, the potential was scanned from 0.2 to 1.4 V at a scan rate of 50 mV s−1. The electro-catalytic reduction responses of Fe(III) at different electrodes were investigated by DPV, which was performed using an initial potential of 0.7 V, a final potential of 0.2 V, an amplitude of 0.05 V, a potential increment of 0.004 V, a pulse width of 0.2 s, a pulse period of 0.5 s and a quiet time of 2 s.Analysis of real samples
Coastal water samples were collected from the Yuniao River that flows into the Bohai Sea (Shandong province, China) at different locations. All the water samples were stored in polyethylene bottles at 4 °C after filtration and acidification. After a certain amount of H2O2 was added, the samples were UV-digested with a 500 W UV lamp to ensure that all ligands bound to Fe were released and that all Fe species were oxidized to Fe(III). Voltammetric measurements were performed using water samples diluted with the 0.1 M HCl supporting electrolyte.Results and discussion
Effect of GO on morphology and electrochemical response of the DAuNs
Most importantly, GO had a critical controlling effect on the morphology of the deposited Au nanostructures. Fig. 1 shows the typical SEM images of Au nanostructures obtained on the electrodes with different GO contents as morphology-controlling agent. Only common AuNPs could be obtained in the absence of GO on the electrode surface (Fig. 1a). However, Au nanocrystals with the rudiments of a dendritic structure were obtained when 0.05 mg mL−1 GO was used (Fig. 1b). When 0.1 mg mL−1 GO acted as the morphology-controlling agent on the electrode, DAuNs were obtained (Fig. 1c). The DAuNs were made up of few branches with gold nanoparticles grown on them. Interestingly, when the GO concentration was increased to 0.5 mg mL−1, almost no nanoparticles were observed on the electrode with large flakes and slightly scrolled edges, which was the typical morphology of GO (Fig. 1d).23,24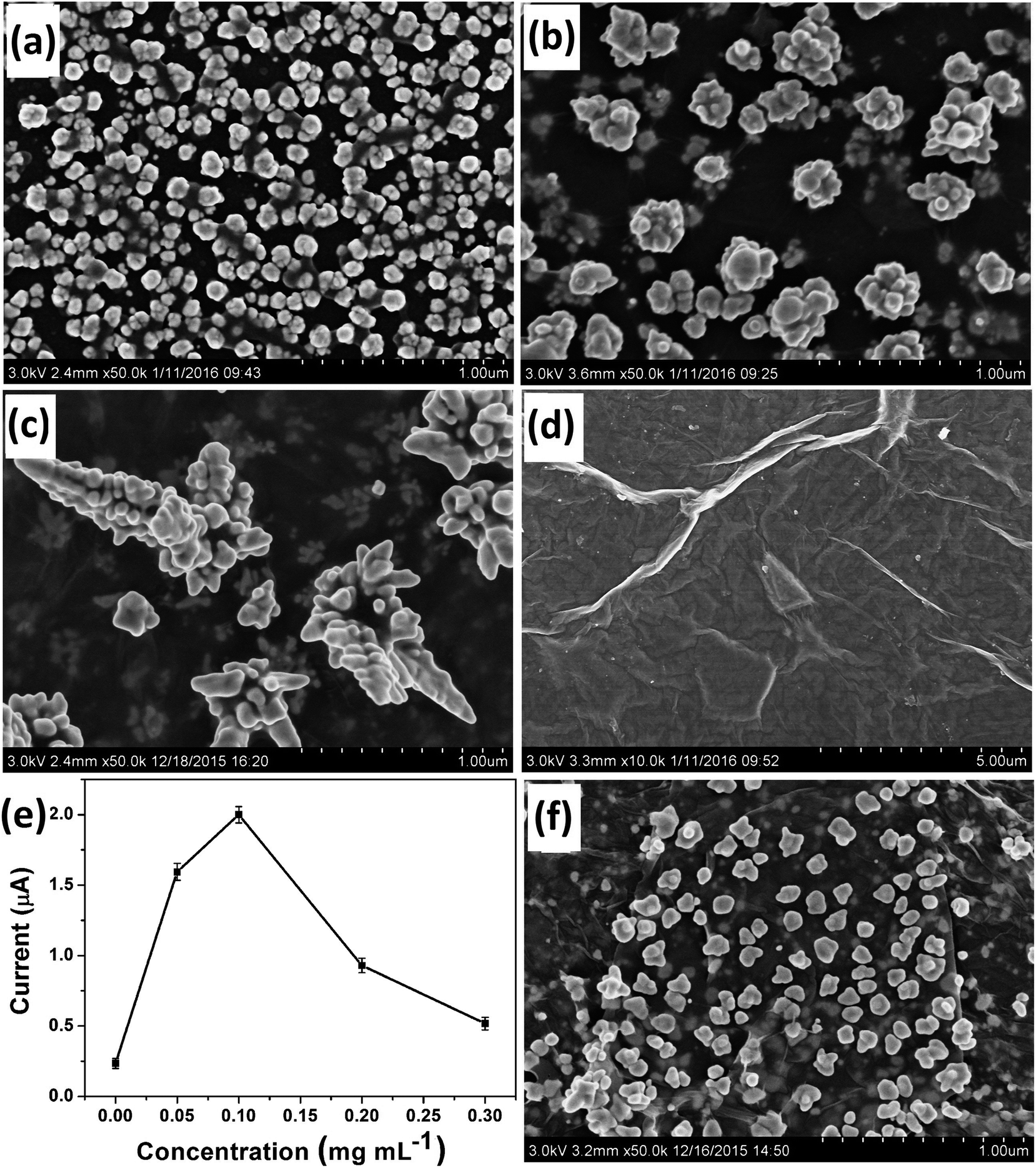 | ||
| Fig. 1 SEM images of Au nanostructures obtained with 0 (a), 0.05 (b), 0.1 (c), 0.5 (d) mg mL−1 of GO (7 μL respectively) as the morphology-controlling agent. (e) Effect of Au morphologies obtained with different contents of GO on the peak current of 5 μM Fe(III) in 0.1 M HCl. (f) SEM image of Au nanostructures obtained with 0.1 mg mL−1 of GR (7 μL) as the morphology-controlling agent. | ||
The failure to obtain DAuNs when the GO concentration was less than 0.1 mg mL−1 might result from an insufficient number of oxygen-containing groups on the GO to direct the formation of DAuNs. However, 0.5 mg mL−1 GO might prevent electron transfer between the precursors and electrode. Thus, GO could act as the morphology-controlling agent for the controlled synthesis of DAuNs only within an appropriate range of GO content and 7 μL of 0.1 mg mL−1 might be the optimal amount. Interestingly, Au nanostructures obtained in this case exhibited the highest current response toward the reduction of Fe(III) (Fig. 1e). In other words, the DAuNs exhibited a higher electrochemical response than other Au nanostructures for iron voltammetric detection.
To conduct the elemental analysis of the nanostructures fabricated with GO as morphology-controlling agent, EDS was performed (Fig. 2). It could be seen that C, Au and O were the major elements. C might come from the GO and GCE, while O was from the oxygen-containing groups of GO. The existence of Au element certainly indicated that the nanostructures of Au were obtained.
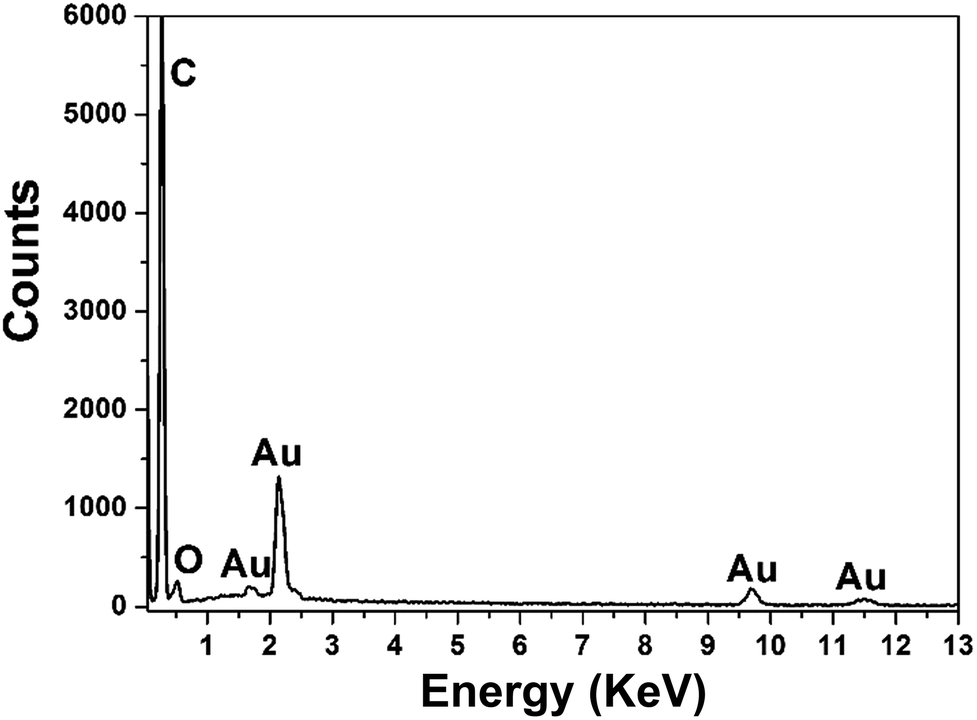 | ||
| Fig. 2 EDS pattern of the typical DAuNs fabricated with GO as morphology-controlling agent. | ||
Mechanism for the controlled synthesis of DAuNs
To verify the assumption that oxygen-containing groups on GO played the dominant role in the controlled synthesis of DAuNs, the performance of GO and GR as the morphology-controlling agent was compared. As expected, the dendritic structure could not be obtained when GR was used; only a nanoparticle structure was obtained (Fig. 1f). When GO was reduced to GR, the content of oxygen-containing groups on GO was substantially reduced. So it was clear that oxygen-containing groups on GO played the dominant role in the controlled synthesis of DAuNs.According to Khalil et al., the functionalities on GO surface (oxygen-containing groups) could cause the attachment of AuCl4− ions through electrostatic interactions.25 After being attached on the surface of GO, the AuCl4− ions were electrochemically reduced. Marques et al. has pointed out that oxygen functionalities at GO surface provided reactive sites for the nucleation and growth of AuNPs.26 Subsequently, oxygen-containing groups served as the links for AuNPs to control the growth of Au nanocrystals.27 Thus, the oxygen-containing groups on GO controlled the diffusion of the AuCl4− ions, the subsequent generation of Au nuclei at the initial beginning stage and the growth direction of the Au nanocrystals in the electrochemical synthesis process. The mechanism of GO as the morphology-controlling agent for the synthesis of DAuNs was also illustrated in Scheme 1.
Effect of deposition time on morphology and electrochemical response of the DAuNs
Deposition time was another important parameter that affected the morphology of the deposited Au nanostructures. SEM images of Au nanostructures obtained with electrodeposition time of 20 s, 40 s, 60 s, 80 s and 100 s are shown in Fig. 3a–e, respectively. Although nanoparticles with the rudiments of a dendritic structure were obtained at 20 s and 40 s, the maturity degree of the obtained Au nanocrystals was inadequate. When the deposition time was increased to 60 s, integral DAuNs were obtained. However, when the electrodeposition was conducted for 80 s or 100 s, the dendritic structure began to be destroyed, because of the accumulation of more nanoparticles on the DAuNs. So, to obtain the DAuNs, 60 s deposition time as the optimal parameter was adopted.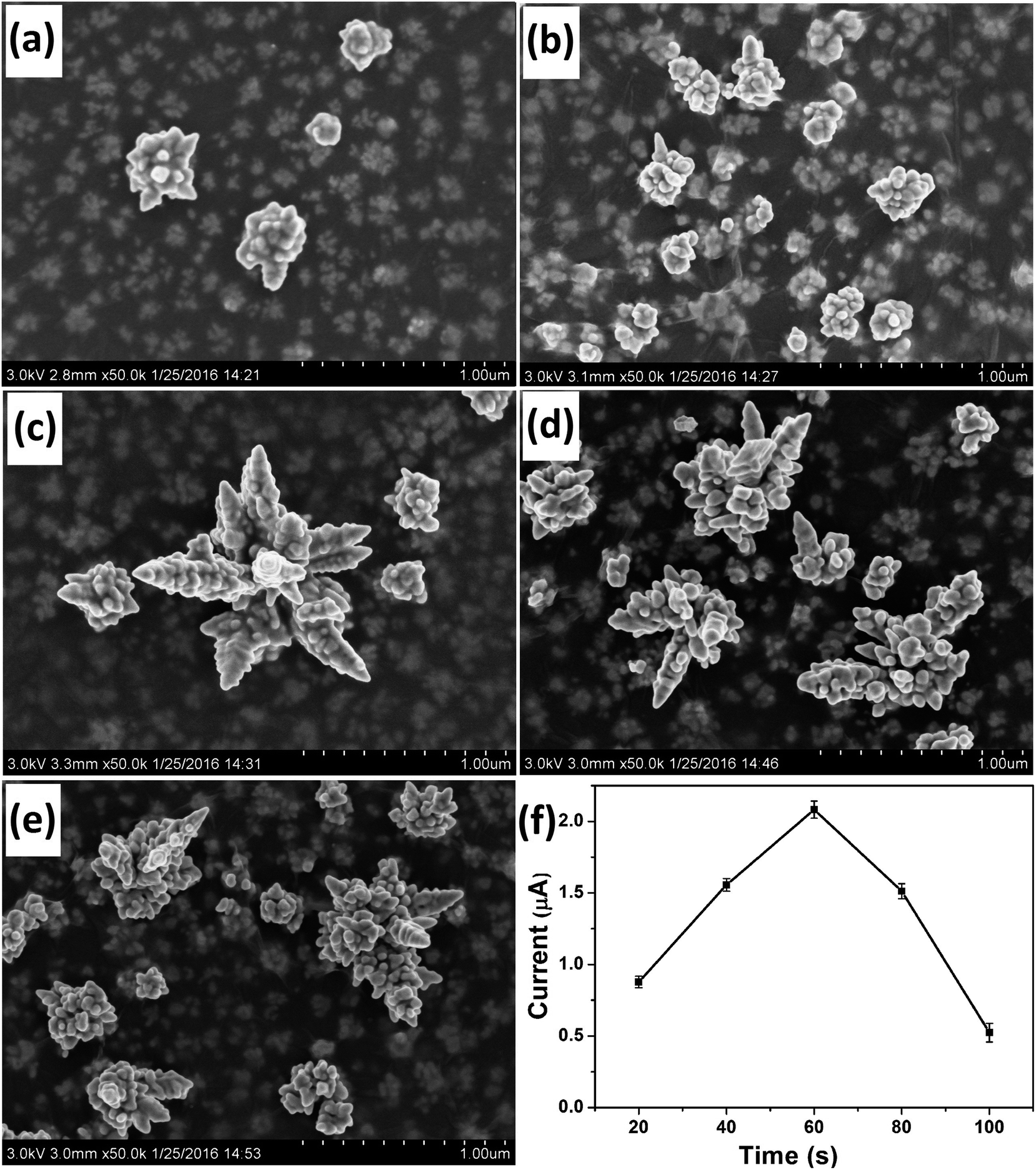 | ||
| Fig. 3 SEM images of Au nanostructures obtained at electrodeposition times of 20 s (a), 40 s (b), 60 s (c), 80 s (d), and 100 s (e) in the presence of parallel GO as the morphology-controlling agent. (f) The effect of Au morphologies obtained with different electrodeposition time on the peak current of 5 μM Fe(III) in 0.1 M HCl. | ||
Fig. 3f shows the effect of Au morphologies obtained using different electrodeposition time on the peak current of 5 μM Fe(III) in 0.1 M HCl. Obviously, Au nanostructures obtained with 60 s of electrodeposition time also exhibited the highest current response toward the reduction of Fe(III). That was, DAuNs exhibited the strongest electrochemical response for the detection of iron.
Effect of precursor concentration on morphology and electrochemical response of the DAuNs
The concentration of the precursor also affected the morphology of the Au nanostructures obtained on the electrode. Fig. 4 shows the SEM images of Au nanostructures obtained with different concentrations of HAuCl4. When 0.1 mM HAuCl4 was used as the precursor, no nanoparticles were observed on the surface of GO and only some small ones were observed under it (Fig. 4a). Very small and few nanoparticles with the rudiments of the dendritic structure were obtained when the concentration of HAuCl4 was 0.5 mM (Fig. 4b). As expected, when the concentration of HAuCl4 was increased to 1 mM, ideal DAuNs were obtained, as expected (Fig. 4c). When the concentration of HAuCl4 was increased to 5 mM, more AuNDs were connected to each other to form leaf-like structures (Fig. 4d and e). | ||
| Fig. 4 SEM images of Au nanostructures obtained in 0.1 (a), 0.5 (b), 1 (c), and 5 (d and e) mM HAuCl4 with parallel GO as the morphology-controlling agent. Insets in panels (a–c) are their magnified images accordingly. (f) The effect of Au morphologies obtained with different HAuCl4 concentrations on the peak current of 5 μM Fe(III) in 0.1 M HCl. | ||
Although the peak current was enhanced with increasing HAuCl4 concentration, when it was greater than 1 mM, the effect became very weak (Fig. 4f). Thus, DAuNs obtained with 7 μL 0.5 mg mL−1 GO as morphology-controlling agent, 60 s deposition time, 1 mM precursor (HAuCl4) concentration were selected for the electrochemical detection of iron. Generally speaking, the electrochemical performance of the Au nanostructures strongly depended on their morphologies, and the dendritic structure gave the best performance. The DAuNs obtained in this way were made up of few branches with top width of 50–70 nm and length of 300–420 nm. A lot of nanoparticles with size of 50 nm grown on the branches were also observed (Fig. 4c).
Electrochemical behaviors of the modified electrodes
CV was performed to characterize the DAuNs modified electrodes. Fig. 5 shows the typical CV curves for the BGCE (curve a), GO/GCE (curve b), and DAuNs/GO/GCE (curve c) recorded between 0.2 V and 1.4 V in 0.5 M H2SO4 at a scan rate of 50 mV s−1. No redox peak was obtained for BGCE. In the case of GO/GCE, the CV curve was very similar to that recorded for the BGCE. However, a sharp reduction peak at 0.95 V and an oxidation peak at 1.1 V due to the redox of DAuNs were observed in the voltammogram of the DAuNs/GO/GCE. It could be expected that the existence of DAuNs would play an important role in facilitating electrons transfer and the electrocatalysis of Fe(III) reduction.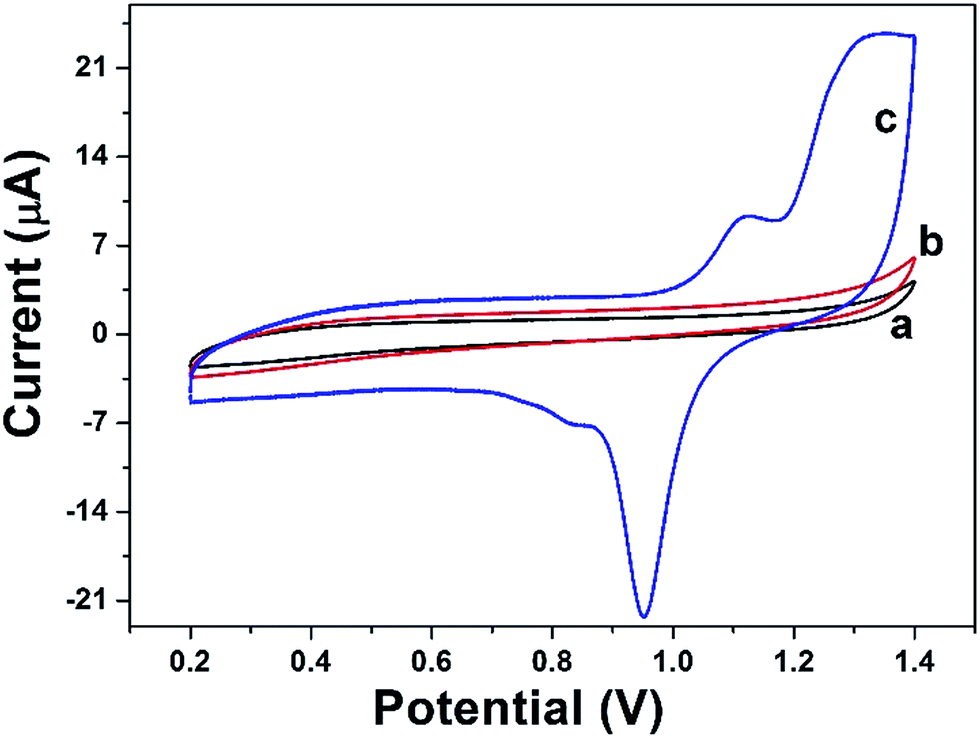 | ||
| Fig. 5 CV curves of BGCE (curve a), GO/GCE (curve b), and DAuNs/GO/GCE (curve c) recorded in 0.5 M H2SO4 with a scan rate of 50 mV s−1. | ||
Electrochemical responses of Fe(III) on the modified electrodes
Fig. 6 shows the DPV curves of BGCE (curve a), GO/GCE (curve b), AuNPs/GCE (curve c), and DAuNs/GO/GCE (curve d) in 0.1 M HCl solution containing 5 μM Fe(III) and their corresponding reproducibility. No signal was obtained when BGCE and GO/GCE were used to detect 5 μM Fe(III). With respect to the AuNPs/GCE and DAuNs/GO/GCE, a small and a large reduction peak of Fe(III) were observed at 0.56 V, respectively. The reduction peak current of Fe(III) obtained at the DAuNs/GO/GCE showed an enhancement as large as 7-fold compared to that at the AuNPs/GCE. Evidently, the electro-catalytic activity of the DAuNs was much greater than that of the general AuNPs toward the reduction of Fe(III) because of their specially dendritic structure.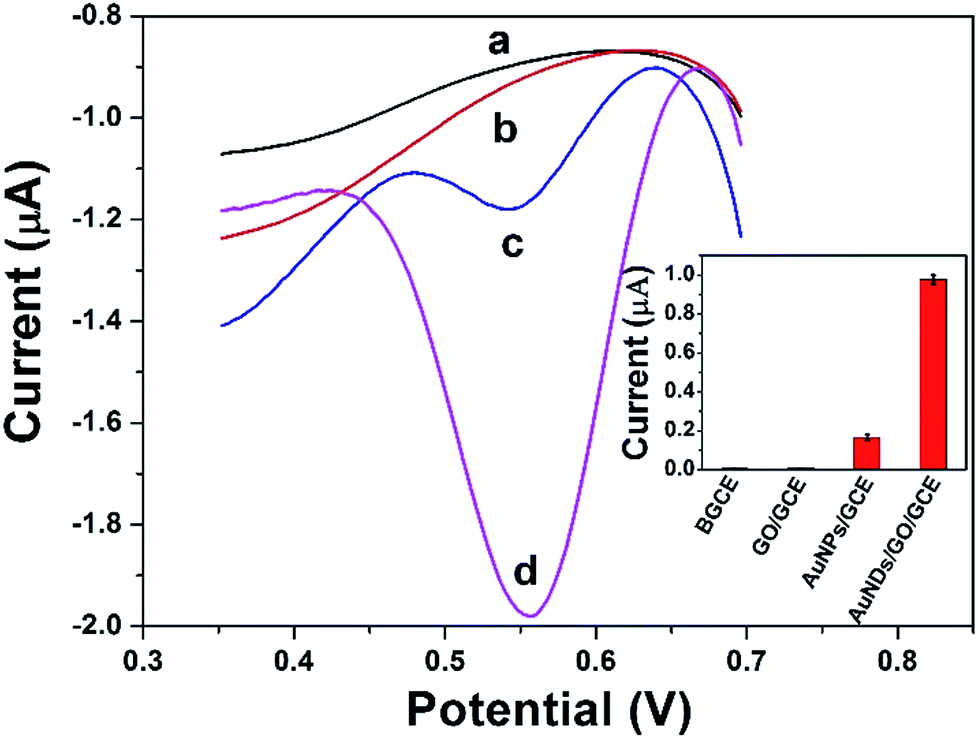 | ||
| Fig. 6 DPV curves of BGCE (curve a), GO/GCE (curve b), AuNPs/GCE (curve c), and DAuNs/GO/GCE (curve d) in 0.1 M HCl solution containing 5 μM Fe(III) and their corresponding reproducibility. | ||
Analytical performance
The calibration curve was constructed from the DPV curves obtained at the DAuNs/GO/GCE in 0.1 M HCl under the conditions previously described (Fig. 7). The peak current increased linearly in the Fe(III) concentration range from 7 nM to 1 μM, with a linear regression equation i = 3.858c + 0.21 (R2 = 0.998) and a LOD of 1.5 nM. The analytical performance of the DAuNs/GO/GCE for Fe(III) detection was superior to that of many other previously reported methods. A comparison of the DAuNs modified electrode with other methods for the determination of Fe(III) is shown in Table 1. So the DAuNs/GO/GCE would certainly be a good choice for the electrochemical determination of iron.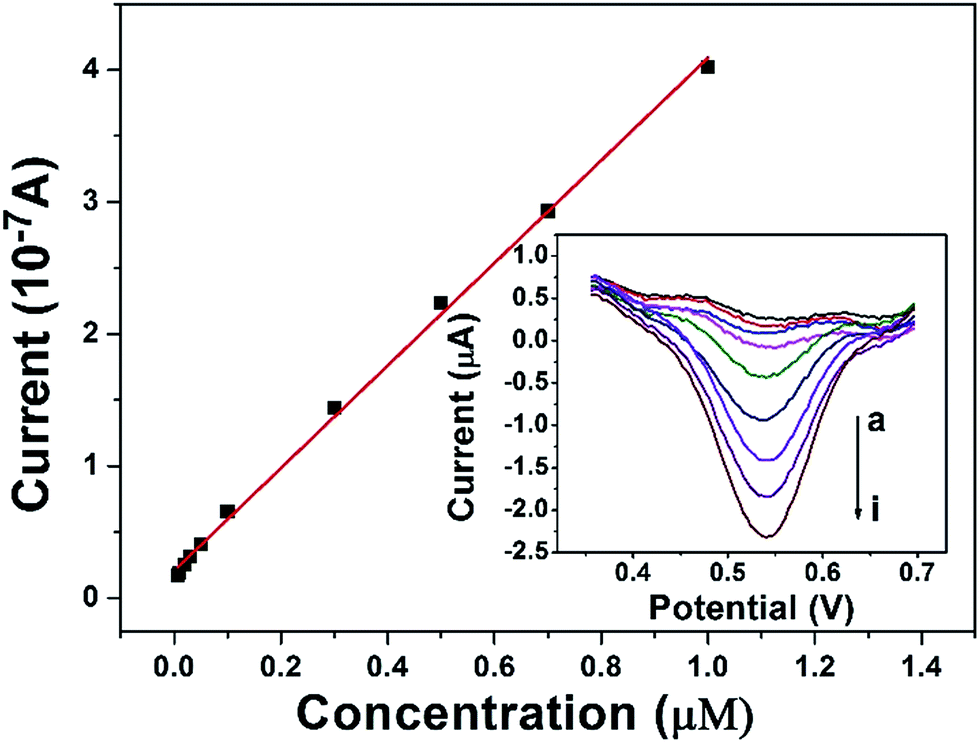 | ||
| Fig. 7 Calibration curve for the determination of Fe(III) and corresponding DPV curves at DAuNs/GO/GCE with different Fe(III) concentrations in 0.1 M HCl. Curves (a) to (i) represent the Fe(III) concentration of 7 nM, 10 nM, 20 nM, 30 nM, 0.1 μM, 0.3 μM, 0.5 μM, 0.7 μM, 1 μM respectively. | ||
| Methods | Electrode | Linear range (μM) | LOD (nM) | Reference |
|---|---|---|---|---|
| a Energy dispersive X-ray fluorescence.b Dispersive liquid–liquid microextraction.c Bismuth film modified glass carbon electrode.d Reduced graphene oxide/methylene blue/gold nanoparticles modified glassy carbon electrode. | ||||
| EDXRFa | — | 40–62![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 500 |
40000 |
28 |
| Fluorescence | — | 1–10 | 50–357 | 29 |
| DLLMEb/UV-vis | — | 0.09–7 | 27 | 30 |
| DPV | BiFEc | 0.025–1 | 7.7 | 31 |
| DPV | rGO/MB/AuNPs/GCEd | 0.3–100 | 15 | 32 |
| DPV | DAuNs/GO/GCE | 0.007–1 | 1.5 | This work |
Reproducibility, repeatability and selectivity
The reproducibility of this method was evaluated using ten independent DAuNs/GO/GCEs for the determination of 5 μM Fe(III), and the relative standard deviation (RSD) was calculated as 3.21%. The repeatability was studied by detecting 5 μM Fe(III) using the same DAuNs/GO/GCE for ten measurements, and the RSD was 2.05%.To verify the anti-interference ability of this electrode, the signal changes (%) were recorded in the presence of possible metal ions (Fig. 8). The current change caused by each of the interfering metal ion was much lower than that recorded in the presence of Fe(III). In another word, the sensor did not respond to any other ions rather than Fe(III).
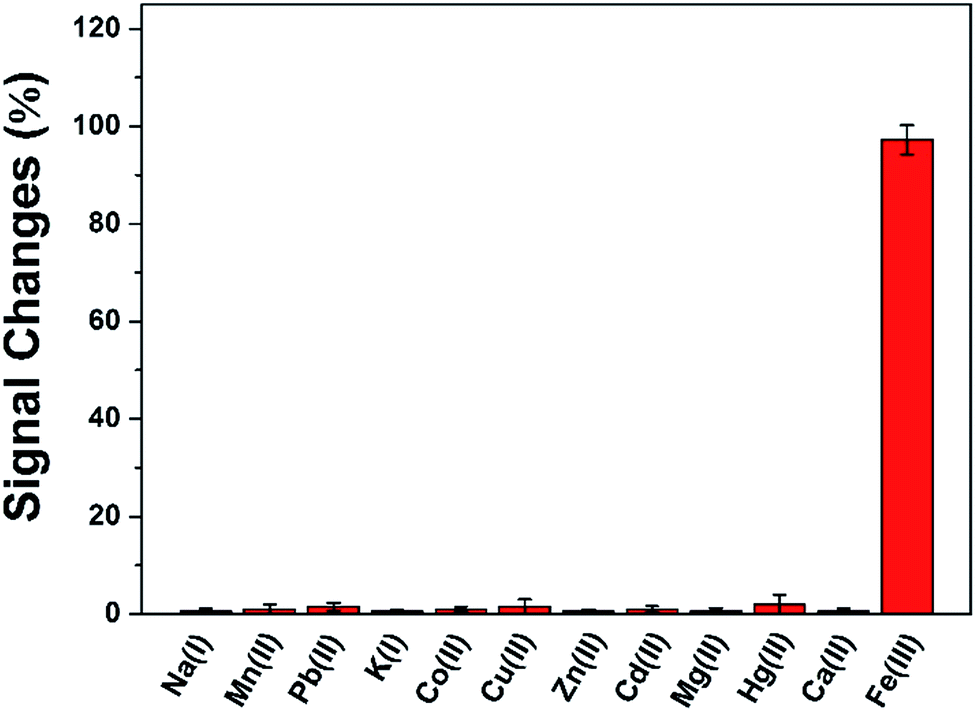 | ||
| Fig. 8 Signal changes (%) recorded in the presence of 50 μM of Na(I), Mn(II), Pb(II), K(I), Co(II), Cu(II), Zn(I), Cd(II), Mg(II), Hg(II) and 5 μM Fe(III). The data were representative of three independent experiments. | ||
Practical application for iron detection in coastal waters
The DAuNs/GO/GCE was successfully used for the analysis of the total dissolved iron in coastal water samples. DPV curves were obtained under optimal conditions discussed above. The concentration of iron was estimated by the standard addition method. Typical DPV curves were obtained after the successive addition of 0, 20, and 40 nM Fe(III), and the linear regression was shown in Fig. 9. The concentration of total dissolved iron was determined to be 14.02 nM in this coastal water sample. The results obtained by the DAuNs/GO/GCE were in agreement with those obtained by ICP-MS; the results were compared in Table 2. The DAuNs/GO/GCE may be a viable functional electrochemical sensor for the determination of total dissolved iron in coastal waters.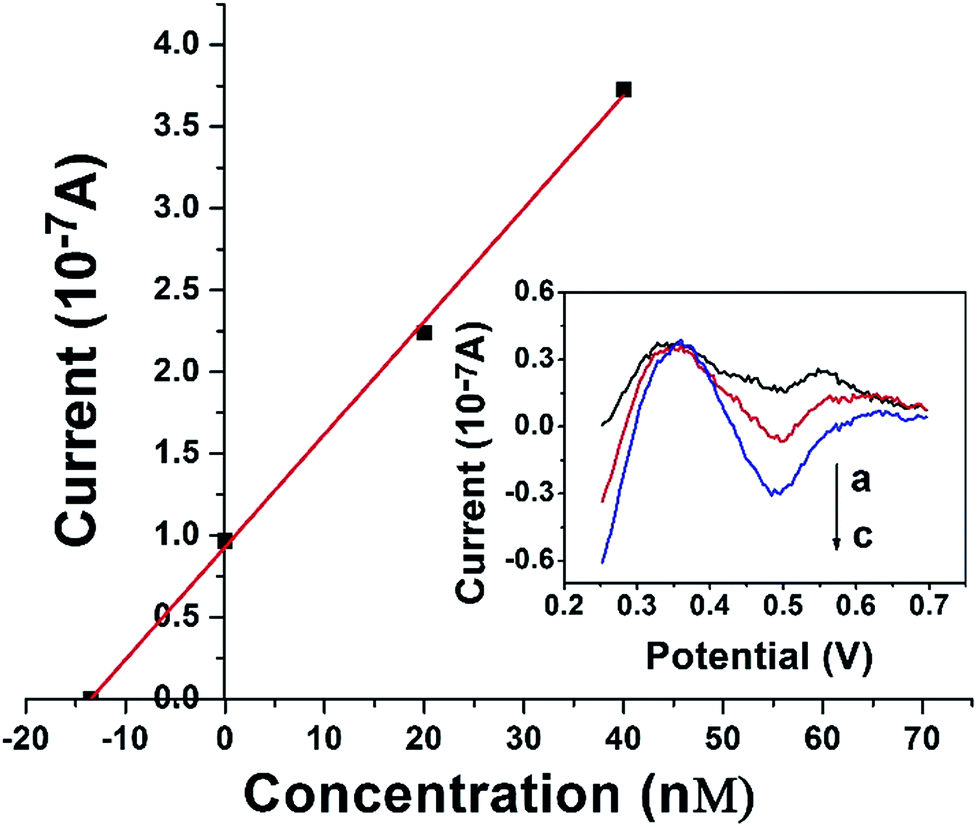 | ||
| Fig. 9 Typical linear analytical curve for the determination of Fe(III) in coastal water samples at DAuNs/GO/GCE by the standard addition method and the corresponding DPV curves (curve a, b, c represent 0, 20 nM, 40 nM Fe(III) respectively). | ||
| Coastal waters | Fe(III) spiked (nM) | Detected by this method (nM) | Detected by ICP-MS (nM) | Recovery (%) |
|---|---|---|---|---|
| Coastal water 1 | 0 | 40.17 ± 0.61 | 39.66 | — |
| Coastal water 2 | 0 | 19.83 ± 0.33 | 20.28 | — |
| 0 | 14.02 ± 0.25 | 13.88 | — | |
| Coastal water 3 | 10 | 23.86 ± 0.32 | — | 98.4 |
| 20 | 34.42 ± 0.35 | — | 102 |
Conclusion
In this paper, the use of GO as an effective morphology-controlling agent on an electrode for the controlled electrodeposition of Au nanostructures was demonstrated. This approach constituted a facile method for the direct electrochemical growth of DAuNs. The morphology-dependent performance of the fabricated electrode for iron detection in coastal waters was investigated. Under the growth mechanism, the oxygen-containing groups on GO controlled the diffusion of the AuCl4− ion, the subsequent generation of Au nuclei at the initial stage and the growth direction of the Au nanocrystals in the electrochemical synthesis process. The electrochemical performance of the Au nanostructures strongly depended on their morphologies, and the dendritic structure exhibited better performance than nanoparticles. The DAuNs/GO/GCE exhibited excellent electrocatalytic activity and could be used for the determination of Fe(III) in real coastal water samples.Acknowledgements
This work was supported by the National Natural Science Foundation of China (41276093); the Youth Innovation Promotion Association (2011170) and the Scientific Research Equipment Development Program of Chinese Academy of Sciences (YZ201558).References
- E. Schmidt, W. Kleist, F. Krumeich, T. Mallat and A. Baiker, Chem.–Eur. J., 2010, 16, 2181–2192 CrossRef CAS PubMed.
- H. Han, D. Pan, M. Lin, X. Hu and F. Li, Int. J. Electrochem. Sci., 2015, 10, 9006–9014 CAS.
- X. Hu, D. Pan, M. Lin, H. Han and F. Li, Microchim. Acta, 2015, 183, 855–861 CrossRef.
- L. Wei, Y.-J. Fan, H.-H. Wang, N. Tian, Z.-Y. Zhou and S.-G. Sun, Electrochim. Acta, 2012, 76, 468–474 CrossRef CAS.
- T. Gan, Z. Shi, Y. Deng, J. Sun and H. Wang, Electrochim. Acta, 2014, 147, 157–166 CrossRef CAS.
- A. K. Das, J. Samdani, H. Y. Kim and J. H. Lee, Electrochim. Acta, 2015, 158, 129–137 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, J. Phys. Chem. B, 2001, 105, 4065–4067 CrossRef CAS.
- B. K. Jena and C. R. Raj, Langmuir, 2007, 23, 4064–4070 CrossRef CAS PubMed.
- H. L. Wu, C. H. Chen and M. H. Huang, Chem. Mater., 2009, 21, 110–114 CrossRef CAS.
- G. Lu, C. Li and G. Shi, Chem. Mater., 2007, 19, 3433–3440 CrossRef CAS.
- C.-J. Huang, Y.-H. Wang, P.-H. Chiu, M.-C. Shih and T.-H. Meen, Mater. Lett., 2006, 60, 1896–1900 CrossRef CAS.
- B. Seo, S. Choi and J. Kim, ACS Appl. Mater. Interfaces, 2011, 3, 441–446 CAS.
- Y.-Y. Yu, S.-S. Chang, C.-L. Lee and C. R. C. Wang, J. Phys. Chem. B, 1997, 101, 66661–66664 Search PubMed.
- H. Chen, P. Kannan, L. Guo, H. Chen and D.-H. Kim, J. Mater. Chem., 2011, 21, 18271 RSC.
- J.-J. Feng, Z.-Y. Lv, S.-F. Qin, A.-Q. Li, Y. Fei and A.-J. Wang, Electrochim. Acta, 2013, 102, 312–318 CrossRef CAS.
- T. N. Huan, T. Ganesh, K. S. Kim, S. Kim, S. H. Han and H. Chung, Biosens. Bioelectron., 2011, 7, 183–186 CrossRef PubMed.
- T. H. Lin, C. W. Lin, H. H. Liu, J. T. Sheu and W. H. Hung, Chem. Commun., 2011, 47, 2044–2046 RSC.
- F. Li, D. Pan, M. Lin, H. Han, X. Hu and Q. Kang, Electrochim. Acta, 2015, 176, 548–554 CrossRef CAS.
- X. Xu, J. Jia, X. Yang and S. Dong, Langmuir, 2010, 26, 7627–7631 CrossRef CAS PubMed.
- J. J. Feng, A. Q. Li, Z. Lei and A. J. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 2570–2576 CAS.
- D.-L. Zhou, R.-Z. Wang, M. Zhang, X. Weng, J.-R. Chen, A.-J. Wang and J.-J. Feng, Electrochim. Acta, 2013, 108, 390–397 CrossRef CAS.
- E. S. Lane, D. M. Semeniuk, R. F. Strzepek, J. T. Cullen and M. T. Maldonado, Mar. Chem., 2009, 115, 155–162 CrossRef CAS.
- S. Jiao, J. Jin and L. Wang, Sens. Actuators, B, 2015, 208, 36–42 CrossRef CAS.
- H. Han, D. Pan, X. Wu, Q. Zhang and H. Zhang, J. Mater. Sci., 2014, 49, 4796–4806 CrossRef CAS.
- I. Khalil, N. Julkapli, W. Yehye, W. Basirun and S. Bhargava, Materials, 2016, 9, 406 CrossRef.
- G. Goncalves, P. A. A. P. Marques, C. M. Granadeiro, H. I. S. Nogueira, M. K. Singh and J. Grácio, Chem. Mater., 2009, 21, 4796–4802 CrossRef CAS.
- G. Park, K. G. Lee, S. J. Lee, T. J. Park, R. Wi and D. H. Kim, J. Nanosci. Nanotechnol., 2011, 11, 6095–6101 CrossRef CAS PubMed.
- S. Biswas, V. H. Rupawate, K. N. Hareendran and S. B. Roy, J. Radioanal. Nucl. Chem., 2015, 306, 543–548 CrossRef CAS.
- K.-W. Cha and K.-W. Park, Talanta, 1998, 46, 1567–1571 CrossRef CAS PubMed.
- B. Peng, Y. Shen, Z. Gao, M. Zhou, Y. Ma and S. Zhao, Food Chem., 2015, 176, 288–293 CrossRef CAS PubMed.
- A. Bobrowski, K. Nowak and J. Zarebski, Anal. Bioanal. Chem., 2005, 382, 1691–1697 CrossRef CAS PubMed.
- M. Lin, H. Han, D. Pan, H. Zhang and Z. Su, Microchim. Acta, 2014, 182, 805–813 CrossRef.
| This journal is © The Royal Society of Chemistry 2017 |