
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The formation of cerium(III) hydroxide nanoparticles by a radiation mediated increase in local pH†
P. Abellan *abc,
T. H. Moserd,
I. T. Lucase,
J. W. Gratec,
J. E. Evansf and
N. D. Browningcg
*abc,
T. H. Moserd,
I. T. Lucase,
J. W. Gratec,
J. E. Evansf and
N. D. Browningcg
aSuperSTEM Laboratory, SciTech Daresbury Campus, Keckwick Lane, Daresbury WA4 4AD, UK. E-mail: pabellan@superstem.org
bInstitute for Materials Research, University of Leeds, Leeds, LS2 9JT, UK
cPhysical and Computational Sciences Directorate, Pacific Northwest National Laboratory, Richland, WA 99352, USA
dDepartment of Mechanical Engineering and Engineering Mechanics, Michigan Technological University, Houghton, MI 49931, USA
eSorbonne Universités, UPMC University of Paris 06, CNRS, UMR 8235, LISE, Paris 75005, France
fEnvironmental Molecular Sciences Directorate, Pacific Northwest National Laboratory, Richland, WA 99352, USA
gDepartment of Materials Science and Engineering, University of Washington, Seattle, WA 98195, USA
First published on 13th January 2017
Abstract
Here we report radiation-induced formation of Ce(III) nanostructures in an in situ liquid cell for the transmission electron microscope (TEM). Small (<5 nm) irregular Ce(OH)3 nanoparticles were identified as the final products from cerium(III) nitrate solutions of initial pH 5.2. Pourbaix diagrams show that solid Ce(OH)3 can only exist above pH 10.4, whereas at lower pH values, Ce(III) should remain soluble as aqueous Ce3+. Reduction of Ce3+ to zerovalent Ce by aqueous electrons followed by hydrolysis is a plausible catalytic mechanism for generating hydroxide. Numerical simulations support that radiolysis of cerium nitrate solutions may lead to pH increases, in contrast to well-known acidification of pure water. Compared to previous radiolytic synthesis routes in aqueous solution for other metal or metal oxide nanoparticles, based on metal ion reduction, for example, the chemical pathways leading to these Ce(III) nanostructures require an increase in the local pH to alkaline conditions where Ce(OH)3 can exist. These results extend the range of chemical conditions that can be induced by radiolysis to form oxidized nanostructures from solution.
Introduction
It is well known that the growth of nanoparticles can be advantageously initiated by the action of ionizing radiation on solutions of metal salts, i.e. by radiolysis.3–6 The energy that is deposited by gamma rays, X-rays, or electron beams generates reactants mainly from the solvent molecules, the most reactive of which (eaq− and OH˙ for the case of water) will mostly interact within or near the irradiated area,7 allowing the location of nucleation and growth to be precisely controlled. In aqueous solutions, the primary reactants are eaq−, H3O+, H˙, OH˙, H2, H2O2 and HO2, and for the case of pure water, the predominant formation of H3O+ over OH− upon irradiation leads to an overall steady state decrease in the local pH.8,9 While exceptional cases of metal oxide nanoparticle growth6,10–12 and metal–organic frameworks (MOFs) growth13 exist, the majority of nanoparticles formed by radiolysis are zero valent metals arising from the reduction of metal ions in solution by the aqueous electrons.The development of in situ liquid stages14–17 for (scanning) transmission electron microscopes (S)TEM has enabled nanoparticle nucleation to be studied with immediate quantitative imaging of the final products.18–20 This in situ (S)TEM approach exploits the chemical reactivity of the radiolytic species formed by the interaction of high-energy imaging electrons with the precursor-containing solution (radiation chemistry), and can avoid large excesses of chemical reagents.
We have an ongoing interest at our laboratory in cerium oxide nanoparticles,21,22 which are actually a mixture of Ce(III) and Ce(IV). Recently, we examined the radiolysis of Ce(III) nitrate solutions by in situ TEM. Because zerovalent cerium is not stable in aqueous solution, we anticipated that cerium oxy or hydroxy species would form. We were surprised to discover that Ce(OH)3 nanoparticles formed, even though this solid is unstable1,2,23 at the initial pH, 5.2, of the starting solutions.
Nanostructures of cerium and oxygen are of interest in a number of applications.24,25 Nanoparticles of cerium oxide containing Ce(III) are of particular interest as potential therapeutics to provide a persistent antioxidant effect in the treatment of a number of medical conditions.21–23,26–32 In this application it has been found that small irregular nanoparticles of less than 5 nm are most effective, while larger more crystalline particles may be inert or even harmful. The desired small particles are produced by a room temperature synthesis involving the action of excess hydrogen peroxide on cerium(III) nitrate, leading initially to Ce(IV)-containing CeO2.21 However, as excess peroxide decomposes, the high surface to volume ratio and surface oxygen vacancies of very small particles lead to high proportions of Ce(III) in the final products, driven by particle size. Although conventionally still called nanoceria, these nanoparticles are not simply CeO2, nor are they mainly Ce(IV). It is further the case that in the production of biomedical nanoparticles, it is desirable to minimize large excesses of chemical reagents.6 For the particular case of biomedical cerium-based nanoparticles, the in situ generation of a suspension of nanoparticles, without gross amounts of reagent to remove, is of great interest due to the strong environmental dependency of nanoceria antioxidant activity.33 Normally, Ce(OH)3 nanostructures are prepared chemically by the action of large excesses of alkali on solutions of cerium(III) nitrate.34 Cerium hydroxide, Ce(OH)3, is used as a protector in corrosion cells24 and has been found to be efficient on modulating the redox properties of the protein hemoglobin (Hb) leading to the preferred stabilization of Hb–Fe(II).25 Ce(OH)3 is also a precursor to Ce(IV) based nanostructures. In air, Ce(OH)3 can oxidize to cubic Ce(OH)4.35 Ce(OH)3 nanoparticles can also be directly oxidized to nanoceria, using either a low-temperature thermal treatment under high oxygen level environment36–38 or by reaction with hydrogen peroxide (H2O2).
Typical electron doses used in the liquid cell are much greater than doses used in conventional radiation chemistry synthesis.9 As such, quantitative control of reactions necessitates careful experimentation and interpretation, since the dose can introduce artefacts and be difficult to control.7,13,18,39 The quasi-2D geometry of a liquid cell provides an ideal scenario for fast diffusion of radicals to the irradiated area20,40 and the presence of membranes mainly dictates the location of particles growth, as repeatedly observed experimentally. These extreme conditions provide opportunities for new synthesis and new scientific insights. As an example, in previous work, we proposed using radiation chemistry and high doses in the STEM to visualize breakdown processes and attack mechanisms in situ, relevant to the electrochemical stability of a number of lithium-ion battery (LIB)-electrolyte solutions in the millisecond timescale,41 while more recently lower dosages outside the STEM have been used to investigate such processes in faster regimes, down to the picosecond time scale.42,43
Here, we present our results for the formation of nanoparticles from solutions of cerium(III) nitrate confined in an in situ liquid cell and irradiated under very high beam currents. As zero valent cerium is unstable in aqueous solutions, metal clusters cannot be expected. Empirically we found Ce(OH)3 nanoparticles in the irradiated zone. Interestingly, with regard to the reaction chemistry taking place in solution, the formation of this solid phase from Ce3+ ions requires a local increase in solution pH (based on the known redox and phase chemistry expressed in the Pourbaix diagram for this system).23 Hence, the formation of Ce(OH)3 represents a new observation for radiolytic nanoparticle growth that requires the induced solution chemistry to be different from more conventional redox processes. Here we describe the identification of the Ce(OH)3 nanoparticles and interpret their formation in terms of a beam-induced increase in pH. Chemical equations are presented whereby hydroxide ions are expected to be generated, entailing initial reduction of Ce3+ by aqueous electrons, followed by hydrolysis reactions producing OH−, molecular hydrogen and regenerating Ce3+. We further support this interpretation with modeling that shows that in contrast to the radiolysis of pure water, where pH becomes more acidic, the radiolysis of solutions containing cerium leads to pH changes in the direction of greater alkalinity.
Experimental
In the experiments performed here, precursor solutions for the in situ TEM investigations were prepared following a procedure described elsewhere.21 The solution consisted of cerium(III) nitrate (Sigma Aldrich), Ce(NO3)3·6H2O, dissolved in deionized (DI) water to a concentration of 0.1 mM. The initial pH was measured to be 5.2. Small volumes of precursor solution (0.5 μL) were loaded in the fluid cell and confined within two silicon nitride (SNx) membranes supported on silicon chips and irradiated using a 300 kV electron beam (see ESI† for details on the experimental methods). The chips were subsequently washed with DI water for post-mortem analysis of the reaction products that were deposited on the SiNx membranes.Results and discussion
In the STEM, the dose rate used for inducing growth can be tuned by setting the beam current, the pixel dwell time and the magnification (irradiation area). Among these parameters, the electron beam current has shown to have the highest impact on formation kinetics of metal particles.18 First, the effect of changing magnification and pixel-dwell time as a way to tune the electron dose, was investigated for a fixed low beam current of 6 pA (in situ experiments not shown) resulting in no apparent particle growth. The effect of beam current on the Ce(III) nitrate solution was subsequently investigated in situ by comparing the low (6 pA) beam current conditions with high (80.5 pA) beam current conditions while keeping the dose rate constant at ∼3 e− per Å2 per frame. We note that the total acquisition time per frame is 3.78 s for all datasets shown in the manuscript; which also accounts for the scan flyback time occurring outside the imaging area. As already mentioned, the lowest beam current provided imaging conditions where no in situ growth was observed in the STEM (current of 6 pA, magnification M = 160![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000×, image size of 1024 × 1024 and pixel dwell time 3 μs). Real-time observation of particle growth was found for the largest current value of 80.5 pA. Fig. 1(a) shows images captured during the in situ experiment, where ∼10 nm sized bright structures grow over time. It is worth noting that, in order to investigate the effect of beam current and make a comparison between in situ experiments we chose to keep the dose rate constant while also keeping unchanged the pixel-dwell time and the number of pixels per image, i.e. the total acquisition time is comparable for the different datasets. The dose variation in this paper was therefore simply achieved by changing the magnification of the images (from M = 160000× to 40000×). Under these conditions, the pixel size was 2.3 nm, which sets a limitation on the precision of in situ measurements we were able to do. Indeed, observation of growth kinetics in the nanometer range was limited (only changes in image intensity of ≥5 nm, corresponding to approximately twice the size of one pixel would be resolved in the in situ data). After disassembling of the cell and rinsing of the chips with DI water, these particles could be resolved on the dried membranes using ex situ high-resolution TEM (HRTEM) Fig. 1(b) and (c). In irradiated areas where particles were still well-separated, a mean diameter of 2.9 nm was measured with particles size ranging from 1 to 4 nm (particle size distribution is shown in Fig. 1(b)). The large bright areas in the in situ movie frames in (a) correspond to regions where a higher abundance of nanometric particles was grown (Fig. 1(c)).
000×, image size of 1024 × 1024 and pixel dwell time 3 μs). Real-time observation of particle growth was found for the largest current value of 80.5 pA. Fig. 1(a) shows images captured during the in situ experiment, where ∼10 nm sized bright structures grow over time. It is worth noting that, in order to investigate the effect of beam current and make a comparison between in situ experiments we chose to keep the dose rate constant while also keeping unchanged the pixel-dwell time and the number of pixels per image, i.e. the total acquisition time is comparable for the different datasets. The dose variation in this paper was therefore simply achieved by changing the magnification of the images (from M = 160000× to 40000×). Under these conditions, the pixel size was 2.3 nm, which sets a limitation on the precision of in situ measurements we were able to do. Indeed, observation of growth kinetics in the nanometer range was limited (only changes in image intensity of ≥5 nm, corresponding to approximately twice the size of one pixel would be resolved in the in situ data). After disassembling of the cell and rinsing of the chips with DI water, these particles could be resolved on the dried membranes using ex situ high-resolution TEM (HRTEM) Fig. 1(b) and (c). In irradiated areas where particles were still well-separated, a mean diameter of 2.9 nm was measured with particles size ranging from 1 to 4 nm (particle size distribution is shown in Fig. 1(b)). The large bright areas in the in situ movie frames in (a) correspond to regions where a higher abundance of nanometric particles was grown (Fig. 1(c)).
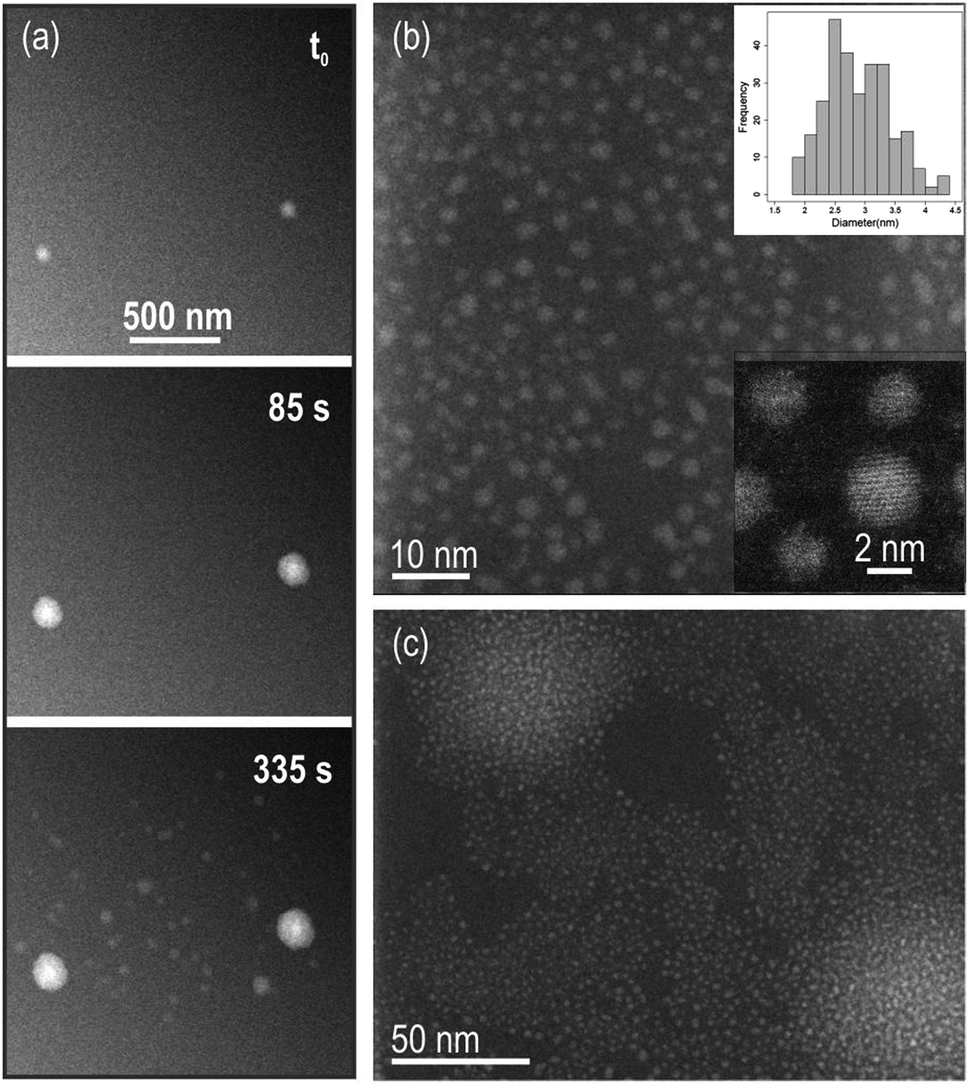 | ||
| Fig. 1 (a) Snapshots of the dark field (DF) STEM in situ Movie S1† showing growth of particles in solution. Beam current was 80.5 pA, magnification 40000× and pixel dwell time 3 μs, to give a dose rate of 3 e− per Å2 per frame. The total acquisition time per frame is 3.78 s and the image size is 1024 × 1024 pixels. (b) Reaction products and particles size distribution including a total of 279 particles (top inset). Mean diameter of 2.9 ± 0.5 nm. Particles are monocrystalline (bottom inset). (c) Brightest areas in the in situ data in (a) are regions with higher density of particles. | ||
Structural analysis using high-resolution (HR) images and electron diffraction patterns (EDP) of these particles allowed us to identify them as Ce(OH)3. See ESI text and Fig. S4–S6† for a detailed characterization of the structure and texture of the reaction products. Fig. 2(a) is a HR TEM image of the observed nanoparticles showing coalesced particles with specific zone axis aligned along the observation direction and their corresponding Fast Fourier Transform (FFT) on the bottom right. Notably, the brightest diffraction rings on the FFT of the overall nanocrystalline area, correspond to the (−121), (2−11), (111) and (2−21), (201), (021) family of planes of the hexagonal structure (with nominal spacing of 2.47 and 2.26 angstroms respectively). A detailed analysis of the images covering individual particles revealed an intermediate situation with randomly distributed particles and a preferential orientation configuration where the above-mentioned planes lie parallel to the membrane surface. Besides high magnification images of the reaction products, lower magnified images were taken showing that the irradiated area on the SNx membrane surface was covered with particles that only slightly extended beyond the window forming a “halo” in the closely surrounding area, (see ESI Fig. S1 and S2† for low magnification images of the irradiated/growth area).
 | ||
| Fig. 2 HR TEM images of the reaction products of the experiments performed for the beam currents indicated on the images. (a) Bottom right inset is a FFT of the differently oriented Ce(OH)3 particles on the HRTEM image showing bright spots corresponding to the (−121), (2−11), (111) and (2−21), (201), (021) family of planes of the hexagonal structure. This HRTEM image indicates that the brightest areas in the in situ data in Fig. 1(a) are regions with a large population of spherical particles that have undergone coalescence and reorientation to specific zone axis aligned along the electron beam direction, see particle in indicated area A. (b) Precipitates observed on the SiNx membranes after rinsing the chips with DI water on a region that had been irradiated using 6 pA beam current. The brightest spots in the FFT correspond to the (111) planes of the fluorite structure. | ||
Ex situ high-resolution TEM (HRTEM) images were also taken after disassembly, rinsing and drying of the chips irradiated only under low beam current conditions of 6 pA, where no in situ growth had been observed. Nanometric crystallites of the cubic fluorite structure of ceria and most likely of its isostructural hydroxide (CeO2 and Ce(OH)4, respectively) were found, see Fig. 2(b). Similar cubic fluorite structures of ceria were found on nonirradiated surfaces of disassembled, rinsed, and dried chips from experiments at both the lower beam current of 6 pA and the higher beam current of 80.5 pA. As a control experiment, a solution of the precursor was drop cast on a holey carbon grid, showing that similar particles covered the grids (see ESI Fig. S7†). These particles are in all cases precipitates that most likely form by oxidation of Ce3+ during the rinsing process. We note that the nanocrystalline films grown during rinsing of the chips are remarkably different from the nanoparticles resulting from the high beam current in situ experiments (Fig. 1 and 2). The hexagonal Ce(OH)3 nanoparticles formed in the beam area at 80.5 pA beam current (Fig. 2(a)) are unambiguously distinguishable from cubic (CeO2/Ce(OH)4) films when using 6 pA (Fig. 2(b)).
Interestingly, in experiments where the high current was used to generate Ce(OH)3, increasing the electron dose per frame from 3 e− Å−2 to 24 e− Å−2, resulted in the previously formed Ce(OH)3 dissolving back into solution (see Fig. 3 and ESI Movie S2†). We discuss all these observations next.
 | ||
| Fig. 3 Frames of a BF STEM in situ movie using a magnification of M = 115000×, pixel-dwell time of 3 μs, the high beam current conditions (80.5 pA) to give an electron dose per frame of 24 e− Å−2. The total acquisition time per frame is 3.78 s and the image size is 1024 × 1024 pixels. Under these conditions, already formed Ce(OH)3 particles dissolved in situ. | ||
Despite the strong potential of in situ experiments to uncover the underlying mechanisms behind nanoparticles growth and their media-dependent stability, an apparent drawback is the lack of information on the pH during the experiment or even after the observations, since suitable nanoscale pH meters have not been developed yet. To aid with this issue, we introduce the use of simplified Pourbaix diagrams which contain information about the pH-dependent stability and reactivity of relevant species with surrounding redox species. Fig. 4 shows a revised and simplified Pourbaix diagram (E–pH diagram) for cerium calculated for the concentration of soluble Ce species at 0.1 mM used in these experiments (see ESI† for details on the calculations). The diagram shows the domains of existence for solid phases like CeO2 or hydrated forms like Ce(OH)4 and Ce(OH)3, and the domains of predominance of soluble cerium species (Ce(IV): [Ce(OH)]3+, [Ce(OH)2]2+ and Ce(III): Ce3+); note that Ce4+ ions are stable only at negative pH, i.e. for [H+] > 1 mol L−1. We have also included in the diagram relevant redox couples for radiation chemical synthesis in water. The Pourbaix diagram can help to examine two issues: (1) the formation of Ce(OH)3, which requires that the local medium become alkaline, and (2) the observation of Ce(IV) precipitates, which requires oxidation.
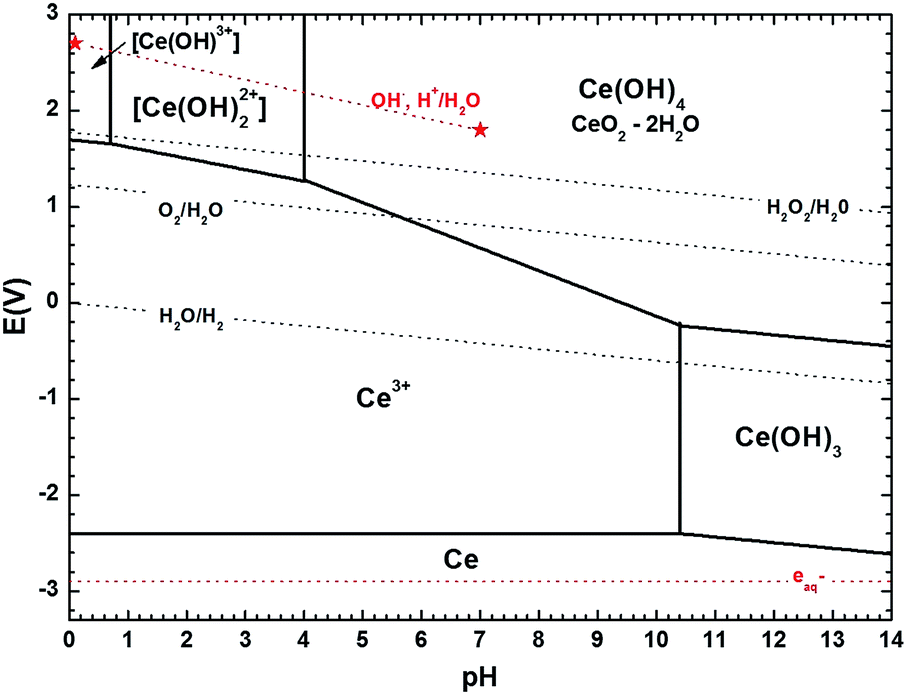 | ||
| Fig. 4 Revised version of the cerium Pourbaix diagram (1974) proposed by Hayes et al. (2006)1 using more recent thermodynamic data on cerium species2 and computed here for a concentration of soluble Ce species of 0.1 mM. Ce(IV) tri-hydroxo and Ce(III) mono-, di-hydroxo complexes were not considered.1 The pH-dependent redox potential of different redox couples associated to water, i.e. O2/H2O, H2O2/H2O, H2O/H2 (dark doted lines) and to radiolytic processes in water, i.e. (OH˙, H+)/H2O, aqueous electrons (red dotted lines) are also represented. All calculations done for plotting the diagram can be found in the ESI.† | ||
When freshly prepared, the precursor solution at pH 5.2 contains only Ce(III) soluble species, mostly Ce3+ ions resulting from dissolution of cerium(III) nitrate salt according to the following reaction:
| Ce(NO3)3·6H2O → Ce3+ + 3NO3− + 6H2O | (1) |
Upon pH increase, Ce3+ can precipitate into Ce(OH)3 according to:
| Ce3+ + 3OH− ⇌ Ce(OH)3 | (2) |
The formation of hexagonal Ce(OH)3 found experimentally requires a horizontal translation on the Pourbaix diagram from Ce3+ at pH 5.2 to Ce(OH)3 at a pH greater than 10.4. While pH or pH changes cannot be currently measured in liquid cells, thermodynamics dictate that such a local pH change must have occurred. This observation is also consistent with chemical synthesis of Ce(OH)3 by mixing cerium(III) nitrate solutions with excess alkali.34 As discussed in the ESI,† we note here that the specific pH threshold at which Ce(OH)3 precipitates (pH > 10.4 in our calculations) as calculated in simplified Pourbaix diagrams would yield a slightly higher or lower value depending on the species considered and on the specific thermodynamic data used from the literature.
The formation of Ce(IV) solids requires oxidation of solution Ce3+ and a vertical translation on the Pourbaix diagram. In aerated solutions (pO2 ∼ 2 × 10−4 Pa, [O2]water = 0.25 mmol L−1) the presence of dissolved oxygen only – without any other oxidizing species – could lead to precipitation of ceria in solution. As seen in Fig. 4, Ce3+ may be oxidized by dissolved O2 to Ce(IV) species of Ce(OH)4 or CeO2·2H2O upon a slight increase of pH to 5.6, (the redox potential of O2/H2O couple is indeed greater than the one of Ce(OH)4/Ce3+ when the pH is greater than 5.6), explaining the “fragile” stability of Ce(III) ion precursor solutions in this pH range. However, as precipitation is not observed in situ, it is therefore more likely that the ceria precipitates formed upon washing and drying in air.44,45
The formation of Ce(OH)3 by electron beam irradiation stands in contrast to conventional radiation-induced growth experiments that generally aim at promoting a reducing environment for metal nanostructure growth by adding scavengers for the OH˙ radicals in water.8,46 An alternative that we have explored in previous work is selecting a solvent that creates only reducing species with no need of scavenging additives.20 In the present research, as seen in Fig. 4, the thermodynamic stability of the Ce(OH)3 phase can be achieved only for pH > 10.4. It is worth noting, that for pure DI water, a decrease in pH during electron beam irradiation would be expected.8,9 Indeed, high energy electron irradiation of DI water typically leads to pH decrease because of the predominant formation of H3O+ over OH− during water radiolysis, after the initial formation of the reductive radical H˙ and oxidizing OH˙. This trend is so for typical radiation sources and has also been confirmed for the higher doses in TEM mode,9 suggesting that Ce(aq)3+ (and possibly NO3(aq)− as discussed below) in the precursor solution must play a role in generating a basic environment that is conducive to the type of growth we observe.
Formation of Ce(OH)3 nanoparticles from Ce(aq)3+ NO3(aq)− precursors, most likely requires the combined effect of aqueous electrons, OH˙ radicals and Ce-species in solution (possibly also nitrate ions) to locally increase the pH. The following redox reactions involving Ce-species in solution are considered as a plausible chemical pathway for the increase of OH− leading to an overall “unexpected” increase in pH:
| Ce3+ + OH˙ ⇌ Ce(IV) + OH− | (3) |
| Ce3+ + 3eaq− ⇌ Ce0 | (4) |
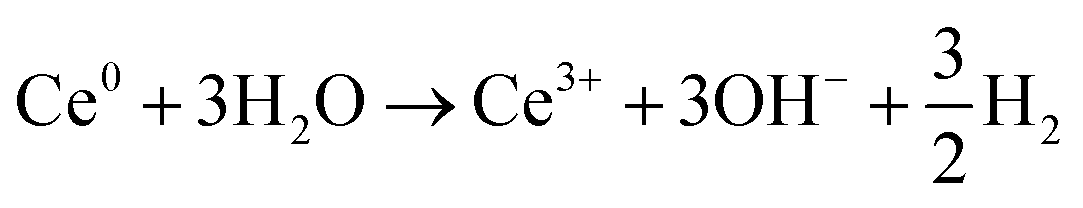 | (5) |
| Ce(IV) + eaq− ⇌ Ce3+ | (6) |
Assuming an initial solution with an abundance of Ce3+ among the Ce-species (initial pH 5.2), Ce(IV) can be produced by the direct scavenging of OH˙ radicals by the Ce3+ ions (eqn (3)) in solution; with Ce(IV) representing Ce(OH)4 precipitates and [Ce(OH)2]2+ soluble species, the ratio of which increases with pH. Ce3+ can be reduced directly to metallic cerium, Ce0, by scavenging of eaq− with highly negative redox potential E0 = −2.9 V vs. SHE.47 However, Ce0 will spontaneously react with water leading to production of OH− and hydrogen gas as shown on eqn (4) and (5). It is worth noting that, radiolytic synthesis of Ce metal nanoparticles is not possible in aqueous solution because of the instability of Ce0 in water, the same as for Li0 and Na0 (E° Ce3+/Ce is highly negative). Note also that, eqn (4) and (5) together constitute a catalytic cycle, where Ce3+ catalyzes the conversion of aqueous electrons to hydroxide, which could alone explain the local pH rise. As the presence of Ce(IV) increases, from the oxidation reaction on eqn (3), Ce(IV) will compete favorably with Ce3+ for the reaction with eaq−. In fact, the rate constant for the first reduction step of Ce3+ (eqn (4)) is k < 1 × 109 L mol−1 s−1,48 versus k = 6.6 × 1010 L mol−1 s−1 for the reduction reaction of Ce(IV) (eqn (6)).49 This means that any Ce(IV) generated by oxidation of Ce(III) with OH˙ should be re-reduced to Ce(III) by aqueous electrons. Overall (eqn (3)–(6)), this means that the combined effect of OH˙ and eaq− on the cerium species can contribute to an increase in pH – through an increase in the concentration of OH− ions – provided that reaction (6) is kinetically favored, and the catalytic cycle of eqn (4) and (5) is occurring.
Additional increase of the pH might originate from the presence of nitrate ions, NO3(aq)−, in the solution and the proximity of the membranes (see details of these reactions S1 and S2 and their possible activation pathways in the ESI†). The electron beam is able to produce a measurable “local drop” of the deposition potential, as has been observed in in situ electrochemical experiments.50 This effect of the electron beam at the membranes, together with a high local concentration of aqueous electrons and nitrate ions could activate additional chemical pathways which might contribute to the local increase of pH51–53 as has been proposed elsewhere and detailed in the ESI.†
In order to demonstrate the viability of the mechanism proposed (eqn (3)–(6)) as a way to raise the pH in situ, computer modeling of the radiation-induced changes9 to both: aqueous solutions containing Ce-species including relevant redox reactions and to pure DI water as a comparison were performed (see ESI† for details on the calculations and a complete list of reactions and results). The radical concentrations have been estimated using a reaction-diffusion model that has already proven useful for explaining in situ trends in the TEM by considering all species present during the experiment.40 The model takes into account the production of radiolysis species in the irradiated area, the effect of reverse reactions and the diffusion of species outside the irradiated area. The model doesn't take into account diffusion of species into the irradiated area and the effect of membranes. STEM radiation conditions have been approximated by considering an irradiation volume of a static single pixel volume. As expected for pure DI water,9 the trend observed in pH evolution upon electron beam irradiation is a decrease of pH which is more pronounced for extended time periods (see Fig. 5) and for higher doses (see ESI Fig. S7†). Modeling results that included the effect of Ce-species are also shown in Fig. 5. In contrast to pH decreases for pure DI water, solutions containing cerium ions are observed to increase their pH, becoming alkaline within a millisecond of constant irradiation. In the simulation, the pH rises to a plateau around 7.23. It is worth noting that although the model used for the calculations qualitatively show the trends proposed, i.e. that an increase of pH is possible by the combined effect of the Ce-species radiolysis in aqueous solution, a quantitative match with results was not expected due to the important approximations that these calculations made, such as neglecting the effect of membranes or assuming a closed system with no inter-exchange of species. We also note that for the particular case of STEM irradiation, interdiffusion will be higher and that the irradiation time (pixel dwell time is in the microsecond time scale) is just at the limit between the inhomogeneous and homogeneous kinetic regimes, which might lead to higher calculation errors. This qualitative agreement rather than a quantitative match is nevertheless consistent with previous modelling efforts using TEM irradiation, where in situ phenomena and general trends have been explained by incorporating in the calculations relevant chemical equations involving the solutes and not only the radiolysis of the solvent.9,40 We believe that also for the case of STEM, these radiation chemistry models can be informative, and greatly aid in the interpretation of new phenomena and dynamic mechanisms observed.
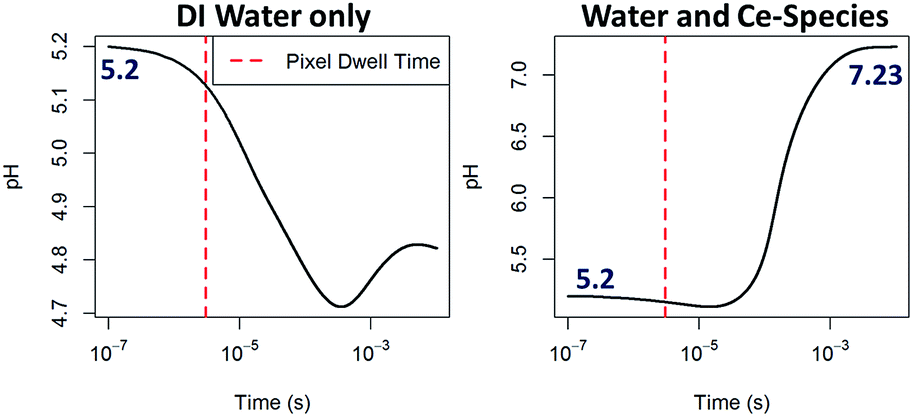 | ||
| Fig. 5 Computing of the pH evolution over time upon continuous e−-beam irradiation within a one pixel interaction volume starting from an initial pH of 5.2, 0.1 mM Ce(NO3)3 solution and dose rate of 106 Gy s−1 for DI water (left) and for a solution considering eqn (3)–(6), right. Further details on these calculations are given in the ESI.† | ||
The apparent stability of the very small nanometric Ce(OH)3 particles in the time frame of our experiments is also of interest. One might expect either re-dissolution to Ce3+ or oxidation to CeO2. For example, Ce(OH)3 formation could produce a drop in OH− concentration, which would decrease the solution pH, translating left on the Pourbaix diagram and promoting dissolution. Alternatively, once formed, Ce(OH)3 could be oxidized by dissolved O2, translating vertically on the diagram. Larger Ce(OH)3 nanostructures have been chemically converted to CeO2 without shape change by chemical means.34 Instead, Ce(OH)3 in the small particle sizes and under conditions of this experiment appear to be chemically stable. Absorbed species, such as nitrate ions, could promote particle stabilization, as previously observed in the stabilization of suspensions of nanometric ceria particles.54 Large electron beam currents might promote the rapid incorporation of ions within the particles during direct irradiation, as compared to lower beam currents. In the field of biomedical nanoceria, it has been shown that very small particles of CeO2 in the 2–5 nm range generated by oxidation with hydrogen peroxide relax to particles containing predominantly Ce(III) when the peroxide has decayed away; this preference for Ce(III) is attributed to small size and surface oxygen vacancies. In any case, the observed nanoparticles, like biomedical nanoceria, containing cerium and oxygen, are in the 2–5 nm range, and are predominantly Ce(III).
The formed particles have been observed to re-dissolve into solution when an eight-fold increase in the electron dose rate is performed in situ while keeping the high beam current conditions (Fig. 3). This increase in electron dose rate is achieved by increasing the magnification (decreasing the irradiation area). We don't believe this process is due to either knock-on damage (energy was kept constant) or to further radiolysis damage of the solution for the reasons discussed above on particles stability after formation has been achieved. We speculate that a different characteristic critical dose might have been surpassed, in this case related to the solid particle. For solid specimens, bond breakage can result in radiolytic decomposition, or mass loss, with a subsequent shrinkage of the specimen.55 By increasing magnification, and thus reducing the area where the beam is rastered, the fraction of broken bonds can be rapidly increased.
Conclusions
In summary, using a fluid stage in combination with STEM, we have observed the formation of cerium oxide nanoparticles Ce(OH)3, starting from slightly acidic precursor solutions. Under the initial conditions of the experiment at pH 5.2, Ce(OH)3 is thermodynamically unstable, according to Pourbaix diagrams1,2 and Ce(III) should instead be dissolved as Ce(aq)3+. Ce(OH)3 is expected to be a stable solid phase only under alkaline conditions. Nevertheless, Ce(OH)3 nanoparticles were identified in these experiments. A cascade of radiolytic and chemical reactions induced by the electron beam, involving cerium species in addition to water molecules, is proposed to induce a local pH increase as required for growth of Ce(OH)3. Reduction of Ce(aq)3+ to zerovalent cerium followed by hydrolysis provides a catalytic mechanism for generating hydroxide ions. Modeling supports the premise that irradiation of Ce(aq)3+ will lead to pH increases rather than the pH decreases conventionally seen upon irradiation of water.Metal oxide nanoparticles formed under radiolytic conditions and reported in previous works arose primarily from redox reactions, while zero valent metal nanoparticles also resulted from reduction reactions.6 The new results reported here are consistent with a mechanism for nanoparticle formation under electron beam irradiation based on a radiolytically-induced pH change in the presence of Ce(aq)3+.
Acknowledgements
The experimental work involving the development and application of in situ liquid stages in the TEM was supported by the Chemical Imaging Initiative; under the Laboratory Directed Research and Development Program at Pacific Northwest National Laboratory (PNNL). PNNL is a multi-program national laboratory operated by Battelle for the U.S. Department of Energy (DOE) under Contract DE-AC05-76RL01830. We thank Yongsoon Shin and Prabhakaran Munusamy for assistance with precursor solutions, and Ajay Karakoti for helpful discussions on solution synthesis chemistry for cerium oxide nanoparticles. A portion of the research was performed using the Environmental Molecular Sciences Laboratory (EMSL), a national scientific user facility sponsored by the Department of Energy's Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory. The SuperSTEM Laboratory is the U.K National Facility for Aberration-Corrected STEM, supported by the Engineering and Physical Sciences Research Council (PA).References
- P. Yu, S. A. Hayes, T. J. O'Keefe, M. J. O'Keefe and J. O. Stoffer, J. Electrochem. Soc., 2006, 153, C74–C79 CrossRef CAS.
- B. Bilal and E. Müller, Zeitschrift für Naturforschung A, 1992, 47, 974–984 CAS.
- A. Abedini, A. R. Daud, M. A. Abdul Hamid, N. Kamil Othman and E. Saion, Nanoscale Res. Lett., 2013, 8, 1–10 CrossRef PubMed.
- J. Belloni, Catal. Today, 2006, 113, 141–156 CrossRef CAS.
- Q. Chen, X. Shen and H. Gao, Adv. Colloid Interface Sci., 2010, 159, 32–44 CrossRef CAS PubMed.
- C. Dispenza, N. Grimaldi, M. A. Sabatino, I. L. Soroka and M. Jonsson, J. Nanosci. Nanotechnol., 2015, 15, 3445–3467 CrossRef CAS PubMed.
- J. M. Grogan, N. M. Schneider, F. M. Ross and H. H. Bau, Nano Lett., 2013, 14, 359–364 CrossRef PubMed.
- J. Belloni, Catal. Today, 2006, 113, 141–156 CrossRef CAS.
- N. M. Schneider, M. M. Norton, B. J. Mendel, J. M. Grogan, F. M. Ross and H. H. Bau, J. Phys. Chem. C, 2014, 118, 22373–22382 CAS.
- A. Abedini, A. R. Daud, M. A. Abdul Hamid and N. Kamil Othman, PLoS One, 2014, 9, e90055 Search PubMed.
- G. Bhavana, A. M. Ambrose, M. Tom, S. Dash and A. K. Tyagi, Mater. Res. Express, 2014, 1, 045507 CrossRef.
- L. M. Alrehaily, J. M. Joseph and J. C. Wren, J. Phys. Chem. C, 2015, 119, 16321–16330 CAS.
- J. P. Patterson, P. Abellan, M. S. Denny Jr, C. Park, N. D. Browning, S. M. Cohen, J. E. Evans and N. C. Gianneschi, J. Am. Chem. Soc., 2015, 137, 7322–7328 CrossRef CAS PubMed.
- H. Zheng, R. K. Smith, Y. W. Jun, C. Kisielowski, U. Dahmen and A. P. Alivisatos, Science, 2009, 324, 1309–1312 CrossRef CAS PubMed.
- J. E. Evans, K. L. Jungjohann, N. D. Browning and I. Arslan, Nano Lett., 2011, 11, 2809–2813 CrossRef CAS PubMed.
- N. de Jonge and F. M. Ross, Nat. Nanotechnol., 2011, 6, 695–704 CrossRef CAS PubMed.
- F. M. Ross, Science, 2015, 350 DOI:10.1126/science.aaa9886.
- T. J. Woehl, J. E. Evans, L. Arslan, W. D. Ristenpart and N. D. Browning, ACS Nano, 2012, 6, 8599–8610 CrossRef CAS PubMed.
- T. J. Woehl, C. Park, J. E. Evans, I. Arslan, W. D. Ristenpart and N. D. Browning, Nano Lett., 2014, 14, 373–378 CrossRef CAS PubMed.
- P. Abellan, L. R. Parent, N. Al Hasan, C. Park, I. Arslan, A. M. Karim, J. E. Evans and N. D. Browning, Langmuir, 2016, 32, 1468–1477 CrossRef CAS PubMed.
- A. S. Karakoti, P. Munusamy, K. Hostetler, V. Kodali, S. Kuchibhatla, G. Orr, J. G. Pounds, J. G. Teeguarden, B. D. Thrall and D. R. Baer, Surf. Interface Anal., 2012, 44, 882–889 CrossRef CAS PubMed.
- A. Kumar, S. Das, P. Munusamy, W. Self, D. R. Baer, D. C. Sayle and S. Seal, Environ. Sci.: Nano, 2014, 1, 516–532 RSC.
- E. Grulke, K. Reed, M. Beck, X. Huang, A. Cormack and S. Seal, Environ. Sci.: Nano, 2014, 1, 429–444 RSC.
- M. van Soestbergen, S. Erich, H. Huinink and O. Adan, Electrochim. Acta, 2013, 110, 491–500 CrossRef CAS.
- L. Wang, Q. Luan, D. Yang, X. Yao and K. Zhou, RSC Adv., 2013, 3, 6339–6342 RSC.
- P. Dutta, S. Pal, M. S. Seehra, Y. Shi, E. M. Eyring and R. D. Ernst, Chem. Mater., 2006, 18, 5144–5146 CrossRef CAS.
- F. Esch, S. Fabris, L. Zhou, T. Montini, C. Africh, P. Fornasiero, G. Comelli and R. Rosei, Science, 2005, 309, 752–755 CrossRef CAS PubMed.
- L. J. Wu, H. J. Wiesmann, A. R. Moodenbaugh, R. F. Klie, Y. M. Zhu, D. O. Welch and M. Suenaga, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69(12), 125415 CrossRef.
- R. W. Tarnuzzer, J. Colon, S. Patil and S. Seal, Nano Lett., 2005, 5, 2573–2577 CrossRef CAS PubMed.
- J. Colon, N. Hsieh, A. Ferguson, P. Kupelian, S. Seal, D. W. Jenkins and C. H. Baker, Nanomedicine, 2010, 6, 698–705 CAS.
- S. M. Hirst, A. Karakoti, S. Singh, W. Self, R. Tyler, S. Seal and C. M. Reilly, Environ. Toxicol., 2013, 28, 107–118 CrossRef CAS PubMed.
- I. Celardo, J. Z. Pedersen, E. Traversa and L. Ghibelli, Nanoscale, 2011, 3, 1411–1420 RSC.
- T. X. T. Sayle, M. Molinari, S. Das, U. M. Bhatta, G. Mobus, S. C. Parker, S. Seal and D. C. Sayle, Nanoscale, 2013, 5, 6063–6073 RSC.
- Z. Ji, X. Wang, H. Zhang, S. Lin, H. Meng, B. Sun, S. George, T. Xia, A. E. Nel and J. I. Zink, ACS Nano, 2012, 6, 5366–5380 CrossRef CAS PubMed.
- E. Abi-aad, R. Bechara, J. Grimblot and A. Aboukais, Chem. Mater., 1993, 5, 793–797 CrossRef CAS.
- N. C. Wu, E. W. Shi, Y. Q. Zheng and W. J. Li, J. Am. Ceram. Soc., 2002, 85, 2462–2468 CrossRef CAS.
- A. I. Y. Tok, F. Y. C. Boey, Z. Dong and X. Sun, J. Mater. Process. Technol., 2007, 190, 217–222 CrossRef CAS.
- S. I. Mutinda, M. Sc. thesis, Youngstown State University, 2013.
- P. Abellan, T. J. Woehl, L. R. Parent, N. D. Browning, J. E. Evans and I. Arslan, Chem. Commun., 2014, 50, 4873–4880 RSC.
- J. H. Park, N. M. Schneider, J. M. Grogan, M. C. Reuter, H. H. Bau, S. Kodambaka and F. M. Ross, Nano Lett., 2015, 15, 5314–5320 CrossRef CAS PubMed.
- P. Abellan, B. L. Mehdi, L. R. Parent, M. Gu, C. Park, W. Xu, Y. Zhang, I. Arslan, J.-G. Zhang, C.-M. Wang, J. E. Evans and N. D. Browning, Nano Lett., 2014, 14, 1293–1299 CrossRef CAS PubMed.
- D. Ortiz, I. Jiménez Gordon, J. P. Baltaze, O. Hernandez-Alba, S. Legand, V. Dauvois, G. Si Larbi, U. Schmidhammer, J. L. Marignier and J. F. Martin, ChemSusChem, 2015, 8, 3605–3616 CrossRef CAS PubMed.
- D. Ortiz, V. Steinmetz, D. Durand, S. Legand, V. Dauvois, P. Maitre and S. Le Caer, Nat. Commun., 2015, 6, 6950 CrossRef PubMed.
- S. A. Hayes, P. Yu, T. J. O'Keefe, M. J. O'Keefe and J. O. Stoffer, J. Electrochem. Soc., 2002, 149, C623–C630 CrossRef CAS.
- B. Bouchaud, J. Balmain, G. Bonnet and F. Pedraza, J. Rare Earths, 2012, 30, 559–562 CrossRef CAS.
- E. Sutter, K. Jungjohann, S. Bliznakov, A. Courty, E. Maisonhaute, S. Tenney and P. Sutter, Nat. Commun., 2014, 5, 4946 CrossRef CAS PubMed.
- D. M. Stanbury, Adv. Inorg. Chem., 1989, 33, 69–138 CrossRef CAS.
- J. Baxendale, E. Fielden and C. Capellos, Nature, 1964, 201 CrossRef CAS.
- A. Gogolev, V. Shilov, A. Fedoseev and A. Pikaev, Int. J. Radiat. Appl. Instrum., Part C, 1991, 37, 531–535 CAS.
- A. J. Leenheer, K. L. Jungjohann, K. R. Zavadil, J. P. Sullivan and C. T. Harris, ACS Nano, 2015, 9, 4379–4389 CrossRef CAS PubMed.
- M. Nobial, O. Devos, O. R. Mattos and B. Tribollet, J. Electroanal. Chem., 2007, 600, 87–94 CrossRef CAS.
- L. Arurault, P. Monsang, J. Salley and R. Bes, Thin Solid Films, 2004, 466, 75–80 CrossRef CAS.
- L. Arurault, B. Daffos and F. X. Sauvage, Mater. Res. Bull., 2008, 43, 796–805 CrossRef CAS.
- M. Nabavi, O. Spalla and B. Cabane, J. Colloid Interface Sci., 1993, 160, 459–471 CrossRef CAS.
- R. F. Egerton, Ultramicroscopy, 2013, 127, 100–108 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, EELS scans used for estimating the thickness of liquid during experiments, the structural characterization of the oxide nanostructures by HRSTEM and HRTEM, calculation of the solubility of oxygen in water, equilibrium constants and thermodynamic calculations for constructing the Pourbaix diagram matching our experimental conditions, rate constants and results of modelling of the radiation-induced changes in solution chemistry including pH evolution in pure DI water and for an aqueous solution containing Ce-species, other chemical pathways involving the possible effect of nitrate ions, ESI Movies S1 and S2. See DOI: 10.1039/c6ra27066b |
| This journal is © The Royal Society of Chemistry 2017 |