
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, structure, toxicology and in vitro testing of three novel agents for Alzheimer’s disease†
José Daniel Figueroa-Villar *
*
NMR and Medicinal Chemistry Group, Department of Chemistry with Syntheses, Medicinal Chemistry, Spectroscopy and Molecular Modeling, Military Institute of Engineering, Rio de Janeiro, Brazil. E-mail: jdfv2009@gmail.com; Fax: +55 21 254607050; Tel: +55 21 2546 7057
First published on 28th April 2017
Abstract
This article describes the synthesis of three novel compounds for the treatment of Alzheimer’s disease (AD). The compounds were formed by the reaction of 3-methoxy-2-hydroxybenzaldehyde (1) with dimedone (2) using water as solvent at room temperature. Subsequently, Na2S or NaCl was added under reflux to obtain (R)-9-mercapto-5-methoxy-3,3-dimethyl-3,4-dihydro-2H-xanthen-1(9H)-one (5) and 9-chloro-3,4-dihydro-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (6), respectively. The final reaction proceeded under reflux with water only to obtain 3,4-dihydro-9-hydroxy-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (7). Compounds 5, 6 and 7 were selected due to their structural similarities to tacrine, which is one of the most effective AChE inhibitors for AD. The compounds were studied by nuclear magnetic resonance (NMR) to assign all chemical shifts and determine their three-dimensional structures. A parallel molecular modeling study was conducted to confirm the NMR results and obtain the energy, dipole moment, area, polar surface area (PSA) and volume of each compound. The PSA values indicated that the new compounds should be able to cross the blood–brain barrier. Compounds 5, 6 and 7 were then tested as inhibitors of human acetylcholinesterase (HuAChe) using docking experiments, and in vitro tests of the compounds as HuAChe inhibitors were performed using the Fig-NMR method with tacrine as a reference. The results confirmed that the new compounds are effective agents for the treatment of AD. Toxicity tests carried out using mice indicated very low toxicity. The findings suggest that the new compounds are better agents than tacrine for the treatment of AD.
Introduction
Alzheimer’s disease (AD) is a chronic neurodegenerative disease that initially includes problems with language, disorientation and memory, as well as mood swings and loss of motivation, followed by death after several years.1–3 Existing compounds used for AD treatment are inhibitors of human acetylcholinesterase (HuAChE) and include tacrine, donepezil, rivastigmine, galantamine and memantine.4–9 Tacrine is one of the most effective HuAChE inhibitors, but it also exhibits some hepatotoxicity.10 For this reason, it is important to design new, effective HuAChE inhibitors with similar structure and activity to tacrine.6 Tacrine displays some toxicity, especially towards hepatocytes, which has been assayed by the reduction of methyl thiazolyl tetrazolium (MTT) and intracellular glutathione (GSH), as well as by albumin synthesis.11 Compounds 5, 6 and 7, however, display very low toxicity compared with tacrine.This article describes the design and synthesis of (R)-9-mercapto-5-methoxy-3,3-dimethyl-3,4-dihydro-2H-xanthen-1(9H)-one (5), 9-chloro-3,4-dihydro-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (6) and 3,4-dihydro-9-hydroxy-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (7), which are structurally similar to tacrine, as novel compounds for the treatment of AD.
The synthesis of new compounds is very important for the preparation of effective pharmacological agents. When new molecules are synthesized, it is critical to determine their three-dimensional structures and assign all chemical shifts using mass spectrometry, nuclear magnetic resonance (NMR) and molecular modelling, to help determine their potential interactions with biological targets and their capacities as drugs. NMR is effective for determining three-dimensional structures and assigning chemical shifts for new molecules because many different types of spectra can be obtained; 1H, 13C, COSY, TOCSY, HSQC, HMBC, NOESY, ROESY and gated decoupling of 13C are all especially effective methods.12–21
This article describes the complete synthesis of three novel compounds and the complete, non-questionable determination of their three-dimensional structures and chemical shift assignments using NMR. The NMR results are confirmed by molecular modeling. The new compounds, which are similar in structure to tacrine, were identified as HuAChE inhibitors by docking, and their inhibition of HuAChE was determined through in vitro testing using Fig-NMR. To confirm that these compounds could be better than tacrine for the treatment of AD, toxicity texts were performed using several mice, and confirmed that these compounds display very low toxicity as drugs.
The design, synthesis, NMR, docking, molecular modeling and Fig-NMR were performed at the Chemistry Department of the Military of the Institute of Engineering (IME), and the toxicity tests were performed at the Laboratory of Environmental Toxicology of the Department of Biological Science and National School of Public Health of the Oswaldo Cruz Foundation (FIOCRUZ).
Overall, the obtained results suggest that these compounds display appropriate capacity as agents for the treatment AD and are preferable to tacrine.
Experimental section
Synthesis of (R)-9-mercapto-5-methoxy-3,3-dimethyl-3,4-dihydro-2H-xanthen-1(9H)-one (5)
The synthesis of (R)-9-mercapto-5-methoxy-3,3-dimethyl-3,4-dihydro-2H-xanthen-1(9H)-one (5) was carried out using 0.608 g of 3-methoxy-2-hydroxybenzaldehyde (1) and 0.560 g of dimedone (2) dissolved in 50 mL of H2O. The reaction was performed at room temperature (25 °C) for 30 min. After the first reaction was terminated, 0.359 g of sodium sulfide (Na2S) was introduced, and the reaction mixture was refluxed for 1 h. When finished, the reaction mixture was maintained at 0 °C for 30 min; the pure product was precipitated and obtained by filtration in 81% yield. Mp: 202–204 °C. IR (cm−1) 3276, 3256, 3201, 1889, 1857, 1760, 1664, 1627, 1517, 1445, 1373, 1339, 1220, 1199, 8.94. 1H NMR (600 MHz, DMSO-d6, Hz) δppm 10.31 (1H, bs), 6.90 (1H, t, J = 8.0), 6.78 (1H, dd, J = 1.3, 8.1), 6.52 (1H, dd, J = 1.3, 7.9), 5.03 (1H, bs), 3.78 (3H, s), 2.52 (1H, d, J = 17.2), 2.34 (1H, d, J = 17.2), 2.22 (1H, d, J = 16.0), 2.02 (1H, d, J = 16.0), 1.04 (3H, s), 0.97 (3H, s). 13C NMR (150 MHz, DMSO-d6) δppm 195.8 (C), 164.6 (C), 146.7 (C), 139.1 (C), 126.2 (C), 123.8 (CH), 119.7 (CH), 110.6 (C), 109.6 (CH), 55.5 (CH3), 50.4 (CH2), 40.7 (CH2), 31.6 (C), 29.1 (CH3), 26.2 (CH3), 24.9 (CH). HRMS (ESI): m/z [M + H]+ calculated for monoisotopic mass 290.0977, found 290.0981.Synthesis of 9-chloro-3,4-dihydro-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (6)
9-Chloro-3,4-dihydro-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (6) was synthesized by dissolving 0.608 g of 3-methoxy-2-hydroxybenzaldehyde (1) and 0.560 g of dimedone (2) in 50 mL of H2O. The reaction was performed at room temperature (25 °C) for 30 min. Subsequently, 0.350 g of NaCl was introduced, and the reaction mixture was refluxed for 1 h. When finished, the reaction mixture was maintained at 0 °C for 30 min; the pure product was precipitated and obtained by filtration in 78% yield. Mp: 206–208 °C. IR (cm−1) 3160, 3119, 1764, 1688, 1641, 1534, 1502, 1494, 1433, 1317, 1269, 1230, 1218, 1108, 1090, 1040, 757, 720, 517. 1H NMR (600 MHz, DMSO-d6, Hz) δppm 10.29 (1H, bs), 6.90 (1H, t, J = 7.9), 6.79 (1H, dd, J = 1.7, 8.2), 6.52 (1H, dd, J = 1.7, 7.7), 5.35 (1H, bs), 3.78 (3H, s), 2.53 (1H, d, J = 17.3), 2.35 (1H, d, J = 17.3), 2.23 (1H, d, J = 15.9), 2.03 (1H, d, J = 15.9), 1.04 (3H, s), 0.98 (3H, s). 13C NMR (150 MHz, DMSO-d6) δppm 195.78 (C), 164.5 (C), 146.7 (C), 139.1 (C), 126.2 (C), 123.8 (CH), 119.7 (CH), 110.6 (C), 109.5 (CH), 55.5 (CH3), 50.4 (CH2), 43.0 (CH), 40.7 (CH2), 31.6 (C), 29.2 (CH3), 26.2 (CH3). HRMS (ESI): m/z [M + H]+ calculated for monoisotopic mass 292.0866, found 292.0868.Synthesis of 3,4-dihydro-9-hydroxy-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (7)
3,4-Dihydro-9-hydroxy-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (7) was synthesized by dissolving 0.608 g of 3-methoxy-2-hydroxybenzaldehyde (1) and 0.560 g of dimedone (2) in 60 mL of water and refluxing for 2 h. The final reaction mixture was maintained at 0 °C for 50 min; the pure product was precipitated and obtained by filtration in 70% yield. Mp: 211–213 °C. IR (cm−1) 3330, 3271, 3208, 1945, 1861, 1792, 1673, 1597, 1454, 1388, 1335, 1279, 1219, 1174, 561, 527. 1H NMR (600 MHz, DMSO-d6, Hz) δppm 9.85 (1H, bs), 6.97 (1H, t, J = 7.8), 6.77 (1H, dd, J = 1.3, 7.9), 6.43 (1H, dd, J = 1.3, 7.8), 7.73 (1H, s), 3.78 (3H, s), 2.56 (1H, d, J = 17.2), 2.36 (1H, d, J = 17.2), 2.26 (1H, d, J = 16.1), 2.10 (1H, d, J = 16.1), 1.01 (3H, s), 0.96 (3H, s). 13C NMR (150 MHz, DMSO-d6) δppm 197.2 (C), 165.1 (C), 146.7 (C), 140.0 (C), 127.5 (C), 124.4 (CH), 120.1 (CH), 111.2 (C), 109.8 (CH), 60.5 (CH), 55.0 (CH3), 49.9 (CH2), 40.8 (CH2), 31.8 (C), 30.0 (CH3), 26.7 (CH3). HRMS (ESI): m/z [M + H]+ calculated for monoisotopic mass 274.1205, found 274.1208.NMR
All NMR spectra were obtained using a Varian 600 MHz Premium Compact NMR spectrometer. Compounds 5, 6 and 7 (15 mg) were dissolved in 600 μL of DMSO-d6 in 5 mm NMR tubes, and the temperature of the NMR spectrometer was 25.0 °C. The general spectral parameters include the number of scans (nt), acquisition time (aq), relaxation delay (d1) and the number of increments (ni). The obtained spectra were: 1H (600 MHz, nt = 16, aq = 1.317 s, d1 = 1 s), 13C (150 MHz, nt = 10.000, aq = 0.865 s, d1 = 1 s), gCOSY (nt = 16, aq = 0.150 s, d1 = 1 s, ni = 128), gHSQC (nt = 16, aq = 0.150 s, d1 = 1 s, ni = 128, t1 increments 128, J1 × h = 145 Hz), gHMBC (nt = 16, aq = 0.150 s, d1 = 1 s, ni = 128, t1 increments 200, J1 min = 130 Hz, J1 max = 165 Hz, Jn × h = 8 Hz) and ROESY-1D (nt = 32, aq = 0.150 s, d1 = 1 s, ni = 200, t1 increments 200, NOE mixing time = 500).Molecular modeling and docking
Density functional theory (DFT) molecular modeling calculations were performed using the Spartan’8 procedure with the M06 method and the 6-311G* basis set.22 For the compounds synthesized in this study, DFT calculations were used to determine the three-dimensional structures, electronic charges of all carbons, energy (au), polar surface area (PSA, Å2), element volume (Å2) and dipole moment (Debye), as well as to confirm the chemical shift assignments obtained by NMR. Docking of these compounds was performed using AutoDock Vina, which assessed their interactions with HuAChE.23HuAChE inhibition tests
HuAChE was obtained from Sigma-Aldrich (1 mg), and the HuAChE inhibitions activities of compounds 5, 6 and 7 were determined by NMR using a 600 MHz spectrometer and 5 mm NMR tubes. The NMR method used for these inhibition tests was the Fig-NMR method, which was previously described in our articles on this topic,24,25 and tacrine was used as the reference material. HuAChE was dissolved in 2 mL of deuterium oxide (D2O) containing 5% phosphate buffer at pH 7.4 in the 5 mm NMR tube; and the concentration of HuAChE was 0.2 mM in the presence of 1% of bovine serum albumin. Acetylcholine (ACh) was dissolved in D2O to a concentration of 0.1 M. To initiate HuAChE activity within the NMR tube, 30 mL of ACh solution was added, and immediately following its introduction, the time for the following spectra was set as time = zero. The introduction of the tube to the NMR machine, including the locking and shimming lasted for 3.5 min and the recording of the first 1H spectrum commenced 5 min after the introduction of the ACh. All the other 1H spectra were obtained 5 min after finalization of the previous 1H spectrum, and all of them were obtained using a single scan (nt = 1). All the required spectra were obtained after scanning for 110 min. The chemical shift of the methyl group in ACh is found at 2.24 ppm, and that from the methyl group in acetic acid is found at 2.16 ppm. The relative concentrations of ACh and Ac determined from the various 1H spectra, a process that provides information on the HuAChE kinetics. Compounds 5, 6 and 7 were used as inhibitors of HuAChE activity and were dissolved in 5.0 μL of D2O at concentrations of 12 μM. The AChE inhibition tests were performed using the same procedure detailed above, but with the initial addition of 5.0 μL of 5, 6 or 7 (12 μM) along with the 30 mL of the ACh, at time = zero. To ensure the correct results, as before, the locking and shimming procedures were also carried out for 3.5 min, the first 1H spectrum started recording at 5 min and subsequent spectra were recorded 5 min after the previous spectra. The pure HuAChE results were compared with the HuAChE results in the presence of compounds 5, 6 and 7 by determining the concentrations of ACh and Ac by integration of the 1H spectra, which were obtained by looking for their characteristic methyl group shifts. The percentage of HuAChE inhibition exhibited by the compounds was determined using Microsoft Excel. All results were obtained in triplicate.In-vivo Toxicity Tests
Toxicity tests were performed in accordance with guidelines and procedures set by the World Health Organization (WHO)26 and the Organization of Economic Cooperation and Development (OECD).27 The mice were purchased using procedures defined by the WHO and in compliance with procedures of the OECD, and were allowed to adapt to the conditions of the animal room for 4 h prior to the start of the study and also evaluation for several years. These mice were obtained from the laboratory of FIOCRUZ, Brazil. All the mice used in this study were cared for in accordance with the principles outlined in the “Used Laboratory Animals” of the Laboratory of Environmental Toxicology, Department of Biological Sciences, National School of Public Health, Oswaldo Cruz Foundation, from Dr Francisco José Roma Paumgartten, FIOCRUZ (Brazil).The environmental conditions were maintained at a temperature of 25 °C ± 1 °C, a relative humidity of 50% ± 5%, and a 10 h light/dark cycle. Over 20 days, all mice were repeatedly exposed to the compounds dissolved in water at 500 mg kg−1 via oral administration. For each compound, 10 mice were administered the compound, and three mice were not. The two groups of mice were compared to assess any toxicity caused by the used compounds. Accumulation in tissues was observed to be dose-dependent, but genetic toxicity-related reactions were not observed in any of the groups.
Results and discussion
As tacrine is an effective agent for HuAChE inhibition, (R)-9-mercapto-5-methoxy-3,3-dimethyl-3,4-dihydro-2H-xanthen-1(9H)-one (5), 9-chloro-3,4-dihydro-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (6) and 3,4-dihydro-9-hydroxy-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (7) were selected as potential compounds that may inhibit HuAChE, while having lower toxicity than tacrine. As discussed in the Experimental section, the compounds 5, 6 and 7 were synthesised in 81%, 78% and 70% yield respectively, according to Scheme 1. To determine the capacities of these compounds to interact with HuAChE, docking was performed,23 and the percentage inhibition of HuAChE was determined using Fig-NMR.24,25 The toxicity was assessed in vivo using mice.26,27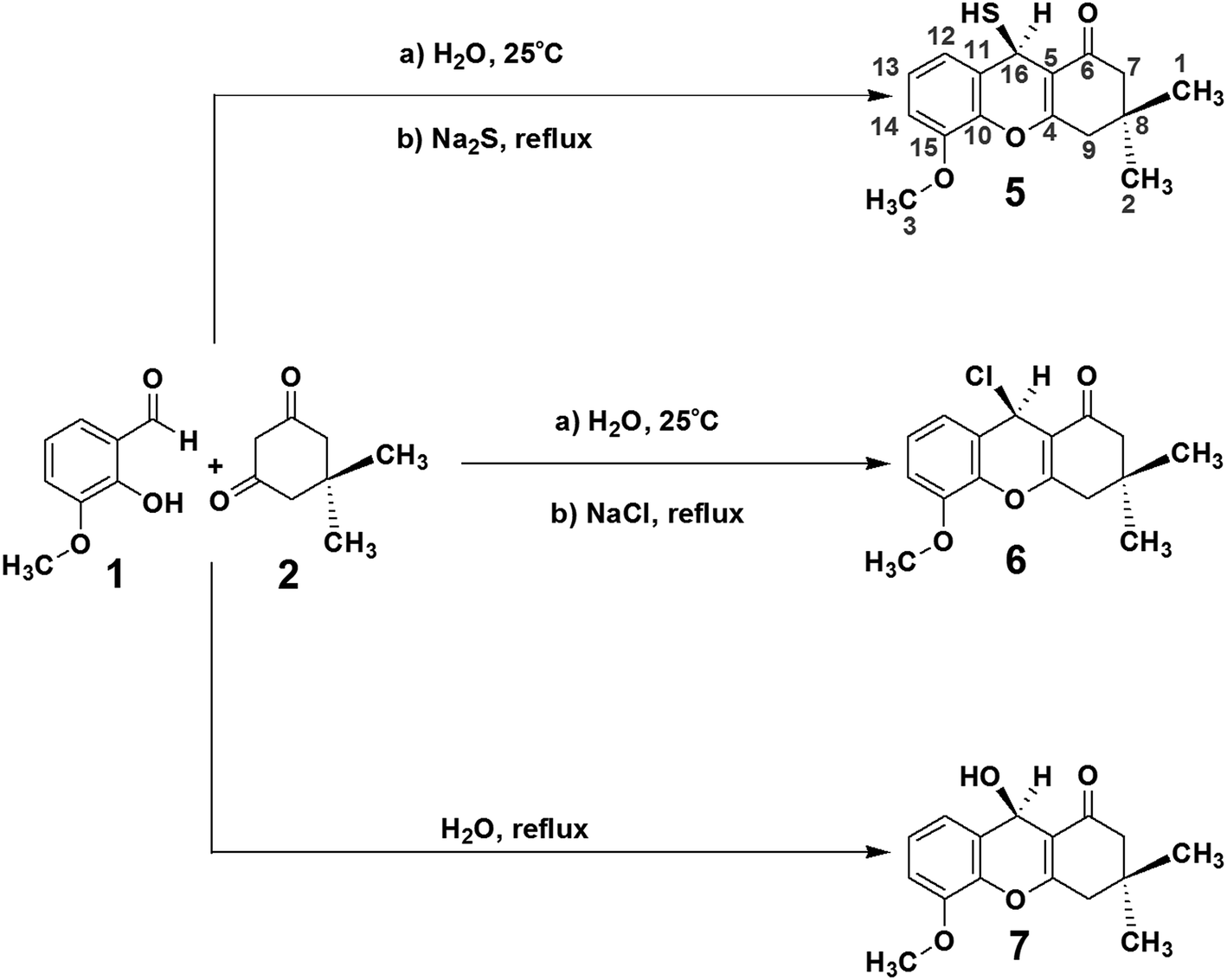 | ||
| Scheme 1 Syntheses of compounds (R)-9-mercapto-5-methoxy-3,3-dimethyl-3,4-dihydro-2H-xanthen-1(9H)-one (5) (81%), 9-chloro-3,4-dihydro-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (6) (78%) and 3,4-dihydro-9-hydroxy-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (7) (70%). | ||
Concerning the synthesis, the reaction of 3-methoxy-2-hydroxybenzaldehyde (1) and dimedone (2) afforded two possible compounds, 3 and 4, as shown in Scheme 2. Compound 3 was initially obtained by the reaction of 3-methoxy-2-hydroxybenzaldehyde (1) with dimedone (2), and it was possible to eliminate the OH group of compound 3 to prepare compound 4. Both 3 and 4 displayed susceptibility to nucleophilic attack with the selected elements being Na2S, NaCl or H2O.
 | ||
| Scheme 2 The mechanism for the formation of compounds 3 and 4 by the reaction of 3-methoxy-2-hydroxybenzaldehyde (1) and dimedone (2) dissolved in H2O at 25 °C for 30 min. | ||
Compounds 3 and 4 were subjected to nucleophilic attack by Na2S, NaCl or water under reflux, affording compounds 5, 6 and 7. The nucleophilic attack mechanisms are shown in Scheme 3. Refluxing Na2S in water afforded HS−, which performed nucleophilic attack on the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C group bound to dimedone to afford compound 5. Similarly, Na+ Cl− also initiates nucleophilic attack on the CC group within compounds 3 or 4 to obtain compound 6. Compound 7 was obtained from compound 3 by the addition of the OH− or H2O, again through nucleophilic attack on the CC group.
C group bound to dimedone to afford compound 5. Similarly, Na+ Cl− also initiates nucleophilic attack on the CC group within compounds 3 or 4 to obtain compound 6. Compound 7 was obtained from compound 3 by the addition of the OH− or H2O, again through nucleophilic attack on the CC group.
 | ||
| Scheme 3 Mechanisms of nucleophilic attack on compounds 3 and 4 by Na2S, NaCl and water to afford compounds 5, 6 and 7. | ||
The complete chemical shift assignments and three-dimensional structures of these new compounds were determined using NMR by collecting 1H, 13C-APT, gCOSY, ROESY-1D, gHSQC and gHMBC spectra.12–21 All the NMR results were confirmed by molecular modeling. The NMR data for compound 5, including the 1H, 13C, gHSQC and gHMBC spectra, are shown in Tables 1 and 2. Copies of the NMR and IR spectra of compounds 5, 6 and 7 are included in the ESI.†
| δH (integration, multiplicity, Hz) | gCOSY | ROESY-1D |
|---|---|---|
| a The emboldened ROESY-1D results are the most effective correlations; and those in italics are the less effective ones. | ||
| 10.31 (1H, bs) | — | 2.23 |
| 6.90 (1H, t, 8.0) | 6.78, 6.52 | 6.79, 6.52, 3.78 |
| 6.78 (1H, dd, 1.3, 8.1) | 6.90 | 6.90, 3.78 |
| 6.52 (1H, d, 7.7) | 6.90 | 6.90, 5.03 |
| 5.03 (1H, bs) | — | 6.52, 3.78 |
| 3.78 (3H, s) | — | 6.90, 6.78, 5.03 |
| 2.52 (1H, d, 17.2) | 2.35 | 2.34, 2.22, 1.04, 0.97 |
| 2.34 (1H, d, 17.2) | 2.52 | 2.52, 2.02, 1.04(+), 0.97(−) |
| 2.22 (1H, d, 16.0) | 2.02 | 2.52, 2.02, 1.04, 0.97 |
| 2.02 (1H, d, 16.0) | 2.22 | 2.34, 2.22, 1.04(−), 0.97(+) |
| 1.04 (3H, s) | — | 2.52, 2.34, 2.22, 2.02, 0.97 |
| 0.97 (3H, s) | — | 2.52, 2.34, 2.22, 2.02, 1.04 |
| δC | APT | gHSQC | gHMBC |
|---|---|---|---|
| a The emboldened gHMBC results are the most effective correlations, and those in italics are the less effective ones. | |||
| 195.8 | C | — | 5.03, 2.22, 2.02 |
| 164.6 | C | — | 5.03, 2.52, 2.34 |
| 146.7 | C | — | 6.90, 6.78, 6.52, 3.78 |
| 139.1 | C | — | 6.90, 6.78, 6.52, 5.03 |
| 126.2 | C | — | 6.90, 6.78, 5.03 |
| 123.8 | CH | 6.90 | 6.78, 6.52 |
| 119.7 | CH | 6.52 | 6.90, 6.78, 5.03, 3.78 |
| 110.6 | C | — | 5.03, 2.52, 2.34, 2.22, 2.02 |
| 109.6 | CH | 6.78 | 6.90, 6.52 |
| 55.5 | CH3 | 3.78 | — |
| 50.4 | CH2 | 2.22, 2.02 | 2.52, 2.34, 1.04, 0.97 |
| 40.7 | CH2 | 2.52, 2.34 | 2.22, 2.02, 1.04, 0.97 |
| 31.6 | C | — | 2.52, 2.34, 2.22, 2.02, 1.04, 0.97 |
| 29.1 | CH3 | 1.04 | 2.52(+), 2.34(−), 2.22(+), 2.02(−), 0.97 |
| 26.2 | CH3 | 0.97 | 2.52(−), 2.34(+), 2.22(−), 2.02(+), 1.04 |
| 24.9 | CH | 5.03 | 6.52 |
The 1H and gCOSY spectra display the chemical shifts of all hydrogens. For example, the peak at 6.90 ppm is a signature triplet, characteristic of a hydrogen occupying position 13 of compound 5, (positions indicated on the structure of 5 in Scheme 1). The peaks at 6.78 and 6.52 ppm, which are double-doublets, correspond to positions 12 and 14 of compound 5. Their environments are further confirmed by gHMBC, which indicates that these hydrogens can interact with particular carbons via a three bond coupling interaction, and that the protons at 6.78 and 6.52 ppm are directly bound to the carbons with shifts 109.6 and 119.7 ppm, respectively. The strength of the coupling between hydrogen atoms and carbon atoms located two bonds away is normally low or sometimes medium strength, and across four bonds the coupling is generally zero. The hydrogens from the CH2 groups display duplicity, meaning that the two hydrogens from one CH2 group have different chemical shifts, with one being at the α-position and the other at the β-position. On the other hand, the hydrogen atoms of the methyl groups in compound 5 do not display coupling.
ROESY-1D data were used to determine the three-dimensional structure of compound 5. The ROESY-1D spectrum shows a strong interaction between the OCH3 hydrogen signals (3.78 ppm) and the signal at 6.78 ppm, indicating that this proton is spatially close to this OCH3 group, corresponding to position 14 of compound 5. The hydrogen associated with the peak at 2.02 ppm interacts spatially with the hydrogen at 2.22 ppm, and both are bound to the same carbon atom (50.4 ppm). The hydrogen at 2.02 ppm also interacts with the one 5.03 ppm, but the other hydrogen at 2.22 ppm does not; this may be due to the aforementioned α- and β-positioning of these two hydrogens. From this result, it can be inferred that the hydrogen atoms 5.03, 2.02 and 0.97 ppm are all in β-positions in compound 5. The hydrogen at 2.22 ppm interacts more strongly with the methyl at 1.04 ppm and slightly less with the one at 0.97 ppm. For this reason, the methyl at 1.04 ppm and the hydrogen at 2.22 ppm are assigned as occupying α-positions in compound 5. The other environments can be inferred from the hydrogens at 2.52 and 2.34 ppm, as the hydrogen at 2.52 ppm interacts well with the methyl at 1.04 ppm, and less with that at 0.97 ppm, it must occupy an α-position on compound 5. The 2.34 ppm hydrogen interacts equally the two methyl groups at 1.04 and 0.97 ppm, but does not interact with the hydrogen at 2.22 ppm, indicating that the 2.34 ppm hydrogen occupies a β-position on compound 5.
The gHSQC spectrum indicates which hydrogen atoms are directly bound to which carbon atoms, while the gHMBC spectrum displays the interactions of hydrogen atoms with carbon atoms over two, three and four bonds. In this case, the most effective interactions are observed between C and H atoms separated by three bonds. The interaction between those separated by four bonds is very low or zero, whilst coupling interactions over two bonds can sometimes be adequate, but are often lower or zero. On this basis, the positions of all the quaternary carbons in compound 5 can be confirmed. For example, the hydrogen at 6.52 ppm interacts well with the carbon atoms at 24.9, 109.6 and 139.1 ppm, indicating that this atom is three bonds from the CH carbon at 24.9 ppm. Using the same constraints with the other hydrogen atoms, the gHMBC results confirm the complete chemical shift assignment of all the hydrogen and carbon atoms in compound 5. The NMR results shown for compound 5 in Tables 1 and 2 confirm the three-dimensional structure and all the 1H and 13C chemical shifts. Fig. 1 shows the complete structures and chemical shift assignments of compounds 5, 6 and 7
 | ||
| Fig. 1 Complete NMR chemical shift assignments and three-dimensional structures of compounds (R)-9-mercapto-5-methoxy-3,3-dimethyl-3,4-dihydro-2H-xanthen-1(9H)-one (5), 9-chloro-3,4-dihydro-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (6) and 3,4-dihydro-9-hydroxy-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (7). | ||
DFT was used to confirm the structures and chemical shift assignments of compounds 5, 6 and 7 using the program Spartan’8 with the M06 method and the 6-311G* basis set.22 The structures of compounds 5, 6 and 7 were determined using equilibrium conformers with Spartan’8, and the equilibrium geometry of each compound was used to determine the energy (au), PSA (Å2), element volume (Å2) and dipole moment (Debye). All this information is shown in Fig. 2.
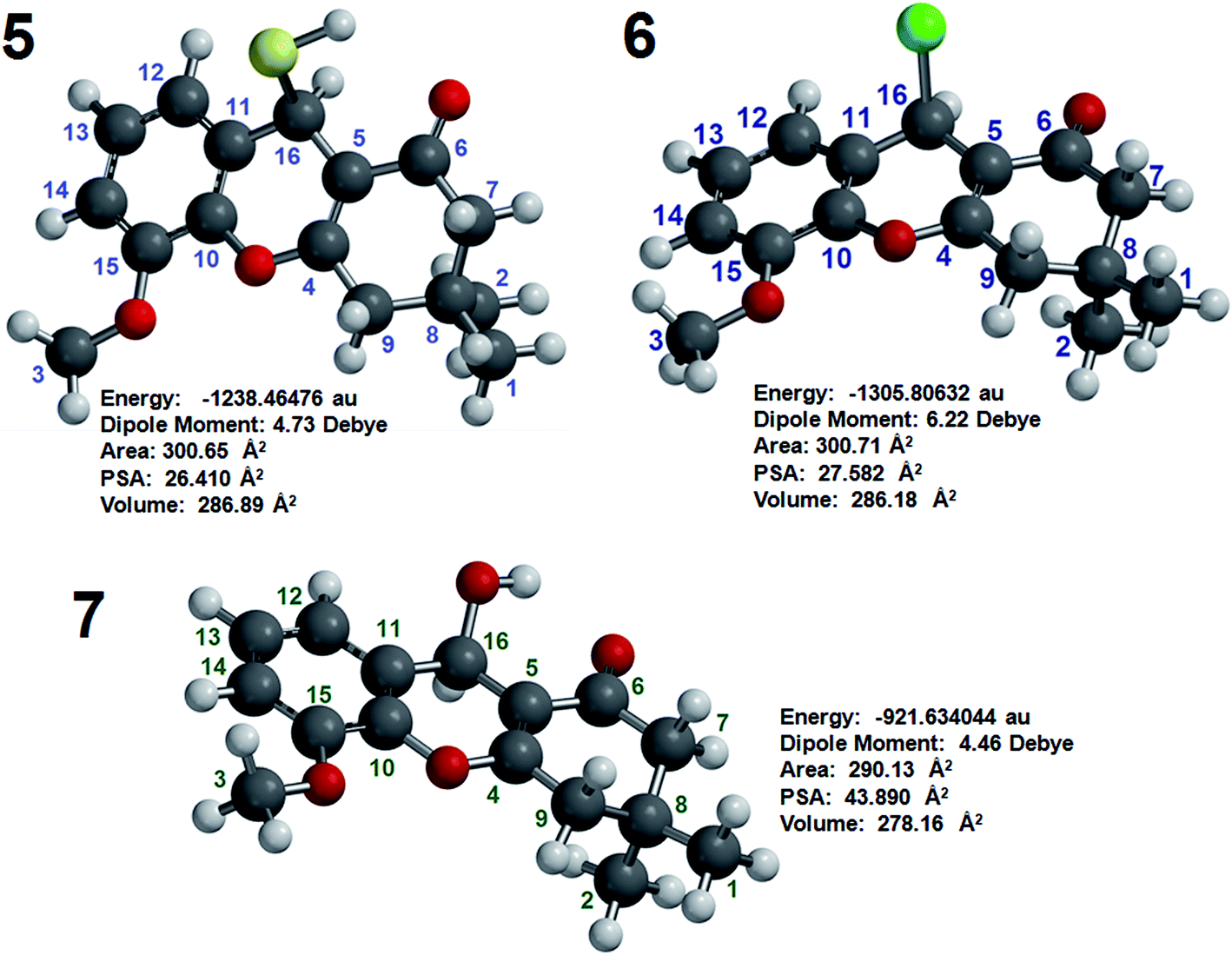 | ||
| Fig. 2 Energy (au), dipole moment (Debye), PSA (Å2) and volume (Å2) of compounds 5, 6 and 7 as determined by molecular modeling. | ||
The chemical shifts of carbon atoms were also calculated by DFT and the results were similar to those obtained by NMR, although some values showed small differences. As an example, the chemical shifts obtained by NMR and molecular modeling are compared for compound 6 in Table 3. The chemical shift of C16 determined by NMR as 43.0 ppm, while that calculated by DFT was 48.5 ppm, a difference of 5.5 ppm. For another carbon (C4), the chemical shifts calculated by NMR and DFT were different by 3.7 ppm (164.5 versus 168.2 ppm, respectively). The difference for C10 was similar (4.0 ppm; 139.1 ppm by NMR and 143.0 by DFT). For some other carbons, the differences were slightly smaller, including C1 (difference of 2.9 ppm), C5 (difference of 3.1 ppm), C13 (difference of 1.3 ppm) and C15 (difference of 1.5 ppm). The NMR and DFT chemical shifts for the other carbon atoms were more equal. The correlation between the NMR and DFT chemical shifts of the carbon atoms in compound 6 confirms the complete chemical shift assignment of this compound.
| Carbon | δC NMR | δC Mol. Mod. |
|---|---|---|
| C1 | 29.1 | 32.1 |
| C2 | 26.2 | 26.0 |
| C3 | 55.4 | 54.6 |
| C4 | 164.5 | 168.2 |
| C5 | 110.5 | 113.6 |
| C6 | 195.8 | 194.6 |
| C7 | 50.4 | 49.9 |
| C8 | 31.6 | 32.8 |
| C9 | 40.7 | 41.5 |
| C10 | 139.1 | 143.0 |
| C11 | 126.2 | 125.3 |
| C12 | 119.7 | 121.8 |
| C13 | 123.7 | 125.1 |
| C14 | 109.5 | 110.7 |
| C15 | 146.7 | 148.1 |
| C16 | 43.0 | 48.5 |
The PSAs of compounds 5, 6 and 7 were 26.409, 27.582 and 43.890 Å2, respectively, indicating that these agents have good properties for penetrating the blood–brain barrier (BBB) and are appropriate for the treatment of AD. The penetration of these compounds into the human brain is important for achieving inhibition HuAChE and thus for potentiality enhancing the memories of humans with AD.
To confirm the potential of these compounds to interact with and inhibit the HuAChE, docking was performed using AutoDock Vina.23 The structure of HuAChE for use in the docking study was obtained from the Protein Data Bank.28 The results are shown in Fig. 3 and indicate that compounds 5, 6 and 7 can interact with HuAChE, in a similar manner to tacrine. The compounds interact well with the amino acids tryptophan-86, tyrosine-337, tyrosine-341 and glycine-121, thus mimicking the interactions required to inhibit HuAChE. These results indicate that the three compounds can interact with and inhibit HuAChE, confirming their use as potential agents for AD.
 | ||
| Fig. 3 Docking of compounds 5, 6 and 7 with HuAChE. | ||
To experimentally confirm that compounds 5, 6 and 7 can inhibit HuAChE in vitro, the Fig-NMR method was performed, as described in two prior articles published by our research group.24,25 The 1H NMR spectrum of a solution containing pure AChE in the presence of ACh was recorded every 5 minutes for 110 minutes, providing 22 spectra. By looking at the relative amounts of ACh and its decomposition product, acetic acid, in the resultant spectra, it was possible to asses the ability of AChE to decompose ACh. These spectra were then compared to the analogous spectra obtained from solutions of AChE, ACh and compound 5, 6 or 7, in order to assess their ability to prevent the decomposition of ACh through inhibition of AChE. All the results were obtained in triplicate and tacrine was used as the reference material for the in vitro testing.
To calculate the percentage of HuAChE inhibition exhibited by compounds 5, 6 and 7, it was necessary to integrate the acyl methyl groups of both the substrate (ACh, 2.24 ppm) and the product acetic acid (Ac, 2.16 ppm). One example of this procedure is shown here for compound 5, while the inhibition results for tacrine and compounds 6 and 7 are shown in the ESI.† The 1H NMR spectra of the methyl groups of ACh and Ac for pure HuAChE (A) and in the presence of compound 5 (B) are shown in Fig. 4. The 1H spectra showing the methyl groups of ACh and Ac for pure HuAChE (A) indicate a decrease in ACh and an increase in Ac. In contrast, in the spectra for HuAChE in the presence of compound 5 (B), the decrease in ACh and increase in Ac are significantly diminished, confirming that compound 5 is an efficient HuAChE inhibitor and thus a potentital drug for the treatment of AD. A graphical representation of the 1H NMR spectra from various control experiments is shown in Fig. 5. The various lines show how the percentage concentration of ACh or Ac change over time.
 | ||
| Fig. 4 1H NMR results of the methyl groups of ACh and Ac for pure HuAChE (A) and in the presence of compound 5 (B). | ||
 | ||
| Fig. 5 Graphical comparison of the 1H NMR spectra from various experiments showing how the concentration of ACh or Ac changes over time, spectra of ACh and Ac with pure HuAChE, as well as ACh and Ac in the presence of compound 5 as the inhibitor, are shown in the figure.. | ||
The relative concentrations of the methyl groups from ACh and Ac were obtained from the from the 1H spectra by integration (Table 4). The relative concentrations of the methyl groups from ACh and Ac for pure HuAChE are shown in columns 2 and 3 of Table 4, while those in the presence of compound 5 are shown in columns 4 and 5.
| Time (min) | Pure AChE | AChE + Comp. 5 | Ac(3) ( = Ac(2) × 100/Ac(1)) | Ac(3) – 5.6579 | ||
|---|---|---|---|---|---|---|
| Ac(1) | ACh(1) | Ac(2) | ACh(2) | |||
| 0 | 0 | 100 | 0 | 100 | — | — |
| 5 | 5.9089 | 94.0911 | 0.4685 | 99.5315 | 7.9287 | 2.2708 |
| 10 | 9.1735 | 90.8265 | 0.6321 | 99.3679 | 6.8905 | 1.2326 |
| 15 | 13.7373 | 86.2627 | 0.7956 | 99.2044 | 5.7915 | 0.1336 |
| 20 | 17.2624 | 82.7376 | 0.9596 | 99.0404 | 5.5589 | 0.0990 |
| 25 | 19.5281 | 80.4719 | 1.1234 | 98.8766 | 5.7527 | 0.0948 |
| 30 | 23.6157 | 76.3843 | 1,2821 | 98.7179 | 5.4290 | 0.2289 |
| 35 | 28.0254 | 71.9745 | 1.4506 | 98.5494 | 5.1760 | 0.4819 |
| 40 | 30.8499 | 69.1501 | 1.6147 | 98.3853 | 5.2340 | 0.4239 |
| 45 | 32.9332 | 67.0668 | 1.7783 | 98.2217 | 5.3997 | 0.2582 |
| 50 | 36.7961 | 63.2039 | 1.9422 | 98.0578 | 5.2783 | 0.3796 |
| 55 | 39.9990 | 60.0020 | 2.1059 | 97.8941 | 5.2650 | 0.3929 |
| 60 | 41.96473 | 58.0353 | 2.2695 | 97.7305 | 5.4081 | 0.2498 |
| 65 | 45.3098 | 54.6902 | 2.4333 | 97.5667 | 5.3704 | 0.2875 |
| 70 | 48.6310 | 51.3690 | 2.5974 | 97.4026 | 5.3410 | 0.3169 |
| 75 | 50.9019 | 49.0981 | 2.7613 | 97.2387 | 5.4247 | 0.2332 |
| 80 | 53.4535 | 46.5465 | 2.9253 | 97.0747 | 5.4726 | 0.1853 |
| 85 | 56.8910 | 43.1090 | 3.0891 | 96.9109 | 5.4299 | 0.2280 |
| 90 | 58.1777 | 41.8223 | 3.2528 | 96.7472 | 5.5911 | 0.0668 |
| 95 | 61.4080 | 38.5920 | 3.4169 | 96.5831 | 5.5643 | 0.0936 |
| 100 | 63.4420 | 36.5580 | 3.5810 | 96.4190 | 5.6445 | 0.0134 |
| 105 | 65.0320 | 34.9680 | 3.7448 | 96.2552 | 5.7584 | 0.1005 |
| 110 | 67.8054 | 32.1946 | 3.9090 | 96.0910 | 5.7650 | 0.1071 |
| Summation | 124.47 | 7.8782 | ||||
| Division by 22 | 5.6579 | 0.3581 | ||||
| Inhibition of HuAChE (%) | 94.34 | ±0.36 | ||||
The concentrations of Ac produced from pure AChE and in the presence of compound 5 were compiled using Microsoft Excel and compared to determine the complete percentage of HuAChE inhibition by compound 5 (Table 4). Initially, the concentration of Ac(2) (from HuAChE + compound 5) was multiplied by 100 and divided by the concentration of Ac(1) (from pure HuAChE) to display the comparative concentration, denoted as Ac(3). The summation of all Ac(3) afforded 124.47, and dividing by the number of time intervals (i.e., 22) afforded 5.6579, which was subtracted from 100 to obtain the percentage inhibition of HuAChE (94.34%, as shown in column 6 of Table 4). The final calculation, shown in column 7 of Table 4 is the difference between individual Ac(3) values and the average Ac(3) value (5.6579). The summation of these values is 7.8782, and dividing this number by 22 affords 0.3581, which was used as 0.36. This indicates the possible variation in the percentage HuAChE inhibition. On this basis, the HuAChE inhibition of compound 5 is confirmed as 94.34 ± 0.36%.
The results shown in Fig. 4 + 5 and Table 4 indicate that compound 5 is able to inhibit HuAChE, confirming that this compound can be used to treat AD. The final HuAChE inhibition percentages for all three compounds are listed in Table 5, the corresponding tables and figures detailing the inhibition of HuAChE by compounds 6 and 7 can be found in the ESI.† When tacrine was previously tested as an HuAChE inhibitor, the Ellman test indicated 100% inhibition, whereas Fig-NMR results indicate 94.64% inhibition, similar to those of compounds 5, 6 and 7. Compounds 5 and 7 displayed the most similar HuAChE inhibition behavior to tacrine, while compound 6 displayed lower activity than tacrine.
| Compound | HuAChE inhibition |
|---|---|
| Tacrine | 94.64 ± 0.76 |
| 5 | 94.35 ± 0.36 |
| 6 | 86.65 ± 4.55 |
| 7 | 92.37 ± 1.64 |
The most effective compound is (R)-9-mercapto-5-methoxy-3,3-dimethyl-3,4-dihydro-2H-xanthen-1(9H)-one (5) (94.35% inhibition of HuAChE), which is as effective as tacrine. Compounds 6 and 7 show similar effectiveness to tacrine for the inhibition of this enzyme because the also limit the decrease in acetylcholine (ACh) levels in the presence of this enzyme. Maintaining the concentration of ACh, which is an important neuron transmitter in the human brain, could enhance the memories of people with AD.
The toxicities of compounds 5, 6 and 7 were tested using 30 mice (10 for each compound). Of the 10 mice in each group, 3 were not administered the new compounds, a process allowing for comparison of the potential toxicity of these agents. The mice were initially tested four hours after administering the compounds, and were continually evaluated for 20 days. They were then compared to the mice that were not administered the compounds. The health of the mice that received the compounds was similar to those that did not receive them, suggesting that compounds 5, 6 and 7 have low toxicity. The in vivo tests performed with these mice included the pentobarbital-induced sleep test, rotarod performance test, open field test and elevated plus maze test. The results indicated that the new agents induce low hypnotic potency and notable anxiolytic potency. The method was based on the determination of the toxin dose capable of killing at least 50% of the animals. The dose was determined by the regression analysis of a dose–response curve. All animals were studied for 20 days after the introduction of compounds 5, 6 and 7. The findings indicated that all the animals were in good condition without any problems after the introduction of these compounds. Thus, as the mice survived with normal behaviour, it can be concluded that the new compounds do not display any notable toxicity but interestingly do display similar HuAChE inhibition to that of tacrine.
Conclusion
3 novel compounds, 5, 6 and 7, were synthesized as AChE inhibitors for the treatment of AD. Complete, non-questionable chemical shift and three-dimensional structural assignments were determined by NMR and confirmed by molecular modeling. Elucidation of these properties is important to determine the abilities of new compounds to interact with biological targets, inhibit enzymes and display pharmacological activity.Molecular modeling calculations indicated PSAs of 26.409 Å2 (5), 27.582 Å2 (6) and 43.890 Å2 (7), indicating that these compounds have appropriate properties for penetrating the BBB, which is necessary for reaching the human brain. Docking tests also indicated that these compounds behave as inhibitors of HuAChE. The in vitro Fig-NMR testing of these compounds’ inhibition of HuAChE indicated that 9(R)-9-mercapto-5-methoxy-3,3-dimethyl-3,4-dihydro-2H-xanthen-1(9H)-one (5) inhibits 94% of the enzyme, 9-chloro-3,4-dihydro-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (6) displays 86% inhibition, and 3,4-dihydro-9-hydroxy-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (7) shows 92% inhibition. These results indicate that the new compounds are important agents that could enhance the memories of people with AD.
Toxicity testing using mice indicated that the new compounds do not display significant toxicity and are thus appropriate for use as pharmacological agents.
The findings of this study indicate that (R)-9-mercapto-5-methoxy-3,3-dimethyl-3,4-dihydro-2H-xanthen-1(9H)-one (5), 9-chloro-3,4-dihydro-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (6) and 3,4-dihydro-9-hydroxy-5-methoxy-3,3-dimethyl-2H-xanthen-1(9H)-one (7) are appropriate, non-toxic drugs for the treatment of AD.
Conflict of interest
The author confirms that this article does not contain any conflicts of interest.Acknowledgements
Financial support from the following companies is acknowledged; CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior), CNPq (Conselho Nacional de Pesquisa), FIOCRUZ (Fundação Oswaldo Cruz) and FAPERJ (Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro).References
- A. Nordberg and A. L. Svensson, Drug Saf., 1998, 19, 465 CrossRef CAS PubMed.
- H. W. Querfurth and F. M. LaFerla, N. Engl. J. Med., 2010, 362, 329 CrossRef CAS PubMed.
- A. Kumar and A. Singh, Pharmacol. Rep., 2015, 67, 195 CrossRef CAS PubMed.
- M. Pohanka, Expert Opin. Ther. Pat., 2012, 22, 871 CrossRef CAS PubMed.
- H. Feldman, S. Gauthier, J. Hecker, B. Vellas, P. Subbiah and E. Whalen, Neurology, 2001, 57, 613 CrossRef CAS PubMed.
- S. A. Eagger, R. Levy and B. J. Sahakian, Lancet, 1991, 337, 989 CrossRef CAS.
- B. Reisberg, R. Doogy, A. Stöffler, F. Schnitt, S. Ferris and H. J. Möbius, N. Engl. J. Med., 2003, 348, 1333 CrossRef CAS PubMed.
- E. Y. Shintani and K. M. Uchida, Am. J. Health-Syst. Pharm., 1997, 54, 2805 CAS.
- K. L. Lanctôt, N. Herrmann, K. K. Yau, L. R. Khan, B. A. Liu, M. M. LouLou and T. R. Einarson, CMAJ, 2003, 169, 6 Search PubMed.
- D. Lagadic-Gossmann, M. Rissel, M. A. Le Bot and A. Guillouzo, Cell Biol. Toxicol., 1998, 14, 361 CrossRef CAS PubMed.
- K. J. Barnham, F. Haeffner, G. D. Ciccotosto, C. C. Curtain, D. Tew, C. Mavros, K. Beyreuther, D. Carrington, C. L. Master, R. A. Cherny, R. Cappal and A. I. Bush, FASEB J., 2004, 18, 1427 CAS.
- S. Balayssac, S. Trefi, V. Gilard, M. Malet-Martino, R. Martino and M. A. Delsuc, J. Pharm. Biomed. Anal., 2009, 50, 602 CrossRef CAS PubMed.
- D. Genest and J. P. Simorre, Magn. Reson. Chem., 1990, 28, 21 CrossRef CAS.
- H. Desvaux, P. Berthault, N. Birlirakis and M. Goldman, J. Magn. Reson., Ser. A, 1994, 108, 219 CrossRef CAS.
- S. J. Gould, V. A. Palaniswamy, H. Bleich and J. J. Wilde, J. Chem. Soc., Chem. Commun., 1984, 16, 1075 RSC.
- G. Kontaxis and J. Keeler, J. Magn. Reson., 1995, 115, 35 CrossRef CAS.
- W. F. Reynolds, S. McLean, L. L. Tay, M. Yu, R. G. Enriquez, D. M. Estwick and K. O. Pascoe, Magn. Reson. Chem., 1997, 35, 455 CrossRef CAS.
- J. D. Figueroa-Villar and L. W. Tinoco, Curr. Top. Med. Chem., 2009, 9, 811 CrossRef CAS PubMed.
- W. Willker and D. Leibfritz, Magn. Reson. Chem., 1995, 33, 632 CrossRef CAS.
- P. Giraudeau and E. Baguet, J. Magn. Reson., 2006, 180, 110 CrossRef CAS PubMed.
- J. D. Figueroa-Villar, Appl. Magn. Reson., 2015, 46, 607 CrossRef CAS.
- Y. Shao, L. F. Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. T. Brown, A. T. B. Gilbert, L. V. Slipchenko, S. V. Levchenko, D. P. O’Neill, J. R. A. DiStasio, R. C. Lochan, T. Wang, G. J. O. Beran, N. A. Besley, J. M. Herbert, C. Y. Lin, T. Van Voorhis, S. H. Chien, A. Sodt, R. P. Steele, V. A. Rassolov, P. E. Maslen, P. P. Korambath, R. D. Adamson, B. Austin, J. Baker, E. F. C. Byrd, H. Dachsel, R. J. Doerksen, A. Dreuw, B. D. Dunietz, A. D. Dutoi, T. R. Furlani, S. R. Gwaltney, A. Heyden, S. Hirata, C.-P. Hsu, G. Kedziora, R. Z. Khalliulin, P. Klunzinger, A. M. Lee, M. S. Lee, W. Z. Liang, I. Lotan, N. Nair, B. Peters, E. I. Proynov, P. A. Pieniazek, Y. M. Rhee, J. Ritchie, E. Rosta, C. D. Sherrill, A. C. Simmonett, J. E. Subotnik III, H. L. W. Woodcock Zhang, A. T. Bell, A. K. D. M. Chakraborty, F. J. Chipman Keil, A. Warshel, W. J. Hehre, H. F. Schaefer, J. Kong, A. I. Krylov, P. M. W. Gill and M. Ead-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 3172 RSC.
- O. Trott and A. J. J. Olson, Comput. Chem., 2010, 31, 455 CAS.
- E. C. Petronilho, M. N. Rennó, N. G. Castro, F. M. Silva, A. C. Pinto and J. D. Figueroa-Villar, J. Enzyme Inhib. Med. Chem., 2016, 31, 1069 CrossRef CAS PubMed.
- S. F. C. X. Soares, A. A. Vierira, R. T. Delfino and J. D. Figueroa-Villar, Bioorg. Med. Chem., 2013, 21, 5923 CrossRef PubMed.
- X. Zhang, General Guidelines for Methodologies on Research and Evaluation of Traditional Medicine, World Health Organization, Geneva, Switzerland, 2000, http://whqlibdoc.who.int/hq/2000/WHO_EDM_TRM_2000.1.pdf?Ua=1 Search PubMed.
- The Organization of Economic Co-operation and Development (OECD), The OECD Guideline for Testing of Chemical: 420 Acute Oral Toxicity, OECD, France, 2001, pp. 1–6 Search PubMed.
- G. Kryger, M. Harel, K. Giles, L. Toker, B. Velan, A. Lazar, C. Kronman, D. Barak, N. Ariel, A. Shafferman, I. Silman and J. L. Sussman, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2000, 56, 1385 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra27042e |
| This journal is © The Royal Society of Chemistry 2017 |