
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
pH-Responsive nanodrug encapsulated by tannic acid complex for controlled drug delivery†
Huan Huang‡
,
Ping Li‡*,
Changliang Liu,
Huailei Ma,
He Huang,
Yuchen Lin,
Chen Wang* and
Yanlian Yang*
CAS Key Laboratory of Standardization and Measurement for Nanotechnology, CAS Key Laboratory of Biological Effects of Nanomaterials and Nanosafety, CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology, Beijing 100190, China. E-mail: lip@nanoctr.cn; wangch@nanoctr.cn; yangyl@nanoctr.cn
First published on 12th January 2017
Abstract
A facile method for encapsulation of insoluble drugs has been developed based on the metal ion/polyphenol complexation and aerosol spraying process. The encapsulated paclitaxel nanodrug (PTX-C) is well-controlled in morphology and pH-responsive. In vivo and in vitro experiments demonstrated the enhanced anti-tumour activity of PTX-C. This study may pave the way for the fabrication of low cost, high efficiency drug delivery systems for cancer therapies.
A variety of nanostructured materials, including micelles, liposomes, dendrimers and polymeric nanoparticles,1–12 have been extensively pursued for improving drug solubility, enhancing permeability and retention (EPR) effect, and tumour tissue targeting. The commercialized nanodrugs, including FDA-approved Doxil and Abraxane (ABI-007) have shown improved drug efficacy for treatment of advanced breast and ovarian cancer. The associated carriers or dispersing agents have contributed significantly to the enhanced drug availability for prolonging the retention time in tumour tissues and controllable drug release.13
On the other hand, synthesized carriers are inevitably accompanied by various organic solvents which may affect the properties of the therapeutic drugs. Therefore, notable efforts have been made to explore natural ingredients as potential carriers for pharmaceutical applications. For example, natural polyphenols and metal ions complexes can be assembled into hollow capsules by various fabrication technologies.14–17 The unique properties, including stimuli-response, selective permeability and enhanced mechanical/thermal stability of these hollow capsules have been intensely studied. The way to fabricate hollow capsules includes two steps: coating sacrificial particulate templates with robust films and template removal. It has been found that the coordination capsules of tannic acid (TA) and metal ions can successfully encapsulate a variety of materials with different shapes and properties, including planar and particulate materials such as gold, polydimethylsiloxane, silica, calcium carbonate, Escherichia coli and Staphylococcus epidermidis.18,19 Researchers have tried to remove the encapsulated CaCO3 to get the empty capsule by using poly(ethylene glycol)–polyphenol as the building block to reduce nonspecific protein adsorption.20 Using the template removing strategy, preliminary studies on drug carrier were performed by coating doxorubicin (DOX)-loaded spherical CaCO3 particle and subsequently removing the CaCO3.21
The above mentioned fabrication strategy using the coordination-triggered rapid deposition of TA/metal ions complexes on different materials could be applied in controllable drug release. The contribution of Frank Caruso group opened up a new direction in drug delivery. If it is possible to obtain nanodrug-loaded capsules by a one-step process based on the above method, the application of this drug delivery system would be more feasible in practical applications. A one-step process would facilitate not only the simple and efficient fabrication but also high availability of the drugs, both of which could greatly reduce the cost of nanodrug fabrication. In order to accomplish these goals, we propose a method to encapsulate prepared nanodrug directly with organic and metal ion complexes. This approach does not involve a step for template removal but involves directly coating the nanodrug without further processing. Furthermore, the capacity for drug loading can be well controlled due to its one-step fabrication procedure. This strategy simplifies the synthesis of capsules and guarantees high efficiency of drug loading.
Paclitaxel (PTX), which is extracted from the bark of the Pacific Yew tree,22 is known to be effective for treating various cancers, especially ovarian and breast cancers.23,24 However, PTX's poor aqueous solubility has greatly hindered its commercial use. Up to now, two types of PTX drugs have been commercially available. One is used together with adjuvants (Cremophor® EL). This formulation was reported to have hypersensitivity reactions caused by its adjuvant.25,26 In addition, rapid metabolism of paclitaxel after intravenous injection could reduce its therapeutic efficiency.27 Another is albumin-bound PTX nanodrug, called ABI-007, which successfully avoids hypersensitivity reactions caused by solvent, however involves notably increased expenses.
In this study, we report a novel one-step fabrication process to obtain paclitaxel nanoparticles (PTX-NPs) encapsulated by thin-film (PTX-C). The PTX-NPs are fabricated by aerosol generators, and the as-prepared PTX-NPs are simultaneously further coated by coordination of polyphenol tannic acid and FeIII. It is worth mentioning that TA and FeIII are generally recognized as safe (GRAS) by the U.S. Food and Drug Administration and are also readily available commercially. The resulting PTX-C nanodrugs are not only stable in water but also pH-responsive for drug release. Moreover, further experiments of the synthesized PTX-C both in vivo and in vitro reveal significant anti-tumour activity.
In light of Caruso's recent studies showing the rapid deposition of complexes of TA and metal ions on various nano-templates triggered by coordination, we use complexes of TA and metal ions as shells to encapsulate PTX nanoparticles. In order to develop a facile method for controlled nanodrug fabrication, herein, the aerosol spray method is used to obtain PTX-NPs as illustrated in Fig. 1. Briefly, commercially available PTX powder was dissolved in ethanol with a concentration of 200 μg mL−1. The solution was subsequently introduced into an atomizer aerosol generator machine. Nitrogen flowed into the generator and created a mass of aerosol with a special spray nozzle equipped inside the machine. Then the obtained aerosol was transferred to deionized water by a continuous nitrogen stream. In this process, dissolved PTX molecules in aerosol precipitated into particles due to the poor water solubility of PTX. The size of the precipitated nanoparticles is restricted by that of the aerosol. After obtaining the suspension of PTX-NPs in water, the prepared PTX-NPs were coated immediately with the natural polyphenols and FeIII. To coat PTX-NPs, FeCl3·6H2O (0.08 mg mL−1) and then TA solution (0.1 mg mL−1) were added into this suspension separately, followed by vigorous stirring. The colour of the suspension immediately became blue after all the chemicals were added. As shown in Fig. 1, PTX-NPs are utilized as a template. The coordination complexes of FeIII and TA formed a thin layer at the interface between PTX-NPs and the solution, resulting in the formation of PTX-C. As reported, TA has been widely used in drug systems.28,29 John K. Jackson has reported to use TA as hydrotrope-like solubilizing agent to improve the aqueous solubility of hydrophobic drugs.30 They also demonstrated the excellent drug transfer of docetaxel into cells in many cell lines. This work offers a clinically relevant alternative for hydrophobic drug formulation. Different from Jackson's work, TA in this paper is used as a component for the formation of TA/FeIII complex as a drug delivery system for PTX-NPs. This method possesses several advantages. TA/FeIII complex coated outside the PTX-NPs can greatly improve the stability of PTX-NPs in aqueous solution. The nanoscale size of the drug particles lead to the huge surface area for controlled release, and the coated nanoparticles could also be further functionalized by versatile surface chemistry. What's more, the special pH sensitive property of TA/FeIII complex endows PTX-C with controlled drug release features. To make sure that this process is applicable to other drugs, quercetin was chosen to be encapsulated by using the same methods, SEM and TEM images (Fig. S8A and B†) suggested that quercetin nanoparticles were successful obtained and encapsulated by TA/FeIII complex, which demonstrate that this method can be applicable to other drugs.
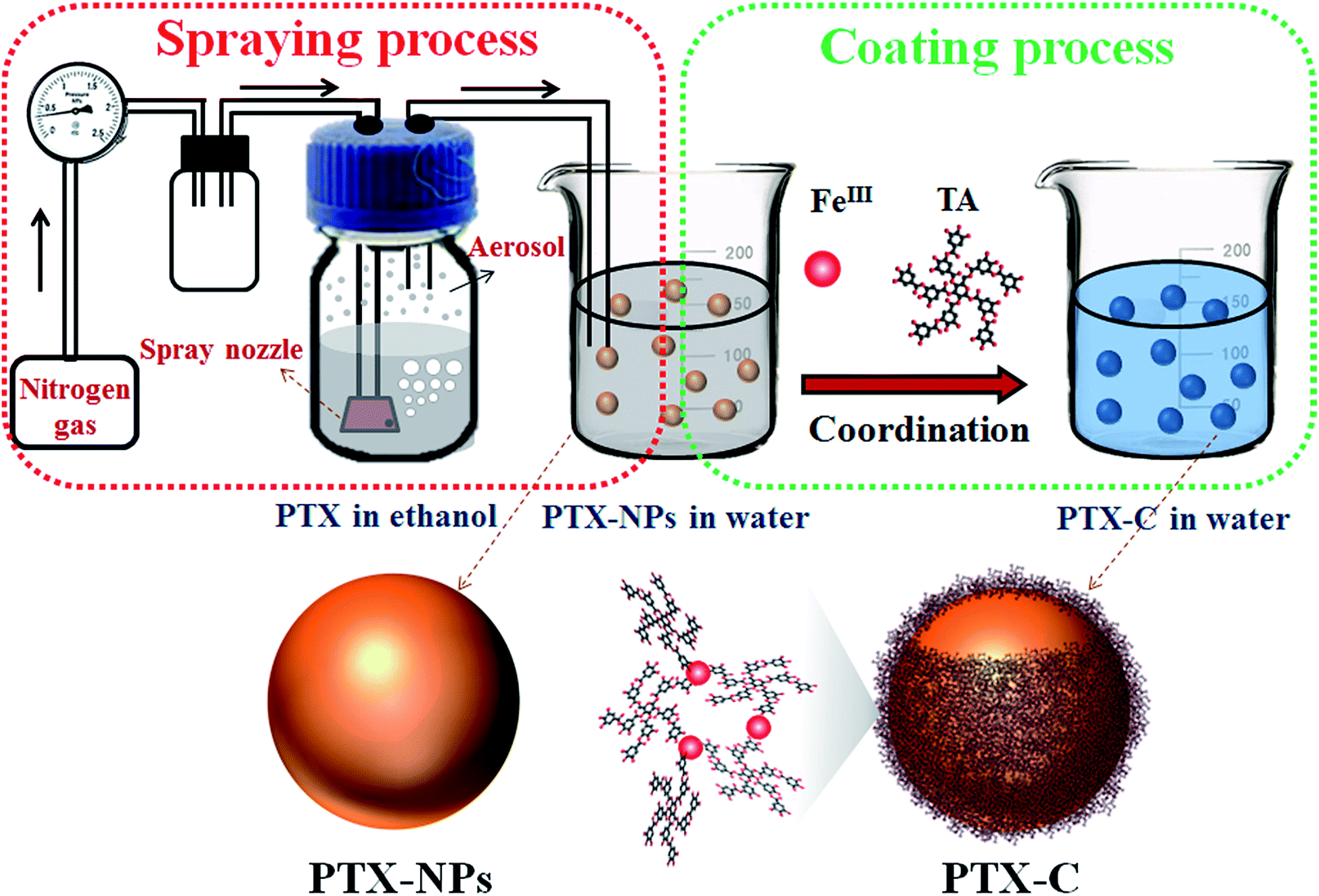 | ||
| Fig. 1 Schematic illustration of the fabrication procedure for PTX-NPs and PTX-C. In the spraying process, the PTX aerosol was generated by using the aerosol spray method, and the nitrogen gas will transport the aerosol into the water for the formation of PTX nanoparticles (PTX-NPs). In the coating process, the as prepared PTX-NPs are encapsulated by the complexes of tannic acid and FeIII. The schematic models for the PTX-NPs and PTX-C particles are also shown at the lower part of the figure. | ||
The morphologies of PTX, PTX-NPs and PTX-C were then characterized by scanning electron microscopy (SEM) and transmission electron microscopy (TEM) in Fig. 2. Due to its poor solubility in water, untreated PTX crystallizes into thick fibres which are micrometres in length as shown in Fig. 2C. The poor solubility of PTX in water, causing large fibres of pristine PTX to form in aqueous solution greatly hinders the direct use of PTX as an anti-cancer drug. In contrast, the morphology and size of PTX crystals fabricated by the aerosol spray method was well-controlled. Different concentration of FeIII (0.08–0.2 mg mL−1) and TA (0.1–0.25 mg mL−1) led to different coating conditions (Fig. 2B, S1A and B†). Among which, 0.08 mg mL−1 FeCl3·6H2O and 0.1 mg mL−1 TA is the optimal concentration. At the optimal condition, few excess complexes could be observed. Slight increasing of TA and FeCl3·6H2O would result in the excess complex which can be observed in the SEM images. SEM (Fig. 2A, B and S1C, D†) and TEM (Fig. 2D and F) images present the particulate morphologies of PTX-NPs and PTX-C. The specific morphologies may be cubic, spherical or amorphous, while no thick fibres can be observed. PTX-NPs (Fig. 2A and D) are uniform in size and about 200 nm in diameter. TEM images of PTX-NPs in high resolution show that these particles are porous (Fig. S1E and F†). The isolated environment of each aerosol droplet may prevent the process of full crystallization and result in formation of the porous crystalline. After being coated by complex compounds (the morphologies of TA/Fe complex without template are shown in Fig. 2E), these nanoparticles are observed to develop amorphous forms with some silk-like film surrounding the surface of particles as shown in Fig. 2F. This can be considered a clear confirmation of the successful encapsulation of the PTX-NPs with the coordination complex. In TEM images of PTX-C, there exist two different forms (Fig. 2F and S3B†) which can be defined as tightly coated and loosely coated. In the former form, PTX-NPs are tightly coated by TA/FeIII complex film with almost no gaps between film and particle (Fig. 2F). In the latter form, obvious gaps between film and PTX-NPs can be observed (Fig. S3B†). There may be more than one nanoparticle encapsulated in either the tightly or loosely coated forms (Fig. S2A and B†). However, the coating effect of all the forms is identical with significantly improved dispersibility and stability.
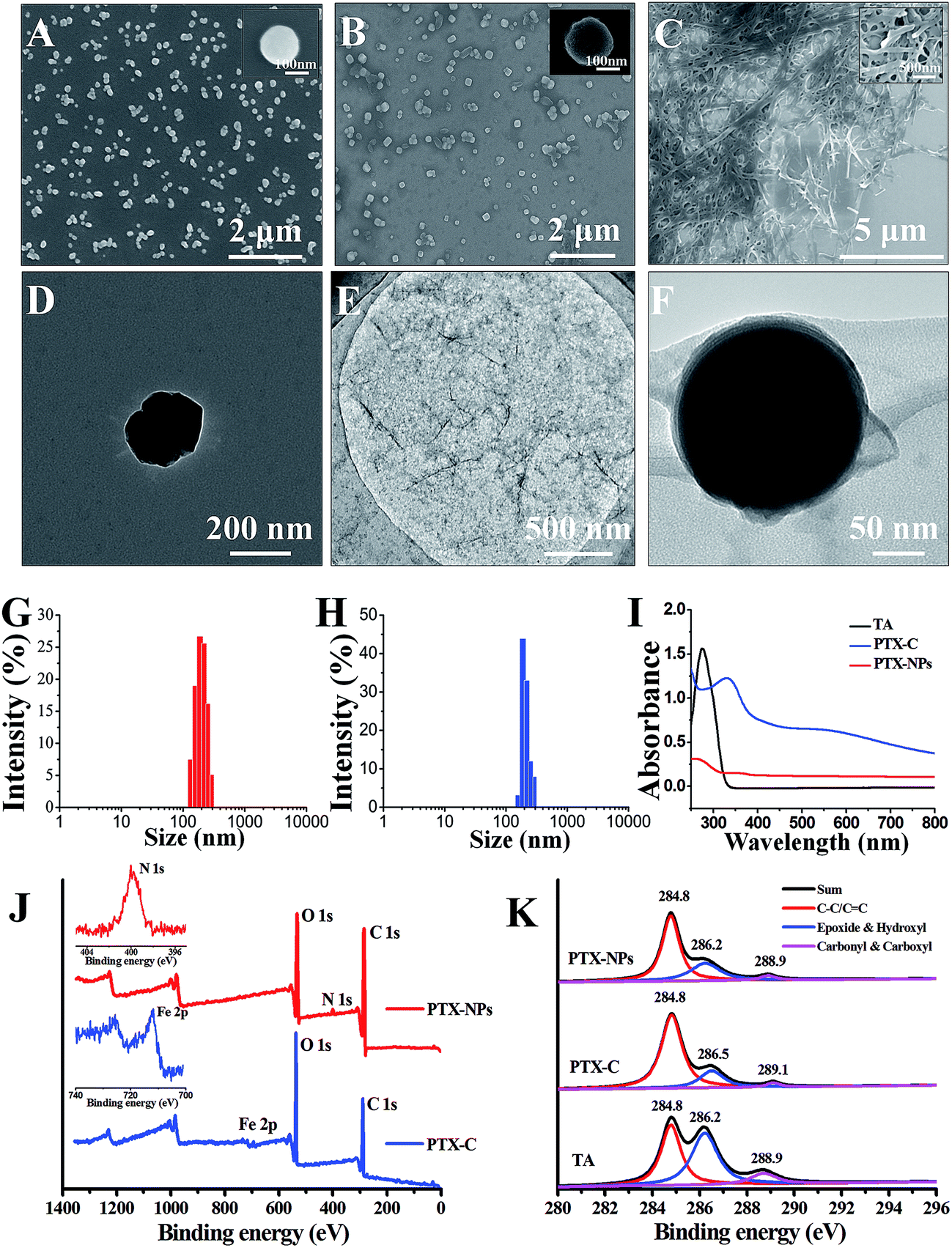 | ||
| Fig. 2 Micrographs of PTX-NPs and PTX-C. SEM images of PTX-NPs (A), PTX-C (B) and PTX (C) in water. The insets in (A), (B) and (C) are corresponding magnified SEM images. TEM images of PTX-NPs (D), TA/FeIII complex (E) and PTX-C (F). (G) and (H) Size distribution of the nanoparticles in PTX-NPs and PTX-C obtained by DLS. (I) UV-Vis absorption spectra of TA, PTX-C and PTX-NPs. Full scan (J) and C1s profiles (K) of the XPS spectra of TA, PTX-C and PTX-NPs, the inset in (J) is the zoomed-in spectra for N1s and Fe2p peaks. | ||
Dynamic light scattering (DLS) was performed to measure size distribution of PTX-NPs before and after coating. The size distribution results in Fig. 2G demonstrate that the diameters of PTX-NPs are 200 ± 100 nm. After coating, the full width at half maximum (FWHM) was greatly reduced from 108 nm to 65 nm, and the population of particles with average diameter at 200 nm increased to about 80% (shown in Fig. 2H). The zeta potential of PTX-C was also measured by DLS to be −10.8 ± 0.3 mV, which is about 5.6 mV higher than that of PTX-NPs (−16.4 ± 0.4 mV). The slight increase of the zeta potential could be ascribed to the positive ferric ions contained in the film.
Ultraviolet to visible (UV-Vis) spectra in Fig. 2I were used to confirm formation of TA/FeIII complex during the coating process. No obvious peak can be observed in the UV-Vis spectrum of PTX-NPs before coating, while a strong peak at 290 nm appears in the spectrum of TA, which is ascribed to the n–π transition. After coating, a new peak at 330 nm was observed for PTX-C, which could be ascribed to the TA/FeIII complex. This confirms the successful coating of TA/FeIII complex on PTX-NPs.
To further confirm the successful coating of the PTX-NPs (Fig. 2F) associated with the TA/FeIII complexes, rather than assembly or aggregation of TA alone, we conducted X-ray photoelectron spectroscopy (XPS) to analyze the element composition on the surface of PTX-NPs and PTX-C. The results are shown in Fig. 2J and K. In the full scan spectrum (Fig. 2J), the N1s peak (399.9 eV) appears in PTX-NPs, which represented the nitrogen element in the PTX molecule. While the PTX-C, N1s peak disappeared, a new peak at 711.7 eV (Fe2p) appeared. Due to the TA/FeIII complex coating, the N1s signal in PTX-NPs is undetectable, while Fe2p signal outside of PTX-C can be easily collected by XPS equipment. The disappearance of N1s signal and appearance of Fe2p signal in PTX-C demonstrated that the outside film is TA/FeIII complex. The binding energy in XPS spectra gives further information regarding the coordination. C1s profiles of PTX-NPs, PTX-C and TA are shown in Fig. 2K, and three distinct peaks are assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C in aromatic rings, C–O (epoxide and hydroxyl) and CO (carbonyl/carboxyl) groups, respectively.31 For the PTX-NPs and TA, the peak locations are identical for all three peaks. The different peak intensities originate from the different chemical compositions of PTX and TA. For PTX-C, the peak corresponding to C–O groups shifts from 286.2 to 286.5 eV, indicating the coordination interaction between hydroxyl groups and Fe ions. The peak corresponding to CO groups shifts from 288.9 to 289.1 eV,32 indicating that the carbonyl groups in TA could also contribute to coordination with Fe ions. Both UV-Vis and XPS spectra indicate that PTX-NPs are successfully coated with the TA/FeIII complex.
C/C–C in aromatic rings, C–O (epoxide and hydroxyl) and CO (carbonyl/carboxyl) groups, respectively.31 For the PTX-NPs and TA, the peak locations are identical for all three peaks. The different peak intensities originate from the different chemical compositions of PTX and TA. For PTX-C, the peak corresponding to C–O groups shifts from 286.2 to 286.5 eV, indicating the coordination interaction between hydroxyl groups and Fe ions. The peak corresponding to CO groups shifts from 288.9 to 289.1 eV,32 indicating that the carbonyl groups in TA could also contribute to coordination with Fe ions. Both UV-Vis and XPS spectra indicate that PTX-NPs are successfully coated with the TA/FeIII complex.
Design of stimuli-responsive drugs is a promising approach to reduce systemic toxicity and enhance therapeutic outcome of the drugs. Thus, it is beneficial to examine the stimuli-responsive capability of our proposed drug delivery system. Caruso has previously reported the pH-dependent behaviour of coordination between FeIII and TA.19 A change in pH can greatly influence the assembly behaviour of the complex, reflected by the colour change of the complex suspension. As expected, for PTX-C suspension, the similar pH-responsive phenomenon is also observed. As pH changes from 1.0 to 8.4, the colours of the suspension vary from colourless to blue and then to red (inset in Fig. 3A). Such colour changes can be ascribed to the transitions between mono-, bis-, and tri-complex states.20 At pH 1.0, the complexes are totally disassembled owing to protonation of all the hydroxyl groups. As the pH increases, protonated hydroxyl groups gradually decrease, leading to the recovery of coordination. As pH reaches 7.4, the coordination can be totally recovered. The UV spectra (Fig. 3A) strongly support the above changes. At low pH (pH 1.0 and pH 3.0), a strong peak can be observed at 290 nm, which is ascribed to the signal of TA. When pH is increased, this peak was gradually shifted to high wavelength and then disappeared. Herein, signal variations in UV along with increasing pH are consistent with the transition between mono-, bis-, and tri-complexation states.
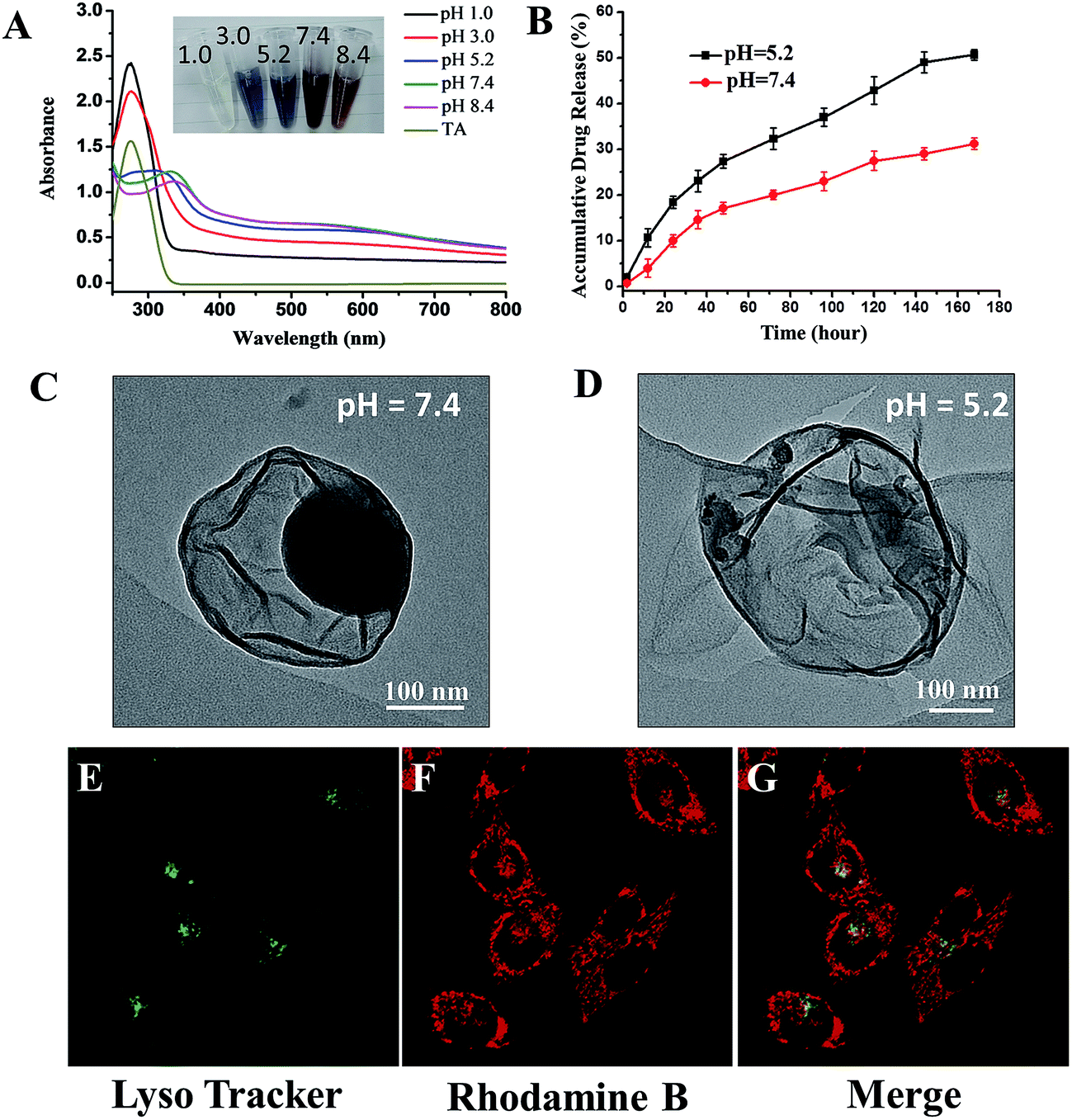 | ||
| Fig. 3 (A) pH-Responsive disassembly of TA/FeIII films coated on PTX-NPs. UV absorption spectra of PTX-C (0.5 mg mL−1) at various pH values. Inset is a photograph of PTX-C at the indicated pH values. (B) The drug release profiles of PTX-C in PBS at 37 °C at pH 7.4 and pH 5.2. (C) and (D) The TEM images of PTX-C containing or without nanoparticles at pH 7.4 and pH 5.2 respectively. (E), (G) and (F) Intracellular localization of PTX-C labelled by Rhodamine B in MCF-7 cells. Cells were incubated with labelled PTX-C at the concentration of 0.5 μg mL−1 for 8 h at 37 °C, followed by labelling with LysoTracker® Green DND-26 for 30 min and then observed by laser confocal microscopy (60×). The images from left to right show (E) labelled lysosomes (green), (G) Rhodamine B fluorescence from PTX-C in cells (red), and (F) overlays of the former two images. | ||
Since the condition of the complex film varies with pH, the release efficiency of PTX may also be affected by pH changes. Drug release experiments were performed at pH 5.2 and 7.4. 100 μg freeze-dried PTX-C were suspended in 1 mL phosphate buffer saline and then dialyzed against 400 mL PBS (pH 7.4 and 5.2 respectively) at 37 °C with gentle stirring. The cumulative released drug could be easily obtained with high performance liquid chromatography (HPLC) by subtraction of the measured drug amount from the initial drug amount in the dialysis bag. Under different pH environment, PTX-C shows a significant difference in drug release behaviours (Fig. 3B). The red and black curves represent the release tendency at pH 5.2 and 7.4. At pH 5.2 and 7.4, drug release efficiency reached 51% and 30% respectively in 7 days. The increased drug release efficiency at the lower pH support the sensitivity of pH responsive encapsulation. In addition, TEM images were obtained for direct observation of the effect of pH on PTX-C morphology (Fig. 3C, D and S3†). It is obvious that the particles were well encapsulated by complex film at pH 7.4 (Fig. 3C, S3A and B†). While at pH 5.2 (Fig. 3D, S3C and D†), hollow film with few residue particles was observed. It can be concluded that weaker coordination of TA and FeIII at pH 5.2 cannot protect PTX-NPs in the capsule. Reliable stability in a neutral pH environment and controlled release at lower pH makes this drug a promising candidate as an anti-cancer drug. Stability at neutral pH can protect the drug when being transported in a drug delivery vessel. After the drug is endocytosed by the target cell and reaches the lysosome, the acidic environment in the lysosome will promote release of the drug.
Cellular uptake and distribution of PTX-C in MCF-7 (human breast cancer cells) cells were further investigated using laser scanning confocal microscope (LSCM). The lysosomes are easily observed under the LSCM by green fluorescence of LysoTracker® Green DND-26 labelling. Rhodamine B and PTX-C were mixed together overnight, after centrifuging the mixture and discarding the supernatant for three times, the fluorescent labelled PTX-C solution was then incubated with cells for 8 hours. It should be noted that the Rhodamine B adsorption method is not full proof of the PTX-C localization, since intracellular processes could possibly displace the dye. Under this treatment, most lysosomal compartments show both red (Rhodamine B) and green fluorescence (Fig. 3E–G). This result indicates that PTX-C could be endocytosed by cells and possibly localized in lysosomes if the dye molecules was not desorbed from the nanoparticles. Control experiments on incubation of pure Rhodamine B with the cells (Fig. S4†) further indicated the possible localization of the PTX-C at lysosomes. The red fluorescence was also observed on cell membrane in Fig. 3F, which could be associated with the non-specific adsorption of Rhodamine B labelled PTX-C or free Rhodamine B molecules.
Fig. 4A shows the in vitro viability of six cell lines (MCF-7, SKBR-3 (human breast cancer cells), HepG2 (human hepatoma cells), PC-3 (human prostate cancer cells), A549 (human lung cancer cells) and Hela (human cervical cancer cells)) after 72 hours incubation with PTX, PTX-NPs, PTX-C, and ABI-007 at a PTX concentration of 0.98–31.2 ng mL−1. As expected, the cell viability of all cell lines decreases with increasing PTX concentration. The in vitro cytotoxicity of the drugs can be quantitatively compared by IC50, the drug concentration at which 50% of cells have been killed. The IC50 values of PTX obtained in different formulations at 72 hours are given in Table S1.† In A549, HepG2, SKBR-3 and PC-3 cells, the IC50 of PTX-C is lower than that of PTX, PTX-NPs and ABI-007. Specifically for A549 cells, PTX-C demonstrates superior anti-tumour activity with much lower IC50 (3.1 ng mL−1) than PTX, PTX-NPs and ABI-007 (5.5 ng mL−1, 31.3 ng mL−1 and 8.6 ng mL−1, respectively). In Hela and MCF-7 cells, the IC50 of PTX-C is lower than PTX-NPs and ABI-007, but is comparable with that of PTX. Based on the above results, PTX-C is generally an efficient anti-tumour drug compared with PTX, PTX-NPs and ABI-007. In the control experiment, the TA/FeIII complex is nearly non-cytotoxic (Fig. S5†), which is consistent with the previous report.18 Thus, the improved inhibitory effect on cancer cells is attributed to PTX-C, but not the influence of the complex materials. The IC50 value of PTX-C is 2–10 times lower than that of PTX-NPs (Table S1†), and specifically the IC50 observed in A549 cells is one-tenth of that for PTX-NPs. This enhanced cytotoxicity can be attributed to the increased stability and dispersity of PTX-C in water, and the enhanced endocytosis because of the nanometre size of PTX-C.33 Moreover, it is noteworthy that the cytotoxicity of PTX-C in vitro is comparable and even better than that of ABI-007, which is a commonly used chemotherapeutic drug. Considering the simple fabrication process of this method and the low price of the TA and Fe ion starting materials, spray-generated and complex-encapsulated PTX-C could be a very effective and promising anti-tumour nanodrug.
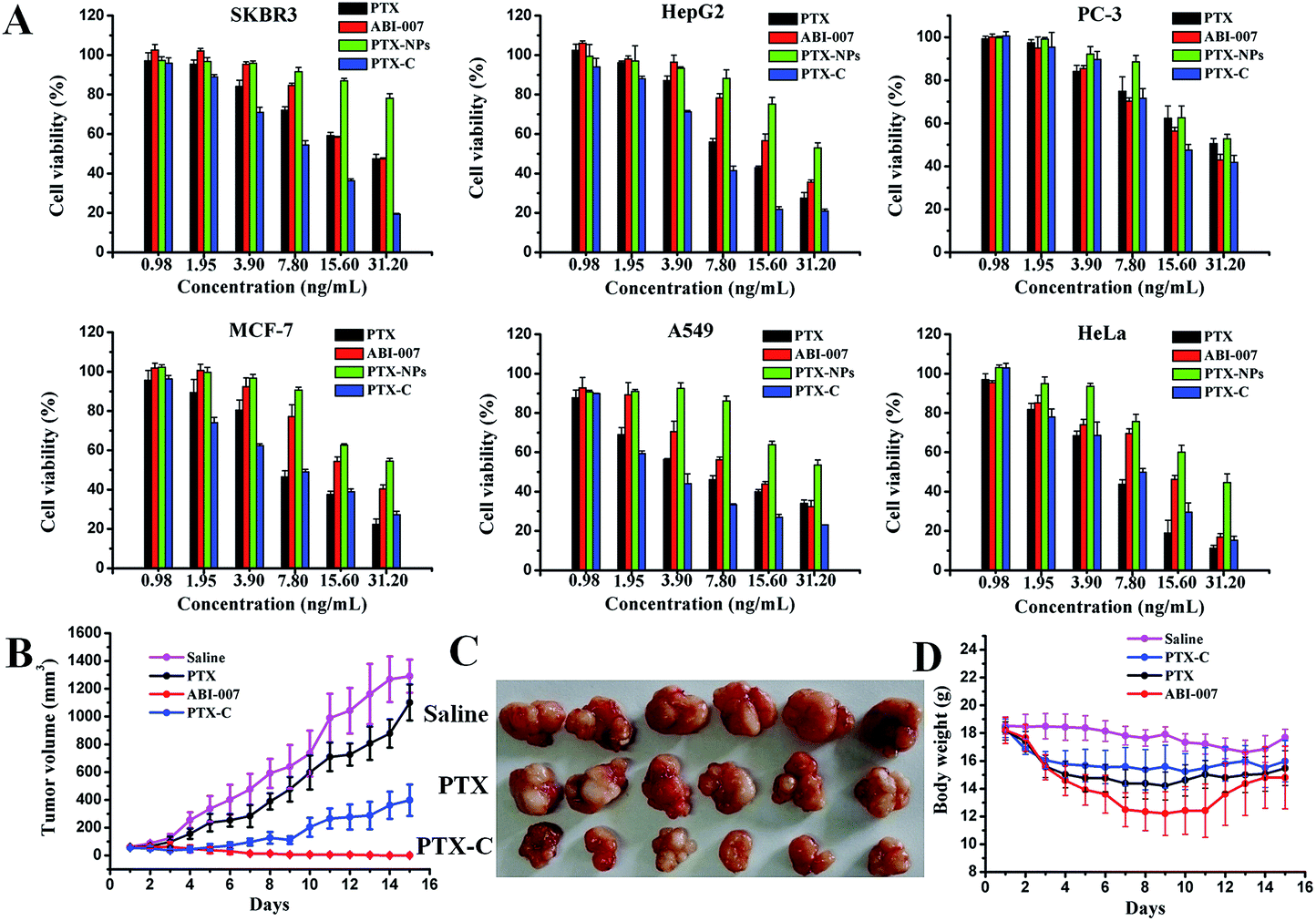 | ||
| Fig. 4 (A) In vitro viabilities of cells after incubations with PTX, ABI-007, PTX-NPs and PTX-C for 72 h. (B) The tumour volume of the mice bearing MCF-7 xenografts for saline (control), PTX, ABI-007 and PTX-C groups. A dosage of 10 mg kg−1 (with respect to PTX) was administrated intravenously for mice, and the tumour volumes were measured at the indicated time points (n = 6–8). (C) Photographs of the tumours taken after 15 days. (D) Effects of PTX, ABI-007 and PTX-C on body weight of mice bearing MCF-7 xenografts. Body weight was measured everyday. | ||
We further investigated the in vivo anti-tumour activity of PTX-C. As shown in Fig. 4B and C, PTX-C leads to a 69.2% tumour growth inhibition rate, compared with 14.7% and 99.9% in the cases of PTX and ABI-007, respectively. Compared to saline and PTX groups, tumour growth is significantly reduced in the group treated with PTX-C. The inhibition effect is also well reflected by the tumour weights (Fig. S6†). These results again demonstrate the greatly enhanced anti-tumour efficacy of PTX-C compared with free PTX drugs. In the control experiment, the TA/FeIII complex showed no inhibition or promotion effects on the tumour growth (data not shown), which demonstrates that the TA/FeIII complex is neutral in the anti-tumour activity.
It should be noted that even though anti-tumour activity of PTX-C is slightly lower than the positive control ABI-007 group, the body weight maintaining effect in PTX-C group was better than that of ABI-007 group (Fig. 4D). Especially from day 4 to day 12, average body weight of ABI-007 group significantly decreases compared with that of PTX-C group (p < 0.05). As body weight change is a clear reflection of systemic toxicity, the above result indicates that the potential toxicity of PTX-C to mice is lower than that of ABI-007. To evaluate the side effects, heart, liver, spleen, lung and kidney were dissected after 15 days of treatment and stained with hematoxylin and eosin (H&E) for pathologic analysis. PTX-C shows no obvious damage to the main organs (Fig. S7†). The in vivo results indicate that PTX-C possesses an excellent anti-tumour ability at lower systematic toxicity.
Conclusions
We have demonstrated a facile approach to encapsulate anti-tumour nanodrugs using metal ions/polyphenol complexation and aerosol spraying process. The PTX nanoparticles encapsulated by TA/FeIII complexes were fabricated with well-controlled morphology and size. The highly efficient pH-responsive drug release, uptake by cells and localization at lysosomes contribute to PTX-C serving as an excellent anti-tumour nanodrug. The IC50 values of PTX-C are similar or significantly lower than the values of PTX and ABI-007 in 6 different cancer cell lines. PTX-C leads to a 69.2% tumour growth inhibition rate, compared with 14.7% and 99.9% in the cases of PTX and ABI-007, respectively. The in vitro and in vivo results demonstrate the greatly improved anti-tumour activity of PTX-C over free PTX drugs, and the body weight maintaining results show the reduced potential toxicity. Together with the readiness of scaling up of this fabrication process, PTX-C could be a novel anti-tumour nanodrug possessing method highly potential in therapeutic applications. This one-step encapsulating method could be applicable for many other hydrophobic drugs.Compliance with ethical standards
The authors declare that they have no conflict of interest. All animal experiments were performed with the approval of the Institutional Animal Care and Use Committee, Institute of Process Engineering, Chinese Academy of Sciences (IACUC, IPE, CAS, IRB number 2014-0002).Acknowledgements
P. L., H. H. contributed equally to this work. This work was supported by the National Natural Science Foundation of China (21273051) and the Chinese Academy of Science (XDA09030306 and YZ201317).References
- W. Mehnert and K. Mader, Adv. Drug Delivery Rev., 2001, 47, 165–196 CrossRef CAS PubMed.
- V. P. Torchilin, J. Controlled Release, 2001, 73, 137–172 CrossRef CAS PubMed.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS PubMed.
- V. P. Torchilin, Adv. Drug Delivery Rev., 2006, 58, 1532–1555 CrossRef CAS PubMed.
- L. Sheihet, K. Piotrowska, R. A. Dubin, J. Kohn and D. Devore, Biomacromolecules, 2007, 8, 998–1003 CrossRef CAS PubMed.
- P. Ghosh, G. Han, M. De, C. K. Kim and V. M. Rotello, Adv. Drug Delivery Rev., 2008, 60, 1307–1315 CrossRef CAS PubMed.
- O. C. Farokhzad and R. Langer, ACS Nano, 2009, 3, 16–20 CrossRef CAS PubMed.
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette, C. Kreuz, J. S. Chang, Y. K. Hwang, V. Marsaud, P. N. Bories, L. Cynober, S. Gil, G. Ferey, P. Couvreur and R. Gref, Nat. Mater., 2010, 9, 172–178 CrossRef CAS PubMed.
- U. Kedar, P. Phutane, S. Shidhaye and V. Kadam, Nanomedicine: Nanotechnology, Biology and Medicine, 2010, 6, 714–729 CrossRef CAS PubMed.
- J. W. Yoo, N. Doshi and S. Mitragotri, Adv. Drug Delivery Rev., 2011, 63, 1247–1256 CrossRef CAS PubMed.
- I. A. Siddiqui, V. M. Adhami, J. C. Chamcheu and H. Mukhtar, Int. J. Nanomed., 2012, 7, 591–605 CAS.
- A. Sanchez-Sanchez, F. Suarez-Garcia, A. Martinez-Alonso and J. M. D. Tascon, Carbon, 2015, 94, 152–159 CrossRef CAS.
- C. Fonseca, S. Simoes and R. Gaspar, J. Controlled Release, 2002, 83, 273–286 CrossRef CAS PubMed.
- M. A. Rahim, H. Ejima, K. L. Cho, K. Kempe, M. Mullner, J. P. Best and F. Caruso, Chem. Mater., 2014, 26, 1645–1653 CrossRef CAS.
- J. J. Richardson, M. Bjornmalm, S. T. Gunawan, J. Guo, K. Liang, B. Tardy, S. Sekiguchi, K. F. Noi, J. Cui, H. Ejima and F. Caruso, Nanoscale, 2014, 6, 13416–13420 RSC.
- J. Guo, X. Wang, D. C. Henstridge, J. J. Richardson, J. Cui, A. Sharma, M. A. Febbraio, K. Peter, J. B. de Haan, C. E. Hagemeyer and F. Caruso, Adv. Healthcare Mater., 2015, 4, 2170–2175 CrossRef CAS PubMed.
- M. A. Rahim, K. Kempe, M. Mullner, H. Ejima, Y. Ju, M. P. van Koeverden, T. Suma, J. A. Braunger, M. G. Leeming, B. F. Abrahams and F. Caruso, Chem. Mater., 2015, 27, 5825–5832 CrossRef CAS.
- H. Ejima, J. J. Richardson, K. Liang, J. P. Best, M. P. van Koeverden, G. K. Such, J. Cui and F. Caruso, Science, 2013, 341, 154–157 CrossRef CAS PubMed.
- J. L. Guo, Y. Ping, H. Ejima, K. Alt, M. Meissner, J. J. Richardson, Y. Yan, K. Peter, D. von Elverfeldt, C. E. Hagemeyer and F. Caruso, Angew. Chem., Int. Ed., 2014, 53, 5546–5551 CrossRef CAS PubMed.
- Y. Ju, J. W. Cui, M. Mullner, T. Suma, M. Hu and F. Caruso, Biomacromolecules, 2015, 16, 807–814 CrossRef CAS PubMed.
- Y. Ping, J. Guo, H. Ejima, X. Chen, J. J. Richardson, H. Sun and F. Caruso, Small, 2015, 11, 2032–2036 CrossRef CAS PubMed.
- M. C. Wani, H. L. Taylor, M. E. Wall, P. Coggon and A. T. Mcphail, J. Am. Chem. Soc., 1971, 93, 2325–2327 CrossRef CAS PubMed.
- K. L. Hennenfent and R. Govindan, Ann. Oncol., 2006, 17, 735–749 CrossRef CAS PubMed.
- I. J. Majoros, A. Myc, T. Thomas, C. B. Mehta and J. R. Baker, Biomacromolecules, 2006, 7, 572–579 CrossRef CAS PubMed.
- R. T. Liggins, W. L. Hunter and H. M. Burt, J. Pharm. Sci., 1997, 86, 1458–1463 CrossRef CAS PubMed.
- H. Gelderblom, J. Verweij, K. Nooter and A. Sparreboom, Eur. J. Cancer, 2001, 37, 1590–1598 CrossRef CAS PubMed.
- F. Li, J. N. Li, X. J. Wen, S. H. Zhou, X. W. Tong, P. P. Su, H. Li and D. L. Shi, Mater. Sci. Eng., C, 2009, 29, 2392–2397 CrossRef CAS.
- T. G. Shutava and Y. M. Lvov, J. Nanosci. Nanotechnol., 2006, 6, 1655–1661 CrossRef CAS PubMed.
- B. Kim, H. Lee, Y. Min, Z. Poona and P. T. Hammond, Chem. Commun., 2009, 4194–4196 RSC.
- J. K. Jackson and K. Letchford, J. Pharm. Sci., 2016, 105, 3143–3152 CrossRef CAS PubMed.
- O. C. Compton, D. A. Dikin, K. W. Putz, L. C. Brinson and S. T. Nguyen, Adv. Mater., 2010, 22, 892–896 CrossRef CAS PubMed.
- S. Park, K. S. Lee, G. Bozoklu, W. Cai, S. T. Nguyen and R. S. Ruoff, ACS Nano, 2008, 2, 572–578 CrossRef CAS PubMed.
- Y. Yan, G. K. Such, A. P. R. Johnston, J. P. Best and F. Caruso, ACS Nano, 2012, 6, 3663–3669 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra26936b |
| ‡ P. L. and H. H. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2017 |