
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantum binding energy features of the T3-785 collagen-like triple-helical peptide†‡
Katyanna S. Bezerraa,
Jonas I. N. Oliveiraa,
José X. Lima Neto a,
Eudenilson L. Albuquerquea,
Ewerton W. S. Caetanob,
Valder N. Freirec and
Umberto L. Fulco*a
a,
Eudenilson L. Albuquerquea,
Ewerton W. S. Caetanob,
Valder N. Freirec and
Umberto L. Fulco*a
aDepartamento de Biofísica e Farmacologia, Universidade Federal do Rio Grande do Norte, 59072-970, Natal, RN, Brazil. E-mail: umbertofulco@gmail.com; Fax: +55-84-32153791; Tel: +55-84-32153793
bInstituto Federal de Educação, Ciência e Tecnologia do Ceará, 60040-531, Fortaleza, CE, Brazil
cDepartamento de Física, Universidade Federal do Ceará, 60455-760, Fortaleza, CE, Brazil
First published on 12th January 2017
Abstract
Collagen-based biomaterials are expected to become a useful matrix substance for various biomedical applications in the future. By taking advantage of the crystallographic data of the triple-helical peptide T3-785, a collagen-like peptide whose homotrimeric structure presents large conformational similarity to the human type III collagen, we present a quantum biochemistry study to unveil its detailed binding energy features, taking into account the inter-chain interaction energies of 90 amino acid residues distributed into three interlaced monomers. Our theoretical model is based on the density functional theory (DFT) formalism within the molecular fragmentation with conjugate caps (MFCC) approach. We predict the individual relevance (energetically) of the amino acid triplets Pro–Hyp–Gly, Ile–Thr–Gly, Ala–Arg–Gly and Leu–Gly–Ala, as well as the influence of the N-terminal, central and C-terminal regions, looking for the integrity of the collagen's triple helix. We found that the amino acid residues comprising the peptide T3-785 have an interaction energy that depends not only on the chemical nature of the side chain, but also the surrounding solvent molecules and inter-chain intermolecular interactions. The energy profile of this collagen-model molecule depicts a character essentially attractive to its conformational stability, encouraging research focusing on the development and synthesis of artificial collagen with high stability for bioengineering applications.
Biomaterial science is an expanding area, which encompasses a wide range of medical knowledge. Collagen, a well-known protein, is regarded as one of the most useful biomaterial in the medical, dental and pharmacological fields. Biomedical applications of collagen require improved methods to synthesize its triple helices feature, essential for the development of artificial structures displaying its natural properties. Its excellent biocompatibility and safety due to its biological characteristics, made it the primary resource in medical applications. Besides, it is the most abundant family of extracellular matrix proteins expressed in connective tissues of animals.1 Human beings have at least 45 distinct collagen genes accounting for 28 protein isoforms, amounting about one quarter of the total protein mass of the human body.2
A single collagen molecule is a structural insoluble fibrillar protein used to set up larger collagen aggregates that performs unique physiological functions in bones, skin, cartilage, ligaments and tendons.3 Also, there has been increasing appreciation of the biological importance of collagen in many cellular process, such as adhesion, proliferation and migration, matrix degradation/remodeling, tissue regeneration and homeostasis.4,5 Furthermore, due to a number of biological properties such as high biocompatibility, rare adverse reactions and weak antigenicity,6 collagens have been widely used as a natural material for diverse biomedical applications, including its clinical use as biomimetic scaffold and drug delivery system.7 Therefore, the understanding of how these properties are derived from its fundamental structural units requires a comprehensive knowledge of the mechanisms underlying its structure and stability.8
Seven types of collagen (I, II, III, V, XI, XXIV and XXVII) are assembled in stable fibrils, forming a complex three-dimensional fibrous superstructure.9 They are composed of individual helices of three polypeptide chains/strands twisted in a right-handed manner and held together by a ladder of intermolecular backbone hydrogen bonds between adjacent strands.10 Each chain shows one or more collagenous domain characterized by a repetition of Xaa–Yaa–Gly motif (or triplet), where Xaa and Yaa are often proline and hydroxyproline residues, respectively. Variations among the collagen family members include differences in assembly of basic polypeptide chain, different length of the triple helix, and difference in termination of the helical domain.11
Collagen type III is usually assembled as a homotrimer by three identical α1(III) chains found in extensible connective tissues such as skin, intestine, and vascular system, frequently in association with collagen type I. In aorta, it is essential for the maintenance of the mechanical strength of the extracellular matrix and to withstand the high blood pressure produced by the heartbeat.12 To give a better understanding about its molecular structure, a biologically relevant sequence of this collagen was modeled in the T3-785 molecule, a collagen-like triple-helical peptide that provided the first visualization of how the sequence of collagen defines distinctive local conformational variations in triple-helical structure.13 Fig. 1 depicts the T3-785 peptide sequence from three different outlooks. In Fig. 1a, we show the amino acids sequence that composes each single chain of this collagen-like structure, each one being comprised by three Pro–Hyp–Gly amino acid repetitions (N-terminal zone), followed by the residues Ile–Thr–Gly–Ala–Arg–Gly–Leu–Ala–Gly (central zone), and ending with four repetitions of Pro–Hyp–Gly (C-terminal zone). This three repetitions are also shown in Fig. 1b and c, displaying a transversal and longitudinal molecular overview of the crystallographic structure, respectively. As it can be seen, the homotrimer is formed by A- (blue), B- (green) and C- (yellow) chains, fragmented in the N-terminal zone (top), central zone (middle) and C-terminal zone (bottom). Variations in the helical symmetry among its different zones indicate that the triple-helical twist is sequence dependent. The 10/3 (7/2) helical pitch of Pro/Hyp-poor regions (Pro–Hyp–Gly triplet) could play a role in the interaction of collagenous domains with other biomolecules (nucleation of collagen fibrils).14 The ESI‡ gives an overview of the identification of the amino acid residues and surrounding water in the A- (Fig. ES1‡), B- (Fig. ES2‡) and C- (Fig. ES3‡) chains of the peptide T3-785, as well as the details of how they are organized into the three structural zones (Fig. ES4‡), namely N-terminal (amino-acid residues comprising three Pro–Hyp–Gly repetitions), central (sequence Ile–Thr–Gly–Ala–Arg–Gly–Leu–Ala–Gly of human type III collagen) and C-terminal (four Pro–Hyp–Gly triplets). Its Fig. ES5‡ depicts also the subsystems AB (A chain interacting with the B chain), BC (B chain interacting with the C chain) and CA (C chain interacting with the A chain).
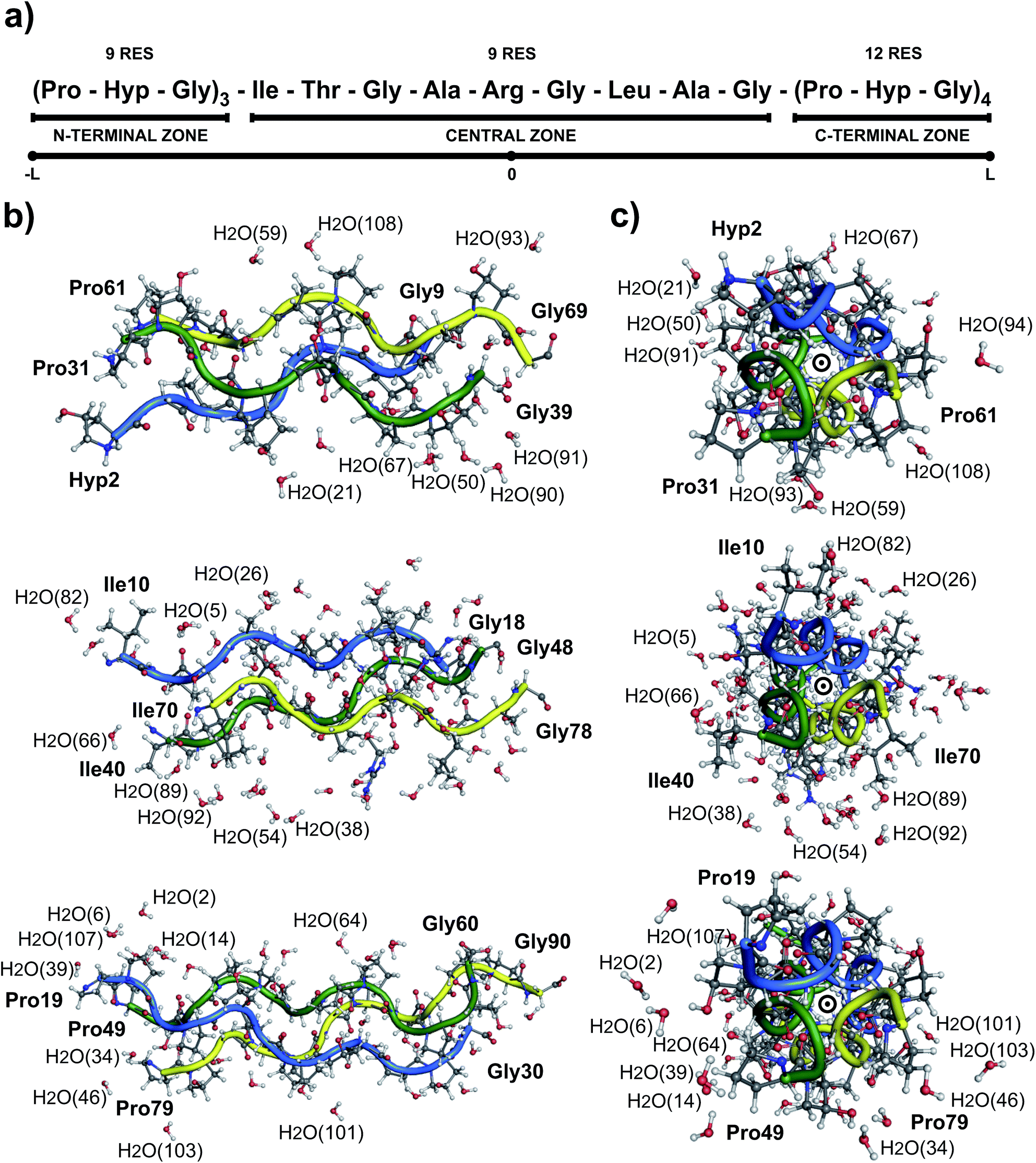 | ||
| Fig. 1 (a) Schematic (b) transversal and (c) longitudinal representation of the three zones which structurally constitute the collagen-like triple helical peptide T3-785. The three monomers were designed by the letters A (blue), B (green), C (yellow). | ||
Despite its simplified sequence and regular structure, the definition of the molecular basis of the collagen triple helix stability has been hitherto proved to be a difficult task.15 Thus, several computational studies have been performed to describe the stability of the collagen molecule, particularly those involving quantum mechanical tools. Among them, the HF/6-31G* and B3LYP/6-31G* calculations of relative and solvation free energies of collagen triplets have revealed that the collagen-like conformation is energetically more stable than its extended one.16 Suitable ab initio models to explore the stability of the implicit17 and explicit18 hydration network of collagen at B3LYP/6-31G(d) level of the theory were developed, whose collagen and β-sheet forming sequences GGG and AAG (PPG and POG) triplets destabilize (stabilize) the collagen triple helix. The semi-empirical PM6 (Parameterization Method 6) model and the CAM-B3LYP functional were carried out to optimize the geometry of close-packed motif (CP) for collagen and the more established 7/2 structure,19 suggesting a possible biological function for molecular hydrogen localized in the cavity of the CP structure. Employing ONIOM (Our own N-layered Integrated molecular Orbital and molecular Mechanics) and AM1 (Austin Model 1) calculations, Tsai et al. evaluated the effect of mutations in collagen-like triple helices, demonstrating the importance of the glycine residue in the repeating triad X–Y–Gly.20
However, the number of atoms that can be treated by quantum mechanics (QM) is still small. Thus, the idea of representing the total energy of macromolecular structures as a combination of fragmented energies has received increasing interest,21,22 and indeed, the ability to perform fragment-based quantum mechanical calculations on large biomolecules could be a very useful approach to predict their energetic properties, like a relative free energy of a collagen-model peptide with respect to its monomer state.23 Recently, Rodrigues et al. developed a computer simulation to describe, at the quantum level, the non-covalent interaction energies among the amino acid residues of T3-785 tropocollagen triple-helical structure.24
The purpose of this work is to analyze the conformational stability of the collagen-like peptide T3-785 (PDB: 1BKV)13 by using molecular quantum chemistry calculations within the MFCC scheme. The MFCC approach is a route to investigate accurately large biological systems with low computational cost.25 It has been employed previously to describe ligand–protein interactions at the quantum level related to hypercholesterolemia,26 anti-inflammatory drugs,27 central nervous system disorders,28 breast cancer,29 and dengue viral infection,30 to cite just a few.
We intend to present an adequate description of the interactions between its subunits, looking for an improvement in the understanding of its physical, chemical and biological properties, necessary to address some of the drawbacks in collagen based applications. Taking into account the entire system (system ABC), residue–residue interaction energy calculations have been undertaken in a peer-to-peer way between the residues of the chains A, B and C with those of B (subsystem AB), C (subsystem BC) and A (subsystem CA), respectively. We identified the inter-chain binding interactions involved in the stability of the structural zones (N-terminal, central and C-terminal) and triplets (Pro–Hyp–Gly, Ile–Thr–Gly, Ala–Arg–Gly and Leu–Ala–Gly) of T3-785. Finally, the importance of the amino acid residues Hyp, Gly, Ile, Thr, Ala, Arg and Leu to collagen triple helix stability were evaluated. All of these quantum binding energies calculations contribute to the development and synthesis of artificial collagenous materials with high stability for biomedicine and nanobiotechnology applications.
Materials and methods
The crystallographic structure of the collagen-like peptide T3-785 was obtained by X-ray diffraction technique with a resolution of 2.0 Å, and is stored in the Protein Data Bank (PDB) files under the code 1BKV.13 The protonation state of the system at physiological pH was obtained using the PDB2PQR server, an automated pipeline for the setup, execution, and analysis of the Poisson–Boltzmann electrostatics calculations.31 Biomolecular structures determined by crystallography rarely include hydrogens explicitly because they have a very low electronic density of state. Thus, while the atomic positions of the non-hydrogen atoms were kept fixed, the hydrogen atomic positions were added to the structure, and optimized by using the parametrization tool for organic molecules CHARMM22 (Chemistry at Harvard Macromolecular Mechanics 22 forcefield).32 The calculations were carried out with convergence tolerances set to 10−5 kcal mol−1 (total energy variation), 0.001 kcal mol−1 Å−1 (RMS gradient) and 1.0 × 10−5 Å (maximum atomic displacement).Afterwards, simulations within the Density Functional Theory (DFT) formalism together with the Local Density Approximation (LDA) approach based on the exchange–correlation functional of Perdew and Wang (DFT/LDA-PWC) were carried out by using the DMOL3 code (for a review see ref. 33). A double numerical plus polarization (DNP) basis set was chosen to expand the electronic Kohn–Sham orbitals taking into account all electrons with unrestricted spin explicitly, being 10 to 100 times faster and in good agreement with those obtained using the large scale Gaussian basis sets, such as 6-311+G(3df,2pd).34,35 The orbital cutoff radius was set to 3.7 Å and the self-consistent field convergence threshold was adjusted to 10−6 Ha, in order to reduce the computation time with little impact on the accuracy of the results. The hydrogen atomic positions of the peptide T3-785 optimized classically were optimized again using the DFT approach.
Unfortunately, the LDA calculations is not the best option to achieve a good description of hydrogen bonds (H-bond) and van der Waals (vdW) forces, because it overestimates weakly non-covalent interactions, widely found in biological systems.36,37 In order to improve the description of these non-covalent interactions, we adopted a reliable semiempirical correction on the description of dispersive, covalent, and ionic bonds within the improved density functional theory (DFT+D) proposed by Ortmann, Bechstedt, and Schmidt.38
Based on these quantum chemistry computations and using the formalism of the MFCC method proposed by Zhang and Zhang,39 we calculated the interaction energies among the amino acid residues belonging to the different chains of T3-785 peptide, forming three subsystems representing interactions between two chains (AB, BC and AC). For each amino acid residue of interest Ri, we draw an imaginary sphere with radius equal to 8.0 Å, and evaluate the interaction energy EI(Ri–Rj) with each residue Rj, considering at least one atom inside the sphere. The residues are identified by their names and corresponding chain. Hence, the scheme to compute the residue–residue interaction energies follows the MFCC approach, in which the interaction energy between residue Ri and other residue Rj was calculated according to:
| EI(RiRj) = E(CiRiCi*–CjRjCj*) − E(CiRiCi*–CjCj*) − E(CiCi*–CjRjCj*) + E(CiCi*–CjCj*) | (1) |
In order to achieve the structural stability of the triple helix collagen promoted by interactions with extended hydration network, all water molecules forming hydrogen bond with a particular residue were included for completeness in the fragments where it appears. The analysis of the binding scenario were based in the following criteria:
(a) conventional and water-mediated H-bonds between the inter-chain residues within the collagen-like peptide T3-783 were defined using geometric and energetic criteria derived after a search of 77![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 378 binding sites from 25096 PDB entries;40
378 binding sites from 25096 PDB entries;40
(b) H-bond donor (D) and the H-bond acceptor (A) atoms distance, dH–A, ranging from 2.6–3.1 Å;
(c) acceptor–hydrogen–donor angle, θA–H–D, not falling below 120°.
Besides classical hydrogen bonds, Derewenda et al. presented unequivocal evidence for the existence of non-conventional H-bonds between CH donor groups and oxygen acceptors in 13 well-defined protein structures.41 Here, non-conventional H-bonds occurred when dH–A ≤ 3.5 Å and θA–H–D ≥ 120°.
Finally, ion–induced dipole interaction, a long-range interaction that may exist between ions and molecules with no permanent dipoles occurring when an external electric field (ion) temporarily distorts the electron cloud of a neutral/non-polar molecule until it forms an induced dipole moments, were implemented mainly following the measures published by Gallivan and Dougherty.42 For the existence of hydrophobic (induced dipole–induced dipole) contacts, three hydrophobic Ri (Rj) atoms, namely carbon atoms with accessible surface and halogens, must lie in the range of the hydrophobic Rj (Ri) side chain. The maximum distance is set to the sum of the van der Waals radii of atoms in question with a tolerance of 0.8 Å.43
Results and discussions
Due to the intrinsic complexity of the collagen molecule, reductionist approaches through the use of peptide models have been widely applied to obtain information on the structure of this complex system.44–47 Following this line, the T3-785 molecule, a collagen-like triple-helical peptide, was chosen as the collagen-model peptide for studying the conformational stability of human collagen. Fig. 1 shows its structure with the interlaced monomers A (1–30 amino acid residues), B (31–60 amino acid residues) and C (61–90 amino acid residues) in a schematic, transverse and longitudinal views. This structure has helical symmetry and displays many of the overall features observed in others collagen-model peptides, like the human collagen type III.10It is important to notice that the Pro–Hyp–Gly triplets of the T3-785 collagen-like peptide are highly inaccurate, as evidenced by low crystallographic quality, particularly poor electron density map and lower average temperature factor of its atoms (see Fig. 2). Therefore, N-terminal (C-terminal) residues Pro31, Pro61 and Hyp2 (Gly30, Gly60 and Gly90) in chains A, B and C, respectively, and hence the sequences Hyp2–Gly3, Pro31–Hyp32–Gly33 and Pro61–Hyp62–Gly63 (Pro28–Hyp29–Gly30, Pro58–Hyp59–Gly60 and Pro88–Hyp89–Gly90) were discarded from our analysis in the same way as done by other studies.13
 | ||
| Fig. 2 Structural representation of the collagen-like peptide T3-785 according to the temperature factors (B-factor) signed for all atoms. Blue-red color change represents the range of B-factor from 10 to 100 Å2. | ||
Energetic aspects of the T3-785 zones
The energetic aspects of the T3-785 zones are summarized in Table 1, which shows the binding energies of the AB (−625.67), BC (−566.12), and CA (−566.31) subsystems, all units in kcal mol−1 (from now on all energy units are given in kcal mol−1). Accordingly, the three chains preserve collagen triple-helical conformation with a pronounced attractive energy value of −1758.10, in which the central zone (N-terminal and C-terminal) is accountable for 42.09% (32.61% and 25.29%) of this binding. The regions N-terminal and C-terminal are composed by three and four Pro–Hyp–Gly triplet, respectively, and this difference account for about 8% of their interaction binding energies. For AB (CA) subsystem, the order of energetic importance in these three zones follows the sequence: central > C-terminal > N-terminal, with the negative energy value of −300.13, −195.77, and −129.77 (−252.51, −170.75, and −143.05), respectively. On the order hand, BC subsystem shows an inversion in the order of the binding energy between the central and C-terminal zones, with the second becoming more attractive (−206.90) than the first one (−187.40), as a result of the small intensity interactions between the amino acid Arg44 and the residues from the C-chain.| Subsystem AB | Subsystem BC | Subsystem CA | Subsystem ABC | |||||
|---|---|---|---|---|---|---|---|---|
| N-Terminal | −129.8 | 20.7% | −171.9 | 30.4% | −143.1 | 25.3% | −444.7 | 25.3% |
| Central | −300.1 | 48.0% | −187.4 | 33.1% | −252.5 | 44.6% | −740.0 | 42.1% |
| C-Terminal | −195.8 | 31.3% | −206.9 | 36.5% | −170.7 | 30.2% | −573.4 | 32.6% |
| Total | −625.7 | 100.0% | −566.1 | 100.0% | −566.3 | 100.0% | −1758.1 | 100.0% |
Energetic aspects of the triplets
The energetic aspects of the triplets in the T3-785 peptide are depicted in Table 2 by means of their average interaction energy within the AB, BC and CA subsystems. For instance, the energy value of −65.11 calculated for the Pro–Hyp–Gly triplet in AB subsystem actually represent the average value of the binding energies of the 15 amino acid residues that compose the seven Pro–Hyp–Gly sequences of the A-chain, namely the residues Pro4, Hyp5, Gly6, Pro7, Hyp8, Gly9, Pro19, Hyp20, Gly21, Pro22, Hyp23, Gly24, Pro25, Hyp26 and Gly27, due to their interactions with the B-chain. Likewise, the (average) energetic values assigned in the last column of Table 2 represent the attractive contribution of the four triplet types for the T3-785 peptide (ABC system) stability. Therefore, one can see that Ala13–Arg14–Gly15 (A-chain), Ala43–Arg44–Gly45 (B-chain) and Ala73–Arg74–Gly75 (C-chain) triplets stand out from the others with a strongly average attractive energy of −130.15, almost twice bigger than any other one. Nevertheless, Pro–Hyp–Gly, Ile–Thr–Gly and Leu–Ala–Gly triplets present a very close attractive contribution (−67.87, −65.23 and −55.12, respectively).| Subsystem AB | Subsystem BC | Subsystem CA | Subsystem ABC | |||||
|---|---|---|---|---|---|---|---|---|
| Pro–Hyp–Gly | −65.1 | 17.3% | −75.7 | 28.8% | −62.8 | 19.9% | −67.9 | 21.3% |
| Ile–Thr–Gly | −74.6 | 19.8% | −61.9 | 23.5% | −59.2 | 18.8% | −65.2 | 20.5% |
| Ala–Arg–Gly | −167.6 | 44.5% | −68.1 | 25.9% | −154.8 | 49.1% | −130.2 | 40.9% |
| Leu–Ala–Gly | −69.6 | 18.5% | −57.4 | 21.8% | −38.5 | 12.2% | −55.2 | 17.3% |
Energetic description of the residues
The energetic affinities between the specific amino acids of chains A–B, B–C and A–C, from a perspective of the relative (average) binding energies of the T3-785 peptide's residues, are shown in Table 3. Analyzing the average energy of residue–residue interactions of each amino acid that compose the T3-785 collagen-like peptide, the energetic importance follows the order: Arg (39.5%) > Hyp (17.60%) > Thr (16.43%) > Ala (11.13%) > Gly (7.80%) > Pro (3.50%) > Leu (2.17%) > Ile (1.87%). These results are consistent with those obtained by Persikov et al. through circular dichroism (CD) spectroscopy of the collagen-model Ac–(Gly–Pro–Hyp)3–Gly–X–Y–(GlyPro–Hyp)4–Gly–Gly–CONH2, asserting that the same residue could display different propensities related to the nonequivalence of these positions in terms of the inter-chain interactions and the solvent exposure.48,51| Subsystem AB | Subsystem BC | Subsystem CA | System ABC | |||||
|---|---|---|---|---|---|---|---|---|
| Pro | −7.7 | 2.9% | −10.9 | 5.8% | −5.9 | 2.4% | −8.2 | 3.5% |
| Hyp | −38.7 | 14.4% | −48.3 | 25.5% | −36.8 | 15.0% | −41.2 | 17.6% |
| Gly | −19.0 | 7.1% | −17.0 | 9.0% | −18.8 | 7.7% | −18.3 | 7.8% |
| Ile | −6.1 | 2.3% | 0.4 | −0.2% | −7.4 | 3.0% | −4.4 | 1.9% |
| Thr | −40.1 | 14.9% | −48.1 | 25.3% | −27.4 | 11.2% | −38.5 | 16.4% |
| Ala | −39.1 | 14.6% | −20.3 | 10.7% | −18.8 | 7.7% | −26.1 | 11.1% |
| Arg | −110.2 | 41.1% | −42.5 | 22.4% | −124.9 | 51.0% | −92.5 | 39.5% |
| Leu | −7.3 | 2.7% | −3.0 | 1.6% | −4.9 | 2.0% | −5.1 | 2.2% |
The thermal stability of collagen-like models with many Gly–Pro–Yaa triplets in the central zone decreases gradually with a number of arginine residues in the Yaa position.52 However, when Glu and Arg amino acids occupy Xaa and Yaa, respectively, inter-chain electrostatic interactions increase the stability of the triple-helix heterotrimer.49 In fact, conformational studies in collagen-like peptides indicate that Arg residues are entropically favorable in this situation.50 In our model, the residues Arg14, Arg44 and Arg74 are in the Yaa position of their respective triplets. Fig. 3 depicts the interaction energy (in kcal mol−1) of the residues Arg14, Arg44 and Arg74 at the AB, BC and CA subsystems, respectively. The residue Arg14 (Arg44) interacts attractively with Ala43, Gly45, Leu46 and Ala47 (Ala73, Gly75, Leu76 and Ala77) residues, with energy values equal to −42.57, −8.50, −54.76 and −12.28 (−33.32, −4.65, −11.35 and −2.54), respectively. Similarly, the residue Arg74 binds with the A-chain residues Gly15 (−1.45), Leu16 (−50.47), Ala17 (−38.27), Gly18 (−7.85), Pro19 (−41.63) and Hyp20 (−12.98).
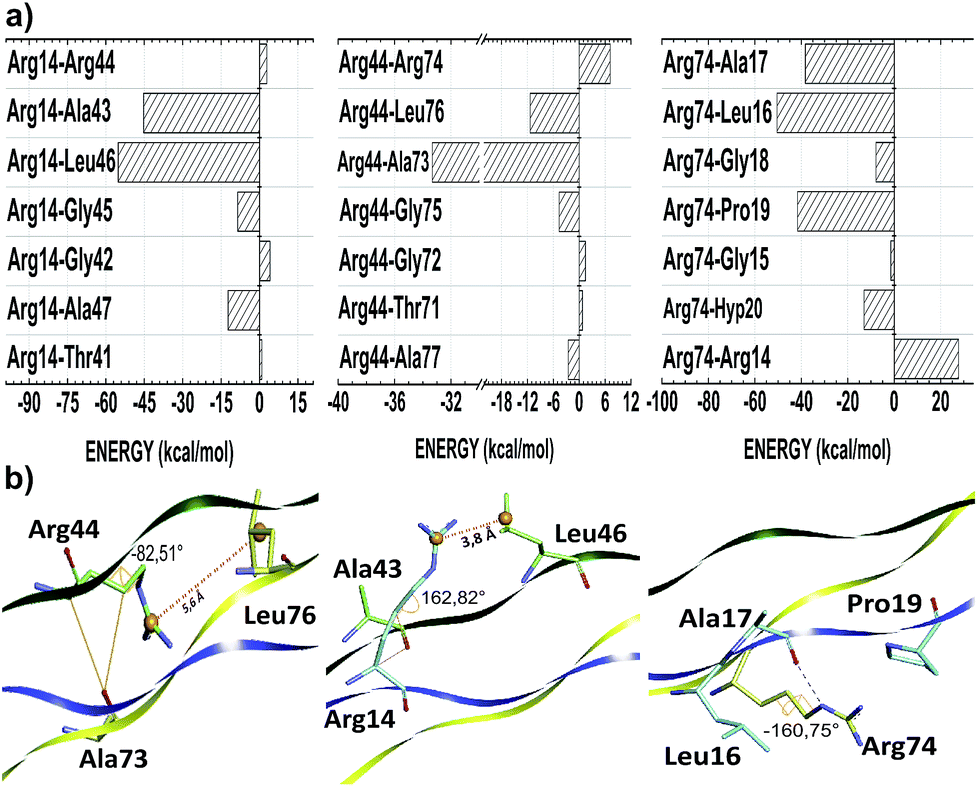 | ||
| Fig. 3 Graphical panel (a) and spatial arrangement (b) of the most relevant interactions involving the Arg residues 14, 44 and 74 in the subsystem AB, BC and CA, respectively. Inter-chain intermolecular contacts are depicted by black dashed line (hydrogen bond), orange solid line (non-conventional hydrogen bond) and dotted line (ion–induced dipole). | ||
As one can see from Table 3, the amino acid arginine is the most relevant to conformational stability of the T3-785 peptide, been accountable for 39.50% of the sum of average total interaction energy per amino acid residue. The residues Arg14, Arg44 and Arg74 are strongly attracted to the B, C and A strands, respectively, with the highest attractive binding energies (−110.23, −42.46 and −124.94, respectively). Although Arg44 is still one of the main residues in the BC subsystem, it has a much lower interaction energy compared to its chemical partners because it has just a single strong interaction with the residue Ala73 (−33.32), while the residue Arg14 (Arg74) shows a high attraction to the Ala43 and Leu46 ones (Leu16, Ala17 and Pro19 ones) with energy values of −42.57 and −54.76 (−50.47, −38.27 and −41.63), respectively. Analyzing their conformational and solvation aspects, one can determine what Arg amino acid is able to interact not only with their side-chain functional groups but also with other nearby amino acid residues. Interestingly, the most significant energetically Arg–residue systems tend to pick up more surrounding water.
Additionally, the trans-side chain conformation adopted for Arg14 (Arg74) residue and characterized by the dihedral angle 162.82° (−160.75°), is responsible for a close approach of its guanidine group with Leu46 (Leu16), strengthening an ion–induced dipole interaction type between them. Furthermore, the bifurcated non-conventional H-bond between Arg14 and Ala43 (Arg14:CαH–OC:Ala43), as well as the conventional H-bond formed between Arg74–Ala17 (Arg74:NεH–OC:Ala17), justify the energy values of −42.57 and −38.17, respectively, with a single oxygen acceptor simultaneously participating in the two hydrogen bonds. Likewise, the high ion–induced dipole interaction between the residues Arg74 and Pro19 is also favored by the trans-conformation ensuring parallel orientation of their side chains.
On the order hand, Arg44 acquire gauche (+) conformation determined by the dihedral angle equal to −82.51°. In this state, there is a small displacement of its protonated guanidine moiety in the opposite direction of their C-chain interacting residues, with the exception of the Ala73 residue. In fact, the distance between guanidine (Arg44) and isobutyl centroids (Leu76) is approximately 5.6 Å, while the Arg14–Leu46 system does not reach 3.8 Å (see Fig. 3). Concerning the Arg44–Ala73 interaction, there is one H-bond (Arg44:CαH–OC:Ala73) very similar to those observed in the Arg14–Ala43 pair, yielding a very close interaction energy. Finally, the repulsion involving the residues Arg74–Arg14, Arg44–Arg74 and Arg14–Arg44 binding is related to a strong charge–charge interactions among the guanidine functional groups of the same charge.
Threonine residues account for an average interaction energy of −38.50, mainly due to the binding energy contribution of the residue Thr11 (Thr41; Thr71) interacting with the B- (C-; A-) chain with an energy contribution of −40.05 (−48.07; −27.39). Thus, the threonine residues are the third most relevant amino acid class to maintain the conformational integrity of the T3-785 peptide. Among all threonine–residue interactions, the two most effective are those in which water molecules mediate the hydrogen bond between the polar group of Thr11 and Thr41 with the α-amino group of Ile40 and Ile70 respectively, particularly the bindings between the residues Thr11–Ile40 (−31.39) and Thr41–Ile70 (−43.86) (see Fig. 4). A third binding interaction, Thr71–Ala13 (−28.77), is characterized by a non-conventional H-bond between Thr71:CαH and Ala13:CO.
 | ||
| Fig. 4 Graphical panel (a) and spatial arrangement (b) of the most relevant interactions involving the Thr residues 11, 41 and 71 in subsystem AB, BC and CA respectively. Inter-chain intermolecular contacts are depicted by black dashed line (hydrogen bond) and orange solid line (non-conventional hydrogen bond). | ||
In collagenous domain characterized by a repetition of Xaa–Yaa–Gly triplets, the Xaa and Yaa positions are frequently occupied by proline (Pro, 28.1%) and hydroxyproline (Hyp, 38.1%) amino acid residues respectively, which together represent approximately 22% of all amino acids of collagen strands.53 The abundance of these residues decreases the entropic cost for collagen folding, although increasing dramatically the thermal stability of the triple helices. It is known that appropriate ring pucker, enforced by a stereoelectronic or steric effect, preorganizes the torsion angles to those required for the triple-helix formation.8
In our calculations, within a radius of 8 Å, Pro4, Pro7, Pro19, Pro22 and Pro25 (A-chain); Pro34, Pro37, Pro49, Pro52 and Pro55 (B-chain); and Pro64, Pro67, Pro79, Pro82 and Pro85 (C-chain), interact with many residues of the B, C and A chains, with a total attractive energy value of −6.58, −1.50, −18.39, −6.26, −5.82; −18.66, −4.78, −6.89, −16.67, −7.75; and −3.33, −5.32, −7.64, −5.67, −7.73, respectively. Consequently, the average interaction energy of prolines from the A-chain with residues from the B-chain (Pro–AB) is equal to −7.71 (2.88%). Similarly, Pro–BC and Pro–CA have binding interaction energy equal to −10.95 (5.77%) and −5.96 (2.42%), respectively (see Table 3).
Although Bhatnagar et al. proposed that the van der Walls interactions between the proline residues are important for stabilization of the triple-helix structures,54 nevertheless among the 35 Pro–Pro interactions of the collagen-like model T3-785 assessed here, just a few has some degree of attractiveness. As it can be seen from Fig. 5, the pyrrolidine rings of the inter-chain parallels prolines are relatively distant to each other (distCγ–Cγ > 7.9 Å), preventing strong Pro–Pro interactions. This is in good agreement with the parameters of the crystal structure of (Pro–Pro–Gly)9 at 1.0 Å resolution.55
 | ||
| Fig. 5 Distances (in Å) of the proline amino acid pairs present in the collagen-like peptide T3-785. | ||
Regarding the Hyp5, Hyp8, Hyp20, Hyp23 and Hyp26 (Hyp35, Hyp38, Hyp50, Hyp53 and Hyp56; Hyp65, Hyp68, Hyp80, Hyp83 and Hyp86) residues within the A- (B-; C-) chain, they are attracted with the B- (C-; A-) chain in a stronger way, specifically with binding energies equal to −31.13, −45.25, −60.68, −29.22, −27.05 (−27.17, −85.63, −44.69, −43.32, −40.69; −32.57, −52.65, −55.36, −21.11, −22.06). As indicated by the average interaction binding energies of −38.66 (Hyp–AB), −48.30 (Hyp–BC) and −36.76 (Hyp–CA), hydroxyprolines are energetically important in inter-strand interaction, mainly as a result of the association of non-conventional H-bonds (see more details in the hydrogen bond patterns sub-section). In fact, our calculations show that the largest residue–residue attractions involving proline and hydroxyproline residues occur in the presence of non-conventional hydrogen bonding. The carbonyl groups of prolines in A (B; C) make a contact of this nature with the CαH protons of glycines in B (C; A), specifically by the residues pairs: Pro4–Gly33, Pro7–Gly36, Pro19–Gly48, Pro22–Gly51, Pro25–Gly54 (Pro34–Gly63, Pro37–Gly65, Pro49–Gly78, Pro52–Gly81, Pro55–Gly84; Pro64–Gly6, Pro67–Gly9, Pro79–Gly21, Pro82–Gly24, Pro85–Gly27).
Unlike the other amino acid residues, the alanines one are in both positions of Gly–Xaa–Yaa triplet. In the Xaa(Yaa) location, Ala13, Ala43 and Ala73 (Ala17, Ala47 and Ala77) interact with average energies of −14.56 (−37.58). Notwithstanding not conformationally distinct from each other, the distances of Ala(Xaa)–residue inter-chain interactions are on average 28% higher than those observed in Ala(Yaa)–residue. Due to the nearness of interacting amino acid residues of different chains, alanine in Yaa position present more favorable binding energies, namely Ala17–Leu46 (−23.05), Ala17–Pro49 (−11.68), Ala47–Leu76 (−13.76), Ala47–Pro79 (−16.39) and Ala77–Pro19 (−11.74) (see Fig. 6). Considering the chemical nature of the amino acids involved, these contacts have probably a strong hydrophobic character (induced dipole–induced dipole forces). Additionally, Ala17–Leu46 and Ala47–Leu76 also make non-conventional hydrogen bonds.
 | ||
| Fig. 6 Graphical panel (a) and spatial arrangement (b) of the most relevant interactions involving the Al residues in Xaa and Yaa positions of the Xaa–Yaa–Gly motif. Inter-chain intermolecular contacts are depicted by orange solid line (non-conventional hydrogen bond). | ||
Regardless of their structural and functional peculiarities, many types of collagen are formed by Xaa–Yaa–Gly triplet repetitions. The packing of the triple-helical coiled-coil structure requires Gly in every third position. Because of its compact structure, assembly of the triple-helix puts this residue at the interior of the helix, and the side chain of the Gly is small enough to fit into the center of the helix.57 Despite the Gly residues do not present a charged (polar) side chain yielding an ion–dipole (dipole–dipole) interaction, as the Arg (Hyp and Thr) ones, they can perform strong intermolecular hydrogen bonds. In fact, in the T3-785 peptide each Gly triplets presents an average binding energy value of −18.27 (−17.02; −18.81) for the AB (BC; CA) subsystem. Therefore, the CO carbonyl (NH and CαH) moiety act as an acceptor (donor) element in a well-defined H-bond patterns, which are essential for the consistency of the triple helix collagen.
According to their distribution into the host–guest triple-helical peptides, Ile and Leu amino acids residues occupy the Xaa position in 11.2% of cases,53 with similar average binding energies equal to −4.38 and −5.08, respectively. The weak attraction between the residues Ile10–Ile40 (−1.15), Leu16–Leu46 (−0.34), Leu16–Ala47 (−0.49), Leu16–Gly48 (−0.33), Leu46–Leu76 (−2.40), Leu46–Ala77 (−0.15), Leu46–Gly78 (−0.26), Ile70–Ile10 (−0.62), Ile70–Ala13 (−1.58), Leu76–Ala17 (−1.76) and Leu76–Pro22 (−0.82), suggest the presence of an induced dipole–induced dipole interaction among their non-polar side-chain atoms (see Fig. 7). More significantly attractions occur when these hydrophobic contacts are replaced by ion–induced dipole forces in Arg–residue contacts, water-mediated H-bonds (Lys9–Ile40: −10.92, Thr11–Ile40: −31.39, Gly15–Leu46: −9.66, Gly39–Ile70: −8.24, Thr41–Ile70: −43.86, Gly45–Leu76: −8.18, Gly66–Ile10: −12.17 and Hyp68–Ile10: −47.50), or even non-conventional H-bonds Gly39–Ile10: −5.96, Gly45–Leu16: −3.88, Gly69–Leu40: −3.83, Gly75–Leu46: −4.20, Gly12–Ile70: −3.29 and Gly18–Leu76: −7.98), consistent with the hydration network description of the collagen-like triple-helical peptides with (Leu–Hyp–Gly)n and (Gly–Leu–Leu)n repeat triplets.56
 | ||
| Fig. 7 Graphical panel (a) and spatial arrangement (b) of the most relevant interactions involving Leu and Ile residues of the collagen-like peptide T3-785. Inter-chain intermolecular contacts are depicted by black dashed line (hydrogen bond) and orange solid line (non-conventional hydrogen bond). | ||
Hydrogen bond pattern
The T3-785 peptide shows a typical Rick and Crick hydrogen bonding pattern,58 where N–H groups of the Gly act as donor and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (carbonyl) groups of the Xaa residues act as acceptors. These inter-chain H-bond, Gly:NH–OC:Xaa, present average distance (angle) approximately equal to 2.88 Å (166 degrees).59 Specifically, 25 out of the 156 Gly–Xaa interactions analyzed (excluding terminal glycines) showed this pattern, mainly those with attractive residues binding energies: Gly6–Pro34 (−4.08), Gly9–Pro37 (−6.54), Gly12–Ile40 (−5.63), Ala43–Gly15 (−1.26), Gly18, Leu46 (−7.63), Gly21–Pro49 (−7.79), Gly24–Pro52 (−11.65), Gly27–Pro55 (−8.94) in the subsystem AB; Gly36–Pro64 (−5.80), Gly39–Pro67 (−8.88), Gly42–Ile70 (−10.70), Gly45–Ala73 (−4.73), Gly48–Leu76 (−9.58), Gly51–Pro79 (−4.16), Gly54–Pro82 (−6.23) and Gly57–Pro85 (−5.10) in the subsystem BC; finally Gly66–Pro7 (−5.20), Gly69–Ile10 (−6.80), Gly72–Ale13 (−10.80), Gly75–Leu16 (−6.57), Gly78–Pro19 (−3.96), Gly81–Pro22 (−5.73), Gly84–Pro25 (−4.87), Gly87–Pro28 (−4.16) in the subsystem CA. The average value of these interaction binding energies is three times higher than that without H-bond.
O (carbonyl) groups of the Xaa residues act as acceptors. These inter-chain H-bond, Gly:NH–OC:Xaa, present average distance (angle) approximately equal to 2.88 Å (166 degrees).59 Specifically, 25 out of the 156 Gly–Xaa interactions analyzed (excluding terminal glycines) showed this pattern, mainly those with attractive residues binding energies: Gly6–Pro34 (−4.08), Gly9–Pro37 (−6.54), Gly12–Ile40 (−5.63), Ala43–Gly15 (−1.26), Gly18, Leu46 (−7.63), Gly21–Pro49 (−7.79), Gly24–Pro52 (−11.65), Gly27–Pro55 (−8.94) in the subsystem AB; Gly36–Pro64 (−5.80), Gly39–Pro67 (−8.88), Gly42–Ile70 (−10.70), Gly45–Ala73 (−4.73), Gly48–Leu76 (−9.58), Gly51–Pro79 (−4.16), Gly54–Pro82 (−6.23) and Gly57–Pro85 (−5.10) in the subsystem BC; finally Gly66–Pro7 (−5.20), Gly69–Ile10 (−6.80), Gly72–Ale13 (−10.80), Gly75–Leu16 (−6.57), Gly78–Pro19 (−3.96), Gly81–Pro22 (−5.73), Gly84–Pro25 (−4.87), Gly87–Pro28 (−4.16) in the subsystem CA. The average value of these interaction binding energies is three times higher than that without H-bond.
As shown in Fig. 8 and 9a, a second network of repetitive hydrogen bonds is observed in the imino acid-free zone. Here, water molecules connecting glycine carbonyl and Xaa amides located on adjacent chains, Xaa:NH–HOH–OC:Gly, creating a set of water-mediated inter-chain H-bonds, as it was proposed in ref. 60. These intermolecular contacts are observed in: Gly9–Ile40 (−10.92), Gly12–Ala43 (−16.01) and Gly15–Leu46 (−9.66) in the subsystem AB; Gly39–Ile70 (−8.24) and Gly45–Leu76 (−8.18) in the subsystem BC; and Gly66–Ile10 (−12.17), Gly69–Ala13 (−7.56) and Gly72–Leu16 (−7.95) in the subsystem CA. Observe that the Ala73:NH–HOH:OC–Gly42 H-bond does not exist since the nearest water molecule in Ala73:NH is 6.43 Å away, explaining the weak interaction (−1.01) calculated for the Gly42–Ala73 residue pair. Those strong attractive interactions confirm the hypothesis that this second network of hydrogen bonds strengthens the triple-helical conformation in imino acid-poor regions of collagen59 and refutes the idea that regions of triple helix conformation without imino acids will be more flexible and dynamic than those where all X/Y positions are occupied by Pro/Hyp residues.10
 | ||
| Fig. 8 3D spatial visualization of the inter-chain water-mediated hydrogen bonds among the amino acid residues of the collagen-like peptide T3-785. | ||
 | ||
| Fig. 9 2D spatial visualization of the inter-chain hydrogen bond patterns, namely conventional (a) and non-conventional (b) H-bonds, of the collagen-like peptide T3-785. | ||
In three situations, water molecules involved in this second set of bonds make an additional contact with the side chain (hydroxyl group) of Yaa residues, namely Ile:NH–HOH–OH:Yaa. In our collagen-model, one water molecule connects Ile40 (Ile70) with Gly9 and Thr11 (Gly39 and Thr41), the latter within the same chain. The importance of Ile40:NH–HOH–OH:Thr11 (Ile70:NH–HOH–OH–Thr41) contact is expressed by the energetic value equal to −31.29 (−43.86), third (first) most important of the subsystem AB (BC). At least, the water molecule between the residues Ile10 and Gly66 also is in contact with Hyp68, which directly affect the Hyp68–Ile10 interaction, the second most attractive in the sub-system CA (−47.50).
In addition, we evaluated the non-conventional hydrogen bonds, Gly:CαH–OC:Xaa (Ile/Leu/Pro) (Fig. 9b). We found six contacts involving Ile/Leu and Gly residues, namely Gly39:CαH–OC:Ile10 (−5.96), Gly45:CαH–OC:Leu16 (−3.88), Gly69:CαH–OC:Ile40 (−3.83), Gly75:CαH–OC:Leu46 (−4.42), Gly12:CαH–OC:Ile70 (−3.29) and Gly18:CαH–OC:Leu76 (−7.98). In addition, 15 Pro–Gly pairs also present this non-covalent forces, namely: Pro4–Gly33 (−5.09), Pro7–Gly36 (−6.12), Pro19–Gly48 (−8.03), Pro22–Gly51 (−4.79), Pro25–Gly54 (−6.58), Pro34–Gly63 (−4.81), Pro37–Gly66 (−4.48), Pro49–Gly78 (−6.13), Pro52–Gly81 (−5.41), Pro55–Gly84 (−7.00), Pro64–Gly6 (−6.61), Pro67–Gly9 (−3.00), Pro79–Gly21 (−4.72), Pro82–Gly24 (−7.39) as well as Pro85–Gly27 (−3.91).
We also highlight the existence of 14 important non-conventional H-bonds, namely Pro:CγH–Oδ:Hyp, involving residues of glycine or proline with other proline or hydroxyproline of adjacent chains, respectively: Hyp5–Pro34 (−20.66), Hyp8–Pro37 (−27.54), Hyp20–Pro49 (−41.01), Hyp23–Pro52 (−11.77), Hyp26–Pro55 (−14.34), Hyp35–Pro64 (−16.54), Hyp38–Pro67 (−22.42), Hyp50–Pro79 (−20.62), Hyp53–Pro82 (−19.19), Hyp56–Pro85 (−25.15), Hyp65–Pro7 (−17.52), Hyp80–Pro22 (−26.48), Hyp83–Pro25 (−13.86), and Hyp86–Pro28 (−10.81). Finally, we distinguished two Ala:CαH–OC:Leu contacts in Ala17–Leu46 (−23.05) and Ala47–Leu76 (−13.76) interactions.
The existence and importance of this non-conventional H-bond for collagen triple-helix stability was initially discussed by Bella and Berman.61 It is believed that CαH–OC network interaction acts to align the three chains and, thereby, cooperatively decreases the total energy of the biological system. In our study, the hydrogen bonds – whether conventional, water-mediated or non-conventional types – are present in 62 residue–residue interactions, which together account approximately for 44% of the total interaction energy between the collagen strands. It is therefore likely that these H-bond patterns contribute to the local stability of the triple helix and help to maintain the triple helical conformation, confirming the water-bonded model for collagen (see ref. 10).
Conclusions
In this paper, we presented a quantum chemistry calculations of the electronic structure of type III collagen-like peptide T3-785, in order to identify the nature of the binding interactions that stabilize its three-dimensional structure. The inter-chain interaction energies of amino acids that compose this collagen reveal the most energetically important regions (triplets) in this system, obeying the sequence: N-terminal zone < C-terminal zone < central zone (Leu–Ala–Gly < Ile–Thr–Gly < Pro–Hyp–Gly < Ala–Arg–Gly). In order to evaluate the individual importance of each amino acid type in this peptide, we compared their average interaction binding energies, giving the following order sequence Arg > Hyp > Thr > Ala > Gly > Pro > Leu > Ile.We also highlighted the importance of intermolecular interactions to keep the conformational stability of collagen. Fig. 10 depicts a condensed graphical view of the 15 most and fewest energetically significant amino acids within the peptide T3-785. Only two (Ala17 and Ala47) of the top 15 amino acid residues do not present a polar side chain, although performing a dipole–dipole interaction (Ala47:CαH–OC:Leu76). On the other hand, the residues with lowest affinity are essentially non-polar, as is the case of C-/N-terminal prolines. The aliphatic amino acid residues Ala, Leu, Ile and Gly jointly account for 22.97% of the sum of average interaction energy per amino acid residue, corroborating the hypothesis that the process of self-organization in triple-helix is also directed by hydrophobic interactions.56
 | ||
| Fig. 10 Pictorial view of the 15 most and fewest energetically significant amino acids within the peptide T3-785. | ||
The strong attractive character observed in the amino acid residues Arg14, Arg44 and Arg74 indicates the importance of ion–induced dipole force for triple-helix stability. Besides them, the conventional H-bonds (Gly:NH–OC:Xaa) are present in stronger interactions involving Gly residues. Non-conventional (Gly:CαH–OC:Xaa) and water-mediated H-bonds (Xaa:NH–HOH–OC:Gly) involving the carbonyl, amine and hydroxyl of residues Gly, Ile and Thr are important for their inter-chain interactions. The binding energy values calculated for residue–residue pairs with mediator water molecules suggest that the solvent network play an important role in the stabilization of the collagen-model T3-785. As a matter of fact, water could determine the subtle balance among driving forces of different physical natures, thus broadening the array of available binding mechanisms in biomolecular associations.62
The results generated from this investigation provided detailed knowledge on the stability and hydration network of collagen triplets, and this will certainly help researchers working in the area of de novo protein design, mainly those related to the synthesis of model collagen-like peptides.
Acknowledgements
This work was partially financed by the Brazilian Research Agencies CAPES (Rede NanoBioTec, PNPD) and CNPq (INCT-Nano(Bio)Simes, Casadinho/Procad).References
- M. C. Smith, N. Burns, J. R. Sayers, J. A. Sorrell, S. R. Casjens and R. W. Hendrix, Science, 1998, 279, 1834 CAS.
- D. J. Prockop and K. I. Kivirikko, Annu. Rev. Biochem., 1995, 64, 403–434 CrossRef CAS PubMed.
- K. Gelse, E. Pöschl and T. Aigner, Adv. Drug Delivery Rev., 2003, 55, 1531–1546 CrossRef CAS PubMed.
- L. A. Staudinger, S. J. Spano, W. S. Lee, N. Coelho, T. J. Moriarty and C. A. McCulloch, Int. J. Biochem. Cell Biol., 2015, 62, 80–87 CrossRef CAS PubMed.
- C. Bonnans, J. Chou and Z. Werb, Nat. Rev. Mol. Cell Biol., 2014, 15, 786–801 CrossRef CAS PubMed.
- F. Pati, P. Datta, B. Adhikari, S. Dhara, K. Ghosh and P. K. Das Mohapatra, J. Biomed. Mater. Res., Part A, 2012, 100, 1068–1079 CrossRef PubMed.
- B. An, Y. S. Lin and B. Brodsky, Adv. Drug Delivery Rev., 2016, 97, 69–84 CrossRef CAS PubMed.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958 CrossRef CAS PubMed.
- J.-Y. Exposito and C. Lethias, Invertebrate and vertebrate collagens, in Evolution of Extracellular Matrix, Springer, Berlin, Heidelberg, 2013, pp. 39–72 Search PubMed.
- J. Bella, Biochem. J., 2016, 473, 1001–1025 CrossRef CAS PubMed.
- M. Schumacher, K. Mizuno and H. P. Bachinger, J. Biol. Chem., 2005, 280, 20397–20403 CrossRef CAS PubMed.
- X. Liu, H. Wu, M. Byrne, S. Krane and R. Jaenisch, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 1852–1856 CrossRef CAS.
- R. Z. Kramer, J. Bella, P. Mayville, B. Brodsky and H. M. Berman, Nat. Struct. Biol., 1999, 6, 454–457 CrossRef CAS PubMed.
- S. P. Boudko, J. Engel, K. Okuyama, K. Mizuno, H. P. Bächinger and M. A. Schumacher, J. Biol. Chem., 2008, 283, 32580–32589 CrossRef CAS PubMed.
- R. Improta, R. Berisio and L. Vitagliano, Protein Sci., 2008, 17, 955–961 CrossRef CAS PubMed.
- R. Parthasarathi, B. Madhan, V. Subramanian and T. Ramasami, Theor. Chem. Acc., 2003, 110, 19–27 CrossRef CAS.
- V. K. Pálfi and A. Perczel, J. Comput. Chem., 2008, 29, 1374–1386 CrossRef PubMed.
- V. K. Pálfi and A. Perczel, J. Comput. Chem., 2010, 31, 764–777 Search PubMed.
- K. J. Jalkanen, K. Olsen, M. Knapp-Mohammady and J. Bohr, EPL, 2012, 100, 28003 CrossRef.
- M. I. Tsai, Y. Xu and J. J. Dannenberg, J. Am. Chem. Soc., 2005, 127, 14130–14131 CrossRef CAS PubMed.
- M. S. Gordon, D. G. Fedorov, S. R. Pruitt and L. V. Slipchenko, Chem. Rev., 2012, 112, 632–672 CrossRef CAS PubMed.
- K. Raghavachari and A. Saha, Chem. Rev., 2015, 115, 5643–5677 CrossRef CAS PubMed.
- E. Suárez, N. Díaz and D. Suárez, J. Chem. Theory Comput., 2009, 5, 1667–1679 CrossRef PubMed.
- C. R. F. Rodrigues, J. I. N. Oliveira, U. L. Fulco, E. L. Albuquerque, R. M. Moura, E. W. S. Caetano and V. N. Freire, Chem. Phys. Lett., 2013, 559, 88–93 CrossRef CAS.
- M. A. Collins and R. P. Bettens, Chem. Rev., 2015, 115, 5607–5642 CrossRef CAS PubMed.
- R. F. da Costa, V. N. Freire, E. M. Bezerra, B. S. Cavada, E. W. S. Caetano, J. L. de Lima Filho and E. L. Albuquerque, Phys. Chem. Chem. Phys., 2012, 14, 1389–1398 RSC.
- D. S. Dantas, J. I. N. Oliveira, J. X. Lima Neto, R. F. da Costa, E. M. Bezerra, V. N. Freire, E. W. S. Caetano, U. L. Fulco and E. L. Albuquerque, RSC Adv., 2015, 5, 49439–49450 RSC.
- J. X. Lima Neto, U. L. Fulco, E. L. Albuquerque, G. Corso, E. M. Bezerra, E. W. S. Caetano, R. F. da Costa and V. N. Freire, Phys. Chem. Chem. Phys., 2015, 17, 13092–13103 RSC.
- K. B. Mota, J. X. Lima Neto, A. H. Lima Costa, J. I. N. Oliveira, K. S. Bezerra, E. L. Albuquerque, E. W. S. Caetano, V. N. Freire and U. L. Fulco, Comput. Theor. Chem., 2016, 1089, 21–27 CrossRef CAS.
- G. S. Ourique, J. F. Vianna, J. X. Lima Neto, J. I. N. Oliveira, P. W. Mauriz, M. S. Vasconcelos, E. W. S. Caetano, V. N. Freire, E. L. Albuquerque and U. L. Fulco, RSC Adv., 2016, 6, 56562–56570 RSC.
- T. J. Dolinsky, J. E. Nielsen, J. A. McCammon and N. A. Baker, Nucleic Acids Res., 2004, 32, W665–W667 CrossRef CAS PubMed.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiòrkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- E. L. Albuquerque, U. L. Fulco, V. N. Freire, E. W. S. Caetano, M. L. Lyra and F. A. B. F. de Moura, Phys. Rep., 2014, 535, 139 CrossRef.
- Y. Inada and H. Orita, J. Comput. Chem., 2008, 29, 225–232 CrossRef CAS PubMed.
- N. A. Benedek, I. K. Snook, K. Latham and I. Yarovsky, J. Chem. Phys., 2005, 122, 144102 CrossRef CAS PubMed.
- P. L. Silvestrelli, J. Phys. Chem. A, 2009, 113, 5224–5234 CrossRef CAS PubMed.
- K. Berland, V. R. Cooper, K. Lee, E. Schröder, T. Thonhauser, P. Hyldgaard and B. I. Lundqvist, Rep. Prog. Phys., 2015, 78, 066501 CrossRef PubMed.
- F. Ortmann, F. Bechstedt and W. G. Schmidt, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 205101 CrossRef.
- D. W. Zhang and J. Z. H. Zhang, J. Chem. Phys., 2003, 119, 3599–3605 CrossRef CAS.
- C. Bissantz, B. Kuhn and M. Stahl, J. Med. Chem., 2010, 53, 5061–5084 CrossRef CAS PubMed.
- Z. S. Derewenda, L. Lee and U. Derewenda, J. Mol. Biol., 1995, 252, 248–262 CrossRef CAS PubMed.
- J. P. Gallivan and D. A. Dougherty, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9459–9464 CrossRef CAS.
- K. Stierand and M. Rarey, ACS Med. Chem. Lett., 2010, 1, 540–545 CrossRef CAS PubMed.
- A. Perczel, Z. Gáspári and I. G. Csizmadia, J. Comput. Chem., 2005, 26, 1155–1168 CrossRef CAS PubMed.
- E. Suàrez, N. Díaz and D. Suàrez, J. Phys. Chem. B, 2008, 112, 15248–15255 CrossRef PubMed.
- M. D. Shoulders and R. T. Raines, J. Biol. Chem., 2011, 286, 22905–22912 CrossRef CAS PubMed.
- N. Keshwani, S. Banerjee, B. Brodsky and G. I. Makhatadze, Biophys. J., 2013, 105, 1681–1688 CrossRef CAS PubMed.
- A. V. Persikov, J. A. Ramshaw, A. Kirkpatrick and B. Brodsky, Biochemistry, 2000, 39, 14960–14967 CrossRef CAS PubMed.
- A. V. Persikov, J. A. Ramshaw, A. Kirkpatrick and B. Brodsky, J. Mol. Biol., 2002, 316, 385–394 CrossRef CAS PubMed.
- T. Koide, Y. Nishikawa and Y. Takahara, Bioorg. Med. Chem. Lett., 2004, 14, 125–128 CrossRef CAS PubMed.
- V. Gauba and J. D. Hartgerink, J. Am. Chem. Soc., 2007, 129, 2683–2690 CrossRef CAS PubMed.
- R. Berisio, A. De Simone, A. Ruggiero, R. Improta and L. Vitagliano, J. Pept. Sci., 2009, 15, 131–140 CrossRef CAS PubMed.
- J. A. Ramshaw, N. K. Shah and B. Brodsky, J. Struct. Biol., 1998, 122, 86–91 CrossRef CAS PubMed.
- R. S. Bhatnagar, N. Pattabiraman, K. R. Sorensen, R. Langridge, R. D. MacElroy and V. Renugopalakrishnan, J. Biomol. Struct. Dyn., 1988, 6, 223–233 CAS.
- C. Hongo, K. Noguchi, K. Okuyama, Y. Tanaka and N. Nishino, J. Biochem., 2005, 138, 135–144 CrossRef CAS PubMed.
- K. Okuyama, H. Narita, T. Kawaguchi, K. Noguchi, Y. Tanaka and N. Nishino, Biopolymers, 2007, 86, 212–221 CrossRef CAS PubMed.
- M. Bhowmick and G. B. Fields, Methods Mol. Biol., 2013, 1081, 167–194 CAS.
- A. Rich and F. H. Crick, J. Mol. Biol., 1961, 3, 483–506 CrossRef CAS PubMed.
- R. Z. Kramer, J. Bella, B. Brodsky and H. M. Berman, J. Mol. Biol., 2001, 311, 131–147 CrossRef CAS PubMed.
- G. N. Ramachandran and R. Chandrasekharan, Biopolymers, 1968, 6, 1649–1658 CrossRef CAS PubMed.
- J. Bella and H. M. Berman, J. Mol. Biol., 1996, 264, 734–742 CrossRef CAS PubMed.
- R. Baron, P. Setny and J. A. McCammon, J. Am. Chem. Soc., 2010, 132, 12091–12097 CrossRef CAS PubMed.
Footnotes |
| † This document is a collaborative effort of all authors. |
| ‡ Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra25206k |
| This journal is © The Royal Society of Chemistry 2017 |