
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydroxyapatite synthesis from a starfish and β-tricalcium phosphate using a hydrothermal method
V. Rodríguez-Lugo*a,
E. Salinas-Rodrígueza,
R. A. Vázqueza,
K. Alemánb and
A. L. Rivera *c
*c
aUniversidad Autónoma del Estado de Hidalgo, Área Académica de Ciencias de la Tierra y Materiales, Carretera Pachuca-Tulancingo Km. 4.5, C.P. 42184, Pachuca, Hidalgo, Mexico. E-mail: ventura.rl65@gmail.com
bUniversidad Autónoma del Estado de Hidalgo, Área Académica de Computación y Electrónica, Carretera Pachuca-Tulancingo Km. 4.5, C.P. 42184, Pachuca, Hidalgo, Mexico
cInstituto de Ciencias Nucleares, Centro de Ciencias de la Complejidad, Universidad Nacional Autónoma de México, Apartado Postal 70-543, Deleg. Coyoacán, Mexico City, C.P. 04510, Mexico. E-mail: anarivera2000@gmail.com
First published on 23rd January 2017
Abstract
Hydroxyapatite HAp, Ca10(PO4)6(OH)2, was successfully synthesized using a hydrothermal method using β-tricalcium phosphate (β-TCP) and CaO from the starfish Mellita eduardobarrosoi sp. nov. The goal of this research was to synthesize a material with better characteristics, such as a high proportion of the HAp phase, homogeneous dimensions, and a good Ca/P ratio. To get it, different temperatures, concentration ratios and reaction times were tested. Synthesized HAp materials were characterized by Scanning Electron Microscopy (SEM), X-ray powder diffraction (XRD), and Fourier transform infrared spectroscopy (FTIR). XRD showed that the proportion of the HAp phase tends to increase as the temperature and reaction times grow, which is corroborated by SEM characterization, where homogeneous in size HAp fibers were identified. Absorption bands related to the functional groups PO4 and OH, as well as the characteristics of HAp, were found by FTIR. From the results obtained, optimal HAp is achieved at a temperature of 250 °C and a reaction time of 48 hours, producing a greater proportion of fibers with very homogeneous lengths and thicknesses, a Ca/P ratio of 1.65 (near stoichiometric), a higher crystalline degree, and a ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 of β-TCP and CaO.
:1 of β-TCP and CaO.
Introduction
Recently, for biomedical applications, there have been numerous efforts to find materials with suitable characteristics for the restoration or replacement of bone tissue in humans,1 and to develop new technologies that can improve the properties of these materials. Amongst the most popular materials for this goal is hydroxyapatite (HAp), the principal inorganic compound of hard tissues (bones and teeth),2 due to its crystalline nature, chemical composition similar to natural bones, structure, biocompatibility, ability to establish chemical exchange and form interfacial bonds with living tissue, thus promoting integration processes and the growth of new bone, its antimicrobial character3 (that can be used for the treatment of dental caries4), and osteo-conduction properties.5 HAp promotes the direct generation of new bones without the use of intervening fibrous tissue; this method is widely accepted in maxillofacial surgery to fill bone defects created by cysts or tumors, traumas, congenital diseases, and other surgeries.6–9 HAp doped with magnetic ions have also been used as an efficient drug deliver for the treatment of tumors and damage cells.10–12 For clinical applications, due to its high biocompatibility, HAp represents the best alternative as a biomaterial in reconstructive surgery, because it can be used successfully as a filler material.13–23 Moreover, due to its high porosity, it is widely used as a catalytic agent.24The term “apatite” is applied to a large group of mineral compounds featuring a general formula:9 A10(BP4)6X2, where A can be Ca, Sr, Ba, Cd, or other rare earth elements; BP4 can be PO43−, VO43−, SiO43−, or AsO43−; and X can take the form of OH−, Cl−, or F−. HAp is a type of apatite9 and its chemical formula is Ca10(PO4)6(OH)2, where 39% of its weight is Ca, 5.18% is P, and 3.38% is OH. HAp crystallizes in a hexagonal system with a space group of P63/m type; the dimensions of the unit cell are a = b = 9.432 Å, c = 6.881 Å.25 The stoichiometric Ca/P ratio of HAp is 1.666 and its density is 3.219 g cm−3. The structure of HAp consists of an array of tetrahedral phosphates (PO43) constituting the “backbone” of the unit cell; 2 O2 molecules are aligned with axis C, and the other 2 are in a horizontal plane.25–27 Within the unit cell, phosphates are divided into two layers of 1/4 and 3/4 heights, which favor the formation of two types of channels along axis C, referred to as type A and type B, respectively.28
HAp can be synthesized by various techniques using different reactants and production routes, that can modify its physicochemical properties, morphology, chemical composition homogeneity, particle sizes, and degree of crystallinity.5 Daubrée, in 1851, was the first who synthesized HAp by passing phosphorus trichloride vapor on red hot lime.29–32 Since then, various methods of synthesis have been employed:33–37 solid state synthesis, synthesis in aqueous phase,38 growth from molten salts, growth in gel, hydrolysis, mechano-chemical, and the hydrothermal method. In the development of this work was employed the hydrothermal technique because it promotes the growth of various crystals, is cheap and simple to implement. In this method, water is used as a solvent at higher energies to accelerate synthesis and to achieve a better balance of the reaction; this means that products with high crystallinity are obtained, and the corrosion and degradation processes are accelerated.39 These features facilitate the growth of fine crystals, which are homogenous in size, shape, and composition. The growing interest in the hydrothermal technique is derived from its numerous advantages, including the high reactivity of the reactants, easy control of the solution, formation of both the meta-stable phases and the unique phases, less air pollution in the reaction, and low power consumption.40
On Mexican beaches a source of environmental pollution are shells from corals, cockle, and in particular from starfish. These shells due to its high content on CaCO3 can be used as a potential source of the calcium required to produce HAp.41–47 Thus, employing starfish as precursor of HAp, not only generates an important biomaterial, but also provides a solution to deal with this ecological waste.
In this work, HAp synthesis is done by a hydrothermal method using β-tricalcium phosphate (β-TCP) and CaO (obtained from the decomposition of CaCO3 of the starfish of the species “Mellita eduardobarrosoi sp. nov.”). Different working conditions were tested, a ratio of 3:1; for temperatures of 230 °C and 250 °C; and reaction times of 20 and 48 hours. The characterization of the precursors and the obtained products was done through Low Vacuum Scanning Electron Microscopy (LV-SEM), elemental chemical analysis (EDS), X-ray diffraction (XRD), and Fourier transform infrared spectroscopy (FTIR); which allowed the identification of crystalline phases, morphology, chemical composition, and molecular arrangements. Using a waste product, low cost, short term reactions, close to room temperatures, simplicity of the method and good quality of the HAp produced are some of the advantages of the methodology used in this work, that can be implemented for HAp synthesis at industrial scale.
Experimental procedure
Materials used and synthesis systems
To carry out the synthesis of HAp, β-TCP reagent grade Fluka and remnants of the echinoidea starfish Mellita eduardobarrosoi sp. nov. were used. This starfish has compositional and structural properties that make it good as precursor for biomaterials.40,48,49 The starfish was collected on the beaches of the northern region of Veracruz state, Mexico. It consists mainly of CaCO3. Regarding the starfish, this was subjected to a prior cleaning process that consisted of placing the samples for in a 30% hypochlorite solution for 7 days to remove organic material both externally and internally; the samples were subsequently dried and placed in an ultrasound system for periods of 10 minutes to remove any sand deposits that may have been within the starfish (see Fig. 1). The synthesis process used in this study to obtain HAp was the hydrothermal method, for which an autoclave “Cortest Hastolly” was used with a capacity of 250 mL. This system can reach a maximum temperature of 300 °C and a pressure of 5.51 MPa.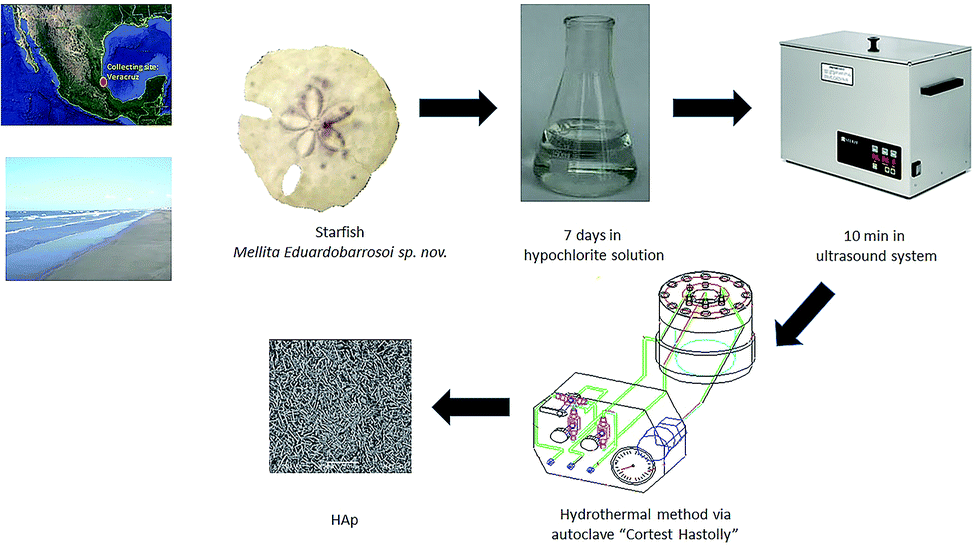 | ||
| Fig. 1 Experimental procedure for HAp synthesis. | ||
Hydrothermal synthesis method
The hydrothermal method was used for the synthesis process. Starfish were subject to treatment at 900 °C for 20 and 48 hours, which led to the formation of calcium oxide (CaO), according to the reaction given by:
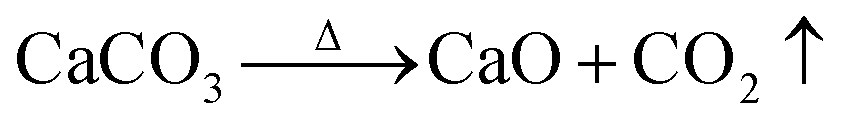 | (1) |
The synthesis process involves mixing in 100 mL of deionized water; 4.5 g of β-TCP, and 0.28 g of CaO of the starfish, which was heat treated. Following this, there was a wait time for the chemical reaction to be effected, which is represented by:
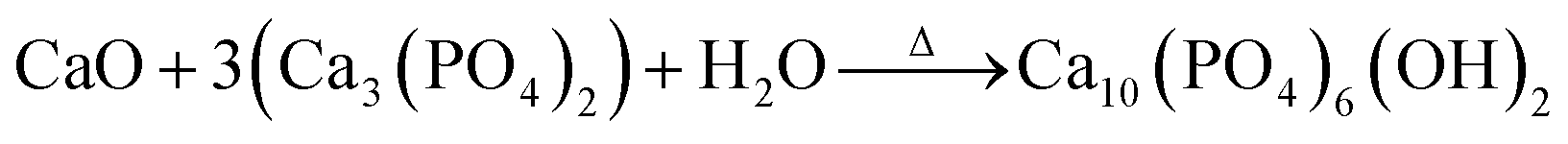 | (2) |
The mixture that was obtained was treated under hydrothermal conditions in an autoclave “Cortest Hastolly”, the conditions are shown in Table 1. Here is reported the synthesis made through the hydrothermal method during 20 h and 48 h. The morphology definition and fiber density is larger for longer reaction times as was shown in previous published papers.17,18,50–53
| Synthesis number | Temperature (°C) | Pressure (MPa) | Reaction time (hours) |
|---|---|---|---|
| 1 | 230 | 3.4 | 20 |
| 2 | 250 | 3.4 | 20 |
| 3 | 230 | 3.4 | 48 |
| 4 | 250 | 3.4 | 48 |
Characterization techniques
For the microstructural characterization process, X-ray diffraction (XRD), low vacuum scanning electron microscopy (LV-SEM), and spectrometry by Fourier transform infrared (FTIR) were used, employing the following equipment:(a) X-ray diffractometer, XRD, for powders (SIEMENS, model D-5000) with an incident beam wavelength of λCu = 1.5405 Å. Said system was adapted with the JCPDS-ICOD database (International Center of Diffraction Data) version 2.16, which was used for phase identification. The working conditions were as follows: 35 kV; 25 mA; with a sweep carried out in the range of 2.5° to 70° in 2θ with a time of 1 s and an increment of 0.005. The different samples were ground in an agate mortar and they were then placed in respective supports to finally be placed in the diffractometer.
(b) LV-SEM (JEOL model JMS 5900-LV) equipped with a probe for elemental chemical analysis, EDS (OXFORD brand). The operating conditions were as follows: a low vacuum mode; an accelerating voltage of 25 kV; a 75 mA emission current; a working distance (WD) of 10 mm; a pressure of 20 Pa; and a spot size of 40–50 mm. On a 10 mm diameter aluminum cylinder, a carbon ribbon was adhered and the test sample was placed in it.
(c) Fourier transform infrared spectrometer (Fourier transform model NEXUS 670 FT-IR ESP, Niclet brand). Each test sample was previously dried at a temperature of 100 °C for 24 hours. Following this, each sample was mixed with potassium bromide (KBr) for final placement in the spectrometer's chamber.
Results
The precursors and the products obtained following the synthesis of HAp using the hydrothermal method for different conditions were characterized by LV-SEM, EDS, XRD, and FTIR spectrometry.Characterization of precursors: starfish
Echinoids like the starfish Mellita eduardobarrosoi sp. nov. have a skeleton composed of calcite (CaCO3), a material of low density and hardness that is comparable to high density limestone.48 This starfish is from the genus of the mellitidae, clypeasteroidea order and equine class of the phylium echinoidermata. In their adult age they have sizes between 10 and 150 mm, their outline is irregular, but has vertical symmetry, the ventral surface is flat and the dorsal surface is thickened, usually presents 5 elongated perforations, its coloration varies from beige, gold and gold-ocher with shades of green (see Fig. 2a). Surface morphology of the starfish is shown in Fig. 2b where it can be seen predominant circular pores with diameters between 6.25 μm and 25 μm. When making a transverse cut in the starfish for analysis, it was observed that the pores are interconnected, as shown in Fig. 2c. The internal pores have dimensions on average ranging from 10 μm to 22.5 μm. Elemental chemical analysis (EDS) of the starfish identify as the main chemical elements that comprise it: O, C, Ca. Low proportions or Mg, Al, Si, and S were also detected (see Table 2). | ||
| Fig. 2 Starfish Mellita eduardobarrosoi sp. nov. (a) bottom and top superficial view, (b) superficial morphology, and (c) internal structure. | ||
| Element | Starfish without treatment | Starfish after thermal treatment at 900 °C for 4 h |
|---|---|---|
| O | 46.50 | 26.05 |
| C | 25.98 | 3.01 |
| Ca | 24.48 | 65.34 |
| Mg | 1.93 | 2.37 |
| Al | 0.47 | — |
| Si | 0.36 | — |
| S | 0.21 | — |
After the grinding process, the morphology of the starfish powder changes, the porous structure has totally disappeared, with particles ranging from 2.5 μm to 22.5 μm (see LV-SEM image, left panel of Fig. 3). Starfish powder after the grinding and cleaning process was also characterized by XRD (upper panel of Fig. 4). Magnesium calcite (CaMg)CO3 is the only phase identified. FTIR spectrum is shown in the bottom of Fig. 4. It can be identified the characteristic peaks of calcite phase localized at 1800 cm−1, 1422 cm−1, 1084 cm−1, 876 cm−1, and 710 cm−1.
 | ||
| Fig. 3 LV-SEM image of the starfish after the grinding process (left panel) and after the thermal treatment (right panel). | ||
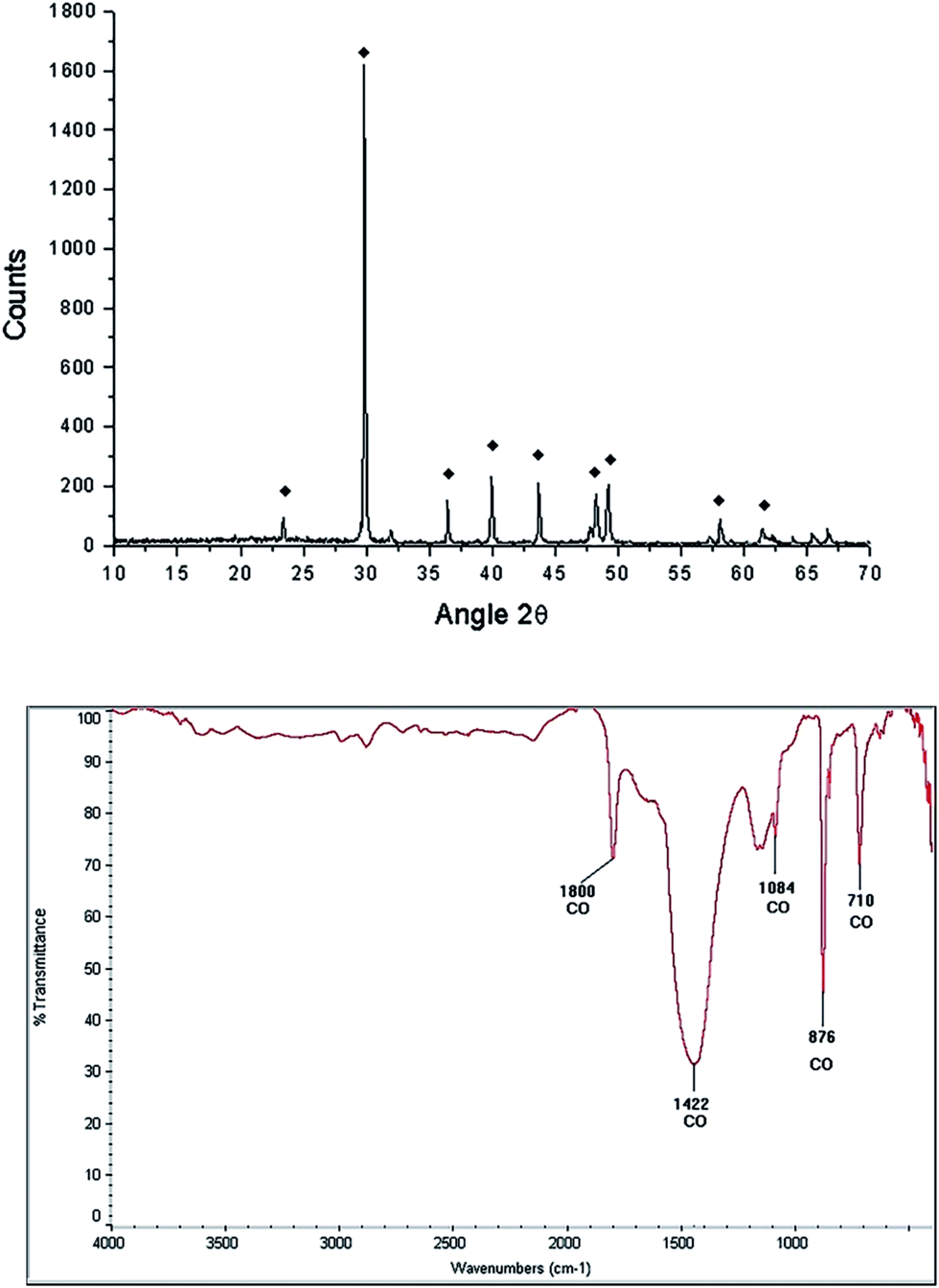 | ||
| Fig. 4 Characterization of the starfish Mellita eduardobarrosoi sp. nov.: diffractogram (upper panel), black marks represent the predominant phase which corresponds to magnesium calcite, and FTIR spectrogram (lower panel). | ||
Characterization of the starfish after the thermal treatment
After the thermal treatment of 900 °C during 4 hours, the resulting product of the starfish changes its morphology, appearing irregular shapes of different dimensions (see LV-SEM image, right panel of Fig. 3). EDS identify Ca and O as main elements, while C and Mg appear in smaller proportions, whereas Al and Si are not detected (see Table 2). When comparing the identified chemical elements of the starfish prior to the thermal treatment, calcium percentage increase, as well as the decrease of carbon, magnesium, and oxygen is notable (Table 2).Starfish product after the thermal treatment was also characterized by XRD (Fig. 5 upper panel). Three phases can be appreciated: calcium oxide [CaO], magnesium oxide [MgO], and portlandite [Ca(OH)2]. The latter could be formed after the treatment when it comes into contact with the ambient humidity according to the following chemical reaction:
| CaO + H2O → Ca(OH)2. |
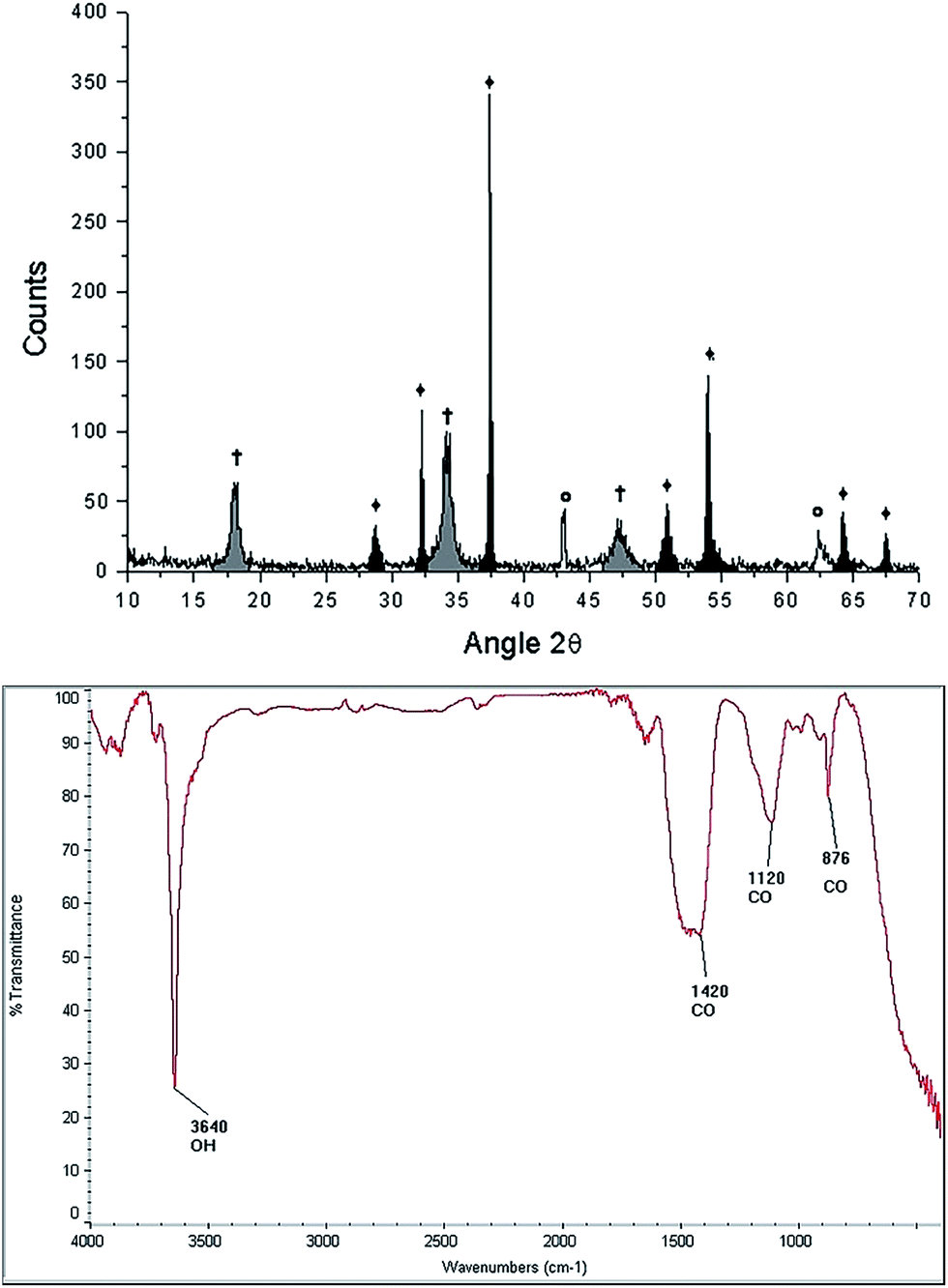 | ||
| Fig. 5 Characterization of the starfish Mellita eduardobarrosoi sp. nov. after the thermal treatment: diffractogram (up) showing the phases: †[Ca(OH)2], ♦[CaO], Θ[MgO], and FTIR spectrogram (bottom). | ||
FTIR spectroscopy of the starfish after thermal treatment (Fig. 5 bottom panel) indicates the presence of a band at 3641 cm−1, which corresponds to a O–H type bond, associated with the presence of the hydroxyl of the portlandite phase. The bands identified at 876 cm−1, 1120 cm−1, 1420 cm−1 correspond to the calcite phase.
Characterization of precursors: β-TCP
β-TCP [Ca3(PO4)2] is a porous calcium phosphate ceramic,54 irregular in shape, biocompatible with hard tissue, it has a molecular weight (wt) of 310.18 g, usually is an amorphous powder, not soluble in water, alcohol or acetic acid and soluble in HCl and HNO3. The characteristic morphology of the β-TCP used in our hydrothermal synthesis is shown in Fig. 6. It can be observed that it is formed of grains, which basically have two sizes, the small ones from 1.76 μm to 4.11 μm, while the larger ones have values of 5.3 μm to 9.41 μm. XRD analysis only shows the β-TCP phase (Fig. 7 upper panel). FTIR spectrogram (Fig. 7 bottom panel) has bands associated to the PO4 ion, localized at 2079 cm−1, 2002 cm−1, 1062 cm−1, 715 cm−1, 605 cm−1, and 654 cm−1. | ||
| Fig. 6 LV-SEM image of the β-TCP. | ||
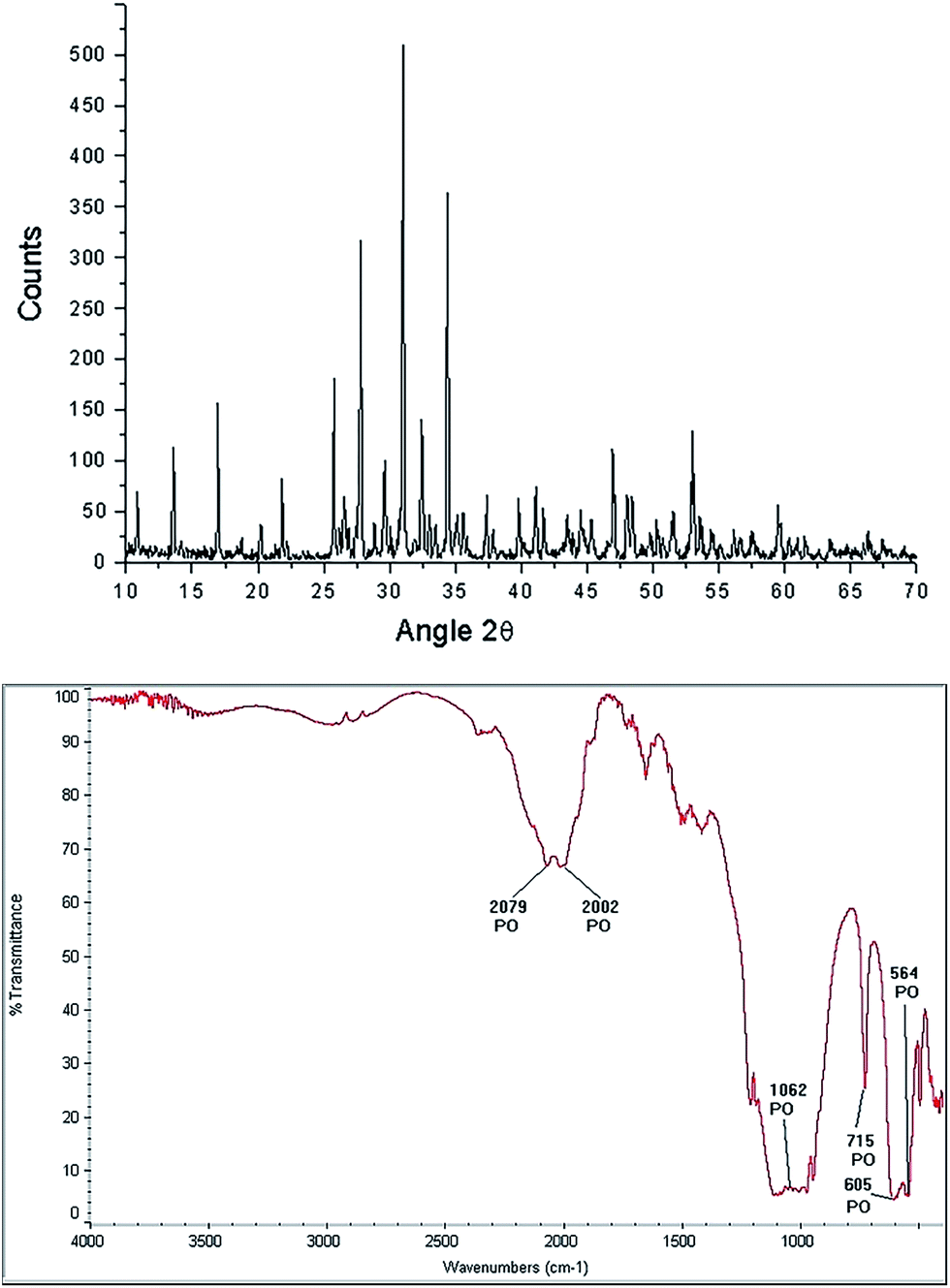 | ||
| Fig. 7 Characterization of the β-TCP phase: diffractogram (up) and FTIR spectrogram (bottom). | ||
Characterization of HAp obtained from the hydrothermal method using LV-SEM
The micrograph of Fig. 8 shows the morphology associated with the product obtained by the hydrothermal method. Fig. 8a is the LV-SEM micrograph of the sample obtained by the hydrothermal method at 230 °C for 20 hours, in which individual grains and agglomerates with sizes ranging from 3.22 μm to 3.87 μm in length are observed; individual grains had dimensions ranging from 2.25 μm to 2.90 μm. The particles exhibiting irregular morphologies are scarce, with dimensions of 1.1 μm (P1) and 14.83 μm (P2). Structures shaped fibers have different lengths ranging from 2.80 μm to 42.10 μm, with diameters ranging from 0.60 μm to 1.60 μm. There was a long fiber (labeled “F1”) that exceed the average size with a length of 34.5 μm and a coarse fiber (denoted by “F2”) which thickness is 2.90 μm, which exceeds by far the average thickness of the other fibers. Chemical composition of the product obtained is given in Table 3, where the presence of O, C, Ca, and P is observed with a Ca/P ratio; in this case, it is 1.65, which is very close to the stoichiometric ratio. | ||
| Fig. 8 LV-SEM micrographs showing the products obtained in the synthesis of HAp by hydrothermal process at (a) 230 °C for 20 h, (b) 250 °C for 20 h, (c) 230 °C for 48 h and (d) 250 °C for 48 h. | ||
| Sample | Synthesis 1: 230 °C for 20 h | Synthesis 2: 250 °C for 20 h | Synthesis 3: 230 °C for 48 h | Synthesis 4: 250 °C for 48 h |
|---|---|---|---|---|
| O | 56.47 | 27.92 | 56.98 | 54.37 |
| 49.11 | ||||
| Ca | 21.64 | 31.91 | 22.21 | 20.59 |
| 19.94 | ||||
| P | 13.58 | 15.72 | 13.39 | 12.98 |
| 11.57 | ||||
| C | 7.97 | 24.44 | 7.03 | 11.24 |
| 19.20 | ||||
| Mg | 0.34 | — | 0.37 | 0.27 |
| Ca/P | 1.58 | 2.03 | 1.66 | 1.59 |
| 1.72 |
Fig. 8b is the micrograph of the HAp sample obtained via hydrothermal technique at 250 °C for 20 hours. Here, it is possible to note greater homogeneity among the abundance of fiber structures. There is a tendency of forming very long fibers with sizes ranging from 36.27 μm to 43.13 μm. Fibers of shorter lengths are still identified, with diameters from 0.98 μm to 1.17 μm. Furthermore, there are smaller structures in the form of cluster of grains with grain sizes ranging from 1.96 μm to 5.88 μm. The morphologies associated with grains are illustrated in the area marked “I” in Fig. 8b, with dimensions of 15.68 μm. The sample is composed of O, Ca, P, and C with minor amounts of Mg and a Ca/P ratio of 1.58 (see Table 3).
In Fig. 8c, the micrograph of the sample obtained at 230 °C for 48 hours is shown. Under these conditions, it can be observed that fiber formation is favored; fibers of different dimensions, which are divided into three groups, are observed. The first group of fibers (I) have diameters of 1.29 μm and average lengths ranging from 12.19 μm to 14.19 μm, the second group of fibers (II) shows lengths ranging from 11.12 μm to 16.12 μm and diameters from 0.64 μm to 0.96 μm and, finally, in the third group (III), which is the most abundant, we can identify fibers with lengths ranging from 7.90 μm to 10.12 μm with an average diameter of 0.32 μm. In the elemental analysis presented in Table 3, it can be seen that, on average, the fibers are composed of O, Ca, P, and C; with a Ca/P ratio of 1.59.
Finally, in Fig. 8d, the LV-SEM micrograph of the sample obtained via the hydrothermal method at 250 °C for 48 hours is observed. The increase in both temperature and time permit the formation of fibers with bigger dimensions (in terms of length and diameter) and with greater homogeneity. It is appreciated that the fibers correspond to the predominant morphology. They are also isolated small particles of sizes ranging from 6.86 μm to 11.76 μm. The fibers have lengths ranging from 41.17 μm to 41.76 μm, diameters varying from 1.96 μm to 2.15 μm. Samples are composed of O, Ca, P, and C, and minor amounts of Mg, with a Ca/P ratio of 1.65 (see Table 3).
XRD characterization of HAp
The characterization performed by XRD using a stoichiometric concentration ratio of 3:1 allows us to identify the presence of HAp, β-TCP, and CaO, as follows. For samples obtained at 230 °C during 20 hours, the phases of HAp, β-TCP, and CaO are identified, as illustrated in the diffractogram of Fig. 9a. β-TCP and CaO are also identified and correspond to the phases belonging precursors. In the case of samples synthesized at 250 °C during 20 hours, the results are shown in the diffractogram of Fig. 9b, in which the major diffraction peaks correspond to the phases of HAp and β-TCP. Greater definition can be observed in the case of HAp when compared with that observed for 230 °C. A decrease in the intensities associated with β-TCP and CaO is also observed.
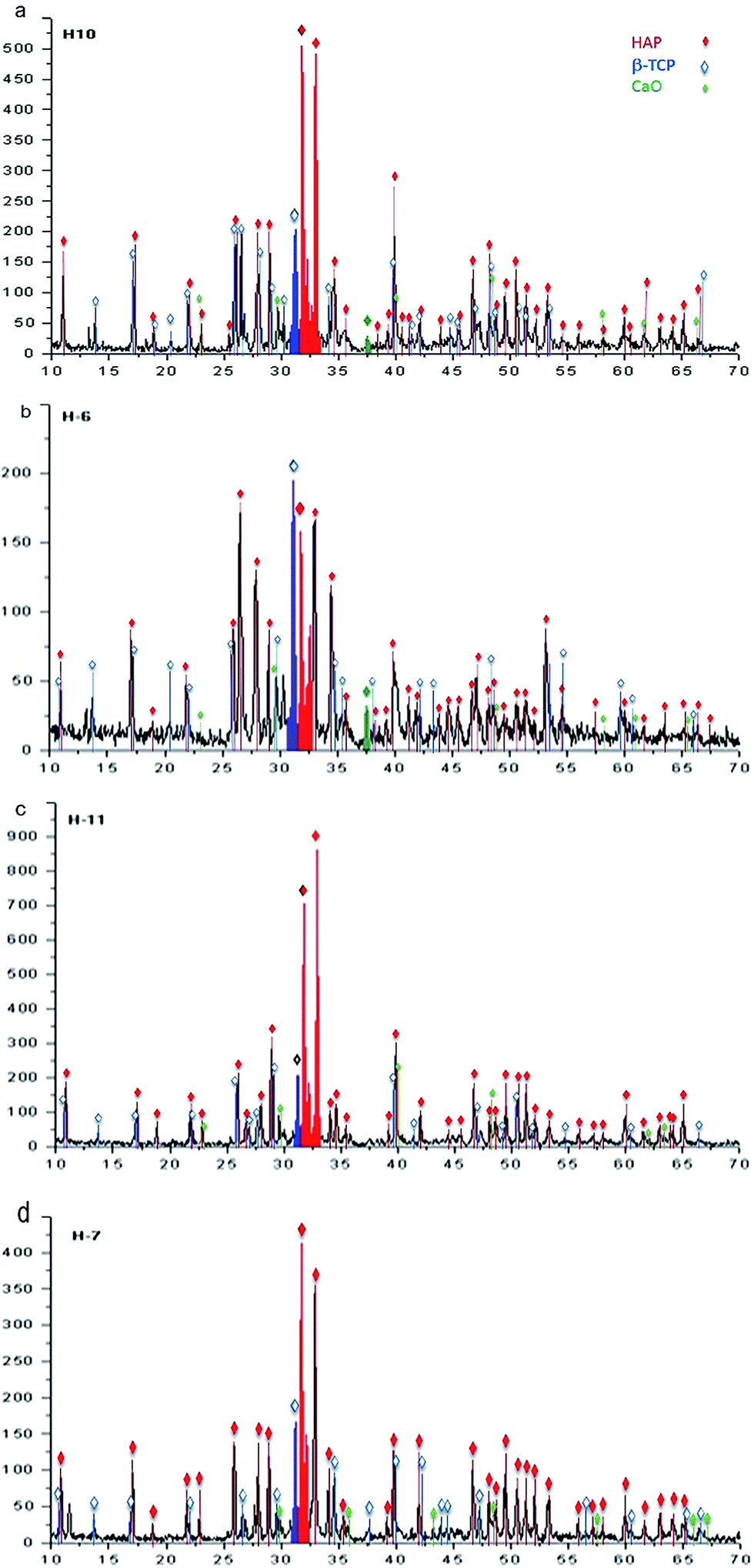 | ||
| Fig. 9 X-ray spectra associated with the hydrothermal synthesis using a stoichiometric concentration ratio of 3:1 of β-TCP:CaO for 20 h at (a) 230 °C, (b) 250 °C, and for 48 h at (c) 230 °C and (d) 250 °C. | ||
For a reaction time of 48 hours, Fig. 9c presents the associated diffraction patterns of the sample synthesized at 230 °C; these patterns are identified only as phases of β-TCP and HAp, where the latter is observed with higher intensity. In the diffractogram of Fig. 9d, the phases of β-TCP and HAp, which were identified after synthesis at a temperature of 250 °C, are observed with a greater signal strength associated to the HAp phase.
Characterization of HAp obtained from the hydrothermal method by FTIR spectrometry
Fig. 10 shows the FTIR spectra obtained from the samples by hydrothermal synthesis performed at: (a) 230 °C for 20 hours; (b) 250 °C for 20 hours; (c) 230 °C for 48 hours; and (d) 250 °C for 48 hours. The bands identified in each reaction are shown in Table 4, where the bands related to the vibrational frequencies of the ions PO4 and OH, characteristic of HAp are identified. Frequencies associated with vibrations of the P–O bond from the PO4 ion of HAp appear at 2079 cm−1, 2009 cm−1, 1093 cm−1, 1062 cm−1, 1036 cm−1, 964 cm−1, 606 cm−1, 654 cm−1, and 741 cm−1. Also, the bands associated with the O–H vibration mode characteristics of HAp at 3568 cm−1 and 631 cm−1 are observed.55,56 In the reaction performed at 230 °C for 20 hours, five of the nine bands of the PO4 ion and one corresponding to the OH ion were initially observed. In all the spectra, it was possible to identify the bands related to the OH and PO4 ions, which are associated with the precursors (β-TCP and CaO). The bands located at 910 cm−1 and 715 cm−1 correspond to the P–O vibration mode of the PO4 ion, which is associated with β-TCP. Furthermore, the formation of a band localized at 3650 cm−1, which may be due to the OH vibration mode by the presence of a hydroxyl, indicates that the formation of Ca(OH)2. Finally, across all spectra, it is possible to identify a band due to the H–O–H mode, which corresponds to moisture located at 3460 cm−1.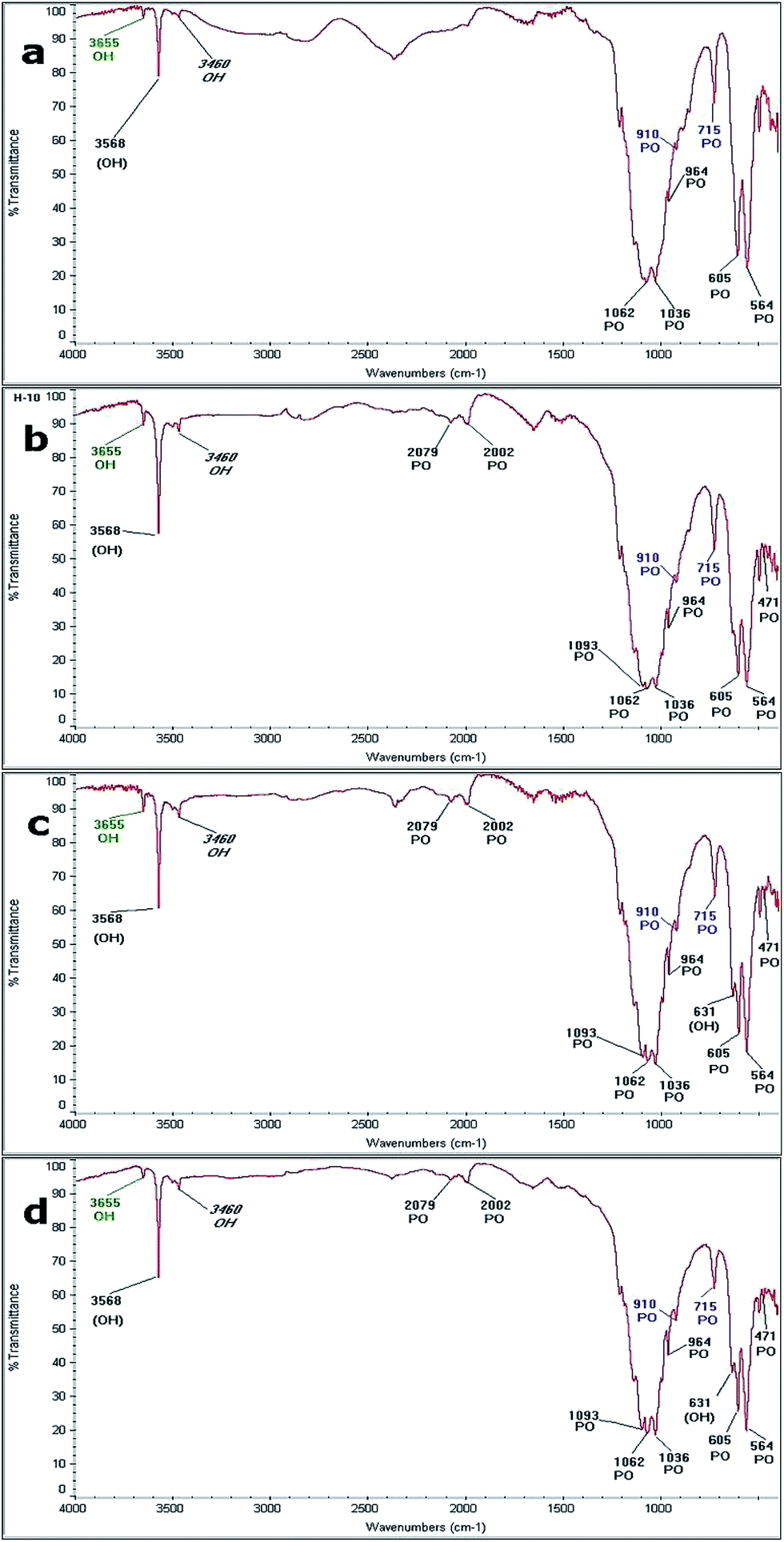 | ||
| Fig. 10 FTIR spectra obtained at: (a) 230 °C for 20 h, (b) 250 °C for 20 h (c) 230 °C for 48 h and (d) 250 °C for 48 h. | ||
| Bond type | Synthesis 1: 230 °C for 20 h | Synthesis 2: 250 °C for 20 h | Synthesis 3: 230 °C for 48 h | Synthesis 4: 250 °C for 48 h |
|---|---|---|---|---|
| PO (HAp) | 2079 | 2079 | 2079 | |
| 2002 | 2002 | 2002 | ||
| 1093 | 1093 | 1093 | ||
| 1062 | 1062 | 1062 | 1062 | |
| 1036 | 1036 | 1036 | 1036 | |
| 964 | 964 | 964 | 964 | |
| 605 | 605 | 605 | 605 | |
| 564 | 564 | 564 | 564 | |
| 471 | 471 | 471 | ||
| OH (HAp) | 3568 | 3568 | 3568 | 3568 |
| 631 | 631 | |||
| PO (TCP) | 910 | 910 | 910 | 910 |
| 716 | 716 | 716 | 716 | |
| OH Ca(OH)2 | 3655 | 3655 | 3655 | 3655 |
| OH humidity | 3640 | 3640 | 3640 | 3640 |
Discussion
In the present work, formation of the HAp phase via a hydrothermal method was achieved. As seen through the evaluation of all the materials parameters, the characteristics of the synthesized HAp products are associated with the precursors used, the method of synthesis employed, and the reaction conditions.XRD analysis served to identify the effect of temperature and reaction time on the synthesized HAp. The proportion of the HAp phase tends to increase as the temperature and reaction times growth. This is corroborated by LV-SEM characterization, where HAp homogeneous fibers were identified with a Ca/P ratio close to the stoichiometric when the reaction is carried out at high temperatures and longer reaction times.
Analysis performed by FTIR spectroscopy allowed the identification of absorption bands related to the functional groups of PO4 and OH, as well as the characteristics of HAp and its precursors employed during synthesis, which also validates the results obtained by XRD.
From the set of results obtained in this research, it can be established that the best synthesis conditions for HAp are a temperature of 250 °C during a reaction time of 48 hours. These parameters yielded the best characteristics of HAp, including a more uniform size, greater amounts, homogeneity with a Ca/P ratio near stoichiometric, a higher crystalline degree, and a greater fiber proportion resulting in a ratio of 3:1 of β-TCP and CaO. By forming a large proportion of fibers with very homogeneous lengths and thickness, it was also possible to achieve a Ca/P ratio of 1.65.
Previous works49 used the same starfish Mellita eduardobarrosoi sp. nov., but the monetite (CaHPO4) as a source of PO43−. There the synthesis is carried out at longer reaction times, generating a low density of fibers with diameters from 1.1 μm to 3.3 μm, and 36.1 μm long, and a Ca/P ratio of 1.24 to 1.50. As in the present method, were obtained portlandite products [Ca(OH)2], calcium oxide [CaO] and magnesium oxide [MgO].49 In contrast, when β-TCP is used (as in this paper) longer reaction times (250 hours) promote the formation of a higher density of fibers having sizes ranging from 2.80 μm to 42.10 μm in length and of diameter from 0.60 μm to 1.60 μm, with a Ca/P ratio of 1.65 (practically stoichiometry), and with a higher homogeneity of the fibers obtained, thus producing a better material for biomedical applications. In past research,47 it has been synthetized hydroxyapatite by the solid state method using a starfish and the β-TCP, producing a porous structure with a Ca/P rate of 1.67 (almost stoichiometry). However, the solid state method is expensive and complicated in comparison with the one proposed here. From the previous papers47,49 and the present methodology proposed here, it is clear than the morphology depends not only on the synthesis technique, but also on the reaction times, temperatures, precursor types and concentrations.
Conclusions
In this study, the synthesis of HAp was obtained from the starfish “Mellita eduardobarrosoi sp. nov.” and β-TCP using the hydrothermal method. The characterization of the obtained products allows to illustrate that temperature, reaction time, and concentration ratios are the most critical parameters for the synthesis of HAp. The fiber size is a function of the synthesis conditions, because as the reaction time and the temperature are increased, better defined fibers with a more homogeneous size are produced.The results indicate that the hydrothermal method encourages the formation of HAp with specific characteristics such as dimensions that maintain high homogeneity, a suitable proportion of that phase, and a Ca/P ratio near to stoichiometric. It is also noted that the increase in temperature and reaction time contributes to the improved the amount of HAp phase produced. This study determined that the best condition for the formation of HAp is achieved at a temperature of 250 °C using a 3:1 ratio of β-TCP and CaO, with a reaction time of 48 hours, which promotes the formation of fibers with homogeneous lengths and thicknesses.
The Ca/P ratio of HAp synthesized by the hydrothermal method was 1.65, which is very close to the stoichiometric. From the results obtained by FTIR spectrometry, absorption bands related to the functional groups PO4 and OH, which are characteristic of synthetic HAp were identified. Moreover, bands corresponding to the PO4 ion coming from the β-TCP precursor were also noted. Finally, a band corresponding to the OH bond was identified, which belongs to the Ca(OH)2 phase and could not be detected by XRD, likely because this phase was present in very low proportions.
Success in the production of HAp from starfish and β-TCP through hydrothermal technique suggested that it is possible to produce in a simple, cheap and reproducible method a material that can be used in diverse biotechnological applications.
Acknowledgements
The authors appreciate the technical support of Marcelo Hernández Ávila and Susana López Ortiz. Analysis were done in Laboratorio de Microscopía Electrónica de Barrido y de Difracción de Rayos X from Instituto Nacional de Investigaciones Nucleares. Financial funding has been provided by the projects: UNAM/DGAPA/PAPIIT IV100116, CONACyT INFR-2015-251767, PROMEP – UAEH – PTC – 6669, and Laboratorio Nacional de Ciencias de la Complejidad. English language editing of this manuscript was provided by Journal Prep.References
- S. Tabassum, S. Zahid, F. Zarif, M. A. Gilani, F. Manzoor, F. Rehman, A. Jamal, A. A. Chaudhry, S. A. Siddiqi and I. ur Rehman, RSC Adv., 2016, 6, 104969 RSC.
- M. González, J. A. Banderas, V. Rodríguez-Lugo and V. M. Castaño, J. Dent., 1999, 27, 595 CrossRef.
- A. Bhowmick, A. Saha, N. Pramanik, S. Banerjee, M. Das and P. P. Kundu, RSC Adv., 2015, 5, 25437 RSC.
- X. Chen, B. Yang, C. Qi, T. W. Sun, F. Chen and J. Wu, et al., Dalton Trans., 2016, 45, 1648 RSC.
- A. A. John, A. P. Subramanian, M. V. Vellayappan, A. Balaji, S. K. Jaganathan, H. Mohandas, T. Paramalinggam, E. Supriyanto and M. Yusof, RSC Adv., 2015, 5, 39232 RSC.
- H. S. Frederick and L. C. David, Biomaterials Science and Biocompatibillity, Springer, New York, 1996 Search PubMed.
- J. L. Lacout, in Biomaterials: hard tissue repair and replacement, ed. D. Muster, Elsevier Science Publisher, North-Holland, 1992, pp. 81–96 Search PubMed.
- V. Rodríguez-Lugo, G. A. Camacho-Bragado and V. M. Castaño, Mater. Manuf. Processes, 2003, 18, 67 CrossRef.
- D. R. Buddy, S. H. Allan, H. J. S. Frederick and E. L. Jack, Biomaterials Science – an introduction to materials in medicine. Academic Press, New York, 1996 Search PubMed.
- V. S. Chandra, K. Elayaraja, K. T. Arul, S. Ferraris, S. Spriano and M. Ferraris, et al., Ceram. Int., 2015, 41, 13153 CrossRef.
- H. Duan, Y. Ma, X. Liu, L. Hao and N. Zhao, RSC Adv., 2015, 5, 83522 RSC.
- M. Manoj, D. Mangalaraj, N. Ponpandian and C. Viswanathan, RSC Adv., 2015, 5, 48705 RSC.
- M. Sivakumar, T. Sampath-Kumar, K. L. Shantha and K. Panduranga-Rao, Biomaterials, 1996, 17, 1709 CrossRef CAS PubMed.
- V. Rodríguez-Lugo, M. Hernández and C. Angeles-Chavez, Mater. Manuf. Processes, 2003, 18, 903 CrossRef.
- S. Recillas, G. Mondragón, V. Rodríguez and V. M. Castaño, Des. Monomers Polym., 2003, 6, 383 CrossRef CAS.
- M. E. Fernández, C. C. Angeles, G. Mondragón-Galicia and V. Rodríguez-Lugo, J. Mater. Sci.: Mater. Med., 2004, 15, 735 CrossRef.
- V. Rodríguez-Lugo, C. Angeles-Chavez, G. Mondragon, S. Recillas-Gispert and V. M. Castaño, Mater. Res. Innovations, 2005, 9, 157 Search PubMed.
- V. Rodríguez-Lugo, J. Sánchez Hernández, M. J. Arellano- Jiménez, P. H. Hernández-Tejeda and S. Recillas-Gispert, Microsc. Microanal., 2005, 11, 516 CrossRef PubMed.
- S. Recillas, J. A. Asecencio, V. Rodríguez-Lugo and V. M. Castaño, NanoTrends, 2009, 2, 5 Search PubMed.
- S. Recillas, V. Rodríguez-Lugo, M. L. Montero, S. Viquez-Cano, L. Hernández and V. M. Castaño, J. Ceram. Process. Res., 2012, 13, 5 Search PubMed.
- V. Rodriguez-Lugo, D. Mendoza-Anaya and R. F. Estrada, Revista Electrónica Ideas CONCYTEG, 2012, vol. 7, p. 87 Search PubMed.
- E. M. Rivera-Muñoz, R. Velázquez and J. L. Cabrera, Mater. Sci. Forum, 2010, 638 Search PubMed.
- O. R. Bingöl and C. Durucan, Am. J. Biomed. Sci., 2012, 4, 50 CrossRef.
- F. Vazquez-Hernandez, C. Mendoza-Barrera, V. Altuzar, M. Melendez-Lira, M. A. SantanaAranda and L. Olvera, Mater. Sci. Eng., B, 2010, 174, 290 CrossRef CAS.
- M. I. Kay, R. A. Young and A. S. Posner, Nature, 1964, 204, 1050 CrossRef CAS PubMed.
- M. E. Londoño, A. Echavarría and F. de la Calle, Rev. EIA, 2006, 5, 109 Search PubMed.
- A. Cuneyt Tas, Powder Diffr., 2001, 16, 102 CrossRef.
- J. Ascencio-Gutierrez, V. Rodríguez-Lugo, C. Angeles, T. Santamaría and V. M. Castaño, Comput. Mater. Sci., 2002, 25, 413 CrossRef.
- M. Yoshimura and H. Suda, J. Mater. Sci., 1994, 29, 3399 CrossRef CAS.
- M. Yoshimura and H. Suda, in Hydroxyapatite and Related Materials, ed. P. W. Brown and B. Constantz, CRC Press, Boca Raton, 1994, pp. 45–72 Search PubMed.
- J. L. Cabrera, R. Velazquez-Castillo and E. M. Rivera-Muñoz, J. Nanosci. Nanotechnol., 2011, 11, 1 CrossRef.
- E. M. Rivera-Muñoz, R. Huirache-Acuña, R. Velazquez, G. Alonzo-Nuñez and S. Eguía-Eguía, J. Nanosci. Nanotechnol., 2011, 1, 5592 CrossRef.
- V. D. Hien, D. Q. Huong and P. T. N. Bich, J. Chem., 2010, 48, 591 CAS.
- K. P. Sanosh, C. Min-Cheol, A. Balakrishnan, T. N. Kim and C. Seong-Jai, Bull. Mater. Sci., 2009, 32, 465 CrossRef CAS.
- R. Sang-Hoon, Biomaterials, 2002, 23, 1147 CrossRef.
- K. Saranya, M. Kowshik and S. R. Ramanan, Bull. Mater. Sci., 2011, 34, 1749 CrossRef CAS.
- A. StrzaBa, W. Simka and M. MarszaBek, Acta Phys. Pol., A, 2012, 121, 561 CrossRef.
- M. Sadat-Shojai, M. T. Khorasani, E. Dinpanah-Khoshdargi and A. Jamshidi, Acta Biomater., 2013, 9, 7591 CrossRef CAS PubMed.
- H. S. Liu, T. S. Chin, L. S. Lai, S. Y. Chiu, K. H. Chung, C. S. Chang and T. M. Liu, Ceram. Int., 1997, 23, 19 CrossRef CAS.
- F. Shouua and X. Ruren, Acc. Chem. Res., 2001, 34, 239 CrossRef.
- A. Takeuchi, T. Tsuge and M. Kikuchi, Ceram. Int., 2016, 42, 15376 CrossRef CAS.
- C. Ma, Z. Wang, X. Lu, J. X. Lu, F. Bai, C. F. Wang, L. Li, S. X. Hou and H. D. Wang, RSC Adv., 2016, 6, 68363 RSC.
- G. Zhu, Y. Hu, Y. Yang, R. Zhao and R. Tang, RSC Adv., 2015, 5, 23958 RSC.
- A. Shavandi, A. E. D. A. Bekhit, A. Ali and Z. Sun, Mater. Chem. Phys., 2015, 149, 607 CrossRef.
- J. Chen, Z. Wen, S. Zhong, Z. Wang, J. Wu and Q. Zhang, Mater. Des., 2015, 87, 445 CrossRef CAS.
- Y. Azis, N. Jamarun, S. Arief and H. Nur, Orient. J. Chem., 2015, 31, 1099 CrossRef CAS.
- J. Kemperl and J. Macek, Int. J. Miner. Process., 2009, 93, 84 CrossRef CAS.
- A. S. Harold and M. Telford, J. Nat. Hist., 1990, 24, 987 CrossRef.
- M. I. Kay, R. A. Young and A. S. Posner, Nature, 1964, 204, 1050 CrossRef CAS PubMed.
- V. Rodríguez-Lugo, C. Angeles-Chavez and M. Hernández, Mater. Manuf. Processes, 2003, 18, 903 CrossRef.
- V. Rodríguez-Lugo, G. A. Camacho and V. M. Castano, Mater. Manuf. Processes, 2003, 18, 67 CrossRef.
- V. Rodríguez-Lugo, J. A. Ascencio, C. Angeles-Chavez, A. Camacho and V. M. Castano, Mater. Technol., 2001, 16, 97 CrossRef.
- L. An, W. Li, Y. Xu, D. Zeng, Y. Cheng and G. Wang, Ceram. Int., 2016, 42, 3104 CrossRef CAS.
- T. J. Blokhuis, M. F. Termaat, F. C. den Boer, P. Patka, F. C. Bakker and J. T. M. Henk, J. Trauma Acute Care Surg., 2000, 48, 179 CrossRef CAS.
- K. C. Blakeslee and A. C. Tobert, J. Am. Ceram. Soc., 1971, 54, 559 CrossRef CAS.
- G. Penel, G. Leroy, C. Rey, B. Sombret, J. P. Huvenne and E. Bres, J. Mater. Sci.: Mater. Med., 1997, 8, 271 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |