
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The impact of an extended nucleobase-2′-deoxyribose linker in the biophysical and biological properties of oligonucleotides†
Alejandro Carnero‡
 a,
Sónia Pérez-Rentero‡bc,
Adele Alagiabc,
Anna Aviñóbc,
Yogesh S. Sanghvid,
Susana Fernándeza,
Miguel Ferrero*a and
Ramon Eritja*bc
a,
Sónia Pérez-Rentero‡bc,
Adele Alagiabc,
Anna Aviñóbc,
Yogesh S. Sanghvid,
Susana Fernándeza,
Miguel Ferrero*a and
Ramon Eritja*bc
aDepartamento de Química Orgánica e Inorgánica, Instituto Universitario de Biotecnología de Asturias, Universidad de Oviedo, 33006-Oviedo, Asturias, Spain. E-mail: mferrero@uniovi.es; Tel: +34 985 105 013
bDpt. Chemical & Biomolecular Nanotechnology, Institute for Advanced Chemistry of Catalonia (IQAC), CSIC, 08034-Barcelona, Spain. E-mail: recgma@cid.csic.es; Tel: +34 934 006 145
cCIBER-BBN Networking Centre on Bioengineering, Biomaterials and Nanomedicine, 08034-Barcelona, Spain
dRasayan, Inc., 2802 Crystal Ridge Road, Encinitas, California 92024-6615, USA
First published on 31st January 2017
Abstract
Interest in artificial DNA mimetics has been triggered by the widespread applications of nucleic acids as they are useful tools for modulation of the biophysical and biological properties of oligonucleotides. In this article, we describe the synthesis and properties of a novel thymine derivative (T*) containing an extended linker between the thymine nucleobase and the 2′-deoxyribose moiety. The modified 2′-deoxyribosyl derivative was prepared via coupling of a functionalized nucleobase to the amino group of 1-aminomethyl-2-deoxyribose, which was synthesized starting from an easily accessible cyano sugar available on a large-scale. Corresponding phosphoramidite and succinyl derivatives have also been incorporated into oligonucleotides at predetermined sites and defined internucleotidic motifs using the solid-phase synthesis approach. This derivative pairs equally well with adenine and guanine and it can be safely introduced at the 3′-end of the siRNAs to generate potent inhibitors of gene expression by the RNA interference mechanism.
Introduction
The discovery of the gene silencing properties of oligonucleotides has triggered significant efforts in the design of novel backbones to increase nuclease resistance while maintaining or improving the hybridization properties of the newly designed DNA/RNA mimetics.1 Artificial nucleoside derivatives have been designed, prepared and tested for the inhibition of gene expression by the “antisense” strategy.2,3 The most outstanding results in terms of improving the hybridization properties were obtained with peptide nucleic acids (PNA)4 and locked nucleic acids (LNA).5 The ability to restrict the conformation of some of these analogues was the key for the improvement of the hybridization properties.5 Additionally, interesting results were obtained with other nucleoside analogues such as hexitol (HNA),6 bicyclo,7 arabino (ANA),8,9 and fluoroarabino (FANA)8,9 nucleic acids. The discovery of RNA interference mechanism provide also an opportunity to the design of novel nucleic acid derivatives with more structural flexibility such as unlocked (UNA),10 acyclic threoninol (aTNA),11 and serinol (SNA)12 nucleic acid derivatives. The presence of these compounds in selected positions of siRNA has shown to improve the silencing properties12,13 as well as diminish off-target effects.14 For all these reasons research on flexible nucleoside analogues has generated an increasing interest for the structural and biological impact of these nucleic acid derivatives. Most of the flexible nucleoside derivatives have been generated by substituting the ribose moiety by an acyclic derivative. In addition, the synthesis and hybridization properties of the methyl and ethyl thymine alkane 2′-deoxyribosides (B in Chart 1) have been described.15,16 These compounds were designed to be used in triplex formation to stabilize triplexes with short polypurine–polypyrimidine tracts as they may be used as a wild card in the interruptions of the polypurine–polypyrimidine tracts by alternate binding.16,17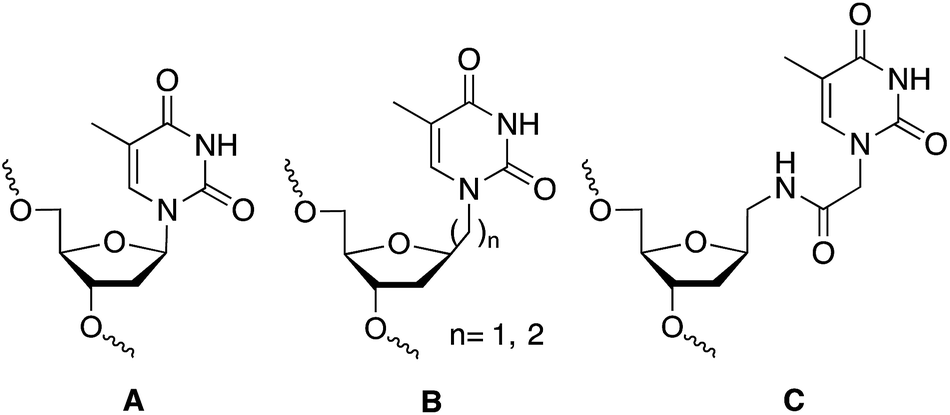 | ||
| Chart 1 Structure of DNA (A) and DNA mimetics (B and C) with extended links between nucleobase and 2′-deoxyribose moiety. (A) Unmodified thymidine; (B) methyl and ethyl derivatives described in ref. 15 and 16; and (C) N-methylacetamido derivative described in this work. | ||
In this communication we described the synthesis of a thymine derivative (T*) with an extended link between the 2′-deoxyribose phosphate backbone and the thymine base (C, in Chart 1). The novel T* derivative has been prepared by the coupling of N1-carboxymethylthymine to 1-aminomethyl-2-deoxyriboside. N1-Carboxymethylthymine is being also used in the preparation of the thymine derivative of PNA, aTNA and SNA. The hybridization properties of modified DNA duplexes and the silencing properties of siRNAs carrying this derivative at the 3′-end of siRNA are described.
Results and discussion
Synthesis of the extended T* derivative monomer unit
For the preparation of thymine derivative we designed a strategy starting with cyano sugar 1 (Scheme 1), which is easily accessible on large-scale.18 Reduction of the cyano group using hydrogen and RANEY® nickel catalyst furnished amino compound 2. Treatment of the later with N1-carboxymethylthymine and propylphosphonic anhydride afforded 3 in 65% yield. Removal of the toluoyl protecting groups in 3 by reaction with KOH in EtOH–H2O followed by neutralization with Dowex 50WX8 gave the diol 4 in excellent yield. Next, protection of the primary alcohol with 4,4′-dimethoxytrityl chloride in the presence of Et3N and pyridine, producing the key intermediate 5, which permits an easy access to the required monomer precursor for oligonucleotide solid-phase synthesis.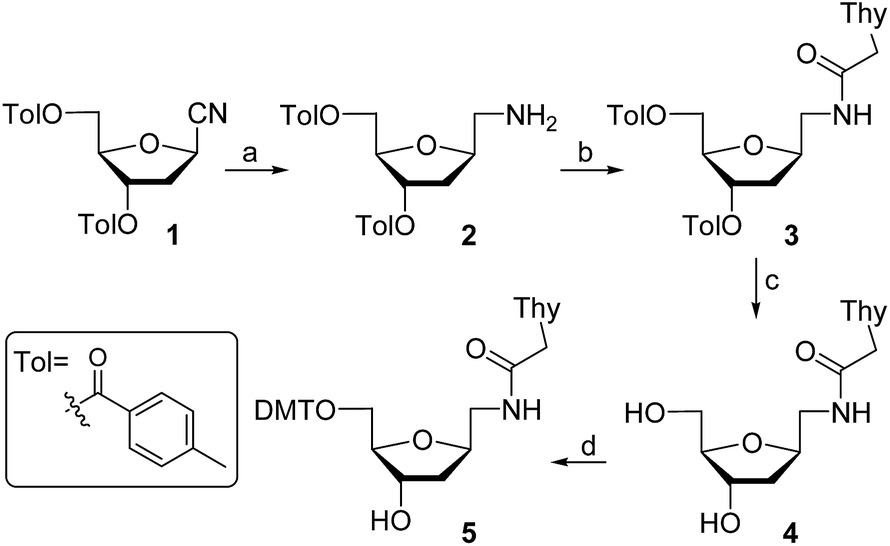 | ||
| Scheme 1 Synthesis of 5′-DMT-T* nucleoside precursor. Reagents and conditions: (a) H2, RANEY® Ni, MeOH, rt, 12 h (70%); (b) Thy-CH2CO2H, propylphosphonic anhydride, iPr2NEt, DMF, rt, 5 h (65%); (c) KOH, EtOH–H2O, rt, 30 min (95%); (d) DMTCl, Et3N, Py, 45 °C, 5 h (69%). | ||
Phosphitylation of DMT-protected compound 5 with 2-cyanoethoxy-N,N-diisopropylaminochlorophosphine in the presence of diisopropylethylamine gave the desired phosphoramidite derivative 6 in 85% yield. This phosphoramidite was used for the introduction of T* at an internal position during oligonucleotide synthesis. To introduce the T* modification at the 3′-end of siRNAs, the derivatized support precursor 8 was prepared by conversion of 5 to the corresponding hemisuccinate 7 via DMAP catalyzed reaction with succinic anhydride (Scheme 2).
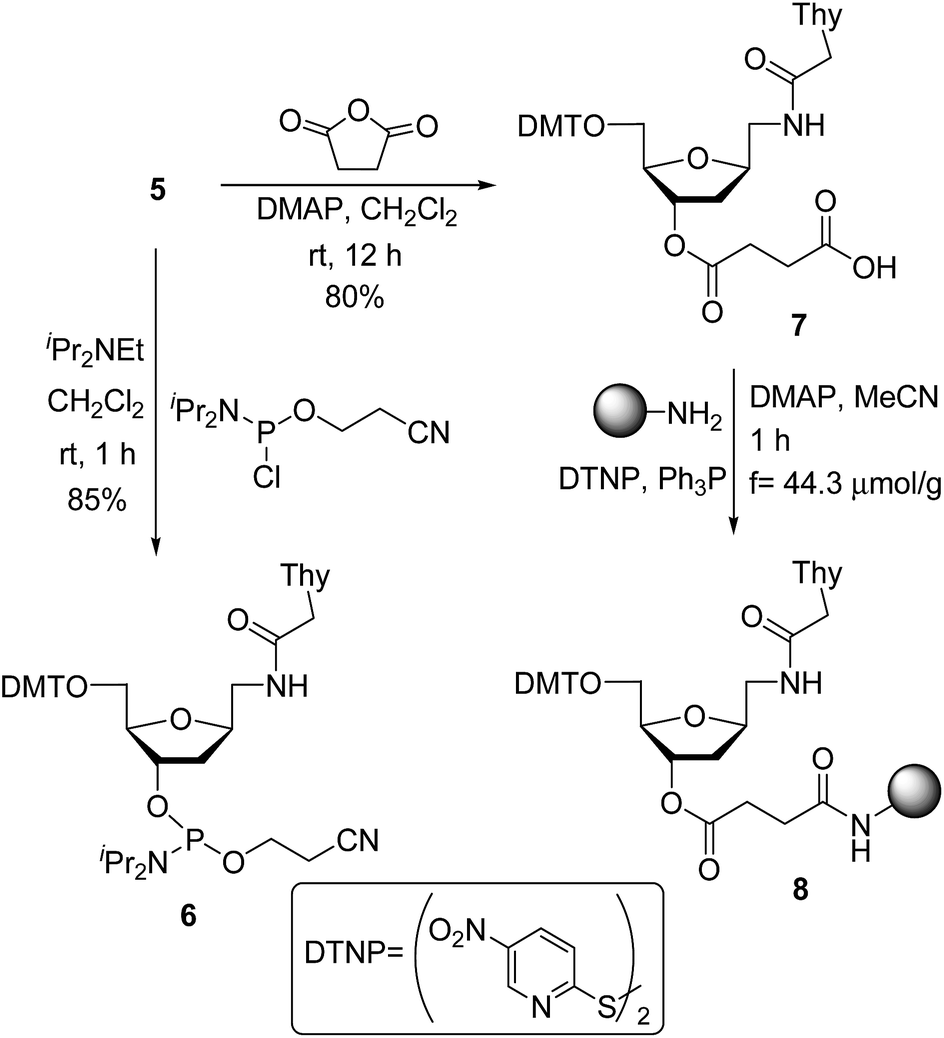 | ||
| Scheme 2 Synthesis of modified nucleoside monomers: phosphoramidite 6 and support-linked hemisuccinate 8. | ||
This hemisuccinate was then used to functionalize controlled pore glass solid-support, yielding derivative 8.
Synthesis of DNA oligonucleotides
Oligodeoxynucleotides used in this study are summarized in Table 1. Modified oligonucleotides 15Mer_T*(01), (02) and (03) were assembled by solid-phase on a DNA synthesizer using polystyrene support. In all cases the coupling yields of the modified phosphoramidite were similar to standard phosphoramidites yielding the desired oligonucleotides that were purified by HPLC and characterized by MALDI-TOF mass spectrometry.| Code | Sequences (5′ → 3′) | MW (calcd) | MWa (found) |
|---|---|---|---|
| a Using MALDI-TOF mass spectrometry. n.d. not determined, since the unmodified oligonucleotides were purchased. | |||
| 15Mer_T*(01) | d(TAG AGG CT*C CAT TGC) | 4639.1 | 4639.2 |
| 15Mer_T*(02) | d(TAG AGG CT*C CAT* TGC) | 4710.2 | 4711.6 |
| 15Mer_T*(03) | d(TAG AGG CTC CAT* T*GC) | 4710.2 | 4709.6 |
| 15Mer_UnMod | d(TAG AGG CTC CAT TGC) | n.d. | n.d. |
| 15MerA | d(GCA ATG GAG CCT CTA) | n.d. | n.d. |
| 15MerG | d(GCA ATG GGG CCT CTA) | n.d. | n.d. |
| 15MerC | d(GCA ATG GCG CCT CTA) | n.d. | n.d. |
| 15MerT | d(GCA ATG GTG CCT CTA) | n.d. | n.d. |
Denaturation studies on DNA oligonucleotides
The effect of the modification (T*) in the duplex stability was analyzed by recording the denaturation curves by UV-visible spectroscopy (Fig. 1).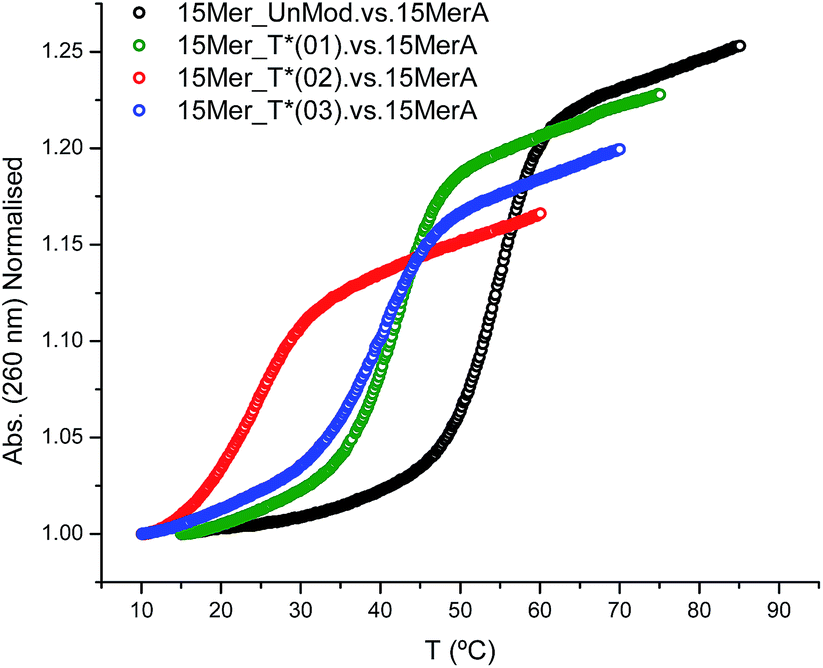 | ||
| Fig. 1 Melting curves of modified oligonucleotides with 15MerA. | ||
Three modified oligonucleotides [15Mer_T*(01), one T* modification; 15Mer_T*(02), two T* modifications at different positions of the sequence; and 15Mer_T*(03), two consecutive T* modifications] were annealed with their corresponding complementary sequence (15MerA, Fig. 1). The results are summarized in Table 2 and represented in Fig. 1. A single modification caused a decrease of 12.5 °C in the Tm. Two consecutive modifications caused a lower destabilization (−15 °C) than two T* modifications located in different places (−30.8 °C). These values are in agreement to the values found for the thymine alkane 2′-deoxyribosides developed by Beaucage and co-workers.15,16 These authors have used a duplex consisting of 24 bases, with two modifications in alternate positions in the middle of the sequence. They observed a decrease of 10 degrees in the Tm for the methyl-T derivative and a decrease of 5 degrees for the ethyl-T derivative.
| Duplex | Tm (°C) | ΔTm (°C) | ΔG° (kcal mol−1) | ΔΔG° (kcal mol−1) |
|---|---|---|---|---|
| a 50 mM NaCl and 10 mM sodium phosphate buffer pH 7.0. | ||||
| 15MerA. vs. 15Mer_UnMod | 54.2 | − | −13.9 | − |
| 15MerA. vs. 15Mer_T*(01) | 41.7 | −12.5 | −9.3 | −4.6 |
| 15MerA. vs. 15Mer_T*(02) | 23.4 | −30.8 | −4.6 | −9.3 |
| 15MerA. vs. 15Mer_T*(03) | 39.2 | −15.0 | −8.3 | −5.6 |
In addition, we studied the base paring properties of the modified T* evaluating the melting temperatures of the duplexes carrying all the possible base pairs. The denaturation curves are shown in Fig. 2 and the results are summarized in Table 3.
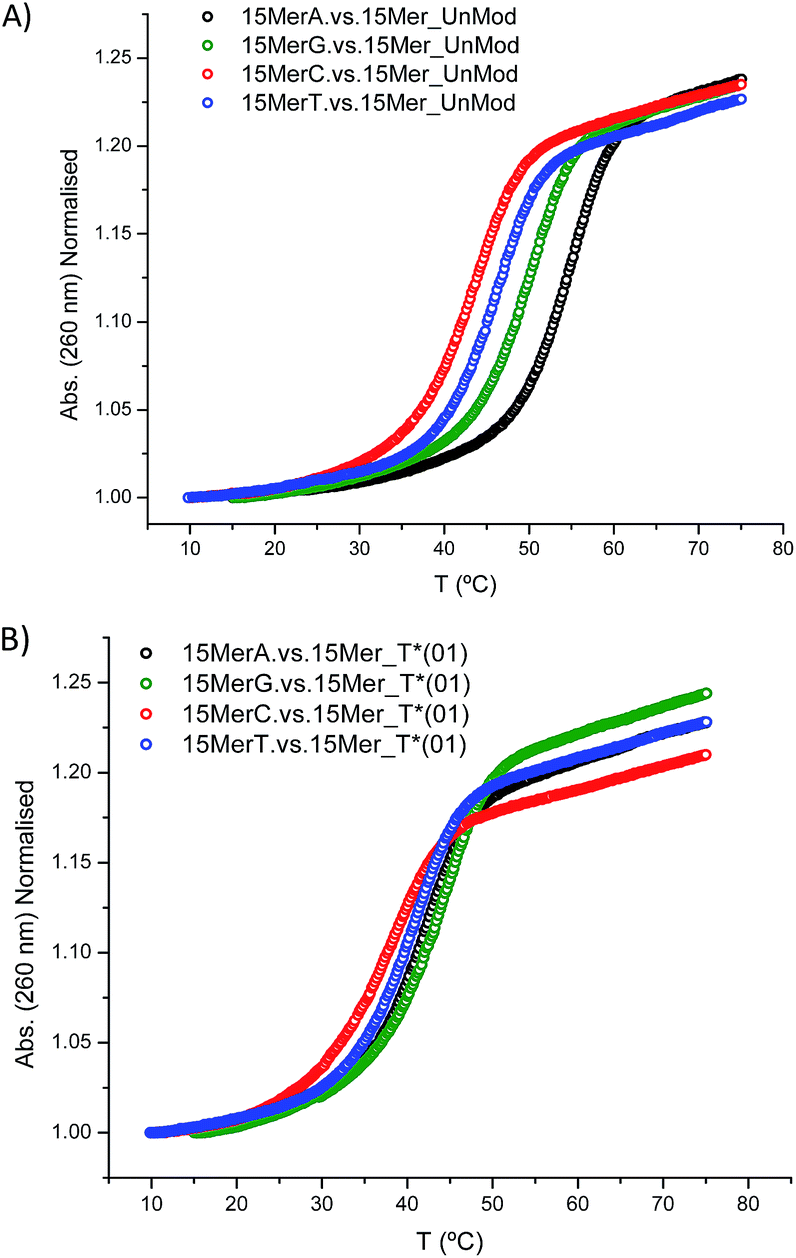 | ||
| Fig. 2 Melting curves of: (A) 15Mer_UnMod vs. 15MerX; (B) 15Mer_T*(01) vs. 15MerX. Note: X = A, G, C, T. | ||
| Duplex | Base pair X:Y | Tm (°C) | ΔTm (1) (°C) | ΔTm (2) (°C) | ΔG° (kcal mol−1) | ΔΔG° (1) (kcal mol−1) | ΔΔG° (2) (kcal mol−1) |
|---|---|---|---|---|---|---|---|
| a ΔTm (1) and ΔΔG° (1): in comparison with 15MerA. vs. 15Mer_UnMod; ΔTm (2) and ΔΔG° (2): in comparison with the corresponding unmodified duplex. | |||||||
| 15MerA. vs. 15Mer_UnMod | A:T | 54.2 | − | − | −13.9 | − | − |
| 15MerG. vs. 15Mer_UnMod | G:T | 49.3 | −4.9 | − | −11.8 | −2.1 | − |
| 15MerC. vs. 15Mer_UnMod | C:T | 42.9 | −11.3 | − | −9.5 | −4.4 | − |
| 15MerT. vs. 15Mer_UnMod | T:T | 45.3 | −8.9 | − | −10.4 | −3.5 | − |
| 15MerA. vs. 15Mer_T*(01) | A:T* | 41.7 | − | −12.5 | −9.3 | − | −4.6 |
| 15MerG. vs. 15Mer_T*(01) | G:T* | 43.2 | − | −6.1 | −9.5 | − | −2.3 |
| 15MerC. vs. 15Mer_T*(01) | C:T* | 37.0 | − | −5.9 | −7.7 | − | −1.8 |
| 15MerT. vs. 15Mer_T*(01) | T:T* | 39.8 | − | −5.5 | −8.6 | − | −1.8 |
As expected, when a mispair was introduced in the unmodified duplex a decrease in the thermal stability was observed. Surprisingly with the 15Mer_T*(01) series, we found that the G:T* base pair had the highest Tm (∼1.5 °C than the A:T* base pair). The insertion of a –CH2CONHCH2– tether between the carbohydrate and the nucleobase in general is not favorable for a good alignment of the nucleobases to form stable Watson–Crick base pairs, but in the case of the G:T* base pair the tether may allow the formation of a G:T* wobble base pair (Chart 2).
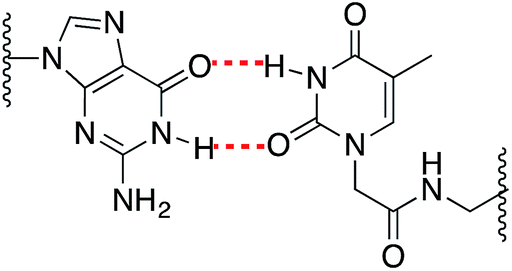 | ||
| Chart 2 Schematic representation of a G:T* wobble base pair. | ||
The T* modification loses the ability of distinguishing between A and G as the melting temperatures of the duplexes carrying A:T* and G:T* base pairs have similar melting temperatures. As a matter of fact, the duplex carrying the G:T* base pair has a higher Tm than the duplex carrying A:T* base pair (1.5 °C higher). This effect has not been described previously. The closest system with Tm values of all the mispairs is the melting study of L-threoninol-T (TL) in a RNA:RNA duplex14 and in this context there was a decrease on Tm for G:TL modified base pair (0.7 °C) compared with A:TL modified base pair.
Preparation of RNA modified oligonucleotides
Next, we have prepared several RNA strands carrying either two natural thymidine or two modified T* units at 3′-end (Table 4). All possible complementary RNA strands were formed (Table 5), and the resulted siRNA duplexes were used to target the Renilla luciferase gene as described.13| siRNA | Code | Sequences | IC50 (pM ± SD) |
|---|---|---|---|
| WT | SSwt | TTAAAAAGAGGAAGAAGUCUA-5′ | 6.3 ± 1.8 |
| ASwt | 5′-UUUUUCUCCUUCUUCAGAUTT | ||
| ST2 | SSwt | TTAAAAAGAGGAAGAAGUCUA-5′ | 6.1 ± 2.6 |
| ASmd | 5′-UUUUUCUCCUUCUUCAGAUT*T* | ||
| ST3 | SSmd | T*T*AAAAAGAGGAAGAAGUCUA-5′ | 3.2 ± 1.3 |
| ASwt | 5′-UUUUUCUCCUUCUUCAGAUTT | ||
| ST4 | SSmd | T*T*AAAAAGAGGAAGAAGUCUA-5′ | 5.0 ± 2.9 |
| ASmd | 5′-UUUUUCUCCUUCUUCAGAUT*T* |
In order to appraise the potency of modified siRNA molecules, we have performed dose–response experiments in HeLa cells. Briefly, we co-transfected Psi-CHECK2 vector with decreasing concentrations of siRNA duplexes (1; 0.3; 0.16; 0.06; 0.016; 0.008; 0.002 nM) and after 24 h of incubation, we measured the expression of Renilla protein.
As shown in Table 5 and Fig. 3 all modified siRNAs were strong inhibitors of Renilla luciferase. Notably, the ST3 siRNA, modified at sense overhang, is significantly more potent compared to unmodified siRNA (WT). The activity of ST2 siRNA, even if retained good inhibitory properties, is comparable to WT siRNA. The different silencing activities between ST2 and ST3 siRNAs basically depend on overhang recognition by the Ago2 protein, the core effectors of the RNAi pathway. The better the interaction between the Ago2's Paz domain and the overhang, the stronger is the inhibition on the target mRNA. Unlike the acyclic threoninol-T modification (structurally close to considered T* modification),13 which exert its best performance when placed at antisense overhang, the T* modification yielded more active siRNA when introduced at sense overhang (ST3). The equivalent potency between antisense modified (ST2) and unmodified (WT) siRNAs suggested that the modification is as well recognized as natural thymidine by the PAZ domain. The application of siRNA-based therapeutics is hampered by their numerous OFF-target effects. One of the most relevant comes from the wrong strand selection by the RISC. Since the strand loading was demonstrated to rely on thermodynamic stability of the two ends of siRNA duplex, we thought to compared the ON-/OFF-target activities of the T* modified siRNAs (ST2 and ST3) with the unmodified siRNA (WT). We used two different psicheck2 reporters, one codifying for an mRNA complementary to antisense strand (AS) and one to the sense strand (SS).
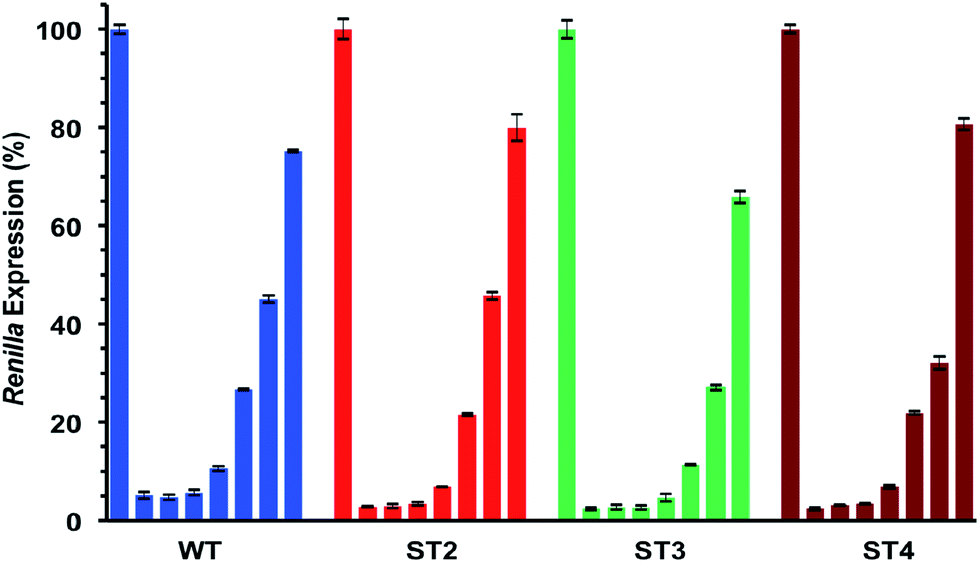 | ||
| Fig. 3 IC50 assessments of siRNA molecules using luciferase assay. Dose–response curves of unmodified siRNA (WT), and T*-modified (ST2, ST3, ST4) siRNAs. Decreasing concentrations (nM): control; 1; 0.3; 0.16; 0.06; 0.016; 0.008; 0.002. n = 3 ± SD. For experimental conditions see the Experimental section. | ||
Thus, we co-transfected HeLa cells with either AS or SS vector and two siRNA concentrations (1 nM and 16 pM). After 24 h incubation we measured the levels of Renilla expression by Dual-luciferase assay system. Such study, assessing the silencing abilities of each strand independently, provided rapid and reliable estimation of the efficiencies of siRNA-mediated inhibition for both the antisense (AS) and sense strand (SS).
As depicted in Fig. 4, all considered siRNAs (WT, ST2 and ST3) at the highest dose (1 nM) disclosed poor balance between ON-/OFF-target activities. However, the best performance owns to the ST3 siRNA.
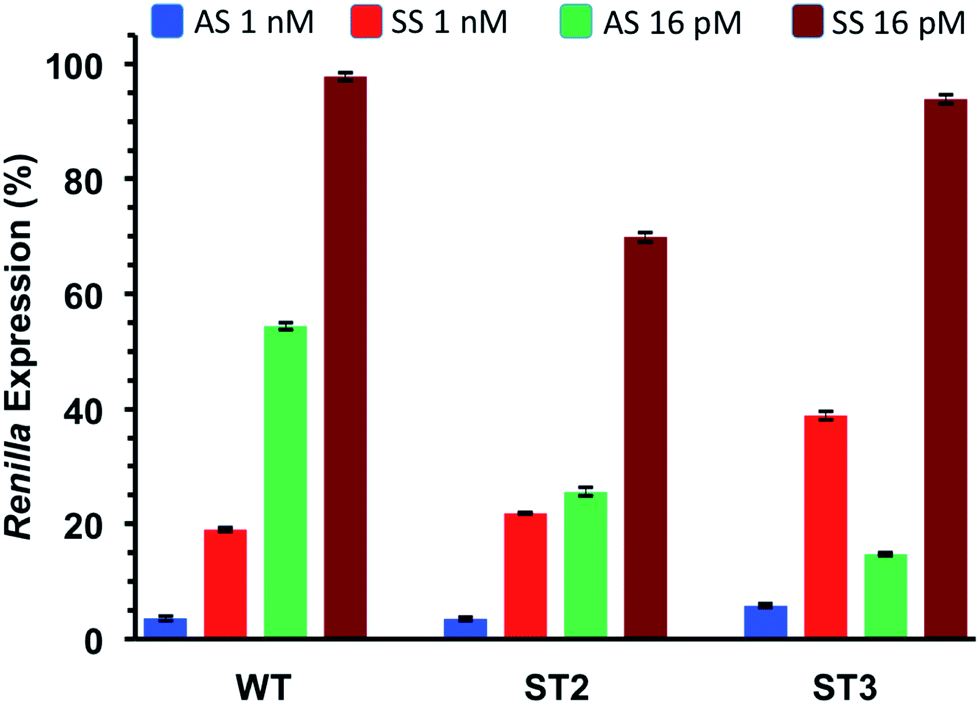 | ||
| Fig. 4 ON-/OFF-target silencing of T*-modified siRNAs at final concentrations of 1 nM and 16 pM, WT, ST2 and ST3 siRNAs were co-transfected with psiCHECK2 sensors (AS and SS) in HeLa cells. n = 3 ± SD. ST2 siRNA carries the T* modification at the antisense strand and ST3 at the sense strand. | ||
At the lowest dose transfected (16 pM), the ST3 siRNA is practically inactive towards the off-target mRNA (SS) while retaining 80% inhibition of the on-target mRNA (AS). Taking all these results together we can conclude than the T* modification is totally compatible with the RNA interference machinery, and it may provide higher efficiency and selectivity than unmodified siRNA when the modification is introduced at the 3′-end of the sense strand.
Conclusions
In the present work the synthesis, binding and gene silencing properties of oligonucleotides carrying the extended nucleobase modification (T*) are described. The novel pseudonucleoside was obtained by the coupling of an amino sugar as the glycosyl donor with N1-carboxymethylthymine. The presence of one single modification in the middle of a DNA duplex (15Mer, low salt conditions) induces a decrease of 12.5 °C (−4.6 kcal mol−1) in the Tm. Two T* modifications in a row are less destabilizing than separated. The T* modification loses the ability of distinguishing between A and G as the melting temperatures of the duplexes carrying A:T* and G:T* base pairs have similar melting temperatures. The introduction of T* at the 3′-end of either or both guide and passenger strand is well tolerated by the RISC machinery giving similar or better IC50 than unmodified siRNA. In the case of having the T* modification at the passenger strand the potential off-target effects are diminished. For these reasons, the introduction of this derivative at the 3′-end of the sense strand generates modified siRNAs that are more potent and selective inhibitors of gene expression by the RNA interference mechanism.Experimental protocols
Synthesis of modified monomer units
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 166.6 (CO) ppm; HRMS (ESI+, m/z): calcd for C22H26NO5 [(M + H)+]: 384.1805, found: 384.1808.
O), 166.6 (CO) ppm; HRMS (ESI+, m/z): calcd for C22H26NO5 [(M + H)+]: 384.1805, found: 384.1808.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). The product precipitated after standing overnight in the fridge and it was collected by filtration. The crude product was purified through column chromatography (2% MeOH/CH2Cl2) to afford 3 in 65% yield. Rf: 0.32 (5% MeOH/CH2Cl2); mp: 183–184 °C; IR (KBr): ν 3551, 3478, 3412, 3333, 2993, 1718, 1665, 1612, 1556 cm−1; 1H NMR (300.13 MHz, CDCl3): δ 1.81 (br s, 1H, NH), 1.91 (s, 3H, Me-C5), 2.12 (m, 2H, H2′), 2.40 (s, 3H, Me-Tol), 2.41 (s, 3H, Me-Tol), 3.44 (dt, 1H, H6′, JHH 14.1, 3.1 Hz), 3.64 (ddd, 1H, H6′, JHH 13.9, 7.5, 3.8 Hz), 4.26 (d, 1H, H7′, JHH 15.8 Hz), 4.35–4.48 (m, 4H, H1′ + H4′ + H5′ + H7′), 4.73 (dd, 1H, H5′, JHH 12.2, 8.1 Hz), 5.42 (d, 1H, H3′, JHH 5.3 Hz), 7.01 (s, 1H, H6′), 7.24 (2d, 4H, Hortho-Me), 7.92 (2d, 4H, Hortho-CO), 8.86 (br s, 1H, NH) ppm; 13C NMR (75.5 MHz, CDCl3): δ 12.5 (CH3-C5), 21.9 (2CH3-Tol), 34.3 (C2′), 41.2 (C6′), 50.7 (C7′), 64.9 (C5′), 77.4 (C3′), 78.4 (C1′), 83.6 (C4′), 110.9 (C5), 126.9 (Cipso-CO), 127.1 (Cipso-CO), 129.3 (2Cortho-Me), 129.4 (2Cortho-Me), 129.8 (2Cortho-CO), 130.0 (2Cortho-CO), 141.1 (C6), 144.4 (2Cipso-Me), 151.1 (C2), 164.2 (C4), 166.3 (OC-C7′), 167.2 (CO), 167.3 (CO) ppm; HRMS (ESI+, m/z): calcd for C29H31N3NaO8 [(M + Na)+]: 572.2003, found: 572.2001.:1) (0.08 mL). The completion of the reaction was reached at 30 min, a cation exchange resin (Dowex® 50WX8, hydrogen form) was added to the basic solution, in small increments, until neutrality according to pH paper. The suspension was filtered of, and the resin carefully washed with EtOH. The crude was subjected to column chromatography (5% MeOH/CH2Cl2) to afford 4 as a white solid in 95% yield. Rf: 0.1 (10% MeOH/CH2Cl2); mp: 133–135 °C; IR (KBr): ν 3427, 2975, 2941, 2676, 1667, 1574, 1476 cm−1; UV (MeOH): λ(max) = 268 nm (ε 3100); 1H NMR (300.13 MHz, MeOH-d4): δ 1.83 (m, 2H, H2′), 1.88 (s, 3H, Me-C5), 3.38 (d, 2H, H6′, JHH 4.8 Hz), 3.53 (dd, 1H, H5′, JHH 11.7, 5.2 Hz), 3.60 (dd, 1H, H5′, JHH 11.7, 4.2 Hz), 3.78 (q, 1H, H4′, JHH 4.3 Hz), 4.22 (m, 2H, H1′ + H3′), 4.40 (s, 2H, H7′), 7.37 (d, 1H, H6, JHH 1.1 Hz) ppm; 13C NMR (75.5 MHz, MeOH-d4): δ 12.2 (Me), 38.8 (C2′), 44.1 (C6′), 51.1 (C7′), 63.7 (C5′), 73.7 (C3′), 78.4 (C1′), 88.7 (C4′), 111.0 (C5), 143.7 (C6), 153.0 (C2), 167.0 (C4), 169.8 (OC-C7′) ppm; HRMS (ESI+, m/z): calcd for C13H19N3NaO6 [(M + Na)+]: 336.1166, found: 336.1166.C-C7) ppm; HRMS (ESI+, m/z): calcd for C34H37N3NaO8 [(M + Na)+]: 638.2473, found: 638.2474.:1 and the gradient used was from EtOAc/hexane 1:1 to pure EtOAc. After the chromatography, a white solid was obtained (198 mg, 85% yield). Rf(EtOAc) = 0.36 and 0.30. 31P NMR (162 MHz, Cl3): δ 148.02 and 147.92; HRMS (ESI+, m/z): calcd for C43H55N5O9P [(M + H)+]: 816.3732, found: 816.3706; calcd for C43H54N5NaO9P [(M + Na)+]: 838.3551, found: 838.3527.:0.5) = 0.20.:3) was added to a solution of 7 (23 mg, 0.03 mmol) and DMAP (8 mg, 0.06 mmol) in MeCN (500 μL). Next, Ph3P (17 mg, 0.06 mmol) was added. The mixture was vortexed for a few seconds and added to the support and allowed to react for 1 h. The support was washed with MeOH, CH2Cl2 and MeCN (2 × 5 mL) and dried under vacuum. The functionality of the resin was determined by DMT quantification (44.3 μmol g−1). Finally, the solid support was treated with a mixture of Ac2O/DMF (1:1, 500 μL) during 30 min to cap free amino groups.
:1). The product precipitated after standing overnight in the fridge and it was collected by filtration. The crude product was purified through column chromatography (2% MeOH/CH2Cl2) to afford 3 in 65% yield. Rf: 0.32 (5% MeOH/CH2Cl2); mp: 183–184 °C; IR (KBr): ν 3551, 3478, 3412, 3333, 2993, 1718, 1665, 1612, 1556 cm−1; 1H NMR (300.13 MHz, CDCl3): δ 1.81 (br s, 1H, NH), 1.91 (s, 3H, Me-C5), 2.12 (m, 2H, H2′), 2.40 (s, 3H, Me-Tol), 2.41 (s, 3H, Me-Tol), 3.44 (dt, 1H, H6′, JHH 14.1, 3.1 Hz), 3.64 (ddd, 1H, H6′, JHH 13.9, 7.5, 3.8 Hz), 4.26 (d, 1H, H7′, JHH 15.8 Hz), 4.35–4.48 (m, 4H, H1′ + H4′ + H5′ + H7′), 4.73 (dd, 1H, H5′, JHH 12.2, 8.1 Hz), 5.42 (d, 1H, H3′, JHH 5.3 Hz), 7.01 (s, 1H, H6′), 7.24 (2d, 4H, Hortho-Me), 7.92 (2d, 4H, Hortho-CO), 8.86 (br s, 1H, NH) ppm; 13C NMR (75.5 MHz, CDCl3): δ 12.5 (CH3-C5), 21.9 (2CH3-Tol), 34.3 (C2′), 41.2 (C6′), 50.7 (C7′), 64.9 (C5′), 77.4 (C3′), 78.4 (C1′), 83.6 (C4′), 110.9 (C5), 126.9 (Cipso-CO), 127.1 (Cipso-CO), 129.3 (2Cortho-Me), 129.4 (2Cortho-Me), 129.8 (2Cortho-CO), 130.0 (2Cortho-CO), 141.1 (C6), 144.4 (2Cipso-Me), 151.1 (C2), 164.2 (C4), 166.3 (OC-C7′), 167.2 (CO), 167.3 (CO) ppm; HRMS (ESI+, m/z): calcd for C29H31N3NaO8 [(M + Na)+]: 572.2003, found: 572.2001.:1) (0.08 mL). The completion of the reaction was reached at 30 min, a cation exchange resin (Dowex® 50WX8, hydrogen form) was added to the basic solution, in small increments, until neutrality according to pH paper. The suspension was filtered of, and the resin carefully washed with EtOH. The crude was subjected to column chromatography (5% MeOH/CH2Cl2) to afford 4 as a white solid in 95% yield. Rf: 0.1 (10% MeOH/CH2Cl2); mp: 133–135 °C; IR (KBr): ν 3427, 2975, 2941, 2676, 1667, 1574, 1476 cm−1; UV (MeOH): λ(max) = 268 nm (ε 3100); 1H NMR (300.13 MHz, MeOH-d4): δ 1.83 (m, 2H, H2′), 1.88 (s, 3H, Me-C5), 3.38 (d, 2H, H6′, JHH 4.8 Hz), 3.53 (dd, 1H, H5′, JHH 11.7, 5.2 Hz), 3.60 (dd, 1H, H5′, JHH 11.7, 4.2 Hz), 3.78 (q, 1H, H4′, JHH 4.3 Hz), 4.22 (m, 2H, H1′ + H3′), 4.40 (s, 2H, H7′), 7.37 (d, 1H, H6, JHH 1.1 Hz) ppm; 13C NMR (75.5 MHz, MeOH-d4): δ 12.2 (Me), 38.8 (C2′), 44.1 (C6′), 51.1 (C7′), 63.7 (C5′), 73.7 (C3′), 78.4 (C1′), 88.7 (C4′), 111.0 (C5), 143.7 (C6), 153.0 (C2), 167.0 (C4), 169.8 (OC-C7′) ppm; HRMS (ESI+, m/z): calcd for C13H19N3NaO6 [(M + Na)+]: 336.1166, found: 336.1166.C-C7) ppm; HRMS (ESI+, m/z): calcd for C34H37N3NaO8 [(M + Na)+]: 638.2473, found: 638.2474.:1 and the gradient used was from EtOAc/hexane 1:1 to pure EtOAc. After the chromatography, a white solid was obtained (198 mg, 85% yield). Rf(EtOAc) = 0.36 and 0.30. 31P NMR (162 MHz, Cl3): δ 148.02 and 147.92; HRMS (ESI+, m/z): calcd for C43H55N5O9P [(M + H)+]: 816.3732, found: 816.3706; calcd for C43H54N5NaO9P [(M + Na)+]: 838.3551, found: 838.3527.:0.5) = 0.20.:3) was added to a solution of 7 (23 mg, 0.03 mmol) and DMAP (8 mg, 0.06 mmol) in MeCN (500 μL). Next, Ph3P (17 mg, 0.06 mmol) was added. The mixture was vortexed for a few seconds and added to the support and allowed to react for 1 h. The support was washed with MeOH, CH2Cl2 and MeCN (2 × 5 mL) and dried under vacuum. The functionality of the resin was determined by DMT quantification (44.3 μmol g−1). Finally, the solid support was treated with a mixture of Ac2O/DMF (1:1, 500 μL) during 30 min to cap free amino groups.Oligonucleotide synthesis and purification
Oligonucleotides used in this study are summarized in Table 1. Modified oligonucleotides 15Mer_T*(01), (02) and (03) were prepared on a DNA synthesizer (Applied Biosystems 3400, Foster City, CA, USA) using 200 nmol scale LV200® polystyrene supports, the T* phosphoramidite described above and commercially available chemicals. In all cases the coupling yields of the modified phosphoramidite were >97%. The unmodified oligonucleotides were synthesized following standard protocols or purchased. The last DMT was left at the end of the synthesis. Then, the resulting supports were treated with aqueous concentrated ammonia at room temperature for 12 h and 1 h at 55 °C to cleave the products from the supports and remove the Bz and Ibu groups. The oligonucleotides were purified by cartridge (Glen-Pak™ DNA purification cartridge) from Glen Research. Then, they were desalted with Sephadex G-25 (IllustraNAP-10 or NAP-5 columns, from GE Healthcare Life Sciences). Finally, they were analyzed by mass spectrometry (MALDI-TOF) and HPLC. Solvent A: 5% MeCN in 100 mM triethylammonium acetate (TEAA) (pH 7) and solvent B: 70% MeCN in 100 mM TEAA (pH 7). Column: XBridgeTM OST C18 column; (4.6 × 50 mm, 2.5 μm). Flow rate: 1 mL min−1. Conditions: 10 min of linear gradient from 0 to 20% of B, for modified 15Mer's.For the synthesis of modified RNA, a solid support carrying 2 units of T* was synthesized on a 1 μmol scale using the modified solid support and the appropriate phosphoramidite. Then the rest of the RNA sequence was assembled using standard RNA synthesis protocols and tert-butyldimethylsilyl (TBDMS) protected RNA monomers (ABz, Gdmf, CAc and U) on 0.2 μmol scale as described.13 The following solutions were used: 0.4 M 1H-tetrazole in MeCN (activation); 3% Cl3CCO2H in CH2Cl2 (detritylation), Ac2O/Py/THF (1:1:8) (capping A), 10% N-methylimidazole in THF (capping B), 0.01 M iodine in THF/Py/H2O (7:2:1) for the oxidation step. The coupling time was 15 min. All oligonucleotides were synthesized in DMT-ON mode. After the solid-phase synthesis, the solid support was transferred to a screw-cap glass vial and incubated at 55 °C for 1 h with 1.5 mL of NH3 solution (33%) and 0.5 mL of EtOH. The vial was then cooled on ice and the supernatant was transferred into a 2 mL eppendorf tube. The solid support and vial were rinsed with 50% EtOH (2 × 0.25 mL). The combined solutions were evaporated to dryness using an evaporating centrifuge. The residue that was obtained was dissolved with 0.15 mL of triethylamine·tris(hydrofluoride)/Et3N/N-methylpyrrolidone (4:3:6) for 2.5 h at 65 °C to remove the TBDMS groups. Oligonucleotides were purified using oligonucleotide purification cartridge (Glen Research) and HPLC. Yields (0.2 μmol scale synthesis) were between 10 and 15 OD units at 260 nm.
Denaturation studies
Melting experiments were performed using a Jasco V-650 instrument equipped with a thermoregulated cell holder. They were performed in duplicate at 3 μM concentration of oligonucleotide. The samples were prepared using a solution 50 mM NaCl and 10 mM sodium phosphate buffer (pH 7.0). Concentrations of all oligonucleotides were estimated by UV-Vis absorption at 50 °C using the ε260 values calculated by the nearest-neighbor method for the DNA coil state. The samples were heated at 90 °C for 5 min, allowed to cool slowly to room temperature and kept overnight in a refrigerator. The melting curves were recorded monitoring the absorbance at 260 nm. The samples were heated with a temperature controller from 10 °C to 75–80 °C at a constant rate of 1 °C min−1 using 1 cm quartz path-length cuvettes with a ground hole at the top to adapt a PTFE stopper to provide a suitable seal to avoid evaporation during the acquisition. When the temperature was below 25 °C nitrogen was flushed to prevent water condensation. At least two different samples were prepared for each melting experiment. Thermodynamic data were calculated from the melting curves by computer fitting using the Meltwin 3.5 software.Cells
HeLa cells (ATCC) were maintained in monolayer culture at exponential growth in high-glucose Dulbecco modified Eagle medium (DMEM) (Gibco, Life Technologies, Carlsbad, CA, USA) supplemented with 10% heat inactivated fetal bovine serum (Gibco, Life Technologies, Carlsbad, CA, USA) and 1× penicillin/streptomycin solution (Gibco, Life Technologies, Carlsbad, CA, USA). Cells were incubated at 37 °C in a humidified environment with 5% CO2 and periodically checked for the presence of mycoplasma contamination. Cell viability was monitored by Trypan Blue exclusion assay and was higher than 95% in all experiments.Transfection and luciferase assay
For siRNA luciferase assay, HeLa cells were plated in 24-well tissue culture plates at density of 1 × 105 cells per well 24 h before transfection. In dose response, ON-/OFF-target assessment 1 μg of psiCHECK2 (AS) or psiCHECK2 and siRNAs at different concentrations were co-transfected using Lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA) in accordance with the manufacturer's instructions. The inhibitory effect of siRNAs on Renilla protein expression was measured on lysates collected 24 h after transfection using the Dual-Luciferase Reporter Assay System (Promega Biotech Iberica, Madrid, Spain) and a GloMax Discover luminometer (Promega Biotech Iberica, Madrid, Spain). The ratios of Renilla luciferase (hRluc) to Photinus luciferase (hluc+) protein activities were normalized to mock transfection and the mock activity was set as 100%. Statistical analysis was performed using GraphPad Prism software (GraphPad, San Diego, CA, USA). IC50 determination was performed using non-linear regression analysis (log[inhibitor] vs. normalized response).Acknowledgements
Financial support by the Spanish Ministerio de Ciencia e Innovación (MICINN) (Projects CTQ2011-24237, CTQ2014-55015-P, and CTQ2014-52588-R) and Principado de Asturias (Project FC-15-GRUPIN14-002) are gratefully acknowledged. CIBER-BBN is an initiative funded by the VI National R + D + i Plan 2008–2011, Iniciativa Ingenio 2010, Consolider Program, CIBER Actions and financed by the Instituto de Salud Carlos III with assistance from the European Regional Development Fund.References
- Y. S. Sanghvi, Chem. Today, 2014, 32, 10–15 CAS.
- Y. S. Sanghvi, in Current Protocols in Nucleic Acid Chemistry, ed. M. Egli, P. Herdewijn, A. Matsuda and Y. S. Sanghvi, 2011, pp. 4.1.1–4.1.22 Search PubMed.
- Y. S. Sanghvi, Future Science, 2013, 6–22 Search PubMed.
- B. Hyrup and P. E. Nielsen, Bioorg. Med. Chem., 1996, 4, 5–23 CrossRef CAS PubMed.
- J. S. Jepsen, M. D. Sørensen and J. Wengel, Oligonucleotides, 2004, 14, 130–146 CrossRef CAS PubMed.
- D. D'Alonzo, A. Van Aerschot, A. Guaragna, G. Palumbo, G. Schepers, S. Capone, J. Rozenski and P. Herdewijn, Chem.–Eur. J., 2009, 15, 10121–10131 CrossRef PubMed.
- M. Bolli, H. U. Trafelet and C. Leumann, Nucleic Acids Res., 1996, 24, 4660–46607 CrossRef CAS PubMed.
- C. J. Wilds and M. J. Damha, Bioconjugate Chem., 1999, 10, 299–305 CrossRef CAS PubMed.
- N. Martín-Pintado, M. Yahyaee-Anzahaee, R. Campos-Olivas, A. M. Noronha, C. J. Wilds, M. J. Damha and C. González, Nucleic Acids Res., 2012, 40, 9329–9339 CrossRef PubMed.
- M. A. Campbell and J. Wengel, Chem. Soc. Rev., 2011, 40, 5680–5689 RSC.
- K. Murayama, H. Kashida and H. Asanuma, Chem. Commun., 2015, 51, 6500–6503 RSC.
- Y. Kamiya, J. Takai, H. Ito, K. Murayama, H. Kashida and H. Asanuma, ChemBioChem, 2014, 15, 2549–2555 CrossRef CAS PubMed.
- A. Alagia, M. Terrazas and R. Eritja, Molecules, 2014, 19, 17872–17896 CrossRef PubMed.
- A. Alagia, M. Terrazas and R. Eritja, Molecules, 2015, 20, 7602–7619 CrossRef CAS PubMed.
- C. L. Scremin, J. H. Boal, A. Wilk, L. R. Phillips and S. L. Beaucage, Bioorg. Med. Chem. Lett., 1996, 6, 207–212 CrossRef CAS.
- J. H. Boal, A. Wilk, C. L. Scremin, G. N. Gray, L. R. Phillips and S. L. Beaucage, J. Org. Chem., 1996, 61, 8617–8626 CrossRef CAS.
- J. Robles, A. Grandas, E. Pedroso, F. J. Luque, R. Eritja and M. Orozco, Curr. Org. Chem., 2002, 6, 1333–1368 CrossRef CAS.
- Pure β-cyano-sugar 1 is available from Sapala Organics Pvt. Ltd., India (http://www.sapalaorganics.com).
Footnotes |
| † Electronic supplementary information (ESI) available: Figures giving 1D and 2D NMR spectral data of modified nucleoside monomers; MALDI-TOF spectra of oligonucleotides. See DOI: 10.1039/c6ra26852h |
| ‡ The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint first authors as they equally contributed to the work. |
| This journal is © The Royal Society of Chemistry 2017 |